
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAccessing spiropiperidines from dihydropyridones through tandem triflation–allylation and ring-closing metathesis (RCM)†
Naresh
Gantasala
a,
Corentin
Fournet
c,
Myriam
Le Roch
c,
Claudia
Lalli
 *c,
Srihari
Pabbaraja
*ab and
Nicolas
Gouault
*c
*c,
Srihari
Pabbaraja
*ab and
Nicolas
Gouault
*c
aCSIR-Indian Institute of Chemical Technology, Uppal Road, Tarnaka, Hyderabad, 500007, TS, India
bAcademy of Scientific and Innovative Research (AcSIR), Ghaziabad-201002, India. E-mail: srihari.iict@gov.in
cUniv. Rennes, CNRS, ISCR (Institut des Sciences Chimiques de Rennes), UMR 6226, F-35000 Rennes, France. E-mail: nicolas.gouault@univ-rennes.fr; Claudia.lalli@univ-rennes.fr
First published on 12th June 2023
Abstract
A novel approach to build 2-spiropiperidine moieties starting from dihydropyridones was developed. The triflic anhydride-promoted conjugate addition of allyltributylstannane onto dihydropyridones allowed for the formation of gem bis-alkenyl intermediates that were converted to the corresponding spirocarbocycles with excellent yields via ring closing metathesis. The vinyl triflate group generated on these 2-spiro-dihydropyridine intermediates could be successfully used as a chemical expansion vector for further transformations namely Pd-catalyzed cross-coupling reactions.
Introduction
Drug discovery typically involves, once an original biological target is identified and validated, screening libraries of compounds or fragments in order to identify potentially active ones that will be further optimized to become drug candidates. The large synthetic libraries available today often suffer from low hit rates in biological assays, in part because of their low degrees of structural complexity and diversity. The development of more complex molecules with increased shape diversity and “three-dimensionality” is therefore of great interest1–9 and would allow for a larger chemical space exploration. Consequently, much effort is directed towards optimizing fragment collections with, in particular the elaboration of original fragments bearing spirocycles, ubiquitous molecules with unique rigidity and a three-dimensional geometry.10–12On the other hand, the piperidine ring is a common structural motif present in natural and/or synthetic products of pharmaceutical importance, and within this class of saturated nitrogen heterocycles, the spiropiperidine moiety, which is capable of exploring and spanning a large binding pocket due to its rigidly defined structure, has been considered a “privileged structure” and thus has become a sought-after motif for medicinal chemists.13–21 (−)-Histrionicotoxin, a potent non-competitive antagonist of nicotinic acetylcholine receptors,22 (+)-nitramine23 and ibutamoren, a potent agonist of the ghrelin receptor,24 are examples of synthetic or natural biologically active spiropiperidines (Fig. 1). Different synthetic approaches have been developed to generate these scaffolds, but their preparation remains challenging. In particular, there are limited methods for the synthesis of 2-spiropiperidines that offer chemical expansion vectors allowing for the generation of additional interactions with a biological target or the optimization of an identified fragment.13,25,26
 | ||
| Fig. 1 Representative bioactive compounds containing spiropiperidine skeletons. | ||
The synthesis of dihydropyridones27 and their use as intermediates for the synthesis of piperidine derivatives has been extensively developed over the last decades.28,29 In the past few years, we have developed a gold-catalyzed approach from the chiral pool of amino acids to build such dihydropyridones with an excellent stereochemical maintenance (Scheme 1a).30 We also demonstrated that these dihydropyridones are valuable intermediates towards various piperidines.31,32
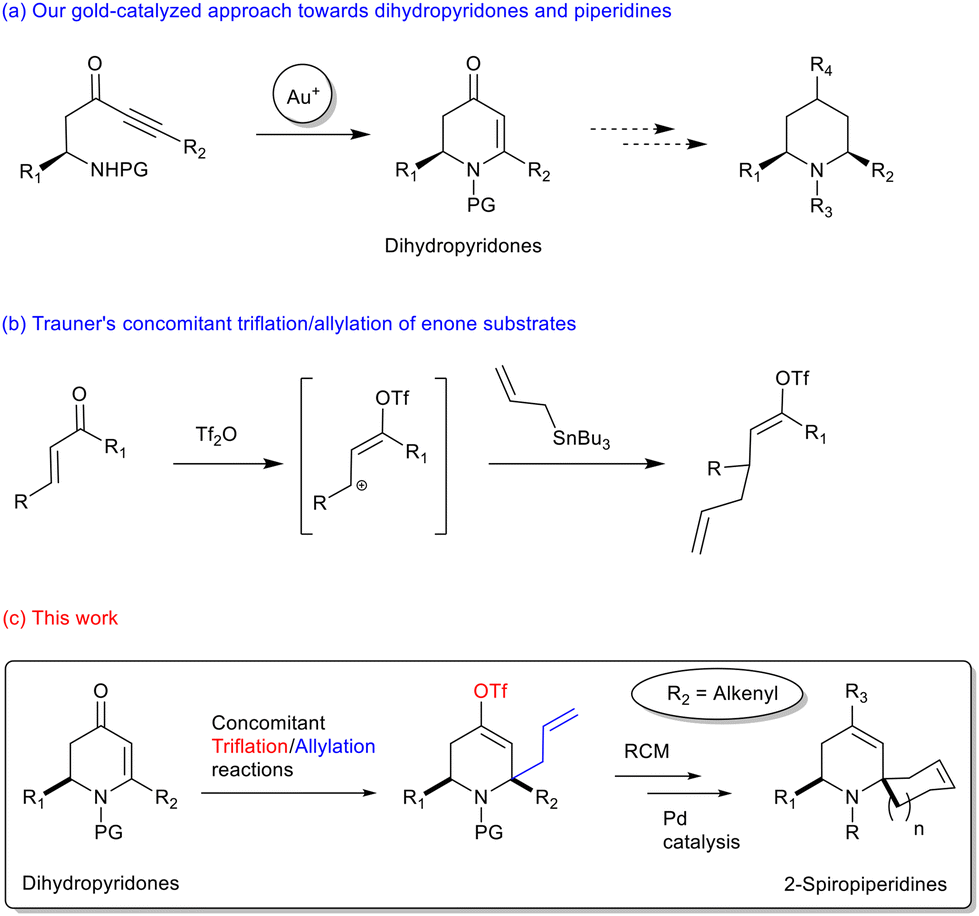 | ||
| Scheme 1 Our approach towards 2-spiropiperidines. | ||
In the same period, the group of Trauner et al. demonstrated in a seminal paper that the triflic anhydride-promoted activation of enone allowed for the formation of an allylic trifloxy cation that can be trapped by the nucleophilic attack of allylstannane (Scheme 1b).33 The scope of this reaction was limited to acyclic and carbocyclic enones and was not extended to heterocyclic enones as their reactivity may be quite challenging. This approach appeared interesting to us because, in a single step, it allows the introduction of (i) an allyl side-chain that may be engaged in a ring-closing metathesis (RCM) reaction to build a spiro-cycle and (ii) a vinyl triflate group that may be used as a chemical expansion vector for further transformations namely Pd-catalyzed cross-coupling reactions. We describe herein a new approach toward 4-substituted-2-spiropiperidines from dihydropyridones.
Results and discussion
To test the feasibility of the triflation/allylation of dihydropyridones, we initiated the exploration of reaction conditions by using N-Boc protected dihydropyridone 1a as the typical substrate (Table 1).34
Such a starting material was selected for two main reasons. The Boc protecting group is probably the most common amine protecting group since it can be easily and quantitatively removed under relatively mild conditions. A dihydropyridone bearing a propyl side chain at position 6 instead of a phenyl ring was preferred since an aromatic in this position may lead to resonance stabilization of the carbenium intermediate, which may be advantageous with regard to the mode of activation.
The reaction of 1a with allyltrimethylsilane (5.0 equiv.) in the presence of triflic anhydride (2.0 equiv.) in dichloromethane at −78 °C was initially examined. Despite complete conversion being observed after 6 h, most of the substrate underwent Boc deprotection under these conditions (Table 1, entry 1). As the Boc protecting group is prone to undergo TMS-mediated cleavage,35 it was replaced with a Cbz moiety (1b). Using this new protecting group, we were able to obtain the desired compound (2b), however, in a low yield (Table 1, entry 2). These first results led us to reconsider the nature of the nucleophile and to replace allyltrimethylsilane with allyltributyltin, which was demonstrated in the studies of Trauner33 and Comins36,37 to exhibit appropriate properties. Notably, good conversion to the desired addition product 2b was observed within 3 h under these novel conditions (Table 1, entry 3). This was also confirmed by using N-Boc protected pyridone 1a as the starting material (Table 1, entry 4).
Finally, the attempt to decrease the nucleophile loading to 2 equivalents resulted in a significant loss of efficiency (Table 1, entry 5). With fewer equivalents of the reagent, the reaction was slower and therefore required a slightly longer reaction time to reach completion.
The substrate scope of dihydropyridones 1 was next investigated under the optimized conditions (Table 1, entry 4) and the results are summarized in Table 2.
| a Isolated yields are mentioned. The dr values were determined by 1H NMR analysis of the crude material. |
|---|

|
Several structural variations were tolerated, including alkyl and aryl substituents at position 6 (1a–h), methyl, phenyl and tert-butyl carboxylate at position 2 (1i–l), and the Cbz protecting group (1b and 1m), and a series of 4-triflate-6-allyl-dihydropyridine derivatives 2a–m could be successfully obtained, in most of the cases, in good to excellent yields. In the case of 2-substituted substrates, the desired compounds (2i–l) were obtained as a mixture of diastereoisomers that we could not separate. The low diastereoselectivities were evaluated by proton NMR analysis of the ethylenic signal near 5.6 ppm or the CH-2 signal near 5.0 ppm and were confirmed by carbon NMR with duplication of some signals in the same proportions.
It should be noted that in the case of 6-aryldihydropyridones (1d–f), in addition to the expected 1,4-addition products (2d–f), compounds 3d–f resulting from the 1,2-addition were also observed (Scheme 2). Such compounds 3 were not observed in the case of dihydropyridones substituted at position 6 with an alkyl side-chain, suggesting that the steric hindrance brought about by the aryl substituent residing out-of-plane favored the observed competition. Moreover, this observation is reinforced when a bulky bromo-substituent is present at the ortho-position on the aromatic ring (3f), as the 1,2-addition is strongly favored in this case. A plausible mechanism for the formation of 3 is proposed in Scheme 2.
 | ||
| Scheme 2 Competition between the 1,2- and 1,4-addition for the triflic anhydride-promoted allylation reaction with 6-aryl-dihydropyridones and the proposed mechanism explaining this competition. | ||
Next, we turned our attention towards the synthesis of the targeted 2-spiropiperidines. To this end, ring-closing metathesis of the intermediates 2g–i with Grubbs’ first-generation catalyst furnished the desired spiro-compounds 4 in excellent yields (Scheme 3).38
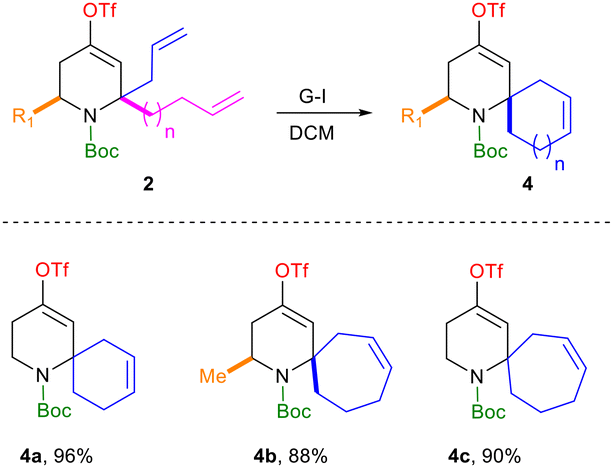 | ||
| Scheme 3 Access to spiro-tetrahydropyridines via ring-closing metathesis. | ||
The 2-spiranic 4-triflate-tetrahydropyridines 4a–c were further functionalized by applying palladium-catalyzed processes such as Suzuki–Miyaura, Heck or Sonogashira cross-coupling reactions (Scheme 4).39–41
 | ||
| Scheme 4 Pd-catalyzed modifications of spiro-piperidines 4. | ||
For instance, compounds 6a–d have been successfully obtained in 97%, 93%, 88% and 91% isolated yields, respectively, by the Suzuki cross-coupling reaction of 4a–c with 4-pyridinylboronic acid or 3,4-dimethoxyphenylboronic acid. Concerning the Heck coupling reaction, two alkenes were chosen, benzyl acrylate and allyl alcohol, respectively, and the reactions were performed starting from vinyl triflate 4a. Products 7a and b were obtained in good to excellent yields, 95% and 81%, respectively. Finally, compounds 8a–c have been successfully obtained in 87%, 78% and 84% isolated yields, respectively, by the Sonogashira cross-coupling reaction of 4a and b with 4-aminophenylacetylene or 2-pyridinylacetylene.
Conclusions
In conclusion, we described a novel approach for the construction of 2-spiropiperidine moieties from dihydropyridones. The gem bis-alkenyl intermediates generated from the triflic anhydride-promoted conjugate addition of allyltributylstannane to dihydropyridones were successfully transformed to the corresponding spirocycles via ring-closing metathesis. The vinyl triflate group generated on these 2-spiro-dihydropyridine intermediates could be successfully used as a chemical expansion vector for further transformations namely Pd-catalyzed cross-coupling reactions.Experimental section
General information
Unless otherwise specified, all commercially available reagents were used as received. Analytical thin layer chromatography (TLC) was carried out on silica gel 60 F254 plates with visualization using ultraviolet light or potassium permanganate dip. Column chromatography was carried out using silica gel 60 (70–200 μm). 1H and 13C NMR spectra were recorded on 300 or 500 MHz instruments. The chemical shifts are given in parts per million (ppm) on the delta scale. The solvent peak was used as the reference value: for 1H NMR, CHCl3 = 7.26 ppm; for 13C NMR, CHCl3 = 77.16 ppm. Infrared spectra were recorded neat. ESI-HRMS were carried out on an Agilent 6510 Q-TOF spectrometer at the CRMPO (Centre Regional de Mesures Physiques de l'Ouest), University of Rennes.General procedure for the synthesis of compounds 2 and 3
To dihydropyridone 1 (1.0 mmol) in dichloromethane (8.0 mL) cooled to −78 °C was added under an argon atmosphere allyltributylstannane (5.0 mmol) followed by triflic anhydride (2.0 mmol) dropwise. The reaction mixture was stirred at −78 °C for 3 h. Upon completion, the reaction was quenched by the addition of 1 M NaOH. After stirring for 30 minutes, the layers were separated and the organic phase was washed successively with 1 M NaOH and brine and then dried over sodium sulfate. Removal of the solvents gave a crude residue that was purified by column chromatography (silica gel, cyclohexane/DCM = 70/30 as the eluent) to give the desired compounds 2a–2m and 3d–f.General procedure for the synthesis of compounds 4 and 5
The diene precursor 2 or 3 (0.25 mmol) was dissolved in freshly distilled and degassed dichloromethane (3 mL) under an argon atmosphere. This solution was degassed again using argon gas for 10 minutes at room temperature; to the solution was added 1st Grubbs catalyst (3 mol%). After 10–14 hours at room temperature, the reaction was complete, as indicated by TLC. The reaction mixture was concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, cyclohexane/EtOAc = 70/30 as eluent) to give the desired compounds 4a–c and 5d–f.General procedure for the Suzuki–Miyaura coupling reaction
A 20 mL dry Schlenk tube was charged with 4 (0.1 mmol) and 1,4-dioxane/H2O (5/1, 2 mL) under an argon atmosphere. Argon was bubbled through the solution for 10 min. To the solution were added boronic acid (0.12 mmol), catalyst (Pd(PPh3)4 – 5 mol%), K2CO3 (0.25 mmol) and LiCl (0.25 mmol) under a gentle flow of argon. The reaction mixture was stirred at 90 °C for 4 h. The resulting mixture was filtered through a pad of Celite. The filtrate was extracted with EtOAc (three times). The reaction mixture was diluted with brine then dried over sodium sulfate. The reaction mixture was concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, cyclohexane/EtOAc = 70/30 as eluent) to give the desired compounds 6a–d.General procedure for the Heck coupling reaction
A 20 mL dry sealed tube was charged with 4 (0.1 mmol) and DMF (1 mL) under an argon atmosphere. To the solution were added the corresponding alkene (0.13 mmol), LiCl (0.15 mmol), and triethylamine (0.2 mmol) under a gentle flow of argon. Then Pd(OAc)2 (5 mol%) was added. The reaction mixture was stirred at 50 °C for 15 h. The resulting mixture was filtered through a pad of Celite. After the addition of 5 mL of satd aq NaHCO3 and extraction with EtOAc (three times), the combined organic phases were washed with brine and then dried over sodium sulfate. The reaction mixture was concentrated under reduced pressure. The crude residue was purified by column chromatography (silica gel, cyclohexane/EtOAc = 70/30 as the eluent) to give the desired compounds 7a–7b.General procedure for the Sonogashira coupling reaction
A 20 mL dry Schlenk tube was charged with 4 (0.1 mmol) and degassed anhydrous ACN (1.5 mL) under an argon atmosphere. To the solution were added the corresponding alkyne (0.12 mmol), triethylamine (0.3 mL), TBAI (0.15 mmol), CuI (5 mol%) and Pd(PPh3)4 (2.5 mol%). The reaction mixture was stirred at room temperature for 10–14 h. The resulting mixture was filtered through a pad of Celite. The filtrate was extracted with EtOAc (three times). The reaction mixture was diluted with brine and then dried over sodium sulfate. The reaction mixture was concentrated under reduced pressure. The crude residue was purified by column chromatography (silica gel, cyclohexane/EtOAc = 70/30 as the eluent) to give the desired compounds 8a–8c.Author contributions
The manuscript was written through contributions from all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge support for this research from the Indo-French “Joint Laboratory for Natural Products and Synthesis towards Affordable Health”. We are especially thankful to Prof. Joël Boustie and Dr René Grée from the University of Rennes and Dr Srivari Chandrasekhar from CSIR-Indian Institute of Chemical Technology, Hyderabad, India. We would like also to thank the University of Rennes for its financial support in the framework of “Défis émergeants”. We are grateful to CRMPO (University of Rennes) for the mass spectra analysis. IICT communication No. IICT/Pubs./2022/327. Part of this work has been performed using the PRISM core facility (Univ Rennes, France) under the guidance of Solenn Ferron.References
- F. Lovering, J. Bikker and C. Humblet, J. Med. Chem., 2009, 52, 6752 CrossRef CAS PubMed.
- F. Lovering, Med. Chem. Commun., 2013, 4, 515 RSC.
- W. Wei, S. Cherukupalli, L. Jing, X. Liu and P. Zhan, Drug Discovery Today, 2020, 25, 1839 CrossRef CAS PubMed.
- K. L. Prosser, R. W. Stokes and S. M. Cohen, ACS Med. Chem. Lett., 2020, 11, 1292 CrossRef CAS PubMed.
- A. R. Hanby, N. S. Troelsen, T. J. Osberger, S. L. Kidd, K. T. Mortensen and D. R. Spring, Chem. Commun., 2020, 56, 2280 RSC.
- D. J. Hamilton, T. Dekker, H. F. Klein, G. V. Janssen, M. Wijtmans, P. O'Brien and I. J. P. de Esch, Drug Discovery Today: Technol., 2020, 38, 77 CrossRef PubMed.
- C. N. Morrison, K. E. Prosser, R. W. Stokes, A. Cordes, N. Metzler-Nolte and S. M. Cohen, Chem. Sci., 2020, 11, 1216 RSC.
- P. Garner, P. B. Cox, U. Rathnayake, N. Holloran and P. Erdman, ACS Med. Chem. Lett., 2019, 10, 811 CrossRef CAS PubMed.
- J. A. Johnson, C. A. Nicolaou, S. E. Kirberger, A. K. Pandey, H. Hu and W. C. K. Pomerantz, ACS Med. Chem. Lett., 2019, 10, 1648 CrossRef CAS PubMed.
- K. Hiesinger, D. Dar'in, E. Proschak and M. Krasavin, J. Med. Chem., 2021, 64, 150 CrossRef CAS PubMed.
- L. T. Lepovitz and S. F. Martin, Tetrahedron, 2019, 75, 130637 CrossRef.
- P. Kostiantyn, K. P. Melnykov, A. N. Artemenko, B. O. Ivanenko, Y. M. Sokolenko, P. S. Nosik, E. N. Ostapchuk, O. O. Grygorenko, D. M. Volochnyuk and S. V. Ryabukhin, ACS Omega, 2019, 4, 7498 CrossRef PubMed.
- Y. Troin and M. E. Sinibaldi, Targets in Heterocyclic Systems: Chemistry and Properties, Italian Chemical Society, Rome, 2009, vol. 13, p. 120 Search PubMed.
- S. D. Griggs, D. T. Tape and P. A. Clarke, Org. Biomol. Chem., 2018, 16, 6620 RSC.
- A. Peneau, P. Retailleau, C. Guillou and L. Chabaud, J. Org. Chem., 2018, 83, 2324 CrossRef CAS PubMed.
- S. D. Griggs, N. Thompson, D. T. Tape, M. Fabre and P. A. Clarke, Chem. – Eur. J., 2017, 23, 9262 CrossRef CAS PubMed.
- T. M. McQueen and S. D. Griggs, Tetrahedron Lett., 2021, 65, 152752 CrossRef CAS.
- C.-W. Lee, R. Lira, J. Dutra, K. Ogilvie, B. T. O'Neill, M. Brod-ney, C. Helal, J. Young, E. Lachapelle, S. Sakya and J. C. Murray, J. Org. Chem., 2013, 78, 2661 CrossRef CAS PubMed.
- U. M. Battisti, S. Corrado, C. Sorbi, A. Cornia, A. Tait, D. Malfacini, M. C. Cerlesi, G. Calo and L. Brasili, MedChemComm, 2014, 5, 973 RSC.
- L. A. Martinez-Alsina, J. C. Murray, L. M. Buzon, M. W. Bundes-mann, J. M. Young and B. T. O'Neill, J. Org. Chem., 2017, 82, 12246 CrossRef CAS PubMed.
- Z. Chen, K. Yan, H. Luo, J. Yan and Y. Zeng, RSC Adv., 2022, 12, 32097 RSC.
- W. Oberthür, P. Muhn, H. Baumann, F. Lottspeich, B. Wittmann-Liebold and F. Hucho, EMBO J., 1986, 5, 1815 CrossRef PubMed.
- N. Y. Novgorodova, S. K. Maekh and S. Y. Yunusov, Chem. Nat. Compd., 1973, 9, 191 CrossRef.
- A. A. Patchett, R. P. Nargund, J. R. Tata, M. H. Chen, K. J. Barakat, D. B. Johnston, K. Cheng, W. W. Chan, B. Butler and G. Hickey, Proc. Natl. Acad. Sci. U. S. A., 1995, 92, 7001 CrossRef CAS PubMed.
- D. L. Comins and X. Zheng, J. Chem. Soc., Chem. Commun., 1994, 2681 RSC.
- J. J. Sahn and D. L. Comins, J. Org. Chem., 2010, 75, 6728 CrossRef CAS PubMed.
- A. K. Chattopadhyay and S. Hanessian, Chem. Commun., 2015, 51, 16437 RSC.
- J. A. Bull, J. J. Mousseau, G. Pelletier and A. B. Charrette, Chem. Rev., 2012, 112, 2642 CrossRef CAS PubMed.
- A. K. Chattopadhyay and S. Hanessian, Chem. Commun., 2015, 51, 16450 RSC.
- N. Gouault, M. Le Roch, A. Cheignon, P. Uriac and M. David, Org. Lett., 2011, 13, 4371 CrossRef CAS PubMed.
- N. Gouault, M. Le Roch, G. Pinto de Campos and M. David, Org. Biomol. Chem., 2012, 10, 5541 RSC.
- T. T. H. Trinh, K. H. Nguyen, P. de Aguiar Amaral and N. Gouault, Beilstein J. Org. Chem., 2013, 9, 2042 CrossRef PubMed.
- E. D. Beaulieu, L. Voss and D. Trauner, Org. Lett., 2008, 10, 869 CrossRef CAS PubMed.
- These substrates were prepared in three steps from N-Boc β-alanine (see the ESI†).
- Z. Liu, N. Yasuda, M. Simeone and R. A. Reamer, J. Org. Chem., 2013, 79, 11792 CrossRef PubMed.
- D. B. Gotchev and D. L. Comins, J. Org. Chem., 2006, 71, 9393 CrossRef CAS PubMed.
- S. V. Tsukanov, L. R. Marks and D. L. Comins, J. Org. Chem., 2016, 81, 10433 CrossRef CAS PubMed.
- In addition to this transformation, intermediates 3 were also submitted to RCM with Grubbs’ first-generation catalyst, affording the corresponding 4-spiro-dihydropyridines 5 in excellent yields. See the ESI.†.
- D. J. Wustrow and L. D. Lawrence, Synthesis, 1991, 993 CrossRef CAS.
- J. H. Lee and F. D. Toste, Angew. Chem., Int. Ed., 2007, 46, 912 CrossRef CAS PubMed.
- S. Peil, A. Gutierrez Gonzalez, M. Leutzsch and A. Furstner, J. Am. Chem. Soc., 2022, 144, 4158 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ob00545c |
| This journal is © The Royal Society of Chemistry 2023 |