
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of 1,4-ketoaldehydes and 1,4-diketones by Mo-catalyzed oxidative cleavage of cyclobutane-1,2-diols†
Sara
Gómez-Gil
 ,
Rubén
Rubio-Presa
,
Raquel
Hernández-Ruiz
,
Samuel
Suárez-Pantiga
,
María R.
Pedrosa
and
Roberto
Sanz
*
,
Rubén
Rubio-Presa
,
Raquel
Hernández-Ruiz
,
Samuel
Suárez-Pantiga
,
María R.
Pedrosa
and
Roberto
Sanz
*
Departamento de Química, Facultad de Ciencias, Universidad de Burgos, Pza. Misael Bañuelos s/n, 09001 Burgos, Spain. E-mail: rsd@ubu.es
First published on 26th April 2023
Abstract
A new two-step procedure for the synthesis of 1,4-dicarbonyls has been developed involving an efficient and clean Mo-catalyzed oxidative cleavage of cyclobutane-1,2-diols with DMSO, which is used as solvent and oxidant. The required starting glycols were prepared by nucleophilic additions of organolithiums and Grignard reagents to easily available 2-hydroxycyclobutanones.
1,4-Dicarbonyls are valuable structural motifs in organic synthesis that are typically employed to prepare five-membered heterocycles and functionalized cyclopentenones.1 In addition, they are common skeletons in natural products possessing interesting biological activities.2 For that reason, several synthetic approaches have been developed for accessing these building blocks, ranging from classical procedures,3 such as the conjugate addition of acyl anions to Michael acceptors, like the Stetter reaction,4 the acylation of homoenolate equivalents,5 the reaction of enolates with α-haloketones,6 or the oxidative coupling of enolates,7 to more recent routes involving metal-catalyzed cross-coupling reactions of different carbonyl derivatives,8 conjugate addition of acyl radicals,9 carbene insertion into 1,3-dicarbonyls,10 hydrocarbonylation reactions,11 as well as different photocatalyzed processes,12 or the organocatalytic redox isomerization of allylic alcohols.13
On the other hand, the oxidative cleavage of 1,2-diols is a fundamental synthetic transformation, early developed by Criegee and Malaprade a century ago.14 Since then, several protocols have been developed trying to improve the efficiency and selectivity of the process and to reduce the drawbacks associated with the use of hazardous reagents and/or conditions.15 In this field our group has reported the employment of DMSO as both solvent and oxidant for the selective oxidative cleavage of a wide variety of glycols under dioxomolybdenum(VI)-catalysis (Scheme 1a).16 These metallic complexes are readily available and have been successfully used by our group and others as efficient catalysts for different processes involving O-atom transfer reactions.17 In addition, DMSO is an inexpensive and widely used solvent with low relative toxicity,18 obtained as a byproduct of the wood industry, which has also been employed as a mild oxidant and as oxygen, one-carbon, or thiomethyl/methiomethyl source in a variety of organic transformations.19
 | ||
| Scheme 1 Dioxomolybdenum(VI)-catalyzed oxidative cleavage of glycols with DMSO (previous work) and proposed synthesis of 1,4-dicarbonyls (this work). | ||
In this context, we envisaged that a simple and straightforward route to access 1,4-dicarbonyl derivatives would be the oxidative cleavage of cyclobutane-1,2-diols (Scheme 1b). This conceptually simple approach has not been previously reported, and only related examples have been scarcely developed, such as the preparation of ortho-keto-functionalized benzaldehyde derivatives from α-siloxybenzocyclobutanone.20 Therefore, to the best of our knowledge, the use of 2-hydroxycyclobutanones for the synthesis of aliphatic 1,4-dicarbonyls has not been previously described.21 These derivatives have been used mainly by Secci et al. for the development of chemoselective methodologies applied to the preparation of different carbo- and heterocyclic scaffolds.22 Herein, we report a new protocol for the preparation of 1,4-dicarbonyls based on the Mo-catalyzed oxidative cleavage of cyclobutane-1,2-diols with DMSO.
2-Hydroxycyclobutanone 1a, easily accessed from commercially available 1,2-bis(trimethylsilyloxy)cyclobutene,23 was selected as starting material for synthesizing cyclobutane-1,2-diol derivatives. Its treatment with an excess of n-BuLi provided glycol 2a in good yields as a mixture of diastereoisomers, which could be isolated independently after column chromatography, and stereochemically assigned from the ready formation of a cyclic acetal from the cis-diol stereoisomer (Scheme 2).24 Then, each diastereoisomer, cis-2a and trans-2a, was evaluated in the Mo-catalyzed oxidative cleavage reaction with DMSO-d6 under microwave irradiation.
 | ||
| Scheme 2 Synthesis of cyclobutane-1,2-diol 2a and its oxidative cleavage with DMSO. | ||
After a brief optimization25 we could establish the conditions for obtaining the corresponding γ-ketoaldehyde 3a in very high yields in both cases, after short reaction times (Scheme 2). Not surprisingly, the temperature required for the cleavage of trans-2a was considerably higher than for cis-2a. In addition, the oxidative cleavage of cis-2a can also be carried out under conventional heating (90 °C) for 1 h with similar yield.
Then, we prepared a wide variety of cyclobutane-1,2-diols 2 from both organolithium and Grignard reagents, which were obtained in good yields as mixtures of cis- and trans-diastereoisomers, being cis-2 the major isomer in all cases (Table 1). Surprisingly, only the reaction with benzylmagnesium chloride proceeded with high diastereoselectivity (entry 6). In most cases both diastereoisomers could be isolated independently. Different organometallics were used successfully, including (cyclo)alkyl (entries 1–5), benzyl (entry 6), (hetero)aryl (entries 7–9), and alkynyl ones (entries 10–13), as well as a lithium enolate (entry 14).
|
|
||||
|---|---|---|---|---|
| Entry | RMet | 2 | drb | Yieldc (%) |
| a Reactions conducted using 2-hydroxycyclobutanone 1a (2 mmol) and the corresponding organometallic (6 mmol) in THF (4 mL) from −78 °C to RT. b Determined by 1H NMR analysis of the crude reaction mixture. c Combined isolated yield of cis- and trans-2 based on starting material 1a. Both diastereoisomers were isolated independently unless otherwise noted. d Only the corresponding cis-2 was isolated. e Similar yield and diastereoselectivity were obtained using PhMgBr. f 5-Methyl-2-thienyl lithium. g 3-Thienylethynyl lithium. | ||||
| 1 | n-BuLi | 2a | 2/1 | 60 |
| 2 | MeLi | 2b | 3/1 | 55 |
| 3 | EtMgBr | 2c | 2/1 | 63 |
| 4 | i-PrMgCl | 2d | 1.2/1 | 58d |
| 5 | c-C6H11MgCl | 2e | 1.2/1 | 63 |
| 6 | PhCH2MgCl | 2f | >20/1 | 61d |
| 7e | PhLi | 2g | 3/1 | 55 |
| 8 | 2-MeOC6H4Li | 2h | 3/1 | 57d |
| 9 | 5-Me-2-ThLif | 2i | 2/1 | 42 |
| 10 | BuC![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CLi CLi |
2j | 2/1 | 67 |
| 11 | PhCCLi |
2k | 2/1 | 66 |
| 12 | 3-ThCCLig |
2l | 2/1 | 61 |
| 13 | PhOCH2CCLi |
2m | 2/1 | 55 |
| 14 | LiCH2CO2t-Bu | 2n | 2/1 | 54d |

Due to the milder conditions required for the oxidative cleavage of cis-2 and their easy isolation, these diastereoisomers were used as starting materials for the synthesis of the corresponding γ-ketoaldehydes 3 (Table 2). Reactions were carried out in DMSO-d6 under microwave irradiation (90 °C, 10 min) and very high yields were achieved for all the essayed substrates, which were obtained in pure form without further purification and properly characterized in the deuterated solvent.25 No significant effect of the R group was observed in the Mo-catalyzed oxidative cleavage. Apart from simple aliphatic γ-ketoaldehydes 3a–f, (hetero)aromatic derivatives 3g–i could also be prepared (entries 7–9) and, more interestingly, alkynones 3j–m, further functionalized with an aldehyde group, were also efficiently synthesized (entries 10–13). Interestingly, tricarbonyl derivative 3n was prepared from glycol 2n (entry 14). Finally, a selection of dicarbonyls 3 were also isolated by simple extraction (entries 1 and 5–7).
|
|
||||
|---|---|---|---|---|
| Entry | 2 | R | Product | Yieldb (%) |
| a Reactions conducted using 0.3 mmol of the corresponding cis-diol 2 in DMSO-d6 (0.6 mL) with [MoO2Cl2(dmso)2] (2 mol%) under microwave irradiation (90 °C, 10 min). b The crude products were pure, as observed by 1H NMR analysis. Yield determined using dibromomethane as internal standard. c Isolated yield after extraction. | ||||
| 1 | 2a | n-Bu | 3a | 92 (86)c |
| 2 | 2b | Me | 3b | 94 |
| 3 | 2c | Et | 3c | 92 |
| 4 | 2d | i-Pr | 3d | 89 |
| 5 | 2e | c-C6H11 | 3e | 92 (87)c |
| 6 | 2f | PhCH2 | 3f | 90 (86)c |
| 7 | 2g | Ph | 3g | 93 (88)c |
| 8 | 2h | 2-MeOC6H4 | 3h | 91 |
| 9 | 2i | 5-Me-2-Th | 3i | 88 |
| 10 | 2j | CCBu |
3j | 94 |
| 11 | 2k | CCPh |
3k | 93 |
| 12 | 2l | CC-(3-Th) |
3l | 92 |
| 13 | 2m | CCCH2OPh |
3m | 90 |
| 14 | 2n | CH2CO2t-Bu | 3n | 89 |

Moreover, the synthesis of γ-ketoaldehydes 3 could also be carried out without purification of glycols 2 (Scheme 3). So, the crude products obtained from the reaction of 1a with selected organometallics were reacted under the Mo-catalyzed conditions. As both diastereoisomers of the corresponding cyclobutane-1,2-diols 2 were present in the crude, the oxidative cleavage with DMSO was performed at 150 °C. After workup, the corresponding ketoaldehydes 3a,g,h were isolated in good overall yields after column chromatography (Scheme 3). In addition, the preparation of 3g was also performed from 1a (8 mmol) in 43% overall yield.
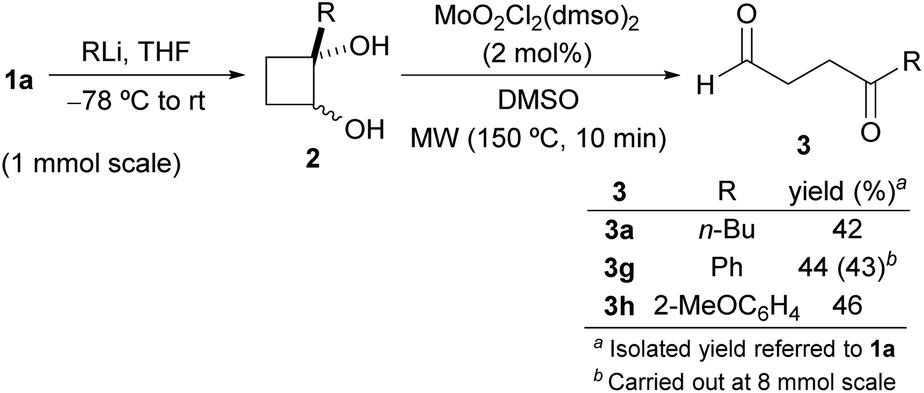 | ||
| Scheme 3 Synthesis of 1,4-ketoaldehydes 3 from 1a (without purification of 2). | ||
Next, we decided to check the suitability of this methodology for the preparation of 1,4-diketones. To achieve this goal, a 2-substituted-2-hydroxycyclobutanone would be required as starting material. This type of compound can be prepared by the Norrish–Yang intramolecular photocyclization of 1,2-diketones.26 Following this methodology, 2-ethyl-2-hydroxycyclobutanone 1b was synthesized from commercially available 3,4-hexanedione (Scheme 4).27 However, its reaction with organolithium reagents did not selectively afford the expected diols 4, as mixtures of diastereoisomers, but also led to formation of glycols 5. These diol derivatives seem to come from a competitive rearrangement of the alkoxide 1b-Li affording lithium 1-propionylcyclopropan-1-olate that undergoes a subsequent nucleophilic addition. Although this undesired pathway is minor,28 glycols 4 and 5 could not be independently isolated by standard column chromatography, except for the case of using lithium phenylacetylide, which allowed the isolation of pure cis-4a. Gratifyingly, this cyclobutane-1,2-diol derivative underwent efficiently the oxidative cleavage with DMSO, under dioxomolybdenum(VI)-catalysis, giving rise to alkynyl diketone 6a (Scheme 5). In addition, we could also isolate diol 5e, arising from the reaction of 1b with PhLi, but when we submitted it to the oxidative cleavage under Mo-catalysis at 150 °C no reaction took place.
 | ||
| Scheme 4 Synthesis of di-tertiary cyclobutanediols 4 from 1b and oxidative cleavage of isolated cis-4a. | ||
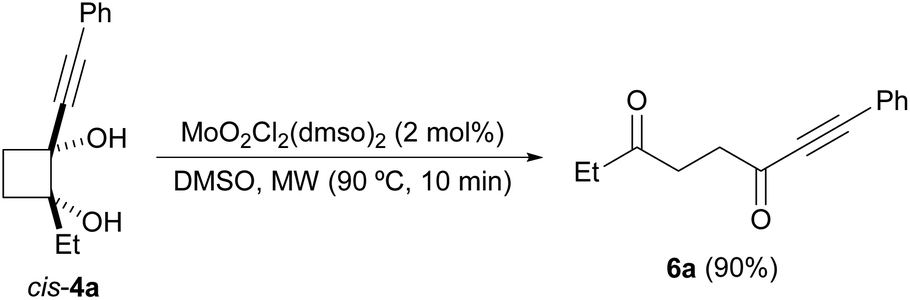 | ||
| Scheme 5 Oxidative cleavage of cis-4a: synthesis of alkynyl diketone 6a. | ||
This result gave us the opportunity to carry out the oxidative cleavage of crude mixtures of di-tertiary cyclobutane-1,2-diols 4 and glycols 5 (Table 3). After column chromatography we were able to obtain and isolate a selection of pure 1,4-diketones 6 in useful overall yields, considering that they are referred to hydroxyketone 1b. In this way γ-diketones bearing alkynyl (entry 1), (cyclo)alkyl (entries 2 and 3), benzyl (entry 4) or (hetero)aryl groups (entries 5 and 6), as substituents of one of the carbonyl groups, could be synthesized.
|
|
||||
|---|---|---|---|---|
| Entry | RMet | Product | R | Yieldb (%) |
| a Reactions conducted starting from 1b (1 mmol) and the corresponding organometallic reagent (3 mmol) in THF (3 mL). The crude mixture is oxidatively cleaved in DMSO (2 mL) under microwave irradiation (150 °C, 10 min). b Isolated yield after column chromatography referred to 2-hydroxycyclobutanone 1b. c 5-Methyl-2-thienyl lithium. | ||||
| 1 | PhCCLi |
6a | CCPh |
48 |
| 2 | n-BuLi | 6b | n-Bu | 42 |
| 3 | c-C6H11MgCl | 6c | c-C6H11 | 39 |
| 4 | PhCH2MgCl | 6d | CH2Ph | 40 |
| 5 | PhLi | 6e | Ph | 38 |
| 6 | 5-Me-2-ThLic | 6f | 5-Me-2-Th | 37 |
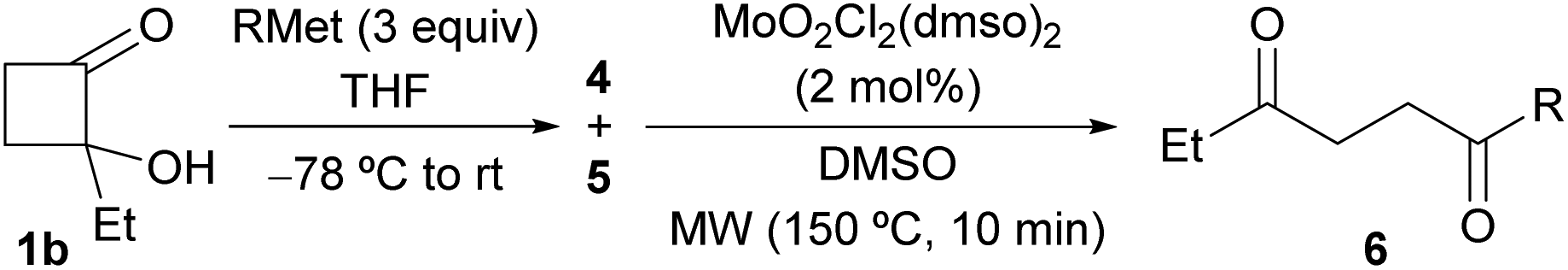
Based on our previous report,16 the reaction mechanism for the oxidative cleavage of cyclobutane-1,2-diols 2 and 4 was proposed as shown in Scheme 6. Firstly, the catalyst [MoO2Cl2(dmso)2] would react with the corresponding cyclobutanediol 2 or 4 leading to diolate complex A, releasing a molecule of water. Then, the oxidative cleavage of the glycolate ligand by the Mo(VI) center would lead to the formation of a new oxomolybdenum(IV) species B. Finally, the weakly coordinated 1,4-dicarbonyl 3 or 6 would be readily displaced by dimethyl sulfoxide affording the unstable Mo(IV) adduct C. Subsequent release of dimethyl sulfide, would eventually regenerate the Mo(VI) catalyst. We further investigated the formation of the proposed diolate complex A.25 According to the 1H-NMR spectra of an equimolecular mixture of diols cis- or trans-2a and the Mo(VI) catalyst in DMSO-d6, recorded at different temperatures, the suggested complex A would likely exist only in the instant of its formation as it could not be detected by NMR. Once the reaction reached the required temperature, ca. 40 °C for cis-2a and 150 °C for trans-2a, only the final 1,4-dicarbonyl and the starting diol were observed by NMR. This study suggests that the diolate generation is the rate-determining step for the oxidative cleavage.29 In addition, we carried out the oxidative cleavage of 2a in the presence of radical scavengers TEMPO and BHT. In both experiments, the addition of these reagents did not have any impact on the reaction outcome. The formation of the ketoaldehyde 3a was not suppressed, thus suggesting that the transformation does not follow a radical pathway.
 | ||
| Scheme 6 Mechanistic proposal for the Mo-catalyzed oxidative cleavage of cyclobutane-1,2-diols 2 and 4 with DMSO. | ||
Finally, we decided to increase the value of our protocol for the synthesis of 1,4-dicarbonyls by their direct transformation into other derivatives. In such manner, the crude DMSO solution of γ-dicarbonyls 3g,e, directly obtained after the Mo-catalyzed oxidative cleavage of selected cyclobutane-1,2-diols 2g,e, was treated with NH4OAc, in the presence of molecular sieves, allowing the isolation of the corresponding pyrroles 7 in good overall yields considering that they are referred to starting glycols 2 (Scheme 6). In addition, when the crude DMSO solution of 3f was reacted with NaBH4, 1,4-diol 8 was obtained in high yield (Scheme 6). Finally, a Wittig ylide was added to the same crude γ-dicarbonyl 3f in DMSO producing ketoester derivative 9 in good yield (Scheme 7).
 | ||
| Scheme 7 Subsequent transformations of DMSO-solutions of crude 1,4-dicarbonyls 3. | ||
Conclusions
In conclusion, we have designed and developed a two-step process for the synthesis of 1,4-ketoaldehydes and 1,4-diketones from readily available 2-hydroxycyclobutanones. The first nucleophilic addition on the cyclobutanone skeleton controls the substituent of the ketone moiety, whereas the C2-substituent of the starting 2-hydroxycyclobutanone is responsible for the generation of γ-ketoaldehydes or γ-diketones. The dioxomolybdenum-catalyzed oxidative cleavage of glycols, using DMSO as solvent and oxidant, allows the clean and high-yielding production of γ-dicarbonyls, which can be employed in a straightforward manner for subsequent useful transformations.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge Ministerio de Ciencia e Innovación and FEDER (PID2020-115789GB-C21), and Junta de Castilla y León and FEDER (BU049P20) for financial support. S. G.-G. and R. H.-R. thank Ministerio de Educación for FPU predoctoral contracts. S. S.-P. thanks Ministerio de Ciencia e Innovación and “NextGenerationEU”/PRTR EU for a Ramón y Cajal contract (RYC2021-031533-I).References
- (a) M. M. Heravi and V. Zadsirjan, Adv. Heterocycl. Chem., 2022, 138, 1–60 CrossRef; (b) A. Balakrishna, A. Aguiar, P. J. M. Sobral, M. Y. Wani, J. A. Silva and A. J. F. N. Sobral, Catal. Rev., 2019, 61, 84–110 CrossRef CAS; (c) S. Khaghaninejad and M. M. Heravi, Adv. Heterocycl. Chem., 2014, 111, 95–146 CrossRef CAS.
- S. S. Davies, V. Amarnath, C. J. Brame, O. Boutaud and L. J. Roberts II, Nat. Protoc., 2007, 2, 2079–2091 CrossRef CAS PubMed.
- For some examples of multistep procedures, see: (a) O. G. Kulinkovich, I. G. Tischenko and V. L. Sorokin, Synthesis, 1985, 1058–1059 CrossRef CAS; (b) N. W. A. Geraghty and N. M. Morris, Synthesis, 1989, 603–607 CrossRef CAS; (c) T. Miyakoshi, Synthesis, 1986, 766–768 CrossRef CAS; (d) O. G. Kulinkovich, I. G. Tischenko and N. V. Masalov, Synthesis, 1984, 886–887 CrossRef CAS; (e) A. Hosomi, H. Hashimoto and H. Sakurai, J. Org. Chem., 1978, 43, 2551–2552 CrossRef CAS.
- For selected examples, see: (a) H. Stetter and H. Kuhlmann, Synthesis, 1975, 379–382 CrossRef CAS; (b) H. Stetter and H. Kuhlmann, Org. React., 1991, 40, 407–495 CAS; (c) A. G. M. Barrett, A. C. Love and L. Tedeschi, Org. Lett., 2004, 6, 3377–3380 CrossRef CAS PubMed.
- B. B. Parida, P. P. Das, M. Niocel and J. K. Cha, Org. Lett., 2013, 15, 1780–1783 CrossRef CAS PubMed.
- See, for instance: (a) M. Yasuda, S. Tsuji, Y. Shigeyoshi and A. Baba, J. Am. Chem. Soc., 2002, 124, 7440–7447 CrossRef CAS PubMed; (b) C.-K. Chan, Y.-L. Chan, Y.-L. Tsai and M.-Y. Chang, J. Org. Chem., 2016, 81, 8112–8120 CrossRef CAS PubMed.
- (a) Y. Ito, T. Konoike and T. Saegusa, J. Am. Chem. Soc., 1975, 97, 2912–2914 CrossRef CAS; (b) R. H. Frazier, Jr. and R. L. Harlow, J. Org. Chem., 1980, 45, 5408–5411 CrossRef. For reviews, see: (c) F. Guo, M. D. Clift and R. J. Thomson, Eur. J. Org. Chem., 2012, 4881–4896 CrossRef CAS PubMed; (d) S. Murarka and A. P. Antonchick, Synthesis, 2018, 50, 2150–2162 CrossRef CAS.
- (a) L. Xu, X. Liu, G. R. Alvey, A. Shatskiy, J.-Q. Liu, M. D. Kärkäs and X.-S. Wang, Org. Lett., 2022, 24, 4513–4518 CrossRef CAS PubMed; (b) P. Fan, Y. Mao and C. Wang, Org. Chem. Front., 2022, 9, 4649–4653 RSC; (c) F. Zhang, P. Du, J. Chen, H. Wang, Q. Luo and X. Wan, Org. Lett., 2014, 16, 1932–1935 CrossRef CAS PubMed; (d) Z.-L. Shen, K. K. K. Goh, H.-L. Cheong, C. H. A. Wong, Y.-C. Lai, Y.-S. Yang and T.-P. Loh, J. Am. Chem. Soc., 2010, 132, 15852–15855 CrossRef CAS PubMed. For a review, see: (e) M. Lemmerer, M. Schupp, D. Kaiser and N. Maulide, Nat. Synth., 2022, 1, 923–935 CrossRef.
- (a) G. Goti, B. Bieszczad, A. Vega-Peñaloza and P. Melchiorre, Angew. Chem., Int. Ed., 2019, 58, 1213–1217 CrossRef CAS PubMed; (b) C. D. Pan, Qi. T. Ni, Y. Fu and J.-T. Yu, J. Org. Chem., 2017, 82, 7683–7688 CrossRef CAS PubMed.
- (a) Z. Liu, X. Zhang, M. Virelli, G. Zanoni, E. A. Anderson and X. Bi, iScience, 2018, 8, 54–60 CrossRef CAS PubMed. For related work, see: (b) M. Ni, J. Zhang, X. Liang, Y. Jiang and T.-P. Loh, Chem. Commun., 2017, 53, 12286–12289 RSC; (c) J. Rong, H. Li, R. Fu, W. Sun, T.-P. Loh and Y. Jiang, ACS Catal., 2020, 10, 3664–3669 CrossRef CAS.
- (a) Y. Xie, L. Huang, Y. Qi, J. Hu, L. Song and H. Feng, Green Chem., 2022, 24, 1978–1982 RSC; (b) Q. Zhang, Y. Huang, L.-W. Zhan, W.-Y. Tang, J. Hou and B.-D. Li, Org. Lett., 2020, 22, 7460–7464 CrossRef CAS PubMed.
- (a) W. H. García-Santos, J. B. Mateus-Ruiz and A. Cordero-Vargas, Org. Lett., 2019, 21, 4092–4096 CrossRef PubMed; (b) T. Morack, C. Mück-Lichtenfeld and R. Gilmour, Angew. Chem., Int. Ed., 2019, 58, 1208–1212 CrossRef CAS PubMed; (c) Y. L. Kuang, K. Wang, X. C. Shi, X. Q. Huang, E. Meggers and J. Wu, Angew. Chem., Int. Ed., 2019, 58, 16859–16863 CrossRef CAS PubMed; (d) J. Liu, L.-Q. Lu, Y. Luo, W. Zhao, P.-C. Sun, W. Jin, X. Qi, Y. Cheng and W.-J. Xiao, ACS Catal., 2022, 12, 1879–1885 CrossRef CAS.
- K. Mondal, B. Mondal and S. C. Pan, J. Org. Chem., 2016, 81, 4835–4840 CrossRef CAS PubMed.
- (a) R. Criegee, Ber. Dtsch. Chem. Ges., 1931, 64, 260–266 CrossRef; (b) L. Malaprade, Bull. Soc. Chim. Fr., 1934, 3, 833–852 Search PubMed.
- For selected examples, see: (a) W. Chen, X. Xie, J. Zhang, J. Qu, C. Luo, Y. Lai, F. Jiang, H. Yu and Y. Wei, Green Chem., 2021, 23, 9140–9146 RSC; (b) Z.-Z. Zhou, M. Liu, L. Lv and C.-J. Li, Angew. Chem., Int. Ed., 2018, 57, 2616–2620 CrossRef CAS PubMed; (c) E. Amadio, J. González-Fabra, D. Carraro, W. Denis, B. Gjoka, C. Zonta, K. Bartik, F. Cavani, S. Solmi, C. Bo and G. Licini, Adv. Synth. Catal., 2018, 360, 3286–3296 CrossRef CAS; (d) V. Escande, C. H. Lam, P. Coish and P. T. Anastas, Angew. Chem., Int. Ed., 2017, 56, 9561–9565 CrossRef CAS PubMed; (e) S. M. Kim, D. W. Kim and J. W. Wang, Org. Lett., 2014, 16, 2876–2879 CrossRef CAS PubMed; (f) R. Wang, P. Sun, W. Jin, Y. Zhang, B. Wang, Y. Xia, F. Xue, A. Abdukader and C. Liu, Org. Chem. Front., 2022, 9, 2664–2670 RSC.
- N. García, R. Rubio-Presa, P. García-García, M. A. Fernández-Rodríguez, M. R. Pedrosa, F. J. Arnáiz and R. Sanz, Green Chem., 2016, 18, 2335–2340 RSC.
- Selected work from our group: (a) N. García, P. García-García, M. A. Fernández-Rodríguez, R. Rubio, M. R. Pedrosa, F. J. Arnáiz and R. Sanz, Adv. Synth. Catal., 2012, 354, 321–327 CrossRef; (b) R. Rubio-Presa, M. R. Pedrosa, M. A. Fernández-Rodríguez, F. J. Arnáiz and R. Sanz, Org. Lett., 2017, 19, 5470–5473 CrossRef CAS PubMed; (c) R. Rubio-Presa, S. Suárez-Pantiga, M. R. Pedrosa and R. Sanz, Adv. Synth. Catal., 2018, 360, 2216–2220 CrossRef CAS; (d) S. Suárez-Pantiga, R. Hernández-Ruiz, C. Virumbrales, M. R. Pedrosa and R. Sanz, Angew. Chem., Int. Ed., 2019, 58, 2129–2133 CrossRef PubMed; (e) R. Hernández-Ruiz, R. Rubio-Presa, S. Suárez-Pantiga, M. R. Pedrosa, M. A. Fernández-Rodríguez, M. J. Tapia and R. Sanz, Chem. – Eur. J., 2021, 27, 13613–13623 CrossRef PubMed. For reviews on dioxomolybdenum-catalysis, see: (f) R. Hernández-Ruiz and R. Sanz, Synthesis, 2018, 50, 4019–4036 CrossRef; (g) S. Suárez-Pantiga and R. Sanz, Org. Biomol. Chem., 2021, 19, 10472–10492 RSC.
- DMSO has been classified as a nontoxic solvent with no risk to human health by the U.S. EPA and belongs to class 3 “solvents with low toxic potential”. See, for instance: (a) I. Soroko, Y. Bhole and A. G. Livingston, Green Chem., 2011, 13, 162–168 RSC; (b) M. Martí, L. Molina, C. Alemán and E. Armelin, ACS Sustainable Chem. Eng., 2013, 1, 1609–1618 CrossRef; (c) Z. Wang, S. M. Richter, J. R. Bellettini, Y.-M. Pu and D. R. Hill, Org. Process Res. Dev., 2014, 18, 1836–1842 CrossRef CAS.
- Swern oxidation: (a) T. T. Tidwell, Synthesis, 1990, 857–870 CrossRef CAS. Kornblum oxidation: (b) N. Kornblum, J. W. Powers, G. J. Anderson, W. J. Jones, H. O. Larson, O. Levand and W. M. Weaver, J. Am. Chem. Soc., 1957, 79, 6562 CrossRef CAS. Sulfenylating agent: (c) X. Gao, X. Pan, J. Gao, H. Jiang, G. Yuan and Y. Li, Org. Lett., 2015, 17, 1038–1041 CrossRef CAS PubMed. One-carbon source: (d) Z. Zhang, Q. Tian, J. Qian, Q. Liu, T. Liu, L. Shi and G. Zhang, J. Org. Chem., 2014, 79, 8182–8188 CrossRef CAS.
- K. Asahina, S. Matsuoka, R. Nakayama and T. Hamura, Org. Biomol. Chem., 2014, 12, 9773–9776 RSC.
- A particular example has been reported by Secci et al. involving a tandem Wittig reaction-ring contraction of 2-hydroxycyclobutanone: F. Cuccu, L. Serusi, A. Luridiana, F. Secci, P. Caboni, D. J. Aitken and A. Frongia, Org. Lett., 2019, 21, 7755–7758 CrossRef CAS PubMed.
- See, for instance: (a) S. Porcu, C. A. Rodriguez, A. Frongia and F. Secci, Synthesis, 2021, 53, 925–932 CrossRef CAS; (b) L. Serusi, F. Cuccu, F. Secci, D. J. Aitken and A. Frongia, Synthesis, 2021, 53, 673–681 CrossRef CAS; (c) S. Porcu, S. Demuro, A. Luridiana, A. Cocco, A. Frongia, D. J. Aitken, F. Charnay-Pouget, R. Guillot, G. Sarais and F. Secci, Org. Lett., 2018, 20, 7699–7702 CrossRef CAS PubMed; (d) A. Martis, A. Luridiana, A. Frongia, M. Arca, G. Sarais, D. J. Aitken, R. Guillot and F. Secci, Org. Biomol. Chem., 2017, 15, 10053–10063 RSC; (e) S. Porcu, A. Luridiana, A. Martis, A. Frongia, G. Sarais, D. J. Aitken, T. Boddaert, R. Guillot and F. Secci, Chem. Commun., 2018, 54, 13547–13550 RSC.
- 1,2-Bis(trimethylsilyloxy)cyclobutene was hydrolyzed with H2O with FeCl3/SiO2 as catalyst (see ref. 21). See, also: J. J. Bloomfield and J. M. Nelke, Org. Synth., 1988, 6, 167 Search PubMed.
- M. J. Brown, T. Harrison, P. M. Herrinton, M. H. Hopkins, K. D. Hutchinson, P. Mishra and L. E. Overman, J. Am. Chem. Soc., 1991, 113, 5365–5378 CrossRef CAS.
- See ESI† for details.
- F. Secci, S. Porcu, A. Luridiana, A. Frongia and P. C. Ricci, Org. Biomol. Chem., 2020, 18, 3684–3689 RSC.
- F. Turnu, A. Luridiana, A. Cocco, S. Porcu, A. Frongia, G. Sarais and F. Secci, Org. Lett., 2019, 21, 7329–7332 CrossRef CAS PubMed.
- Selectivity in favor of cyclobutanediols 4 ranged from 2/1 to 6/1.
- Our results also suggest that cis- and trans-2 could be easily separated by the selective oxidative cleavage of cis-2 to the corresponding dicarbonyl 3, leaving intact trans-2.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, characterization data, NMR spectra of all products. See DOI: https://doi.org/10.1039/d3ob00436h |
| This journal is © The Royal Society of Chemistry 2023 |