
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFlow photolysis of aryldiazoacetates leading to dihydrobenzofurans via intramolecular C–H insertion†
Katie S.
O'Callaghan
a,
Denis
Lynch
a,
Marcus
Baumann
 b,
Stuart G.
Collins
*a and
Anita R.
Maguire
*ac
b,
Stuart G.
Collins
*a and
Anita R.
Maguire
*ac
aSchool of Chemistry, Analytical and Biological Chemistry Research Facility, Synthesis and Solid State Pharmaceutical Centre, University College Cork, Ireland. E-mail: a.maguire@ucc.ie; stuart.collins@ucc.ie
bSchool of Chemistry, University College Dublin, Belfield, Dublin 4, Ireland
cSchool of Pharmacy, University College Cork, Ireland
First published on 23rd May 2023
Abstract
Flow photolysis of aryldiazoacetates 3–5 leads to C–H insertion to form dihydrobenzofurans 6–8 in a metal-free process, using either a medium pressure mercury lamp (250–390 nm) or LEDs (365 nm or 450 nm) with comparable synthetic outcomes. Significantly, addition of 4,4′-dimethoxybenzophenone 9 results in an increased yield and also alters the stereochemical outcome leading to preferential isolation of the trans dihydrobenzofurans 6a–8a (up to 50% yield), while the cis and trans diastereomers of 6–8 are recovered in essentially equimolar amounts in the absence of a photosensitiser (up to 26% yield).
Introduction
α-Diazocarbonyl compounds are synthetically versatile intermediates that have been widely studied since Curtius first reported the synthesis of ethyl diazoacetate in 1883.1,2 These compounds can undergo various carbon–carbon bond forming transformations such as cyclopropanation,3 ylide formation,4 aromatic addition5 and C–H insertion6–9 under mild conditions. The most salient reactions of α-diazocarbonyl compounds involve the use of transition-metal catalysts, facilitating highly chemoselective and stereoselective outcomes. However, replacing these metals with photolysis can lead to a more sustainable, greener process, as evidenced by the resurgence of interest in this area in recent years due to advances in technology for photochemistry.10–14Developments in continuous flow chemistry have enhanced the synthetic potential of photochemistry and its practical use at scale, due to its ability to overcome issues associated with batch reactors, such as non-uniform light penetration leading to over-irradiation and increased side-product formation.15–25 Recently, photochemistry in flow has gained much interest as it enables better control over reaction conditions, facilitating improved reproducibility and scalability.15–25 Continuous flow processing also offers the possibility to improve the safety profile associated with hazardous conditions, reactants and intermediates, such as α-diazocarbonyl compounds, due to in-line reaction monitoring, efficient heat and mass transfer, as well as the ability to generate hazardous reagents in situ in small quantities, without the need for their handling or isolation.26–34 Continuous flow photolysis of aryldiazoacetates leading to cyclopropanation have been reported.35–37
While photochemical reactions of α-diazocarbonyl compounds have been explored for decades, the first example of a photolytic intramolecular C–H insertion was reported by Corey and Felix in 1965 for the synthesis of methyl 6-phenylpenicillinate from an α-diazoamide.38 Since then, the photochemical intramolecular C–H functionalisation of aryldiazoacetates 1a and 1b leading to 5,5-dimethyl-3-phenyldihydrofuran-2(3H)-one 2a and 4,4-dimethyl-3-phenyloxetan-2-one 2b has been reported by Jurberg and Davies (Scheme 1).39 Notably, these transformations required extended reaction times.
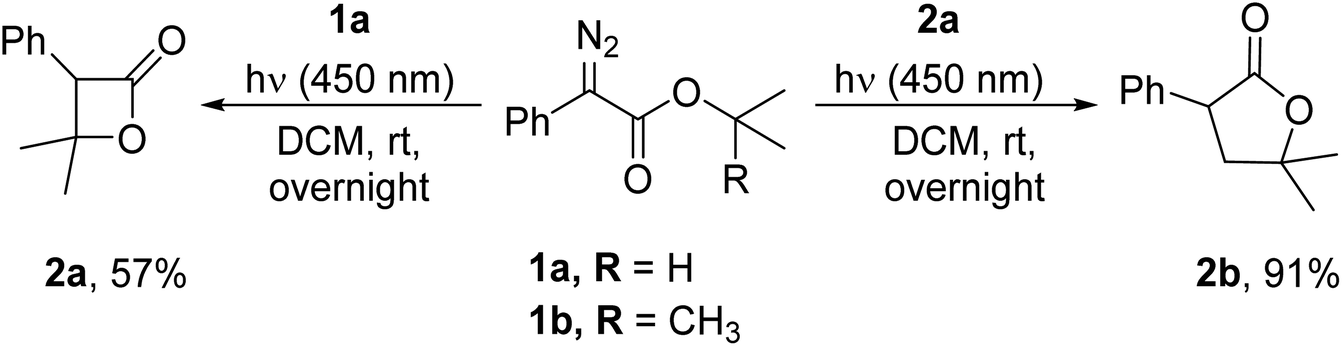 | ||
| Scheme 1 Intramolecular C–H insertion of aryldiazoacetates (Davies et al.).39 | ||
In this work, we investigate the application of continuous flow photochemistry to achieve a metal-free intramolecular C–H insertion of different aryldiazoacetates for the synthesis of a dihydrobenzofuran scaffold, a motif found in a variety of biologically active compounds.40–49 Previously, formation of this moiety has been explored through a number of pathways including, among others, metal catalysed C–H insertion of an aryldiazoacetate,50,51 or of an aryldiazomethane56 and photochemical C–H insertion of an acyl silane.52
Enantioselective rhodium carboxylate catalysed C–H insertions of aryldiazoacetate 3 to form dihydrobenzofurans leading to selective formation of the cis diastereomer, have been investigated by Hashimoto54 and Davies,55 with excellent enantioselectivity through appropriate choice of the rhodium catalyst. Having recently demonstrated that we can provide access to either dihydrobenzofuran 6a or 6b as a major product depending on the catalyst employed, with good enantiocontrol (Scheme 2),53 we wished to explore if a similar transformation could be affected photochemically, providing the dihydrobenzofurans via a metal-free transformation.
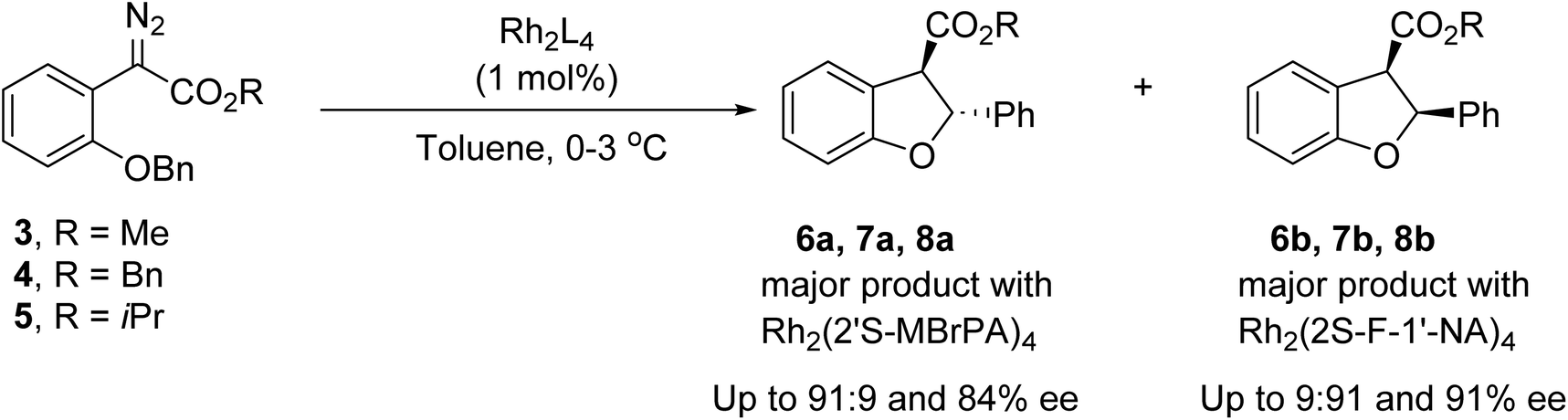 | ||
| Scheme 2 Rhodium-catalysed C–H insertion of aryldiazoacetates 3, 4 and 5 (Maguire et al.).53 | ||
Results and discussion
Photochemical transformations of aryldiazoacetates 3, 4 and 5 were investigated in continuous flow to establish the synthetic utility of this transformation in the absence of a metal catalyst. As summarised in Table 1, the initial studies were conducted in continuous flow with methyl aryldiazoacetate 3, using a photochemical reactor and a medium pressure mercury lamp. The impact of different wavelength filters, concentrations, temperatures and solvents on the efficiency of the C–H insertion were initially explored. The outcomes of the reactions were monitored by recording 1H NMR spectra of the crude products, to estimate the efficiency of the C–H insertion process and the diastereomeric ratio of the resulting dihydrobenzofurans. While no side-products could be isolated and characterised, signals consistent with azine formation were detected, in line with reported azine formation in the metal-catalysed C–H insertion reactions.54|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Wavelength filter (nm) | Temperature (°C) | Solvent | Molarity (mM) | Ratiob6a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :6b (trans:cis) :6b (trans:cis) |
Dihydrobenzofuranc (%) |
|
a Reactions were conducted using Method A for a 5 mL solution of aryldiazoacetate 3 at stated molarity.
b The ratio of trans:cis was determined by the relative integration of C(2)H signals at 6.12 (1H, d) and C(3)H signals at 5.99 (1H, d) ppm, respectively, in the 1H NMR spectrum of the crude reaction mixture.
c The percentage was estimated by comparing the relative integration of signals in the aromatic region (6.6–8.0 ppm) in the 1H NMR spectrum of the crude reaction mixtures to the signals for dihydrobenzofurans 6a and 6b. While there are limitations in the quantitative accuracy of this, this approach proved useful in providing an indication of the relative efficiency of different experiments.
|
||||||
| 1 | No light | 28 | MeCN | 40 | — | 0 |
| 2 | 300–2000 | 28 | MeCN | 40 | 1.0:1.0 |
39 |
| 3 | 109–2000 | 28 | MeCN | 40 | 1.0:1.0 |
29 |
| 4 | 250–390 | 28 | MeCN | 40 | 1.0:1.0 |
36 |
| 5 | 250–390 | 28 | MeCN | 7 | 1.0:1.0 |
30 |
| 6 | 250–390 | 28 | MeCN | 70 | 1.0:1.0 |
32 |
| 7 | 250–390 | 28 | MeCN | 150 | 1.0:1.0 |
24 |
| 8 | 250–390 | 0 | MeCN | 70 | 1.0:1.0 |
14 |
| 9 | 250–390 | 28 | Ethyl acetate | 40 | 1.0:1.0 |
57 |
| 10 | 250–390 | 28 | DCM | 40 | 1.0:1.0 |
57 |
| 11 | 250–390 | 28 | THF | 40 | 1.0:1.0 |
16 |
| 12 | 250–390 | 28 | Toluene | 40 | 1.0:1.0 |
53 |
| 13 | 250–390 | 28 | TBME | 40 | 1.0:1.0 |
66 |
| 14 | 250–390 | 28 | Acetone | 40 | 1.5:1.0 |
45 |
| 15 | 250–390 | 28 | TBME/MeCN (80:20) |
40 | 1.0:1.0 |
60 |

As anticipated, in the absence of light there was no evidence of C–H insertion with only unreacted aryldiazoacetate 3 starting material remaining (Table 1, entry 1), while using any one of three wavelength filters (300–2000 nm, 109–2000 nm or 250–390 nm) led to essentially the same outcome, with total consumption of the aryldiazoacetate 3 and approximately 30–40% formation of the dihydrobenzofurans 6a and 6b within 10 minutes of photolysis (Table 1, entries 2–4). Investigation of the impact of concentration (Table 1, entries 4–7) showed that the extent of C–H insertion decreased somewhat at higher concentrations (0.15 M), presumably due to competing intermolecular reactions such as azine formation.54 When the temperature was reduced to 0 °C the efficiency of the C–H insertion decreased significantly (Table 1, entry 8 cf. entry 6). In practice, due to the background heating effect of the lamp, the reaction coil stabilises at ca. 28 °C with ambient air cooling, and this was used for all subsequent experiments. Notably, as demonstrated in Table 1, entry 1, there is no evidence of thermolytic C–H insertion at this temperature. Utilising a fixed concentration of aryldiazoacetate 3 (0.04 M), a solvent screen (Table 1, entries 9–15 cf. entry 4) indicated that the efficiency of the C–H insertion was greatest in TBME and poorest in THF.
When acetone, a known photosensitiser, was employed as the reaction solvent a diastereomeric ratio of 1.5:1.0 of 6a:6b was observed (Table 1, entry 14); this unanticipated observation led to the investigation of the effect of triplet photosensitisers on the stereochemical outcome of the photochemical C–H insertion.56–58 Six triplet photosensitisers were investigated, which were each added individually, at the same concentration of 0.0013 M, to the starting solutions prior to photolysis (Table 2, entries 1–6). While the presence of tetraphenylporphyrin (TPP) had no detectable effect on the diastereomeric ratio, use of rose bengal, methylene blue, benzophenone, 4,4′-dichlorobenzophenone and 4,4′-dimethoxybenzophenone 9 (Fig. 1) each resulted in alteration of the diastereomeric ratio observed in the crude product mixtures, favouring the trans isomer 6a. As use of 4,4′-dimethoxybenzophenone 9 gave the best diastereomeric ratio at the initial amount, this photosensitiser was investigated further. Increasing the concentration of 9 added to the starting solution of aryldiazoacetate 3 (0.04 M) prior to photolysis resulted in an increase in the diastereomeric ratio of trans:cis for dihydrobenzofurans 6a:6b to a maximum ratio of 3.3:1.0 of 6a:6b, achieved at 0.05 M in acetonitrile (Table 2, entries 6–9). Increasing the concentration or relative amount of the photosensitiser 9 beyond 0.05 M did not result in any further increase in the diastereomeric ratio (Table 2, entry 10). While the initial results from Table 1 showed that TBME was the best solvent choice, a mixture of TBME/acetonitrile (80:20) was ultimately chosen as our optimal solvent due to the limited solubility of 4,4′-dimethoxybenzophenone 9 in TBME alone, leading to the highest diastereomeric ratio (Table 2, entry 11).
 | ||
| Fig. 1 Photosensitisers employed in this investigation. | ||
|
|
|||||
|---|---|---|---|---|---|
| Entry | Solvent | Photosensitiser | Photosensitiser molarity (mol L−1) | Photosensitiser mol% | Ratiob6a:6b (trans:cis) |
|
a Reactions were conducted using Method B for a 5 mL solution of aryldiazoacetate 3 (0.04 M).
b The ratio of trans:cis was determined by the relative integration of C(2)H signals at 6.12 (1H, d) and C(3)H signals at 5.99 (1H, d) ppm, respectively, in the 1H NMR spectrum of the crude reaction mixture.
|
|||||
| 1 | DCM | TPP | 0.0013 | 4 | 1.0:1.0 |
| 2 | MeCN | Rose bengal | 0.0013 | 4 | 1.3:1.0 |
| 3 | MeCN | Methylene blue | 0.0013 | 4 | 1.3:1.0 |
| 4 | MeCN | Benzophenone | 0.0013 | 4 | 1.3:1.0 |
| 5 | DCM | 4,4′-Dichlorobenzophenone | 0.0013 | 4 | 1.3:1.0 |
| 6 | MeCN | 4,4′-Dimethoxybenzophenone 9 | 0.0013 | 4 | 1.6:1.0 |
| 7 | MeCN | 4,4′-Dimethoxybenzophenone 9 | 0.0100 | 28 | 2.6:1.0 |
| 8 | MeCN | 4,4′-Dimethoxybenzophenone 9 | 0.0250 | 67 | 3.1:1.0 |
| 9 | MeCN | 4,4′-Dimethoxybenzophenone 9 | 0.0500 | 125 | 3.3:1.0 |
| 10 | MeCN | 4,4′-Dimethoxybenzophenone 9 | 0.1000 | 280 | 3.3:1.0 |
| 11 | TBME/MeCN (80:20) |
4,4′-Dimethoxybenzophenone 9 | 0.0500 | 125 | 3.5:1.0 |

Investigation of the impact of residence time revealed that the photochemical transformation of aryldiazoacetate 3 in TBME/acetonitrile (80:20) was more rapid in the presence of the photosensitiser 9, with complete consumption within 3 minutes of photolysis (Table 3, entry 4), whereas unreacted aryldiazoacetate 3 was detectable in the absence of the photosensitiser 9 at this residence time (Table 3, entry 5). Significantly, in the 1H NMR spectra of the crude product mixtures, integration of characteristic signals showed that the diastereomeric ratio of 6a:6b was notably altered in the photosensitised reactions (≥2.7:1.0 cf. 1.0:1.0 in the absence of the photosensitiser 9). While 3 minutes photolysis in the presence of the photosensitiser provided the optimum conditions for generation of 6a and 6b the impact of prolonged photolysis was also explored. It was evident that following 3 minutes residence time the diastereomeric ratio seen in the presence of the photosensitiser 9 was ∼2.7:1.0, while extending the radiation time led to an increased diastereomeric ratio of 6a:6b. On closer investigation, it was clear that the dihydrobenzofurans 6a and 6b degrade under prolonged photolysis, and the increased diastereomeric ratio was as a result of the cis isomer 6b degrading faster than the trans isomer 6a (Table 3, entries 1–8), (Fig. 2). Experiments with very short exposure to photolysis (10 s and 1 min) (Table 3, entries 1 and 2) were undertaken and while both contained unreacted aryldiazoacetate 3, it was clear that the diastereomeric ratio was reduced as shown in Fig. 2.
 | ||
| Fig. 2 Impact of residence time/photolysis on the diastereomeric ratio (trans:cis) observed for aryldiazoacetate 3 to 6a and 6b. | ||
|
|
||||
|---|---|---|---|---|
| Entry | Residence time (min) | Ratiob3:6a:6b |
Yield trans6ac (%) | Yield cis6bc (%) |
|
a Reactions were conducted using Method B for a 25 mL solution of aryldiazoacetate 3 (0.04 M); each entry is an individual experiment.
b The ratio of trans:cis was determined by the relative integration of C(2)H signals at 6.12 (1H, d) and C(3)H signals at 5.99 (1H, d) ppm, respectively, in the 1H NMR spectrum of the crude reaction mixture.
c Isolated after purification by column chromatography.
d Isolated as a mixture of the cis isomer 6b and aryldiazoacetate starting material 3. By integration of the 1H NMR spectrum, the isolated product mixture consisted of 73% dihydrobenzofuran 6b and 25% 3.
e Reaction conducted without the addition of a photosensitiser using Method A.
f Reaction not purified by column chromatography.
|
||||
| 1 | 0.17 | 24.9:1.7:1.0 |
—f | —f |
| 2 | 1 | 1.1:2.4:1.0 |
—f | —f |
| 3 | 2 | 0.4:2.7:1.0 |
47 | 28d |
| 4 | 3 | 0:2.7:1.0 |
50 | 20 |
| 5e | 3 | 1.9:1.0:1.0 |
—f | —f |
| 6 | 4 | 0:2.9:1.0 |
45 | 9 |
| 7 | 10 | 0:3.5:1.0 |
42 | 7 |
| 8 | 60 | 0:4.5:1.0 |
39 | 4 |

From a synthetic perspective examination of the 1H NMR spectra of the crude product mixtures show that the C–H insertion in the presence of the photosensitiser 9 provides a much cleaner product mixture than the reactions in the absence of the photosensitiser 9. Accordingly, use of the photosensitiser 9 leads to significantly increased product recovery of the trans dihydrobenzofuran 6a (up to 50% yield) relative to the reaction in the absence of the photosensitiser 9 (up to 26% yield), (Table 4, entries 1 and 4). The recovery of the cis dihydrobenzofuran 6b is comparable both in the presence and absence of the photosensitiser 9 (up to 21% yield), (Table 4, entries 1 and 4). Interestingly, the reactions in the absence of the photosensitiser 9 require 10 minutes to ensure complete consumption of the aryldiazoacetate 3 but there is no alteration of the diastereomeric ratio from 1.0:1.0 in these experiments. Clearly the increased diastereomeric ratio due to selective degradation of the cis dihydrobenzofuran 6b is enhanced in the presence of the photosensitiser 9.
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | R | Residence time (minutes) | Photosensitiser | Ratio (trans:cis) |
Yield trans (%) | Yield cis (%) |
| a Reactions were conducted using Method A for a 25 mL solution of aryldiazoacetate 3, 4 or 5 (0.04 M). b Reactions were conducted using Method B for a 25 mL solution of aryldiazoacetate 3, 4 or 5 (0.04 M). | ||||||
| 1 | Me (3, 6) | 10 | Nonea | 1.0:1.0 |
26 | 21 |
| 2 | Bn (4, 7) | 10 | 1.0:1.0 |
18 | 14 | |
| 3 | iPr (5, 8) | 10 | 1.0:1.0 |
17 | 16 | |
| 4 | Me (3, 6) | 3 | 4,4′-Dimethoxybenzophenone 9 (0.05 M)b | 2.8:1.0 |
50 | 20 |
| 5 | Bn (4, 7) | 3 | 2.7:1.0 |
46 | 17 | |
| 6 | iPr (5, 8) | 3 | 2.6:1.0 |
44 | 15 | |
Interestingly, when the photolysis was conducted in TBME/acetonitrile (80:20) degradation of the photosensitiser 9 was evident from 1H NMR spectra of the crude product mixtures, but not when the photolysis was conducted in acetonitrile alone. Degradation of the photosensitiser was evident following 5 minutes of irradiation and increased with longer residence times but only occurred to a very limited extent in the 3 minutes optimised reactions, and therefore has no impact on the synthetic utility of the photochemical C–H insertion (Table 3, entry 2). Among the degradation products was the dimer 10, which was isolated in agreement with an earlier report by Nagorny (Scheme 3).59 To confirm the origin of this dimer, a sample of the photosensitiser 9 was irradiated in TBME/acetonitrile in the absence of the aryldiazoacetate, leading to isolation of the dimer in 34% yield after column chromatography, presumably formed via dimerisation of the excited state biradical intermediate.
 | ||
| Scheme 3 1,1,2,2-Tetrakis(4-methoxyphenyl)ethane-1,2-diol 10, isolated from photolysis of photosensitiser 9 alone in TBME/MeCN. | ||
Further work to explore the effect of the triplet photosensitiser was carried out in acetonitrile, to avoid complications due to reaction of the photosensitiser 9 when the photolysis is conducted in TBME. A solution containing 4,4′-dimethoxybenzophenone 9 (0.05 M, optimised concentration) and a 1.0:1.0 mixture of the trans and cis isomers 6a and 6b in acetonitrile was exposed to the photolysis conditions for 10 minutes to establish if cis–trans interconversion was occurring on this time scale. However, it was clear that the alteration in diastereomeric ratio on prolonged photolysis is due to faster degradation of the cis dihydrobenzofuran 6b than that of trans6a, rather than interconversion (Fig. 3). This pattern continued when the resulting mixture was re-exposed for another 10 minutes and then 30 minutes of photolysis conditions. The extent of alteration of the diastereomeric ratio in the absence of the photosensitiser 9 is much less. The degradation of the dihydrobenzofurans 6a and 6b on photolysis leads to a complex mixture of unidentified products. A genuine sample of the benzofuran 11 (Fig. 4) was prepared for comparison and there is little evidence in the 1H NMR spectra for the presence of this within the complex mixture.60
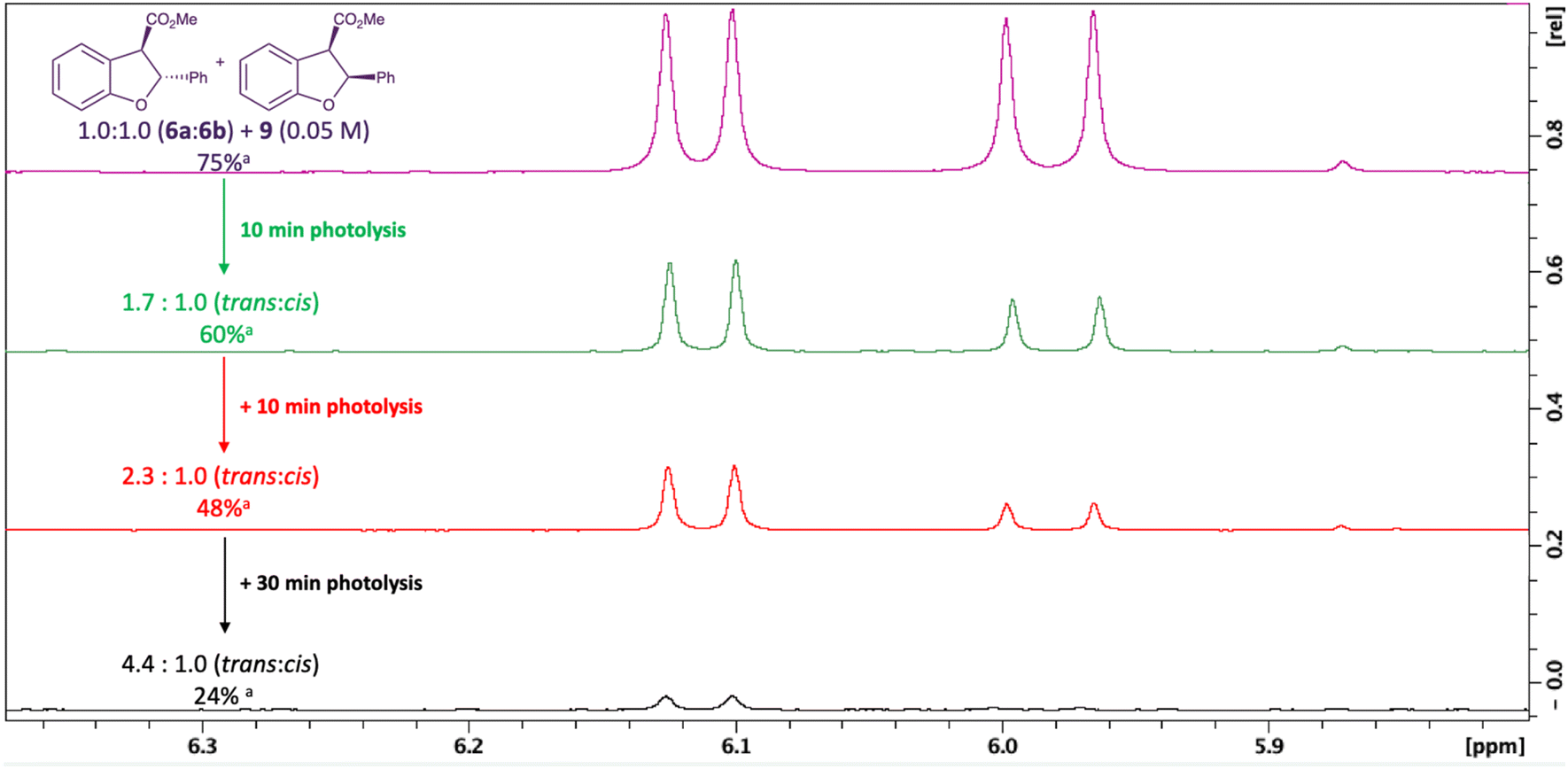 | ||
| Fig. 3 Degradation of the trans and cis dihydrobenzofurans 6a and 6b with repeated exposure to photolysis conditions. aThe percentage was estimated by comparing the relative integration of signals in the aromatic region (6.6–8.0 ppm) in the 1H NMR spectrum of the crude reaction mixtures to the signals for dihydrobenzofurans 6a and 6b, excluding the integration for the signals for the photosensitiser 9. | ||
 | ||
| Fig. 4 Methyl 2-phenylbenzofuran-3-carboxylate 11. | ||
With the optimised conditions established, the substrate scope was extended to include benzyl aryldiazoacetate 4 and isopropyl aryldiazoacetate 5.53 In the absence of any photosensitiser with 10 minutes of photolysis, as seen in the transformation of 3 to 6a and 6b, an equimolar mixture of the cis and trans dihydrobenzofurans 7a:7b and 8a:8b was observed, with the trans and cis diastereomers being isolated in comparable yields after column chromatography (Table 4, entries 1–3). In the presence of 4,4′-dimethoxybenzophenone 9 (0.05 M) and with a shorter residence time of 3 minutes, the diastereomeric ratio of 6a:6b, 7a:7b and 8a:8b increased to ∼2.7:1.0. Addition of the triplet photosensitiser 9 to the starting solution of aryldiazoacetate 3, 4 or 5 consistently resulted in increased diastereomeric ratios in the crude product mixture leading to improved isolated yields of the trans dihydrobenzofuran products 6a, 7a and 8a (Table 4, entries 4–6). Interestingly, the alteration in the stereochemical outcome was consistent across the series with no detectable impact by the nature of the substituent on the ester moiety.
In the reactions of the substrates 4 and 5, the β-lactones 12 and 13 were formed as side products in the photolysis reactions both in the presence and absence of the photosensitiser 9 (Scheme 4). β-Lactone 13 was isolated (37%) and characterised from the reaction in the presence of the photosensitiser 9. Thus, C–H insertion into the benzyl C–H or the isopropyl C–H of the ester competes with insertion into the benzyl ether C–H bond to form the dihydrobenzofuran. Davies has demonstrated C–H insertion of aryldiazoacetates to form β-lactones both photochemically39 and using rhodium catalysts.61 Lowe has reported photochemical C–H insertion of α-diazoamides and α-diazoesters to form lactones.62
 | ||
| Scheme 4 3-(2-(Benzyloxy)phenyl)-4-phenyloxetan-2-one 12 and 3-(2-(benzyloxy)phenyl)-4,4-dimethyloxetan-2-one 13. | ||
Photolysis using LEDs was explored to establish if narrower wavelength ranges would lead to better outcomes in terms of selectivity and efficiency.17,23,24 Photolysis of aryldiazoacetates 3, 4 and 5 was undertaken using LEDs (365 nm and 450 nm). Interestingly, in the presence of the photosensitiser 9, use of the 365 nm LEDs led to essentially identical outcomes to those seen with the medium pressure mercury lamp, without any apparent difference in the product mixture either in terms of yield or diastereomeric ratio, for the photolysis of aryldiazoacetate 3 (Table 5, entries 1 and 2). While the objective was to explore if use of narrower wavelengths could lead to decreased product degradation and, accordingly, higher yields of dihydrobenzofurans, in practice, there was no observable difference.
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | R | Light source | Residence time (minutes) | Temperature (°C) | Ratio 6a:6b:3 |
Yield trans (%) | Yield cis (%) |
| a Reactions were conducted using Method B for a 25 mL solution of aryldiazoacetate 3 (0.04 M). b Reactions were conducted using Method C for a 25 mL solution of aryldiazoacetate 3, 4 or 5 (0.04 M). c Reactions were conducted using Method D for a 25 mL solution of aryldiazoacetate 3, 4 or 5 (0.04 M). d Isolated as a mixture of cis dihydrobenzofuran and aryldiazoacetate starting material. | |||||||
| 1 | Me (3, 6) | Medium pressure Hg lamp (75 W)a | 5 | 28 | 3.0:1.0:0 |
33 | 6 |
| 2 | Me (3, 6) | 365 nm LED (50 W)b | 5 | 33 | 2.8:1.0:0 |
37 | 12 |
| 3 | Bn (4, 7) | 5 | 33 | 3.8:1.0:0 |
30 | 5 | |
| 4 | iPr (5, 8) | 5 | 33 | 4.1:1.0:0 |
32 | — | |
| 5 | Me (3, 6) | 450 nm LED (12 W)c | 60 | 28 | 1.0:1.0:0.6 |
25 | 23d |
| 6 | Bn (4, 7) | 60 | 28 | 1.0:1.0:0.8 |
14 | 15d | |
| 7 | iPr (5, 8) | 60 | 28 | 1.0:1.0:0.6 |
10 | 13d | |

Photolysis of benzyl and isopropyl aryldiazoacetates 4 and 5 was also undertaken using the 365 nm LEDs in the presence of the photosensitiser 9 (0.05 M), resulting in preferential isolation of the trans dihydrobenzofurans 7a and 8a (Table 5, entries 3 and 4). While the trans dihydrobenzofurans 7a and 8a were isolated in modest yields, the cis dihydrobenzofuran 7b was isolated in poor yield. The cis dihydrobenzofuran 8b was not isolated, but the β-lactone 13 was isolated from the photolysis of isopropyl aryldiazoacetate 5. Photolysis of aryldiazoacetate 3 in the absence of photosensitiser 9 was also trialled using the 365 nm LEDs. Full consumption of the aryldiazoacetate 3 was observed within 5 minutes of photolysis in acetonitrile leading to 32% formation of the dihydrobenzofurans 6a and 6b in a ratio of 1.1:1.0 (products not isolated).
In contrast, when 3 was exposed to 450 nm LEDs in the absence of the photosensitiser 9, reactions were slower with decreased reaction efficiency and some aryldiazoacetate 3 remaining unreacted (1.0:1.0:0.6, 6a:6b:3) following 60 min of photolysis conditions (Table 5, entry 5), presumably due to the lower input power of the LEDs employed relative to the mercury lamp (12 W 450 nm LEDs; 75 W mercury lamp). Similar results were seen for aryldiazoacetates 4 and 5 (Table 5, entries 6 and 7). Notably, photolysis using the blue LEDs in the presence or absence of the photosensitiser led to an equimolar mixture of dihydrobenzofurans 6a and 6b; this was the first time we observed no alteration of the diastereomeric ratio in the presence of the photosensitiser. This altered behaviour can be rationalised by examination of the UV/Vis spectra of the aryldiazoacetates and the photosensitiser 9; while the aryldiazoacetates absorb at 450 nm leading to C–H insertion, the photosensitiser 9 does not absorb at this wavelength. In contrast, both compounds absorb at 365 nm (see ESI† for details). Thus, it is clear that the altered diastereomeric ratio seen at shorter wavelengths is associated with the presence of the photosensitiser 9. Recent reports of the impact of photosensitisers on stereochemical outcome are relevant to this work,63,64 although in this instance the altered diastereomeric ratio is principally due to selective degradation.
To demonstrate the practical synthetic utility of this photochemical C–H insertion, 2 g of aryldiazoacetate 3 in TBME/acetonitrile (80:20) was irradiated in the presence of the photosensitiser 9 with a residence time of just 3 minutes leading to isolation of dihydrobenzofurans 6a and 6b in 47% and 11% yield, respectively. The efficiency of the photochemical transformation was comparable at this scale to that seen at smaller scale as evidenced by comparing the 1H NMR spectra of the crude product mixtures (see Fig. S1 in the ESI†).
Conclusion
In conclusion, flow photolysis of aryldiazoacetates 3, 4 and 5 leads to metal-free C–H insertion to form dihydrobenzofurans 6, 7 and 8 in moderate yields. Significantly, while the cis and trans diastereomers of 6, 7 and 8 are formed in essentially equimolar amounts, addition of the photosensitiser 9 results in preferential recovery of the trans dihydrobenzofurans 6a, 7a and 8a (dr approximately ≥2.7:1.0). The photolysis can be effected using a mercury lamp or LEDs with comparable synthetic outcomes, and is readily scaled to multigram quantities without impacting on reaction efficiency or selectivity. Effecting C–H insertion in the absence of a metal catalyst is notable from a green chemistry perspective.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from Synthesis and Solid State Pharmaceutical Centre (SSPC) supported by Science Foundation Ireland and co-funded under the European Regional Development Fund (K. S. O. C., D. L., SFI SSPC3 Pharm5 12/RC/2275_2), and equipment provided though a SFI Research Infrastructure award (ProSpect) (SFI 15/RI/3221) is gratefully acknowledged.References
- T. Curtius, Ueber Die Einwirkung von Salpetriger Säure Auf Salzsauren Glycocolläther, Ber. Dtsch. Chem. Ges., 1883, 16(2), 2230–2231, DOI:10.1002/cber.188301602136.
- A. Ford, H. Miel, A. Ring, C. N. Slattery, A. R. Maguire and M. A. McKervey, Modern Organic Synthesis with α-Diazocarbonyl Compounds, Chem. Rev., 2015, 115(18), 9981–10080, DOI:10.1021/acs.chemrev.5b00121.
- H. Lebel, J. F. Marcoux, C. Molinaro and A. B. Charette, Stereoselective Cyclopropanation Reactions, Chem. Rev., 2003, 103(4), 977–1050, DOI:10.1021/cr010007e.
- A. Padwa and S. F. Hornbuckle, Ylide Formation from the Reaction of Carbenes and Carbenoids with Heteroatom Lone Pairs, Chem. Rev., 1991, 91, 263–309, DOI:10.1021/cr00003a001.
- S. E. Reisman, R. R. Nani and S. Levin, Buchner and Beyond: Arene Cyclopropanation as Applied to Natural Product Total Synthesis, Synlett, 2011, 2437–2442, DOI:10.1055/s-0031-1289520.
- H. M. L. Davies and K. Liao, Dirhodium Tetracarboxylates as Catalysts for Selective Intermolecular C–H Functionalization, Nat. Rev. Chem., 2019, 3(6), 347–360, DOI:10.1038/s41570-019-0099-x.
- M. P. Doyle, R. Duffy, M. Ratnikov and L. Zhou, Catalytic Carbene Insertion into C–H Bonds, Chem. Rev., 2010, 110(2), 704–724, DOI:10.1021/cr900239n.
- H. M. L. Davies and R. E. J. Beckwith, Catalytic Enantioselective C–H Activation by Means of Metal-Carbenoid-Induced C–H Insertion, Chem. Rev., 2003, 103(8), 2861–2903, DOI:10.1021/cr0200217.
- H. M. L. Davies and J. R. Manning, Catalytic C–H Functionalization by Metal Carbenoid and Nitrenoid Insertion, Nature, 2008, 451(7177), 417–424, DOI:10.1038/nature06485.
- Ł. W. Ciszewski, K. Rybicka-Jasińska and D. Gryko, Recent Developments in Photochemical Reactions of Diazo Compounds, Org. Biomol. Chem., 2019, 17(3), 432–448, 10.1039/c8ob02703j.
- N. Candeias and C. Afonso, Developments in the Photochemistry of Diazo Compounds, Curr. Org. Chem., 2009, 13(7), 763–787, DOI:10.2174/138527209788167231.
- O. S. Galkina and L. L. Rodina, Photochemical Transformations of Diazocarbonyl Compounds: Expected and Novel Reactions, Russ. Chem. Rev., 2016, 85(5), 537–555, DOI:10.1070/rcr4519.
- J. Durka, J. Turkowska and D. Gryko, Lightening Diazo Compounds?, ACS Sustainable Chem. Eng., 2021, 8895–8918, DOI:10.1021/acssuschemeng.1c01976.
- Z. Yang, M. L. Stivanin, I. D. Jurberg and R. M. Koenigs, Visible Light-Promoted Reactions with Diazo Compounds: A Mild and Practical Strategy Towards Free Carbene Intermediates, Chem. Soc. Rev., 2020, 49(19), 6833–6847, 10.1039/d0cs00224k.
- M. Baumann, T. S. Moody, M. Smyth and S. Wharry, A Perspective on Continuous Flow Chemistry in the Pharmaceutical Industry, Org. Process Res. Dev., 2020, 24(10), 1802–1813, DOI:10.1021/acs.oprd.9b00524.
- K. Loubière, M. Oelgemöller, T. Aillet, O. Dechy-Cabaret and L. E. Prat, Continuous-Flow Photochemistry: A Need for Chemical Engineering, Chem. Eng. Process., 2016, 104, 120–132, DOI:10.1016/j.cep.2016.02.008ï.
- M. di Filippo, C. Bracken and M. Baumann, Continuous Flow Photochemistry for the Preparation of Bioactive Molecules, Molecules, 2020, 25(2), 356, DOI:10.3390/molecules25020356.
- K. Gilmore and P. H. Seeberger, Continuous Flow Photochemistry, Chem. Rec., 2014, 14(3), 410–418, DOI:10.1002/tcr.201402035.
- F. Politano and G. Oksdath-Mansilla, Light on the Horizon: Current Research and Future Perspectives in Flow Photochemistry, Org. Process Res. Dev., 2018, 22(9), 1045–1062, DOI:10.1021/acs.oprd.8b00213.
- K. Donnelly and M. Baumann, Scalability of Photochemical Reactions in Continuous Flow Mode, J. Flow Chem., 2021, 11, 223–241, DOI:10.1007/s41981-021-00168-z/Published.
- C. Sambiagio and T. Noël, Flow Photochemistry: Shine Some Light on Those Tubes!, Trends Chem., 2020, 2(2), 92–106, DOI:10.1016/j.trechm.2019.09.003.
- J. P. Knowles, L. D. Elliott and K. I. Booker-Milburn, Flow Photochemistry: Old Light Through New Windows, Beilstein J. Org. Chem., 2012, 8, 2025–2052, DOI:10.3762/bjoc.8.229.
- Y. Su, N. J. W. Straathof, V. Hessel and T. Noël, Photochemical Transformations Accelerated in Continuous-Flow Reactors: Basic Concepts and Applications, Chem. – Eur. J., 2014, 20(34), 10562–10589, DOI:10.1002/chem.201400283.
- L. Buglioni, F. Raymenants, A. Slattery, S. D. A. Zondag and T. Noël, Technological Innovations in Photochemistry for Organic Synthesis: Flow Chemistry, High-Throughput Experimentation, Scale-up, and Photoelectrochemistry, Chem. Rev., 2022, 122(2), 2752–2906, DOI:10.1021/acs.chemrev.1c00332.
- T. H. Rehm, Flow Photochemistry as a Tool in Organic Synthesis, Chem. – Eur. J., 2020, 26(71), 16952–16974, DOI:10.1002/chem.202000381.
- B. Gutmann, D. Cantillo and C. O. Kappe, Continuous-Flow Technology – A Tool for the Safe Manufacturing of Active Pharmaceutical Ingredients, Angew. Chem., 2015, 127(23), 6788–6832, DOI:10.1002/ange.201409318.
- D. Webb and T. F. Jamison, Continuous Flow Multi-Step Organic Synthesis, Chem. Sci., 2010, 1(6), 675–680, 10.1039/c0sc00381f.
- I. R. Baxendale, L. Brocken and C. J. Mallia, Flow Chemistry Approaches Directed at Improving Chemical Synthesis, Green Process. Synth., 2013, 2(3), 211–230, DOI:10.1515/gps-2013-0029.
- J. C. Pastre, D. L. Browne and S. V. Ley, Flow Chemistry Syntheses of Natural Products, Chem. Soc. Rev., 2013, 42(23), 8849–8869, 10.1039/c3cs60246j.
- R. Porta, M. Benaglia and A. Puglisi, Flow Chemistry: Recent Developments in the Synthesis of Pharmaceutical Products, Org. Process Res. Dev., 2016, 20(1), 2–25, DOI:10.1021/acs.oprd.5b00325.
- R. L. Hartman, J. P. McMullen and K. F. Jensen, Deciding Whether to Go with the Flow: Evaluating the Merits of Flow Reactors for Synthesis, Angew. Chem., Int. Ed., 2011, 50(33), 7502–7519, DOI:10.1002/anie.201004637.
- M. B. Plutschack, B. Pieber, K. Gilmore and P. H. Seeberger, The Hitchhiker's Guide to Flow Chemistry, Chem. Rev., 2017, 117(18), 11796–11893, DOI:10.1021/acs.chemrev.7b00183.
- M. Movsisyan, E. I. P. Delbeke, J. K. E. T. Berton, C. Battilocchio, S. V. Ley and C. V. Stevens, Taming Hazardous Chemistry by Continuous Flow Technology, Chem. Soc. Rev., 2016, 45(18), 4892–4928, 10.1039/c5cs00902b.
- B. J. Deadman, S. G. Collins and A. R. Maguire, Taming Hazardous Chemistry in Flow: The Continuous Processing of Diazo and Diazonium Compounds, Chem. – Eur. J., 2015, 21(6), 2298–2308, DOI:10.1002/chem.201404348.
- R. Hommelsheim, Y. Guo, Z. Yang, C. Empel and R. M. Koenigs, Blue-Light-Induced Carbene-Transfer Reactions of Diazoalkanes, Angew. Chem., 2019, 131(4), 1216–1220, DOI:10.1002/ange.201811991.
- V. Klöpfer, R. Eckl, J. Floß, P. M. C. Roth, O. Reiser and J. P. Barham, Catalyst-Free, Scalable Heterocyclic Flow Photocyclopropanation, Green Chem., 2021, 23(17), 6366–6372, 10.1039/d1gc01624e.
- C. Empel and R. M. Koenigs, Continuous-Flow Photochemical Carbene Transfer Reactions, J. Flow Chem., 2020, 10(1), 157–160, DOI:10.1007/s41981-019-00054-9.
- E. J. Corey and A. M. Felix, A New Synthetic Approach to the Penecillans, J. Am. Chem. Soc., 1965, 87(11), 2518–2519, DOI:10.1021/ja01089a055.
- I. D. Jurberg and H. M. L. Davies, Blue Light-Promoted Photolysis of Aryldiazoacetates, Chem. Sci., 2018, 9(22), 5112–5118, 10.1039/c8sc01165f.
- S. Tewtrakul, H. Miyashiro, N. Nakamura, M. Hattori, T. Kawahata, T. Otake, T. Yoshinaga, T. Fujiwara, T. Supavita, S. Yuenyongsawad, P. Rattanasuwon and S. Dej-Adisai, HIV-1 Integrase Inhibitory Substances from Coleus Parvifolius, Phytother. Res., 2003, 17(3), 232–239, DOI:10.1002/ptr.1111.
- I. S. Abd-Elazem, H. S. Chen, R. B. Bates, R. Chih and C. Huang, Isolation of Two Highly Potent and Non-Toxic Inhibitors of Human Immunodeficiency Virus Type 1 (HIV-1) Integrase from Salvia Miltiorrhiza, Antiviral Res., 2002, 55, 91–106, DOI:10.1016/s0166-3542(02)00011-6.
- J. H. Lee, Y. G. Kim, S. Y. Ryu, M. H. Cho and J. Lee, Resveratrol Oligomers Inhibit Biofilm Formation of Escherichia Coli, O157:H7 and Pseudomonas Aeruginosa, J. Nat. Prod., 2014, 77(1), 168–172, DOI:10.1021/np400756g.
- C. Billard, J. C. Izard, V. Roman, C. Kern, C. Mathiot, F. Mentz and J. P. Kolb, Comparative Antiproliferative and Apoptotic Effects of Resveratrol, ε-Viniferin and Vine-Shots Derived Polyphenols (Vineatrols) on Chronic B Lymphocytic Leukemia Cells and Normal Human Lymphocytes, Leuk. Lymphoma, 2002, 43(10), 1991–2002, DOI:10.1080/1042819021000015952.
- G. Lemi, M. Gao, A. de Groot, R. Dommisse, J. Lepoivre, L. Pieterd and V. Buss, 3′,4-Di-O-Methylcedrusin: Synthesis, Resolution and Absolute Configuration, J. Chem. Soc., Perkin Trans. 1, 1995, 1775–1779 Search PubMed.
- K. Sawasdee, T. Chaowasku, V. Lipipun, T. H. Dufat, S. Michel and K. Likhitwitayawuid, New Neolignans and a Lignan from Miliusa Fragrans, and Their Anti-Herpetic and Cytotoxic Activities, Tetrahedron Lett., 2013, 54(32), 4259–4263, DOI:10.1016/j.tetlet.2013.05.144.
- R.-B. An, G.-S. Jeong and Y.-C. Kim, Flavonoids from the Heartwood of Dalbergia Odorifera, and Their Protective Effect on Glutamate-Induced Oxidative Injury in HT22 Cells, Chem. Pharm. Bull., 2008, 56(12), 1722–1724, DOI:10.1248/cpb.56.1722.
- M. Gregson, W. David Ollis, B. T. Redhan, I. O. Sutherland, H. H. Dietrichs and O. R. Gottlieb, Obtusastyrene and obtustyrene, cinnamylphenols from Dalbergia retusa, Phytochemistry, 1978, 17, 1395–1400, DOI:10.1016/S0031-9422(00)94596-5.
- M. Pereira, D. E. Campos, V. C. Filho, R. Zanoni, D. A. Silva, R. A. Yunes, S. Zacchino, S. Juarez, R. Cé, B. Cruz and A. B. Cruz, Evaluation of Antifungal Activity of Piper solmsianum C. DC. Var. solmsianum (Piperaceae), Biol. Pharm. Bull., 2005, 28(8), 1527–1530, DOI:10.1248/bpb.28.1527.
- S. Apers, A. Vlietinck and L. Pieters, Lignans and Neolignans as Lead Compounds, Phytochem. Rev., 2003, 2, 201–217, DOI:10.1023/B:PHYT.0000045497.90158.d2.
- Z. Chen, M. Pitchakuntla and Y. Jia, Synthetic Approaches to Natural Products Containing 2,3-Dihydrobenzofuran Skeleton, Nat. Prod. Rep., 2019, 36(4), 666–690, 10.1039/c8np00072g.
- H. Wang, G. Li, K. M. Engle, J. Q. Yu and H. M. L. Davies, Sequential C–H Functionalization Reactions for the Enantioselective Synthesis of Highly Functionalized 2,3-Dihydrobenzofurans, J. Am. Chem. Soc., 2013, 135(18), 6774–6777, DOI:10.1021/ja401731d.
- A. Bunyamin, C. Hua, A. Polyzos and D. L. Priebbenow, Intramolecular Photochemical [2 + 1]-Cycloadditions of Nucleophilic Siloxy Carbenes, Chem. Sci., 2022, 13(11), 3273–3280, 10.1039/d2sc00203e.
- A. M. Buckley, D. C. Crowley, T. A. Brouder, A. Ford, U. B. Rao Khandavilli, S. E. Lawrence and A. R. Maguire, Dirhodium Carboxylate Catalysts from 2-Fenchyloxy or 2-Menthyloxy Arylacetic Acids: Enantioselective C–H Insertion, Aromatic Addition and Oxonium Ylide Formation/Rearrangement, ChemCatChem, 2021, 13(20), 4318–4324, DOI:10.1002/cctc.202100924.
- H. Saito, H. Oishi, S. Kitagaki, S. Nakamura, M. Anada and S. Hashimoto, Enantio- and Diastereoselective Synthesis of cis-2-Aryl-3-Methoxycarbonyl-2,3-Dihydrobenzofurans via the Rh(II)-Catalyzed C–H Insertion Process, Org. Lett., 2002, 4(22), 3887–3890, DOI:10.1021/ol0267127.
- H. M. L. Davies, M. V. A. Grazini and E. Aouad, Asymmetric Intramolecular C–H Insertions of Aryldiazoacetates, Org. Lett., 2001, 3(10), 1475–1477, DOI:10.1021/ol0157858.
- R. F. Borkman and D. R. Kearns, Triplet-State Energy Transfer in Liquid Solutions. Acetone-Photosensitized cis–trans Isomerization of Pentene-2, J. Am. Chem. Soc., 1966, 88(5), 3467–3475, DOI:10.1021/ja00967a001.
- G. Campari, M. Fagnoni, M. Mella and A. Albini, Diastereoselective Photosensitised Radical Addition to Fumaric Acid Derivatives Bearing Oxazolidine Chiral Auxiliaries, Tetrahedron: Asymmetry, 2000, 11, 1891–1906, DOI:10.1016/S0957-4166(00)00138-5.
- H. B. Wang, B. C. Zhai, W. J. Tang, J. Y. Yu and Q. H. Song, Photosensitized Z–E Isomerization of Cinnamate by Covalently Linked 2-(3′,4′-Dimethoxybenzoyl)Benzyl Moiety via Triplet–Triplet Energy Transfer, Chem. Phys., 2007, 333, 229–235, DOI:10.1016/j.chemphys.2007.02.003.
- S. Kim, Y. Khomutnyk, A. Bannykh and P. Nagorny, Synthesis of Glycosyl Fluorides by Photochemical Fluorination with Sulfur(VI) Hexafluoride, Org. Lett., 2021, 23(1), 190–194, DOI:10.1021/acs.orglett.0c03915.
- B. Kang, M. H. Lee, M. Kim, J. Hwang, H. B. Kim and D. Y. Chi, Transition-Metal-Free Synthesis of 2-Substituted Methyl Benzo[b]Furan-3-Carboxylates, J. Org. Chem., 2015, 80(16), 8254–8261, DOI:10.1021/acs.joc.5b01311.
- L. Fu, H. Wang and H. M. L. Davies, Role of Ortho-Substituents on Rhodium-Catalyzed Asymmetric Synthesis of β-Lactones by Intramolecular C–H Insertions of Aryldiazoacetates, Org. Lett., 2014, 16(11), 3036–3039, DOI:10.1021/ol5011505.
- G. Lowe and J. Parker, Photochemical Conversion of α-Diazo-Amides and -Esters into β-Lactams and β- and γ-Lactones, J. Chem. Soc. D, 1971, 577–578, 10.1039/C29710000577.
- J. Mateos, F. Rigodanza, P. Costa, M. Natali, A. Vega-Peñaloza, E. Fresch, E. Collini, M. Bonchio, A. Sartorel and L. Dell'Amico, Unveiling the Impact of the Light Source and Steric Factors on [2 + 2] Heterocycloaddition Reactions, Nat. Synth., 2022, 2(1), 26–36, DOI:10.1038/s44160-022-00191-5.
- S. Yakubov, W. J. Stockerl, X. Tian, A. Shahin, M. J. P. Mandigma, R. M. Gschwind and J. P. Barham, Benzoates as Photosensitization Catalysts and Auxiliaries in Efficient, Practical, Light-Powered Direct C(sp3)–H Fluorinations, Chem. Sci., 2022, 13(47), 14041–14051, 10.1039/d2sc05735b.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ob00541k |
| This journal is © The Royal Society of Chemistry 2023 |