
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Mono- and three-tailed sugar and iminosugar decorated benzenesulfonamide carbonic anhydrase inhibitors†
Maria Giulia
Davighi
 a,
Camilla
Matassini
a,
Andrea
Goti
a,
Marta
Ferraroni
a,
Andrea
Angeli
b,
Claudiu T.
Supuran
*b and
Francesca
Cardona
*a
a,
Camilla
Matassini
a,
Andrea
Goti
a,
Marta
Ferraroni
a,
Andrea
Angeli
b,
Claudiu T.
Supuran
*b and
Francesca
Cardona
*a
aDipartimento di Chimica “Ugo Schiff” DICUS, Università degli Studi di Firenze, Via della Lastruccia 3-13, 50019 Sesto Fiorentino (FI), Italy. E-mail: francesca.cardona@unifi.it
bDipartimento Neurofarba, Sezione di Scienze Farmaceutiche e Nutraceutiche, Università degli Studi di Firenze, 50019 Sesto Fiorentino (FI), Italy. E-mail: claudiu.supuran@unifi.it
First published on 11th May 2023
Abstract
A collection of novel mono- and three-tailed derivatives based on a sugar (glucose) or an iminosugar (trihydroxy piperidine) featuring a terminal benzenesulfonamide were synthesized to investigate the so-called “sugar” and “azasugar” approach with the aim of exploring the activity and selectivity towards the inhibition of human carbonic anhydrases (hCAs). The synthetic approach relies on a general copper(I)-catalyzed azide–alkyne cycloaddition (CuAAC) reaction followed by an amine–isothiocyanate coupling. Biological assays were used to collect subtle information on the role of these single or multiple hydrophilic chains. Among the sugar-based inhibitors, the single-tailed compound 10 was identified as a better inhibitor than the reference compound (AAZ) towards three different hCAs, while, among the three sugar tailed derivatives, potent and selective inhibition was found for compounds 25 and 26. A promising and selective inhibitory activity was discovered for the iminosugar single-tailed compound 31 towards hCA VII (Ki = 9.7 nM).
Introduction
Carbonic anhydrases (CAs, EC 4.2.1.1) are ubiquitous metalloenzymes that catalyze the reversible hydration of carbon dioxide (CO2) to give bicarbonate (HCO3−) and a proton (H+),1 a fundamental reaction related to many important physiological processes such as respiration and transport of CO2/HCO3−, pH regulation and CO2 homeostasis, electrolyte secretion, bone resorption, calcification, biosynthetic reactions and tumorigenesis.2At physiological pH, this reaction is too slow and requires to be catalyzed. Eight different genetic families, named α-, β-, γ-, δ-, ζ-, η-, θ-, and ι-CA classes, are known to be present in living organisms (higher vertebrates, green plants, algae, bacteria, and archaea) and they differ for metal ions in the active site (e.g. zinc, iron, cadmium, and cobalt). Human carbonic anhydrases (hCAs) all belong to the α-family. Fifteen different α-CA isoforms have been identified and characterized and exhibit different enzyme kinetics, expression levels and locations within the cell and tissues.3 Five hCA isozymes are cytosolic (hCAs I–III, hCA VII, and hCA XIII), two are mitochondrial (hCAs VA and VB), four are membrane-bound or transmembrane proteins (hCAs IV, IX, XII and XIV) and one is secreted into the saliva and milk (hCA VI).4 CA isozymes have become drug targets for biomedical applications. For example, hCAs I, II, and IV isoforms have been used to develop antiglaucoma agents, hCAs VA and VB isoforms are targets for obtaining anti-obesity drugs, while hCA VII is implicated in neuropathic pain and in the development of anticonvulsant drugs. Moreover, CAs IX and XII show close association with hypoxic tumours such as those of the lung, breast, colon, esophagus and cervix in which they are overexpressed in tissues and are absent under physiological conditions.5 Therefore, the inhibition (or activation) of hCA activity is employed to treat a wide range of acquired and inherited diseases.2 Nevertheless, the large number of hCA isoforms requires new increasingly selective inhibitors to avoid side effects due to the indiscriminate inhibition of isoforms that are not involved in a certain pathology.6 The so-called “sugar-tail approach”, suggested by Winum and co-workers,5a is emerging as a promising strategy to differently interact with the transmembrane protein (i.e., hCA IX) and the physiologically dominant cytosolic isozymes hCAs I and II. The introduction of a sugar moiety permitted the development of more selective inhibitors with polar or charged tails, thus impairing their ability to diffuse through lipid membranes. The best candidates for this function are carbohydrates, which are widespread in biologically active compounds, influencing their pharmacokinetics, drug targeting, and mechanism of action. Moreover, carbohydrates have good solubility in water, a high degree of polyfunctionality and hydrophilicity and their stereochemical arrangement can potentially allow the differentiation of subtle differences in CA active site topology.7 Several inhibitors with a sulfonamide moiety, the most used and effective functional group for enabling CA inhibition, directly connected to the anomeric carbon (compounds 1, Fig. 1) or to the C-6 hydroxyl group of the sugar (compounds 2, Fig. 1) or N-β-glycosyl sulfamides were reported.5a Wilkinson and co-workers synthesized 1,4-disubstituted-1,2,3-triazole sulfonamide glycoconjugates via the CuAAC (“copper(I)-catalyzed azide–alkyne cycloaddition”) reaction,8 exploiting the versatility of the click chemistry methodology (compounds 3, Fig. 1).9 A promising related strategy developed by our group for targeting more selective carbonic anhydrase inhibitors (CAIs) is the so-called “iminosugar or azasugar approach”, where carbohydrates are replaced by glycomimetics in which a nitrogen atom replaces the ring oxygen or the anomeric carbon of carbohydrates, respectively.10 These compounds have a basic nitrogen atom which can be protonated at physiological pH, establishing different interactions with the enzyme active site or proximal residues with respect to the corresponding carbohydrate.11
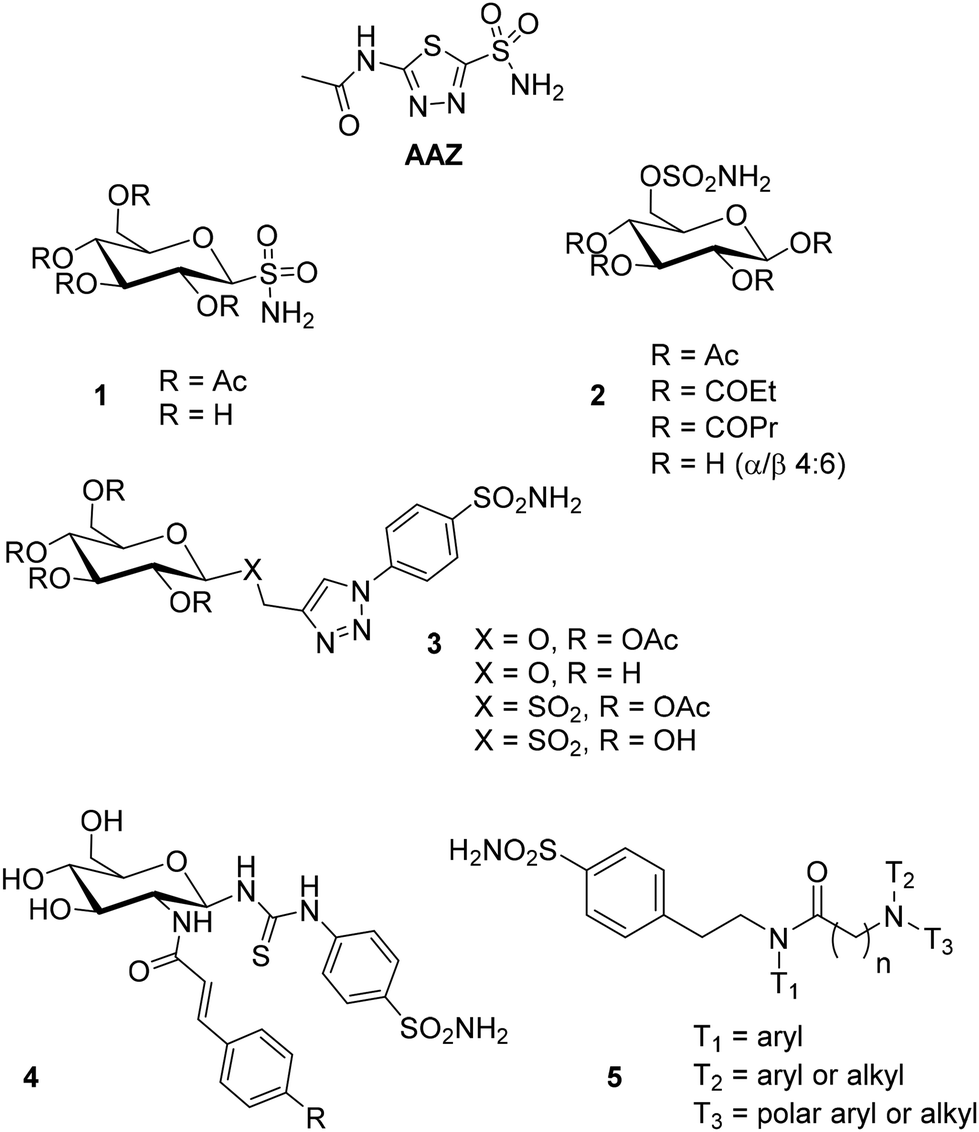 | ||
| Fig. 1 Examples of hCA inhibitors containing a sugar moiety and dual- and three-tailed compounds, and the reference compound AAZ. | ||
We have recently reported the synthesis of chimeric compounds obtained by conjugating a piperidine iminosugar, alkylated at the endocyclic nitrogen with a six carbon atom chain ending with a triazole, to several benzenesulfonamides through various linking moieties (ureido, amido or thioureido), obtaining encouraging results for strong inhibition in the nanomolar range, especially towards hCAs II and VII isoforms.12
The crystallographic structures of CA II and CA IX showed that the active site cavity of these enzymes is composed of two different conserved sub-pockets containing hydrophobic and hydrophilic amino acids, respectively.13 Based on these results, Tanpure et al. first introduced the dual-tail approach as a strategy to further improve the selectivity and specificity of CA inhibitors by addressing both the hydrophobic and hydrophilic parts of the CA active site and conserving the zinc binding group.14 They synthesized dual-tailed compounds combining phenyl moieties (hydrophobic tails) and glucosyl ones (hydrophilic tails) and their corresponding single-tailed compounds as reference compounds. The compounds designed by the dual-tail approach showed less inhibitory activity with respect to the reference compound acetazolamide (AAZ), probably because the relative positions of the two tails were not optimal for synergistic effects. In contrast, the corresponding single-tailed compounds resulted in more potent inhibitors than AAZ. However, the dual-tailed compounds were tested in vitro only on hCAs I and II, suggesting that the information from this preliminary study is limited. Afterwards, Hou et al. revisited the dual-tail approach by designing novel CAIs with an amino glucosamine as a hydrophilic moiety and a cinnamamide fragment as a hydrophobic portion, differently connected to each other. Nine target compounds 4 were synthesized by this approach which revealed very good inhibition values towards CA II and IX isozymes with respect to their corresponding single-tailed compounds, thus proving the validity of the dual-tail approach.15 Nevertheless, recent X-ray crystallographic studies16 led to a more satisfactory knowledge of the active site composition for each isoform (except for CAs VA and VB) revealing that various CA isoforms do not present an exact distinction between hydrophobic and hydrophilic moieties because of many accessory pockets existing in each hCA isoform. Therefore, the simple hydrophobic/hydrophilic division of the isoform binding pocket may be inadequate. Following these data, Bonardi et al. reported the synthesis of 32 benzenesulfonamide derivatives incorporating three tails (5 types, Fig. 1) to increase the matching of the target–ligand interaction within the different hCA active sites.17 They found that the introduction of a third tail changed the inhibition profiles in terms of potency and selectivity of action. In particular, the presence of three lipophilic tails in the 5 structures did not lead to significant advantages, while a great variability of potency and selectivity was observed by increasing the polarity of at least one tail. In the general structure of 5, T1 is an aromatic moiety (e.g. phenyl, furyl, or naphthyl), T2 can be an alkyl or an aryl group bearing two to eight carbon atoms, while T3 is an alkyl or aryl moiety that may contain a more polar group (e.g. cyano, amino or carboxylic acid). As a result, they demonstrated through X-ray crystallography studies and in silico tools that bulky 5 derivatives occupied the binding cavities with a great variability among the isoforms, thus contributing to the development of improved selectivity of action.17
With these premises, we proposed to synthesize a series of benzenesulfonamides bearing multiple hydrophilic tails and screen them as CA inhibitors. Given our experience, we focused on the synthesis of three-tailed compounds containing the sulfonamide group as a zinc binding function and sugars or iminosugars as polar tails (Scheme 1). The synthesized compounds were screened against many different hCAs and compared with the corresponding single-tailed compounds. The results, as well as considerations on the differences in the behaviour of iminosugar vs. sugar moieties, are reported and discussed in this work.
 | ||
| Scheme 1 Aim of the work. | ||
Results and discussion
The synthesis of sugar- and azasugar-linked azides or amines for coupling with benzenesulfonamides functionalized with alkyne or isothiocyanate moieties, respectively, was planned.We started with the synthesis of derivative 7 with a relatively short linker between the sulfonamide and the sugar moiety, since in our previous studies12 we did not find favourable interaction of a long linker within the active site. Thus, in this work, we wanted to bring the sulfonamide closer to the sugar portion. Azide 7 was first obtained from D-glucose in 16% overall yield by following a five step procedure reported in the literature (route I, Scheme 2).18 Alternatively, the same azide 7 was obtained by performing the glycosylation reaction directly on β-D-glucose pentaacetate 6 which reacted with 2-chloroethanol in the presence of BF3·OEt2 and 3 Å molecular sieves in dry CH2Cl2 to give the corresponding 8 in 60% yield (route II, Scheme 2). The treatment of 8 with NaN3 in DMF at 50 °C for three days furnished 7 in 87% yield. With this synthetic strategy (route II), azide 7 was obtained with fewer steps and a higher overall yield (38%, Scheme 2).
 | ||
| Scheme 2 (a) Ac2O, NaOAc, reflux, 74%; (b) 1,2-diaminoethane, AcOH, dry THF, rt, 7 h, 86%; (c) Cl3CCN, DBU, dry CH2Cl2, 0 °C to rt, 5 h, 65%; (d) 2-chloroethanol, BF3·OEt2, 3 Å MS, dry CH2Cl2, – 76 °C, 2 h, 51%; (e) NaN3, dry DMF, 50 °C, 3 d, 87%; (f) 2-chloroethanol, BF3·OEt2, 3 Å MS, dry CH2Cl2, 0 °C to rt, 4 h, 60%. | ||
Initial attempts to obtain benzenesulfonamide thiourea 10 by the copper catalyzed azide–alkyne cycloaddition (CuAAC) reaction between sugar derived azide 7 and alkyne 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 19 were unsuccessful (Scheme 3).
19 were unsuccessful (Scheme 3).
 | ||
| Scheme 3 Initial attempts to obtain 10. | ||
Despite several attempts, varying the reaction time, temperature, reductant/catalyst ratio and solvent mixture (from THF/water to water/t-BuOH), only trace amounts (<10%) of 10 were collected, with the recovery of azide 7. We wondered whether the presence of a preformed thiourea bond might be responsible for the failure of the CuAAC reaction. However, CuAAC reactions are reported in the literature,20 where a substituted thiourea moiety acts both as a reductant and a ligand. We then modified our synthetic strategy by performing the CuAAC first and successively the amine–isothiocyanate coupling.
The CuAAC reaction8 of 7 with propargylamine in the presence of TBTA (tris((1-benzyl-4-triazolyl)methyl)amine), CuSO4 and sodium ascorbate yielded amine 11 in 38% yield (Scheme 4), after treatment with the copper scavenger resin Quadrasil MP® and purification by flash column chromatography (FCC). The TBTA ligand is necessary because it stabilizes the copper(I)-oxidation state ensuring that it does not complex the propargylamine during the reaction, while the Quadrasil MP® resin allows the removal of traces of copper residues from the reaction crude product. Subsequently, 11 was reacted with benzenesulfonamide isothiocyanate 13 and NEt3 in CH3CN and EtOH to afford the single-tailed thiourea 10 in 53% yield (Scheme 4). Deacetylation of 10 with MeONa in MeOH followed by treatment with the acid resin Amberlyst 15 gave 14 in 57% yield (Scheme 4).
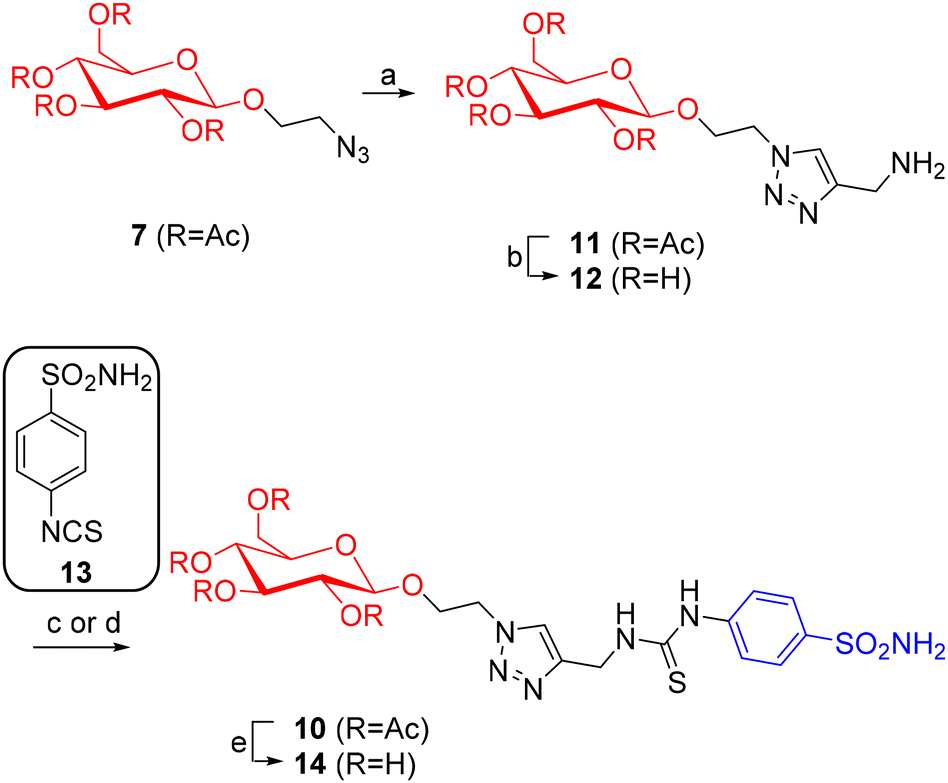 | ||
| Scheme 4 (a) Propargylamine, CuSO4, TBTA, sodium ascorbate, THF:H2O 2:1, rt, 15 h, 38%; (b) Na2CO3, MeOH, rt, 18 h, quantitative; (c) starting from 11, reaction with sulfonamide 13, NEt3, CH3CN:EtOH 1:1, 80 °C, 18 h, 53%; (d) starting from 12, reaction with sulfonamide 13, EtOH, rt, 20 h, 14%; (e) MeONa, MeOH, rt, 2 h, 57%. | ||
Performing the two steps for obtaining 14 from 11 the other way around, that is, first deprotection to 12 and then coupling with the sulfonamide 13, was less efficient (14% overall yield, Scheme 4). While deacetylation of 11 to 12 occurred quantitatively, the following coupling was unsatisfactory, likely due to a sluggish reaction under the conditions required by hydrophilic 12 (EtOH at room temperature).
With the single-tailed 14 in hand, the synthesis of the corresponding triple-tailed benzenesulfonamide 20 was attempted by employing the same synthetic strategy. The trivalent amine 16 was obtained from tris(hydroxymethyl)aminomethane 15 in three steps with a 25% overall yield.21 The CuAAC reaction of 16 with the sugar-derived azide 7 in THF/H2O with CuSO4 and sodium ascorbate under MW irradiation at 80 °C yielded the functionalized amine 17 (89%, Scheme 5), after treatment with the copper scavenger resin Quadrasil MP® and purification by FCC. The reaction of 17 with isothiocyanate 13 in the presence of NEt3 in CH3CN/EtOH provided the triple-tailed benzenesulfonamide 19 in a moderate 36% yield, which can be ascribed to the bulkiness of amine 17. Deprotection of 19 with Na2CO3 in MeOH led to the final triple-tailed compound 20 in 68% yield (Scheme 5). As previously mentioned, the reaction of unprotected 18 with sulfonamide 13 yielded compound 20 in a much lower yield (17%) due to poor solubility in the solvent mixture.
 | ||
| Scheme 5 (a) Boc2O, t-BuOH/MeOH, rt, 18 h, 74%; (b) propargyl bromide, KOH, DMF, 0 °C to 35 °C, 24 h, 35%; (c) TFA, dry CH2Cl2, rt, 2 h, 95%; (d) 7, CuSO4, sodium ascorbate, THF:H2O 2:1, MW 80 °C, 1 h 45 min, 89%; (e) Ambersep 900-OH, MeOH, rt, 16 h, 96%; (f) from 17, reaction with sulfonamide 13, CH3CN:EtOH 1:1, 80 °C, 5 d, 36%; (g) from 18, reaction with sulfonamide 13, EtOH, MeOH, 50 °C, 20 h, 17%; (h) Na2CO3, MeOH, rt, 18 h, 68%. | ||
Since the triple-tailed compounds 19 and 20 resulted in much poorer inhibitors than 10 and 14 (see biological results), the synthesis of congeners 25 and 26 (Scheme 6) was planned, speculating that spacing the sulfonamide moiety from the polar tails would allow it to enter the active site of hCAs more easily. The trivalent alkyne 22 was obtained in 61% yield by coupling 16 with the protected amino acid 21, which was carried out in the presence of 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloride (EDC), 1-hydroxybenzotriazole (HOBt) and diisopropylethylamine (DIPEA) in CH2Cl2 at room temperature. A CuAAC reaction of 22 with the sugar-derived azide 7 under MW irradiation at 80 °C gave the protected amine 23 in 79% yield (Scheme 6), after treatment with the copper scavenger resin Quadrasil MP® and purification by FCC. Deprotection of 23 in trifluoroacetic acid (TFA) furnished the free amine 24 (94%), which was added to benzenesulfonamide 13 in the presence of NEt3 in CH3CN/EtOH to afford the three-tailed compound 25 in 89% yield, after treatment over Sephadex LH-20 resin and purification by FCC. The final hydrolysis of acetates, performed with Na2CO3 in MeOH in order to prevent hydrolysis of amide which might occur under more basic conditions, gave the desired benzenesulfonamide 26 quantitatively (Scheme 6).
 | ||
| Scheme 6 (a) EDC·HCl, HOBt, DIPEA, CH2Cl2, rt, 3 d, 61%; (b) 7, CuSO4, sodium ascorbate, THF:H2O 2:1, MW 80 °C, 1 h 15 min, 79%; (c) TFA, rt, 2 h, 94%; (d) NEt3, CH3CN:EtOH 1:1, 80 °C 4 d, 89%; (e) Na2CO3, MeOH, rt, 18 h, quantitative. | ||
With the sugar derivatives in hand, our attention was focused on the synthesis of related iminosugar-linked benzenesulfonamides, in order to investigate the role of a sugar mimetic in the potency and selectivity of the inhibitors towards the different isoforms of hCAs.
3,4,5-Trihydroxypiperidine iminosugars recently showed interesting biological properties.22
Azide 28,23 the precursor of all the new compounds, was synthesized from aldehyde 27, derived in turn from inexpensive D-mannose in five steps (85% overall yield), through a double reductive amination procedure (DRA) as reported (Scheme 7).24 Compound 28 was selected for having a linker between the azide and the iminosugar moiety roughly of the same length as in the sugar derivative 7. A CuAAC reaction of 28 with benzenesulfonamide 30 bearing an alkyne moiety in the presence of CuSO4 and sodium ascorbate yielded the protected compound 31 in 91% yield (Scheme 7), after treatment with the copper scavenger resin Quadrasil MP® and purification by FCC. Concurrently, deprotection of azide 28 to trihydroxypiperidine 29,23 followed by CuAAC with benzenesulfonamide 30 afforded the corresponding deprotected 32 in 70% yield after treatment with the copper scavenger resin Quadrasil MP® and purification by FCC.
 | ||
| Scheme 7 (a) 3-Azido-1-propanamine, NaBH3CN, CH3COOH, dry MeOH, rt, 7 d, 64%; (b) HCl 12 M, MeOH, rt, 18 h, 93%; (c) CuSO4, sodium ascorbate, THF/H2O 2:1, rt, 18 h, 91% from 28, 70% from 29. | ||
Triazole-iminosugar 34, prepared from azide 28 through the protected intermediate 33 according to the literature25 (Scheme 8), was reacted with benzenesulfonamide 13 in MeOH/EtOH at 50 °C to give thiourea 35 in 15% yield (Scheme 8). Again, the high hydrophilicity of 34 which is not soluble in the typical solvents used for the coupling reaction may be responsible for the low yield of the product.
 | ||
| Scheme 8 (a) Propargylamine, CuSO4, sodium ascorbate, THF/H2O 2:1, rt, 16 h, 96%; (b) HCl 1 M, MeOH, rt, 16 h, DOWEX 50WX8-200, 77%; (c) EtOH, MeOH, 50 °C, 20 h, 15%. | ||
A more satisfactory synthetic route to 35 was developed starting from azide 36, obtained by acetylation of 29 in 90% yield (Scheme 9). The direct CuAAC reaction of 36 with propargylamine in the presence of TBTA gave only traces of the desired triazole 38. Thus, a two-step procedure was employed, reacting 36 with N-Cbz-propargylamine in the presence of CuSO4 and sodium ascorbate in THF/H2O (Scheme 9). The treatment of the resulting protected amine 37 (54%) under an H2 atmosphere in the presence of Pd(OH)2/C led to the free amine 38 in 93% yield. A reaction with benzenesulfonamide 13 in the presence of NEt3 in CH3CN/EtOH at 80 °C afforded the protected compound 39 (59%), which was deacetylated with Na2CO3 in MeOH to the final benzenesulfonamide 35 in 38% yield. This strategy, besides increasing the yield of 35, also allowed access to the corresponding triacetylated 39 to be compared in the biological assays.
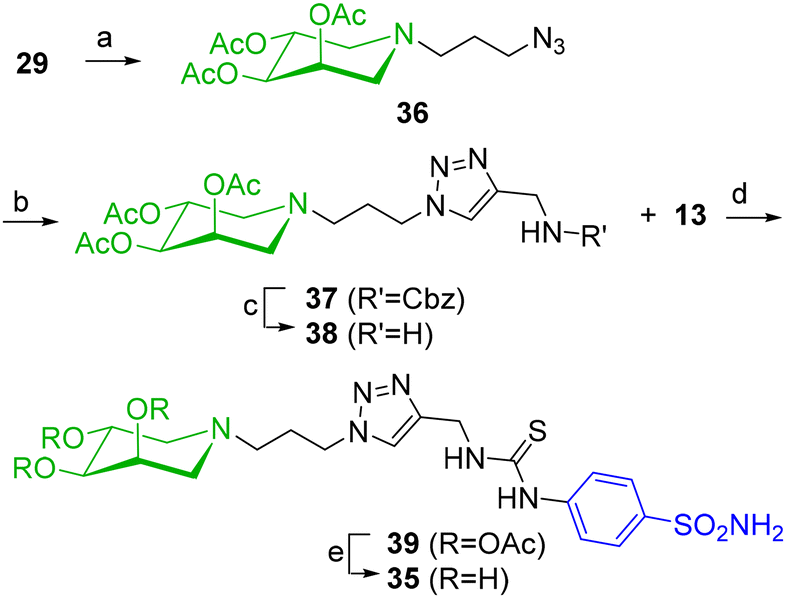 | ||
| Scheme 9 (a) Ac2O, dry pyridine, rt, 18 h, 90%; (b) N-Cbz-propargylamine, CuSO4, sodium ascorbate, THF/H2O 2:1, rt, 18 h, 54%; (c) H2, Pd(OH)2/C, MeOH, rt, 18 h, 93%; (d) NEt3, CH3CN:EtOH, 80 °C, 18 h, 59%; (e) Na2CO3, MeOH, rt, 18 h, 38%. | ||
The synthesis of three-tailed iminosugar benzenesulfonamide analogues was then performed. Compound 43, an analogue of 26 with the iminosugar moieties replacing the sugar ones, was chosen as the target, taking into account the better biological profile of 26 (the three-tailed compound with a longer linker between the sulfonamide moiety and the sugar tails) with respect to 20.
Starting from the acetonide-protected trihydroxypiperidine 28, the target three-tailed benzenesulfonamide 43 was recovered only in traces.
Much better results were obtained by starting with the azido-armed fully acetylated iminosugar 36. Its CuAAC reaction with the trialkyne scaffold 22 in the presence of CuSO4 and sodium ascorbate under MW irradiation at 80 °C yielded the protected compound 40 (96%, after treatment with the copper scavenger resin Quadrasil MP® and purification by FCC, Scheme 10). Deprotection of carbamate with TFA in dry CH2Cl2 furnished the free amine 41 (92%), which was reacted with benzenesulfonamide 13 in the presence of NEt3 in CH3CN/EtOH to give the protected compound 42 in 68% yield. Final deacetylation with Na2CO3 in MeOH and passage over Bio-Beads_SX8 resin gave the triple-tailed 43 in 55% yield (Scheme 10).
 | ||
| Scheme 10 (a) CuSO4, sodium ascorbate, THF/H2O 2:1, MW 80 °C, 1 h, 96%; (b) TFA, dry CH2Cl2, rt, 2 h, 92%; (c) NEt3, CH3CN, EtOH, 80 °C, 3 d, 68%; (d) Na2CO3, MeOH, rt, 18 h, 55%. | ||
The newly synthesized single-tailed sugar (10 and 14) and iminosugar (31, 32, 35, and 39), and triple-tailed sugar (19, 20, 25, and 26) and iminosugar (42 and 43) benzenesulfonamides were tested against different isoforms of hCAs (I, II, III, IV, VA, VB, VI, VII, IX, XII and XIII) using a Stopped Flow CO2 Hydrase assay26 in order to evaluate the role of multivalent presentation of the sugar/iminosugar compared to the corresponding monovalent compounds and also to explore the role of a sugar mimetic (iminosugar) in the selectivity of the inhibitor towards the different isoforms of hCAs. Acetazolamide (AAZ) was used as a reference.27 The inhibition data are reported in Table 1.
| a Mean from three different assays, by a stopped flow technique (errors were in the range of ±5–10% of the reported values). |
|---|

|
The investigated compounds showed weak inhibitory properties against the cytosolic isoforms hCAs I and II (Ki > 200 nM), with the exception of 10, which strongly inhibited hCA I with Ki = 65.2 nM (3.83 times lower than AAZ) and hCA II with Ki = 5.7 nM (2.12 times lower than AAZ). This single-tailed protected sugar 10 also showed powerful inhibition of hCA IX (Ki = 5.1 nM), i.e., 5.06 times lower than that of AAZ. In general, the introduction of an iminosugar moiety did not increase the inhibition with respect to the reference compound AAZ. However, the protected three-tailed sugar compound 25 and the single-tailed iminosugar 31 showed very good selectivity towards hCA VII with low inhibition constant values (Ki = 9.7 nM for both compounds). The deprotected compound 26 featuring three sugar tails is a 1.58 times better inhibitor than AAZ towards hCA VB (Ki = 34.2 nM). Concerning the differences among protected and deprotected compounds in the inhibition of hCAs, 10 was found to be a better inhibitor than 14 mainly on hCAs I, II and IX (Ki = 65.2, 5.7 and 5.1 nM vs. 312.8, 205.2 and 45.8 nM). The three-tailed compounds 19 and 20 with a shorter linker between the sulfonamide group and the sugar moieties are weak to moderate inhibitors against all the hCAs (Ki = 70.3–9595 nM). Probably, the more hindered compounds 19 and 20 experience adverse steric effects when the sulfonamide moiety approaches the hCA active site. However, the protected 19 showed good selectivity for hCA VII (Ki = 70.3 nM), the isoform of interest for the treatment of neuropathic pain. As a matter of fact, the sugar three-tailed compounds 25 and 26 bearing a longer linker between the sulfonamide and the tails resulted in better inhibitors of hCA VII (Ki = 9.7 and 30.0 nM, respectively) with respect to 19 and 20. While acetylated 25 also inhibited the tumour associated hCA XII isoform (Ki = 31.0 nM), deprotected 26 was a 1.58 times stronger inhibitor of hCA VB (Ki = 34.2 nM) as compared to AAZ. Concerning the single-tailed iminosugar compounds 31, 32, 39 and 35, they all inhibited the tumour associated hCA XII with Ki = 21.7–62.1 nM. The protected 31 showed strong inhibition and selectivity (Ki = 9.7 nM) towards hCA VII, in contrast to deprotected 32. Peracetylated 39 showed good inhibition (Ki = 47.6 nM) of hCA IX, while the corresponding deacetylated 35 showed great selectivity towards hCA XII (Ki = 21.7 nM). The three-tailed iminosugar derivatives neither showed impressive levels of inhibition nor particular selectivity. It is worth noting the inverse behaviour of deprotected 43 and its corresponding peracetylated 42 towards hCA VA (43, Ki = 96.5 nM; 42, Ki = 906.1 nM) and hCA VI (43, Ki = 211.9 nM; 42, Ki = 80.4 nM).
According to the data presented in Table 1, compound 31 exhibited weak inhibition against hCA II. To investigate the molecular basis of CA inhibition using this derivative, we used X-ray crystallography to determine its complex with hCA II at a resolution of 1.3 Å (Fig. 2).
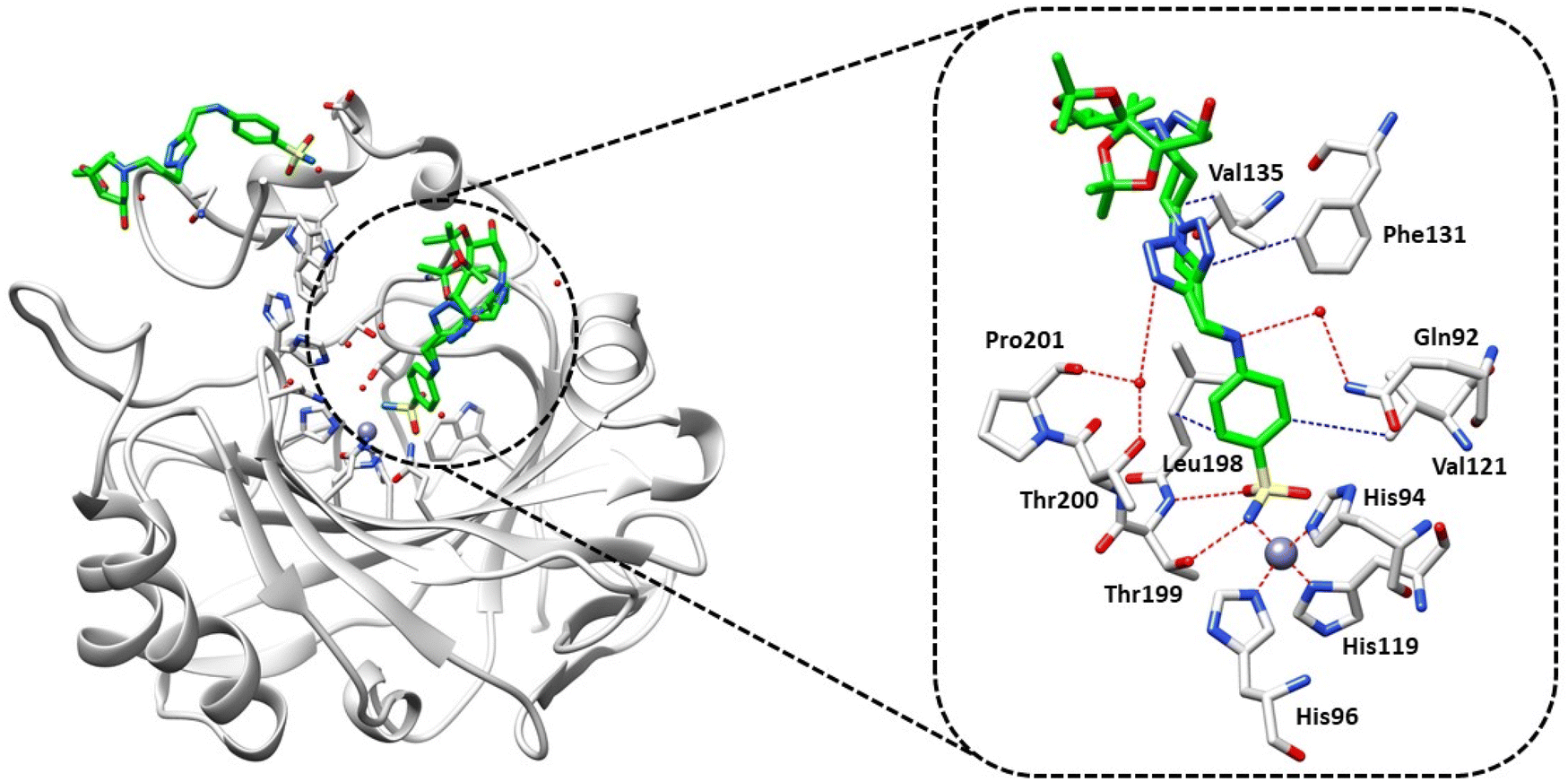 | ||
| Fig. 2 X-ray crystal structure of hCA II bound with compound 31 (PDB: 8OGF). Residues involved in the binding of inhibitors are also shown; the grey sphere represents the zinc ion in the active site of the proteins. | ||
Although 31 is not a very potent hCA II inhibitor, we chose this derivative for crystallographic experiments because in many cases this isoform may be not a drug target but an off-target. There are in fact hundreds of highly potent hCA II inhibitors for which the crystal structure of adducts with this isoform has been reported,3,13 but very few weak inhibitors were characterized by this technique. Understanding the factors that are connected with less efficient hCA II inhibition can in fact be useful for drug design purposes.
Complex hCAII/31 revealed two molecules bound to the protein, with one located inside the active site responsible for the activity, and the other bound in a cleft (N-terminal) and not involved in the mechanism of inhibition, as shown in Fig. 2; this finding is in agreement with previous reports from some of us.28,29
Analysis of the electron density maps (Fig. S1 in the ESI†) revealed a clear density for the portion of the inhibitor which includes the benzenesulfonamide group and the triazole ring, confirming its binding to the enzyme. However, the triazole ring showed double conformation leading probably to the disorder of the tail that resulted in a poor electron density map, so that zero occupancy was attributed to the atoms forming the inhibitor tail. Compound 31 showed the classical binding mode of sulfonamide inhibitors by directly interacting with the zinc ion and forming a hydrogen bond with Thr199. The benzenesulfonamide moiety established hydrophobic interactions with the side chains of Val121 and Leu198, further stabilizing the complex within the active site. Additionally, a water bridge was observed between Gln92 and the secondary amine of the aminobenzenesulfonamide portion. One of the conformations of the triazole ring formed a water bridge with Thr200 and Pro201, while hydrophobic interactions were observed with Phe131. However, the second conformation lacked these interactions, providing a possible explanation for the weak potency of this inhibitor against hCA II.
Experimental
General methods
Commercial reagents were used as received. All reactions were carried out under magnetic stirring and monitored by TLC on 0.25 mm silica gel plates (Merck F254). Column chromatography was carried out on Silica Gel 60 (32–63 μm) or on silica gel (230–400 mesh, Merck). Yields refer to spectroscopically and analytically pure compounds unless otherwise stated. 1H-NMR and 13C-NMR spectra were recorded on a Varian Gemini 200 MHz, a Varian Mercury 400 MHz or on a Varian INOVA 400 MHz instrument at 25 °C. Chemical shifts are reported relative to CDCl3 (13C: δ = 77.0 ppm, 1H: 7.26 ppm), or CD3OD (13C: δ = 49.0 ppm, 1H: 3.31 ppm). Integrals are in accordance with assignments, coupling constants are given in Hz. For detailed peak assignments 2D spectra were measured (COSY, HSQC). Small scale microwave assisted syntheses were carried out in a microwave apparatus for synthesis (CEM Discover) with an open reaction vessel and external surface sensor. IR spectra were recorded with an IRAffinity-1S SHIMADZU system spectrophotometer. Optical rotation measurements were performed on a JASCO DIP-370 polarimeter. ESI-MS spectra were recorded with a Thermo Scientific™ LCQ fleet ion trap mass spectrometer. Elemental analyses were performed with a Thermoscientific FlashSmart Elemental Analyzer CHNS/O.Only the synthesis and characterization of the most relevant compounds are described in this section. For the other compounds described in this work, see the ESI.† For practical reasons, the assignment of H and C atoms in NMR characterization studies reflects the numbering of chemical structures in the ESI.†
Synthesis of compound 10
Benzenesulfonamide 13 (20 mg, 0.09 mmol) and NEt3 (6 μl, 0.04 mmol) were added to a solution of 11 (40 mg, 0.09 mmol) in CH3CN (1 ml) and EtOH (1 mL) and the mixture was stirred at 80 °C for 15 hours until a TLC control attested the disappearance of the starting material 11 (Rf = 0.23, CH2Cl2:MeOH:NH4OH (6%) 10:1:0.1). Then, the crude product was evaporated under vacuum and purified by flash column chromatography on silica gel (CH2Cl2:MeOH:NH4OH (6%) 25:1:0.1) to give 31 mg of 10 (Rf = 0.39, CH2Cl2:MeOH:NH4OH (6%) 10:1:0.1, 0.05 mmol, 53%) as pale yellow oil.
10: [α]20D = −12.50 (c = 0.75, MeOH). 1H-NMR (400 MHz, CD3OD) δ ppm: 7.97 (s, 1H, triazole), 7.84 (d, J = 8.8 Hz, AB system, 2H, Ar), 7.69 (d, J = 8.7 Hz, AB system, 2H, Ar), 5.23 (t, J = 9.5 Hz, 1H, H-3), 5.00 (t, J = 9.8 Hz, 1H, H-4), 4.89 (br s, 2H, H-9), 4.87–4.84 (m, 1H, H-2), 4.68 (d, J = 7.8 Hz, 1H, H-1), 4.60 (t, J = 5.2 Hz, 2H, H-8), 4.26 (dd, J = 4.7, 12.4 Hz, 1H, Ha-6), 4.22–4.16 (m, 1H, Ha-7), 4.15–4.10 (m, 1H, Hb-6), 4.02 (quint, J = 5.3 Hz, 1H, Hb-7), 3.89–3.84 (m, 1H, H-5), 2.05 (s, 3H, OAc), 2.00 (s, 3H, OAc), 1.96 (s, 3H, OAc), 1.95 (s, 3H, OAc). 13C-NMR (50 MHz, CD3OD) δ ppm: 182.8 (s, 1C, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S), 172.3 (s, 1C, CO), 171.6 (s, 1C, CO), 171.3 (s, 1C, CO), 171.2 (s, 1C, CO), 145.7 (s, 1C, triazole), 144.1 (s, 1C, Ar), 140.2 (s, 1C, Ar), 128.0 (d, 2C, Ar), 125.4 (d, 1C, triazole), 123.7 (d, 2C, Ar), 101.6 (d, 1C, C-1), 74.1 (d, 1C, C-3), 72.9 (d, 1C, C-5), 72.6 (d, 1C, C-2), 69.8 (d, 1C, C-4), 69.0 (t, 1C, C-7), 63.0 (t, 1C, C-6), 51.4 (t, 1C, C-8), 40.5 (t, 1C, C-9), 20.7 (q, 2C, CH3) 20.6 (q, 2C, CH3). IR (CDCl3): ṽ = 3343, 3034, 2959, 1755, 1531, 1365, 1333, 1203, 1163, 1056, 1042 cm−1. MS-ESI (m/z, %) = 709.08 (100) [M + Na]+. C26H34N6O12S2 (686.71): calcd C, 45.47; H, 4.99; N, 12.24; S, 9.18. found C, 45.10; H, 5.28; N, 12.30; S, 9.25.
S), 172.3 (s, 1C, CO), 171.6 (s, 1C, CO), 171.3 (s, 1C, CO), 171.2 (s, 1C, CO), 145.7 (s, 1C, triazole), 144.1 (s, 1C, Ar), 140.2 (s, 1C, Ar), 128.0 (d, 2C, Ar), 125.4 (d, 1C, triazole), 123.7 (d, 2C, Ar), 101.6 (d, 1C, C-1), 74.1 (d, 1C, C-3), 72.9 (d, 1C, C-5), 72.6 (d, 1C, C-2), 69.8 (d, 1C, C-4), 69.0 (t, 1C, C-7), 63.0 (t, 1C, C-6), 51.4 (t, 1C, C-8), 40.5 (t, 1C, C-9), 20.7 (q, 2C, CH3) 20.6 (q, 2C, CH3). IR (CDCl3): ṽ = 3343, 3034, 2959, 1755, 1531, 1365, 1333, 1203, 1163, 1056, 1042 cm−1. MS-ESI (m/z, %) = 709.08 (100) [M + Na]+. C26H34N6O12S2 (686.71): calcd C, 45.47; H, 4.99; N, 12.24; S, 9.18. found C, 45.10; H, 5.28; N, 12.30; S, 9.25.
Synthesis of compound 25
Benzenesulfonamide 13 (18 mg, 0.08 mmol) was added to a solution of 24 (64 mg, 0.04 mmol) and NEt3 (6 μL, 0.04 mmol) in EtOH (1 mL) and CH3CN (1 mL) and the mixture was stirred at 80 °C for 4 days until a TLC control attested the disappearance of the starting material 24 (Rf = 0.27, CH2Cl2:MeOH 10:1). Then, the reaction mixture was evaporated under vacuum. The crude product was purified by size exclusion chromatography by employing Sephadex LH-20® resin and eluting with MeOH and then by flash column chromatography on silica gel (CH2Cl2:MeOH 8:1) to give 65 mg of 25 (Rf = 0.46, CH2Cl2:MeOH 8:1, 0.04 mmol, 89%) as a white waxy solid.
25: [α]22d = −10.90 (c = 0.8, CHCl3). 1H-NMR (400 MHz, CD3OD) δ ppm: 7.95–7.81 (m, 5H, triazole, Ar), 7.70–7.61 (m, 2H, Ar), 5.24 (t, J = 9.5 Hz, 3H, H-3), 5.01 (t, J = 9.6 Hz, 3H, H-4), 4.88–4.84 (m, 3H, H-2), 4.71 (d, J = 8 Hz, 3H, H-1), 4.65–4.51 (m, 12H, H-8, H-9), 4.28 (dd, J = 4.4, 12.4 Hz, 3H, Ha-6), 4.25–4.17 (m, 3H, Ha-7), 4.14 (d, J = 12.0 Hz, 3H, Hb-6), 4.05–3.95 (m, 3H, Hb-7), 3.93–3.85 (m, 3H, H-5), 3.85–3.74 (m, 6H, H-10), 3.66–3.54 (m, 2H, H-13), 2.32–2.23 (m, 2H, H-11), 2.05 (s, 9H, OAc), 2.00 (s, 9H, OAc), 1.98–1.84 (m, 20H, OAc, H-12). 13C-NMR (50 MHz, CD3OD) δ ppm: 182.1 (s, 1C, CS), 175.6 (s, 1C, HN-CO), 172.3 (s, 3C, O![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) CH3), 171.6 (s, 3C, OCH3), 171.3 (s, 3C, OCH3), 171.1 (s, 3C, OCH3), 145.8 (s, 3C, triazole), 143.9 (s, 1C, Ar), 140.1 (s, 1C, Ar), 128.1 (d, 2C, Ar), 125.7 (d, 3C, triazole), 123.8 (d, 2C, Ar), 101.6 (d, 3C, C-1), 74.1 (d, 1C, C-3), 73.0 (d, 3C, C-5), 72.5 (d, 3C, C-2), 69.8 (t, 3C, C-10), 69.6 (d, 3C, C-4), 69.0 (t, 3C, C-7), 65.4 (t, 3C, C-8), 63.1 (t, 3C, C-6), 61.4 (s, 1C, HN(CH2O)3-), 51.3 (t, 3C, C-9), 45.0 (t, 1C, C-13), 34.9 (t, 1C, C-11), 26.2 (t, 1C, C-12), 20.8 (q, 6C, OAc), 20.6 (q, 6C, OAc). IR (CDCl3) ṽ = 3405, 3010, 2960, 2912, 1750, 1532, 1460, 1368, 1230, 1160, 1038 cm−1. MS-ESI (m/z, %) = 915.84 (100) [M + 2Na]2+, 1807.87 (48) [M + Na]+. C72H99N13O36S2 (1785.58): calcd C, 48.40; H, 5.58; N, 10.19; S, 3.59; found C, 48.28; H, 5.75; N, 10.10; S, 3.65.
CH3), 171.6 (s, 3C, OCH3), 171.3 (s, 3C, OCH3), 171.1 (s, 3C, OCH3), 145.8 (s, 3C, triazole), 143.9 (s, 1C, Ar), 140.1 (s, 1C, Ar), 128.1 (d, 2C, Ar), 125.7 (d, 3C, triazole), 123.8 (d, 2C, Ar), 101.6 (d, 3C, C-1), 74.1 (d, 1C, C-3), 73.0 (d, 3C, C-5), 72.5 (d, 3C, C-2), 69.8 (t, 3C, C-10), 69.6 (d, 3C, C-4), 69.0 (t, 3C, C-7), 65.4 (t, 3C, C-8), 63.1 (t, 3C, C-6), 61.4 (s, 1C, HN(CH2O)3-), 51.3 (t, 3C, C-9), 45.0 (t, 1C, C-13), 34.9 (t, 1C, C-11), 26.2 (t, 1C, C-12), 20.8 (q, 6C, OAc), 20.6 (q, 6C, OAc). IR (CDCl3) ṽ = 3405, 3010, 2960, 2912, 1750, 1532, 1460, 1368, 1230, 1160, 1038 cm−1. MS-ESI (m/z, %) = 915.84 (100) [M + 2Na]2+, 1807.87 (48) [M + Na]+. C72H99N13O36S2 (1785.58): calcd C, 48.40; H, 5.58; N, 10.19; S, 3.59; found C, 48.28; H, 5.75; N, 10.10; S, 3.65.
Synthesis of compound 26
Sodium carbonate (4 mg, 0.04 mmol) was added to a solution of 25 (15 mg, 0.01 mmol) in MeOH (1 mL) and the mixture was stirred at room temperature for 18 hours. The mixture was filtered and the solvent was removed under vacuum to give 10 mg of 26 (Rf = 0.06, CH2Cl2:MeOH:NH4OH (6%) 10:1:0.1, 0.01 mmol, quantitative) as a white waxy solid.
26: [α]24D = −3.25 (c = 0.4, MeOH). 1H-NMR (400 MHz, D2O) δ ppm: 7.97 (s, 3H, triazole), 7.79 (d, J = 8.2 Hz, AB system, 2H, Ar), 7.35 (d, J = 8.4 Hz, AB system, 2H, Ar), 4.61–4.53 (m, 6H, H-8), 4.50 (s, 6H, H-9), 4.35 (d, J = 7.8 Hz, 3H, H-1), 4.25–4.16 (m, 3H, Ha-7), 4.05–3.96 (m, 3H, Hb-7), 4.30 (d, J = 12.0 Hz, 3H, Ha-6), 3.68–3.57 (m, 9H, Hb-6, H-10), 3.49–3.31 (m, 8H, H-3, H-4, H-13), 3.30–3.23 (m, 3H, H-5), 3.16 (t, J = 8.8 Hz, 3H, H-2), 2.19 (br s, 2H, H-11), 1.75 (br s, 2H, H-12). 13C-NMR (50 MHz, D2O) δ ppm: 175.6 (s, 1C, CS), 163.5 (s, 1C, HN-CO), 143.8 (s, 3C, triazole), 141.0 (s, 2C, Ar), 126.7 (d, 2C, Ar), 125.6 (d, 3C, triazole), 125.0 (d, 2C, Ar), 102.4 (d, 3C, C-1), 75.9 (d, 3C, C-3), 75.6 (d, 3C, C-4), 72.9 (d, 3C, C-2), 69.5 (d, 3C, C-5), 68.0 (t, 3C, C-7), 67.5 (t, 3C, C-10), 63.5 (t, 3C, C-9), 60.7 (t, 3C, C-6), 59.7 (s, 1C, HN(CH2O)3-), 50.3 (t, 3C, C-8), 43.8 (t, 1C, C-13), 33.1 (t, 1C, C-11), 24.1 (t, 1C, C-12). MS-ESI (m/z, %) = 639.58 (100) [M − 2H]2−. C48H75N13O24S2 (1282.31): calcd C, 44.96; H, 5.90; N, 14.20; S, 5.00; found C, 44.70; H, 6.16; N, 14.30; S, 5.26.
Synthesis of compound 31
Sulfonamide 30 (46 mg, 0.22 mmol), CuSO4 (10 mg, 0.06 mmol) and sodium ascorbate (24 mg, 0.12 mmol) were added to a solution of azide 28 (51 mg, 0.20 mmol) in THF (2 mL) and milliQ water (1 mL). The reaction mixture was stirred at room temperature for 18 hours, until a TLC control attested the disappearance of the starting material 28 (Rf = 0.39, CH2Cl2:MeOH:NH4OH (6%) 10:1:0.1). The mixture was filtered through Celite®, the solvent was removed under vacuum and subsequently the crude product was treated with the “Quadrasil MP®” resin keeping the mixture under stirring at room temperature in the minimum amount of MeOH for 1 hour (1 g of resin for each mmol of copper). The crude product was purified by flash column chromatography on silica gel (gradient eluent from CH2Cl2:MeOH:NH4OH (6%) 15:1:0.1 to 10:1:0.1) to give 85 mg of 31 (Rf = 0.13, CH2Cl2:MeOH:NH4OH (6%) 15:1:0.1, 0.18 mmol, 91%) as a white waxy solid.
31: [α]22D = −6.10 (c = 1.00, MeOH). 1H-NMR (400 MHz, CD3OD) δ ppm: 7.89 (s, 1H, triazole), 7.61 (d, J = 7.2 Hz, AB system, 2H, Ar), 6.68 (d, J = 7.4 Hz, AB system, 2H, Ar), 4.45 (s, 2H, 10), 4.42 (t, J = 6.4 Hz, 2H, H-9), 4.25–4.19 (m, 1H, H-3), 3.86–3.81 (m, 1H, H-4), 3.81–3.74 (m, 1H, H-5), 2.76 (d, J = 12.6 Hz, 1H, Ha-2), 2.68–2.60 (m, 1H, Ha-6), 2.36 (dd, J = 3.2, 12.8 Hz, 1H, Hb-2), 2.33–2.16 (m, 2H, H-7), 2.10–1.92 (m, 3H, H-8, Hb-6), 1.46 (s, 3H, Me), 1.33 (s, 3H, Me). 13C-NMR (100 MHz, CD3OD) δ ppm: 152.6 (s, 1C, Ar), 146.9 (s, 1C, triazole), 131.4 (s, 1C, Ar), 128.4 (d, 2C, Ar), 124.6 (d, 1C, triazole), 112.8 (d, 2C, Ar), 110.1 (s, 1C, O(CH3)2), 80.0 (d, 1C, C-4), 74.4 (d, 1C, C-3), 70.5 (d, 1C, C-5), 57.1 (t, 1C, C-6), 55.0 (t, 1C, C-2), 54.6 (t, 1C, C-7), 49.8 (t, 1C, C-9), 39.4 (t, 1C, C-10), 28.5 (q, 1C, OOC(H3)2), 28.1 (t, 1C, C-8), 26.6 (q, 1C, OOC(H3)2). IR (CDCl3): ṽ = 3345, 2990, 2941, 2830, 2641, 2363, 2344, 2328, 2297, 2189, 1998, 1743, 1601, 1510, 1429, 1327, 1327, 1244, 1221, 1196, 1163, 1146 cm−1. MS-ESI (m/z, %) = 465.20 (93) [M − H]−, 930.72 (100) [2M − H]−. C20H30N6O5S (466.55): calcd C, 51.49; H, 6.48; N, 18.01; S, 6.87; found C, 51.28; H, 6.69; N, 17.92; S, 6.80.
Carbonic anhydrase inhibition
An applied photophysics stopped-flow instrument has been used for assaying CA-catalyzed CO2 hydration activity.26 Phenol red (at a concentration of 0.2 mM) was used as an indicator, working at the absorbance maximum of 557 nm, with 20 mM HEPES (pH 7.5) as buffer, and 20 mM Na2SO4 (for maintaining the ionic strength constant), following the initial rates of the CA-catalyzed CO2 hydration reaction for a period of 10–100 s. The CO2 concentrations ranged from 1.7 to 17 mM for the determination of the kinetic parameters and inhibition constants. For each inhibitor, at least six traces of the initial 5–10% of the reaction were used for determining the initial velocity. The uncatalyzed rates were determined in the same manner and subtracted from the total observed rates. Stock solutions of the inhibitor (0.1 mM) were prepared in distilled-deionized water and dilutions up to 0.01 nM were done thereafter with the assay buffer. Inhibitor and enzyme solutions were preincubated together for 15 min at room temperature prior to assay, in order to allow the formation of the E–I complex. The inhibition constants were obtained by non-linear least-squares method using PRISM 3 and the Cheng–Prusoff equation, as reported earlier30 and represent the mean from at least three different determinations. All CA isoforms were recombinant ones obtained in-house as reported earlier.30Crystallization and X-ray data collection
The crystals of hCA II were obtained using the hanging drop vapor diffusion method using a 24 well Linbro plate. 2 μl of 10 mg ml−1 solution of hCA II in Tris-HCl 20 mM pH 8.0 were mixed with 2 μl of a solution of 1.5 M sodium citrate and 0.1 M Tris pH 8.0 and were equilibrated against the same solution at 296 K. The complexes were prepared by soaking the hCA II native crystals in the mother liquor solution containing the inhibitors at a concentration of 10 mM for two days. All crystals were flash-frozen at 100 K using a solution obtained by adding 15% (v/v) glycerol to the mother liquor solution as a cryoprotectant. Data of the crystals of the complexes were collected using synchrotron radiation at the XRD2 beamline at Elettra Synchrotron (Trieste, Italy) with a wavelength of 1.000 Å and a DECTRIS Pilatus 6M detector. Data were integrated and scaled using the XDS program.31 Data processing statistics are shown in the ESI.†Structure determination
The crystal structure of hCA II (PDB accession code: 4FIK) without solvent molecules and other heteroatoms was used to obtain initial phases using Refmac5.32 5% of the unique reflections were selected randomly and excluded from the refinement data set for Rfree calculations. The initial |Fo–Fc| difference electron density maps unambiguously showed the inhibitor molecules. The inhibitor was introduced in the model with 1.0 occupancy. Refinements were performed using normal protocols of positional and anisotropic atomic displacement parameters alternating with the manual building of the models using COOT.33 The quality of the final models was assessed with COOT and RAMPAGE.34 Crystal parameters and refinement data are summarized in the ESI.† Atomic coordinates were deposited in the Protein Data Bank (PDB accession code: 8OGF). Graphical representations were generated with Chimera.35Conclusions
Twelve mono- and three-tailed derivatives based on a sugar (glucose) or a iminosugar (trihydroxy piperidine) containing a terminal benzenesulfonamide as the zinc binding function have been synthesized with the aim of developing potent and selective carbonic anhydrase inhibitors and collecting information on the behaviour of compounds bearing multiple hydrophilic chains. Among the investigated synthetic strategies, the preferred ones involved, in general, a CuAAC reaction followed by an amine–isothiocyanate coupling and final deprotection of the hydroxy groups. The novel compounds (two sugar single tailed, four iminosugar single tailed, four sugar triple tailed and two iminosugar triple tailed) were screened against eleven hCA isoforms. Among the isoforms screened in the biological assays, the following are particularly relevant for addressing biomedical problems: hCAs I, II and IV involved in some forms of glaucoma, hCAs VA and VB that are targets for antiobesity drugs, and hCA VII is implicated in neuropathic pain and in the development of anticonvulsant drugs. Moreover, hCAs IX and XII show close association with hypoxic tumours such as those of the lung, breast, colon, esophagus and cervix. The collected inhibition data do not indicate clear and general trends for the structure/activity relationship and suggest that information must be drawn case by case. The protected mono-tailed sugar derivative 10 was found to be a more powerful hCA inhibitor than the reference compound AAZ, showing lower inhibition constants towards hCAs I, II and IX (Ki = 65.2, 5.7 and 5.1 nM, respectively). In general, the introduction of an iminosugar moiety did not furnish better inhibitors than the reference AAZ; however, increased selectivity may be achieved, as shown by the promising protected iminosugar derivative 31 which is a good and selective inhibitor of hCA VII (Ki = 9.7 nM). X-Ray analysis of hCA II in complex with the iminosugar compound 31 revealed two molecules bound to the active site, one located within the active site and responsible for the activity, and the other bound outside the active site and not involved in inhibition. A typical binding mode of sulphonamide inhibitors was found for compound 31, with the benzenesulfonamidate moiety directly interacting with the zinc ion within the active site. A double conformation was found for the triazole and the iminosugar parts of the inhibitor, suggesting a certain disorder of the tail which is consistent with the relatively weak potency of this inhibitor against hCA II. The introduction of three hydrophilic tails is generally detrimental for the inhibition of hCAs. However, high selectivity towards a specific isoform coupled to powerful inhibition has been occasionally observed. In particular, the three-tailed peracetylated sugar 25 was a potent and selective inhibitor of hCA VII (Ki = 9.7 nM) and its deprotected analogue 26 was a better inhibitor of hCA VB (Ki = 34.2 nM) than AAZ.Author contributions
Project design by F. Cardona and C. Supuran. Synthesis by M. G. Davighi and characterization by M. G. Davighi and C. Matassini. Biological assays and crystallographic experiments and analyses by A. Angeli. The manuscript was written by M. G. Davighi, F. Cardona and A. Goti, with edits from all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by Fondazione CR Firenze (grant number 2017.0734). We also thank MIUR-Italy (“Progetto Dipartimenti di Eccellenza 2018–2022” allocated to the Department of Chemistry “Ugo Schiff”). We gratefully acknowledge Elettra and XRD2 beamline for providing beamtime and support under proposal 20220596References
- T. H. Maren, Carbonic Anhydrase: Chemistry, Physiology, and Inhibition, Physiol. Rev., 1967, 47, 595–781 CrossRef CAS PubMed.
- C. T. Supuran, Carbonic anhydrases: novel therapeutic applications for inhibitors and activators, Nat. Rev. Drug Discovery, 2008, 7, 168–181 CrossRef CAS PubMed.
- (a) S. Pastorekova, S. Parkkila, J. Pastorek and C. T. Supuran, Carbonic anhydrases: current state of the art, therapeutic applications and future prospects, J. Enzyme Inhib. Med. Chem., 2004, 19, 199–229 CrossRef CAS PubMed; (b) C. T. Supuran, Exploring the multiple binding modes of inhibitors to carbonic anhydrases for novel drug discovery, Expert Opin. Drug Discovery, 2020, 15, 671–686 CrossRef CAS PubMed.
- A. Aspatwar, M. E. E. Tolvanen, H. Barker, L. Syrjänen, S. Valanne, S. Purmonen, A. Waheed, W. S. Sly and S. Parkkila, Carbonic anhydrases in metazoan model organisms: molecules, mechanisms, and physiology, Physiol. Rev., 2022, 102, 1327–1383 CrossRef CAS PubMed.
- (a) J.-Y. Winum, P. A. Colinas and C. T. Supuran, Glycosidic carbonic anhydrase IX inhibitors: A sweet approach against cancer, Bioorg. Med. Chem., 2013, 21, 1419–1426 CrossRef CAS PubMed; (b) Y. Lou, P. C. McDonald, A. Oloumi, S. Chia, C. Ostlund, A. Ahmadi, A. Kyle, U. auf dem Keller, S. Leung, D. Huntsman, B. Clarke, B. W. Sutherland, D. Waterhouse, M. Bally, C. Roskelley, C. M. Overall, A. Minchinton, F. Pacchiano, F. Carta, A. Scozzafava, N. Touisni, J. Y. Winum, C. T. Supuran and S. Dedhar, Targeting tumor hypoxia: suppression of breast tumor growth and metastasis by novel carbonic anhydrase IX inhibitors, Cancer Res., 2011, 71, 3364–3376 CrossRef CAS PubMed.
- K. D'Ambrosio, F. Z. Smaine, F. Carta, G. De Simone, J.-Y. Winum and C. T. Supuran, Development of Potent Carbonic Anhydrase Inhibitors Incorporating Both Sulfonamide and Sulfamide Groups, J. Med. Chem., 2012, 55, 6776–6783 CrossRef PubMed.
- D. Macmillan and A. M. Daines, Recent Developments in the Synthesis and Discovery of Oligosaccharides and Glycoconjugates for the Treatment of Disease, Curr. Med. Chem., 2003, 10, 2733–2773 CrossRef CAS PubMed.
- (a) H. C. Kolb, M. G. Finn and K. B. Sharpless, Click Chemistry: Diverse Chemical Function from a Few Good Reactions, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 ( Angew. Chem. , 2001 , 113 , 2056–2075 ) CrossRef CAS; (b) V. C. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, A Stepwise Huisgen Cycloaddition Process: Copper(I)-Catalyzed Regioselective “Ligation” of Azides and Terminal Alkynes, Angew. Chem., Int. Ed., 2002, 41, 2596–2599 ( Angew. Chem. , 2002 , 114 , 2708–2711 ) CrossRef CAS; (c) C. W. Tornøe, C. Christensen and M. Meldal, Peptidotriazoles on Solid Phase: [1,2,3]-Triazoles by Regiospecific Copper(I)-Catalyzed 1,3-Dipolar Cycloadditions of Terminal Alkynes to Azides, J. Org. Chem., 2002, 67, 3057–3064 CrossRef PubMed.
- (a) B. L. Wilkinson, A. Innocenti, D. Vullo, C. T. Supuran and S. A. Poulsen, Inhibition of Carbonic Anhydrases with Glycosyltriazole Benzene Sulfonamides, J. Med. Chem., 2008, 51, 1945–1953 CrossRef CAS PubMed; (b) A. Angeli and C. T. Supuran, Click chemistry approaches for developing carbonic anhydrase inhibitors and their applications, J. Enzyme Inhib. Med. Chem., 2023, 38, 2166503 CrossRef PubMed.
- D. Pratesi, C. Matassini, A. Goti, A. Angeli, F. Carta, C. T. Supuran, R. Spanevello and F. Cardona, Glycomimetic Based Approach toward Selective Carbonic Anhydrase Inhibitors, ACS Med. Chem. Lett., 2020, 11, 727–731 CrossRef CAS PubMed.
- Iminosugars: from synthesis to therapeutic applications, ed. P. Compain and O. R. Martin, Wiley VCH, New York, 2007 Search PubMed.
- D. Pratesi, A. Sodini, C. Matassini, F. Cardona, A. Angeli, F. Carta, M. Ferraroni, C. T. Supuran and A. Goti, Synthesis of Azasugar–Sulfonamide conjugates and their Evaluation as Inhibitors of Carbonic Anhydrases: the Azasugar Approach to Selectivity, Eur. J. Org. Chem., 2021, 2604–2614 CrossRef CAS.
- V. Alterio, A. Di Fiore, K. D'Ambrosio, C. T. Supuran and G. De Simone, Multiple Binding Modes of Inhibitors to Carbonic Anhydrases: How to Design Specific Drugs Targeting 15 Different Isoforms?, Chem. Rev., 2012, 112, 4421–4468 CrossRef CAS PubMed.
- R. P. Tanpure, B. Ren, T. S. Peat, L. F. Bornaghi, D. Vullo, C. T. Supuran and S. A. Poulsen, Carbonic Anhydrase Inhibitors with Dual-Tail Moieties To Match the Hydrophobic and Hydrophilic Halves of the Carbonic Anhydrase Active Site, J. Med. Chem., 2015, 58, 1494–1501 CrossRef CAS PubMed.
- Z. Hou, B. Lin, Y. Bao, H. Yan, M. Zhang, X. Chang, X. Zhang, Z. Wang, G. Wei, M. Cheng, Y. Liu and C. Guo, Dual-tail approach to discovery of novel carbonic anhydrase IX inhibitors by simultaneously matching the hydrophobic and hydrophilic halves of the active site, Eur. J. Med. Chem., 2017, 132, 1–10 CrossRef CAS PubMed.
- (a) M. A. Said, W. M. Eldehna, A. Nocentini, A. Bonardi, S. H. Fahim, S. Bua, D. H. Soliman, H. A. Abdel-Aziz, P. Gratteri, S. M. Abou-Seri and C. T. Supuran, Synthesis, biological and molecular dynamics investigations with a series of triazolopyrimidine/triazole-based benzenesulfonamides as novel carbonic anhydrase inhibitors, Eur. J. Med. Chem., 2020, 185, 111843–111856 CrossRef CAS PubMed; (b) A. Pustenko, A. Nocentini, P. Gratteri, A. Bonardi, I. Vozny, R. Žalubovskis and C. T. Supuran, The antibiotic furagin and its derivatives are isoform-selective human carbonic anhydrase inhibitors, J. Enzyme Inhib. Med. Chem., 2020, 35, 1011–1020 CrossRef CAS PubMed.
- A. Bonardi, A. Nocentini, S. Bua, J. Combs, C. Lomelino, J. Andring, L. Lucarini, S. Sgambellone, E. Masini, R. McKenna, P. Gratteri and C. T. Supuran, Sulfonamide Inhibitors of Human Carbonic Anhydrases Designed through a Three-Tails Approach: Improving Ligand/Isoform Matching and Selectivity of Action, J. Med. Chem., 2020, 63, 7422–7444 CrossRef CAS PubMed.
- (a) N. Michihata, Y. Kaneko, Y. Kasai, K. Tanigawa, T. Hirokane, S. Higasa and H. Yamada, High-Yield Total Synthesis of (−)-Strictinin through Intramolecular Coupling of Gallates, J. Org. Chem., 2013, 78, 4319–4328 CrossRef CAS PubMed; (b) Q. Sun, Q. Yang, S. Gong, Q. Fu and Q. Xiao, Synthesis and enzymatic evaluation of phosphoramidon and its β anomer: Anomerization of α-l-rhamnose triacetate upon phosphitylation, Bioorg. Med. Chem., 2013, 21, 6778–6787 CrossRef CAS PubMed; (c) M. van Scherpenzeel, R. J. B. H. N. van den Berg, W. E. Donker-Koopman, R. M. J. Liskamp, J. M. F. G. Aerts, H. S. Overkleeft and R. J. Pieters, Nanomolar affinity, iminosugar-based chemical probes for specific labeling of lysosomal glucocerebrosidase, Bioorg. Med. Chem., 2010, 18, 267–273 CrossRef CAS PubMed; (d) P. Y. Chong and P. A. Petillo, Synthesis of Carbamate-Containing Cyclodextrin Analogues, Org. Lett., 2000, 2, 1093–1096 CrossRef CAS PubMed.
- A. Nocentini, D. Vullo, G. Bartolucci and C. T. Supuran, N-Nitrosulfonamides: A new chemotype for carbonic anhydrase inhibition, Bioorg. Med. Chem., 2016, 24, 3612–3617 CrossRef CAS PubMed.
- S. Wang, K. Jia, J. Cheng, Y. Chen and Y. Yuan, Dual roles of substituted thiourea as reductant and ligand in CuAAC reaction, Tetrahedron Lett., 2017, 58, 3717–3721 CrossRef CAS.
- (a) Y. M. Chabre, C. Contino-Pépin, V. Placide, T. C. Shiao and R. Roy, Expeditive Synthesis of Glycodendrimer Scaffolds Based on Versatile TRIS and Mannoside Derivatives, J. Org. Chem., 2008, 73, 5602–5605 CrossRef CAS PubMed; (b) M. Segura, F. Sansone, A. Casnati and R. Ungaro, Synthesis of Lower Rim Polyhydroxylated Calix[4]arenes, Synthesis, 2001, 2105–2112 CrossRef CAS.
- K. Pichard, D. Campkin, N. O'Brien, A. Kato, G. W. J. Fleet and M. Simone, Biological activities of 3,4,5-trihydroxypiperidines and their N-and O-derivatives, Chem. Biol. Drug Des., 2018, 92, 1171–1197 CrossRef PubMed.
- C. Matassini, S. Mirabella, X. Ferhati, C. Faggi, I. Robina, A. Goti, E. M. Clavijo, A. J. M. Vargas and F. Cardona, Polyhydroxyamino-Piperidine-Type Iminosugars and Pipecolic Acid Analogues from a D-Mannose-Derived Aldehyde, Eur. J. Org. Chem., 2014, 5419–5432 CrossRef CAS.
- (a) C. Matassini, S. Mirabella, A. Goti and F. Cardona, Double Reductive Amination and Selective Strecker Reaction of a D-Lyxaric Aldehyde: Synthesis of Diversely Functionalized 3,4,5-Trihydroxypiperidines, Eur. J. Org. Chem., 2012, 3920–3924 CrossRef CAS; (b) F. Clemente, C. Matassini and F. Cardona, Reductive Amination Routes in the Synthesis of Piperidine IminoSugars, Eur. J. Chem., 2020, 4447–4462 CrossRef CAS.
- C. Matassini, S. Mirabella, A. Goti, I. Robina, A. J. Moreno-Vargas and F. Cardona, Exploring architectures displaying multimeric presentations of a trihydroxypiperidine iminosugar, Beilstein J. Org. Chem., 2015, 11, 2631–2640 CrossRef CAS PubMed.
- R. G. Khalifah, The Carbon Dioxide Hydration Activity of Carbonic Anhydrase: I. STOP-FLOW KINETIC STUDIES ON THE NATIVE HUMAN ISOENZYMES B AND C, J. Biol. Chem., 1971, 246, 2561–2573 CrossRef CAS PubMed.
- B. Kayser, L. Dumont, C. Lysakowski, C. Combescure, G. Haller and M. R. Tramèr, Reappraisal of Acetazolamide for the Prevention of Acute Mountain Sickness: A Systematic Review and Meta-Analysis, High Alt. Med. Biol., 2012, 13, 82–92 CrossRef CAS PubMed.
- D. Tanini, S. Carradori, A. Capperucci, L. Lupori, S. Zara, M. Ferraroni, C. Ghelardini, L. Mannelli, L. Micheli, E. Lucarini, F. Carta, A. Angeli and C. T. Supuran, Chalcogenides-incorporating carbonic anhydrase inhibitors concomitantly reverted oxaliplatin-induced neuropathy and enhanced antiproliferative action, Eur. J. Med. Chem., 2021, 225, 113793 CrossRef CAS PubMed.
- D. Tanini, L. Ricci, A. Capperucci, L. Di Cesare Mannelli, C. Ghelardin, T. S. Peat, F. Carta, A. Angeli and C. T. Supuran, Synthesis of novel tellurides bearing benzensulfonamide moiety as carbonic anhydrase inhibitors with antitumor activity, Eur. J. Med. Chem., 2019, 181, 111586 CrossRef PubMed.
- (a) G. Annunziato, A. Angeli, F. D'Alba, A. Bruno, M. Pieroni, D. Vullo, V. De Luca, C. Capasso, C. T. Supuran and G. Costantino, Discovery of New Potential Anti-Infective Compounds Based on Carbonic Anhydrase Inhibitors by Rational Target-Focused Repurposing Approaches, ChemMedChem, 2016, 11, 1904–1914 CrossRef CAS PubMed; (b) A. Angeli, F. Carta, G. Bartolucci and C. T. Supuran, Synthesis of novel acyl selenoureido benzensulfonamides as carbonic anhydrase I, II, VII and IX inhibitors, Bioorg. Med. Chem., 2017, 25, 3567–3573 CrossRef CAS PubMed; (c) A. Angeli, T. S. Peat, G. Bartolucci, A. Nocentini, C. T. Supuran and F. Carta, Intramolecular oxidative deselenization of acylselenoureas: a facile synthesis of benzoxazole amides and carbonic anhydrase inhibitors, Org. Biomol. Chem., 2016, 14, 11353–11356 RSC; (d) C. B. Mishra, S. Kumari, A. Angeli, S. M. Monti, M. Buonanno, M. Tiwari and C. T. Supuran, Discovery of Benzenesulfonamides with Potent Human Carbonic Anhydrase Inhibitory and Effective Anticonvulsant Action: Design, Synthesis, and Pharmacological Assessment, J. Med. Chem., 2017, 60, 2456–2469 CrossRef CAS PubMed; (e) A. Angeli, G. Abbas, S. Del Prete, F. Carta, C. Capasso and C. T. Supuran, Acyl selenoureido benzensulfonamides show potent inhibitory activity against carbonic anhydrases from the pathogenic bacterium Vibrio cholerae, Bioorg. Chem., 2017, 75, 170–172 CrossRef CAS PubMed.
- A. G. W. Leslie and H. R. Powell, Processing diffraction data with mosflm, In Evolving methods for macromolecular crystallography, ed. R. J. Read and J. L. Sussman, NATO Science series, Springer, Dordrecht, 2007, vol. 245, pp. 41–51 Search PubMed.
- G. N. Murshudov, A. A. Vagin and E. J. Dodson, Refinement of macromolecular structures by the maximum-likelihood method, Acta Crystallogr., Sect. D: Biol. Crystallogr., 1997, 53, 240–255 CrossRef CAS PubMed.
- P. Emsley, B. Lohkamp, W. Scott and K. Cowtan, Features and development of Coot, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2010, 66, 486–501 CrossRef CAS PubMed.
- S. C. Lovell, I. W. Davis, W. B. Arendall III, P. I. W. de Bakker, J. M. Word, M. G. Prisant, J. S. Richardson and D. C. Richardson, Structure validation by Cα geometry: ϕ,ψ and Cβ deviation, Proteins, 2003, 50, 437–450 CrossRef CAS PubMed.
- E. F. Pettersen, T. D. Goddard, C. C. Huang, G. S. Couch, D. M. Greenblatt, E. C. Meng and T. E. Ferrin, UCSF Chimera—a visualization system for exploratory research and analysis, J. Comput. Chem., 2004, 25, 1605–1612 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: 1H NMR and 13C NMR spectra of the newly synthesized compounds, and detailed preparation and characterization of compounds 11, 12, 14, 17–20, 22–24, 32 and 35–44. See DOI: https://doi.org/10.1039/d3ob00529a |
| This journal is © The Royal Society of Chemistry 2023 |