
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSeven-membered ring nucleobases as inhibitors of human cytidine deaminase and APOBEC3A†
Harikrishnan M.
Kurup
 ab,
Maksim V.
Kvach
a,
Stefan
Harjes
a,
Geoffrey B.
Jameson
ab,
Elena
Harjes
*ab and
Vyacheslav V.
Filichev
*ab
ab,
Maksim V.
Kvach
a,
Stefan
Harjes
a,
Geoffrey B.
Jameson
ab,
Elena
Harjes
*ab and
Vyacheslav V.
Filichev
*ab
aSchool of Natural Sciences, Massey University, Private Bag 11 222, Palmerston North 4442, New Zealand. E-mail: e.harjes@massey.ac.nz; v.filichev@massey.ac.nz
bMaurice Wilkins Centre for Molecular Biodiscovery, Auckland 1142, New Zealand
First published on 7th June 2023
Abstract
The APOBEC3 (APOBEC3A-H) enzyme family as a part of the human innate immune system deaminates cytosine to uracil in single-stranded DNA (ssDNA) and thereby prevents the spread of pathogenic genetic information. However, APOBEC3-induced mutagenesis promotes viral and cancer evolution, thus enabling the progression of diseases and development of drug resistance. Therefore, APOBEC3 inhibition offers a possibility to complement existing antiviral and anticancer therapies and prevent the emergence of drug resistance, thus making such therapies effective for longer periods of time. Here, we synthesised nucleosides containing seven-membered nucleobases based on azepinone and compared their inhibitory potential against human cytidine deaminase (hCDA) and APOBEC3A with previously described 2′-deoxyzebularine (dZ) and 5-fluoro-2′-deoxyzebularine (FdZ). The nanomolar inhibitor of wild-type APOBEC3A was obtained by the incorporation of 1,3,4,7-tetrahydro-2H-1,3-diazepin-2-one in the TTC loop of a DNA hairpin instead of the target 2′-deoxycytidine providing a Ki of 290 ± 40 nM, which is only slightly weaker than the Ki of the FdZ-containing inhibitor (117 ± 15 nM). A less potent but notably different inhibition of human cytidine deaminase (CDA) and engineered C-terminal domain of APOBEC3B was observed for 2′-deoxyribosides of the S and R isomers of hexahydro-5-hydroxy-azepin-2-one: the S-isomer was more active than the R-isomer. The S-isomer shows resemblance in the position of the OH-group observed recently for the hydrated dZ and FdZ in the crystal structures with APOBEC3G and APOBEC3A, respectively. This shows that 7-membered ring analogues of pyrimidine nucleosides can serve as a platform for further development of modified ssDNAs as powerful A3 inhibitors.
1. Introduction
The enzymes of the polynucleotide cytidine deaminase family of APOBEC3 (A3A-H except E) form an important part of the immune system. By hypermutating cytosine to uracil in single-stranded (ss)DNA and RNA (Fig. 1A), A3 enzymes target viruses1 and retrotransposons.2 However, several enzymes, particularly A3A, A3B, A3H and A3G, can deaminate cytosine in human nuclear and mitochondrial genomes.3 Viruses and cancer cells use this mutational activity of A3 proteins to increase the rates of mutagenesis, which allows them to escape adaptive immune responses, and become drug resistant4–8 leading to poor clinical outcomes. Of the seven A3 enzymes, three (A3A, A3B and A3H) are localised in the cell nucleus and are genotoxic in unfavourable situations. Overexpression of A3A and A3B leads to tumourigenesis in transgenic mouse models.9,10 Initially, A3B was identified as the primary source of DNA mutations in breast and other cancers,6,7,11 but more recent research also points to a prominent role of A3A in cancers.12–14 Since A3A and A3B are not essential to primary human metabolism and A3B deletion is prevalent in some populations,15 their inhibition offers a useful strategy to suppress cancer evolution and thereby make the existing anti-cancer therapies more efficient.7,16 As A3 enzymes deaminate predominantly ssDNA, we have been exploring the possibilities of ssDNA-based inhibitors that can be used as conjuvants to existing cancer therapies.17–19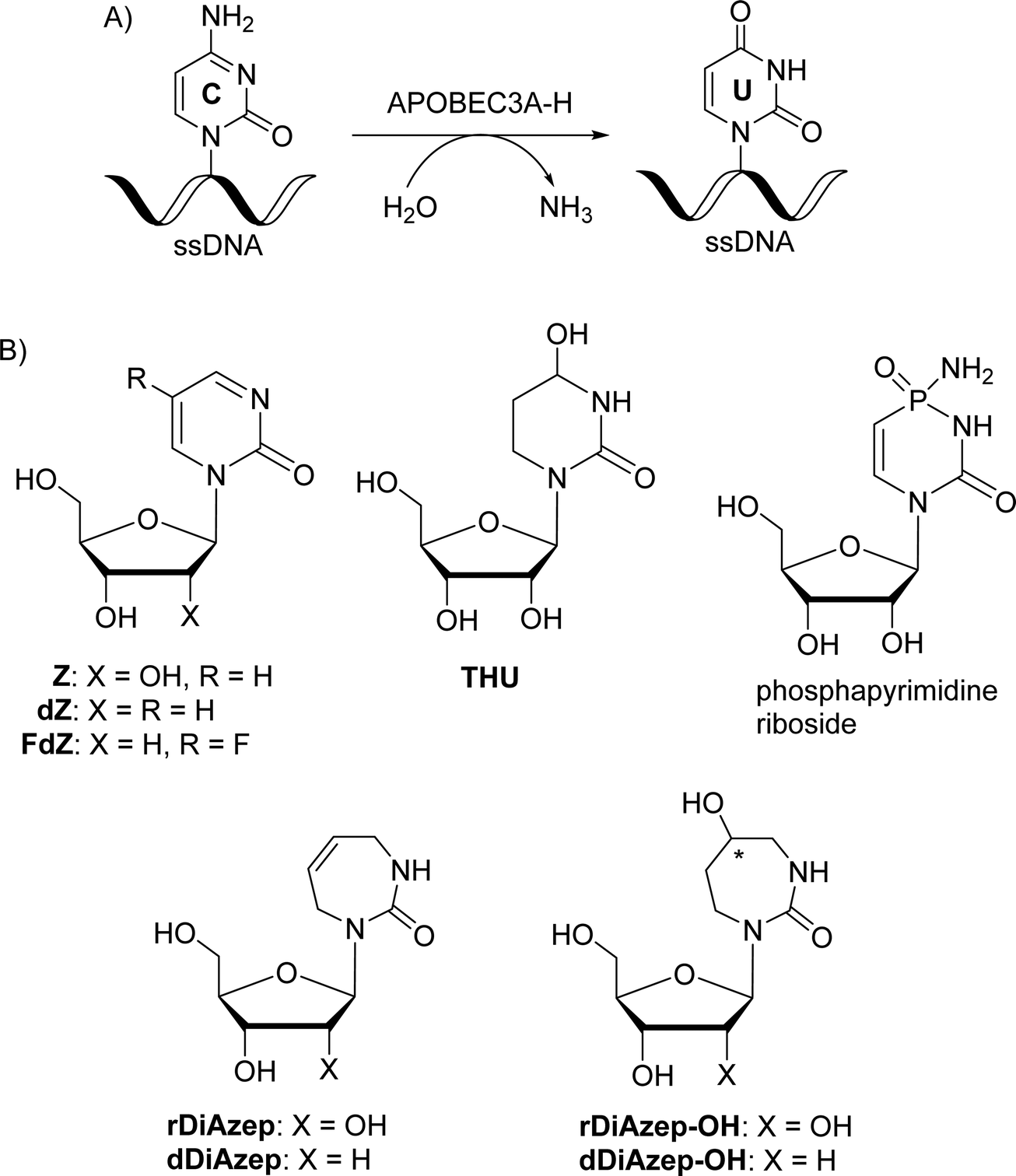 | ||
| Fig. 1 (A) Deamination of dC in ssDNA by A3 enzymes. (B) Previously described powerful CDA inhibitors. | ||
Despite low sequence identity, cytidine deaminase (CDA) and A3 share a similar overall structural topology and close structural homology for the Zn2+-containing active site. Consequently, these enzymes are believed to have a similar mechanism of cytosine deamination. The major difference between them is that CDA accepts only individual nucleosides as substrates, whereas A3s act mainly on ssDNA having at least four nucleotides, one of which is 2'-deoxycytidine.20
CDA is a key enzyme of primary metabolism that catalyses the deamination of cytidine or 2′-deoxycytidine to uridine or 2′-deoxyuridine, respectively.21,22 The spontaneous deamination of cytidine is a very slow process; the accelerated rate of deamination by the enzyme is through the formation of a hydrated intermediate species.23 CDA forms a part of the pyrimidine salvage pathway and full inhibition of CDA leads to accumulation of toxic pyrimidine catabolism intermediates.24,25 However, several cytosine-based chemotherapeutics such as cytarabine,26 gemcitabine27 and decitabine28 have been shown to be deaminated by CDA in the liver and spleen, which reduces their potency. Local and temporary inhibition of CDA in these organs is therefore a useful strategy that can mitigate detrimental degradation of chemotherapeutics. In July 2020, a combination of the known CDA inhibitor cedazuridine, (4R)-2′-deoxy-2′,2′-difluoro-3,4,5,6-tetrahydrouridine,29 and decitabine as an oral pill (C-DEC or ASTX727) developed by Astex Pharmaceuticals was approved by the US Food and Drug Administration (FDA) for the treatment of patients with intermediate/high-risk myelodysplastic syndromes (MDS) and chronic myelomonocytic leukemia (CMML).30
The most powerful inhibitors of CDA based on nucleosides described so far are 1,3-diazepin-2-one riboside (rDiAzep, Fig. 1B),31–36 phosphapyrimidine riboside (unstable in water),37 tetrahydrouridine (THU)38 and zebularine (Z).39,40 We demonstrated that the use of 2′-deoxy analogues of zebularine (dZ) and 5-fluorozebularine (FdZ) instead of dC in the preferred ssDNA substrate motif resulted in competitive inhibitors of A3.17–19 Recently, we have shown that cross-linking of two distant nucleotides at +1 and −2 positions resulted in a more powerful dZ-containing A3A inhibitor than the linear ssDNA.41 DNA hairpins with short loops have also been shown to provide selective inhibitors of A3A when the target dC in the loop was changed to dZ-derivatives.42–44 We also demonstrated that dZ and THU as free nucleosides did not inhibit A3 enzymes, which indicates that ssDNA delivers dZ into the active site of A3.17
In the past, the diazepinone riboside was described as a more powerful inhibitor of CDA than dZ (Ki = 25 nM for 1,3-diazepin-2-one31–35 and 2.9 μM for dZ).45 Hence, we decided to expand a series of 2′-deoxy nucleoside analogues of CDA inhibitors31,34,46 used in ssDNA for A3 inhibition and investigate 1,3-diazepin-2-one riboside (dDiAzep) and its hydrated derivatives (dDiAzep-OH)31–35 in this context.
2. Results and discussion
2.1. Structural considerations for the use of 1,3-diazepin-2-one nucleoside as an A3 inhibitor
The difference in the inhibitory potential of dZ and FdZ against A3BCTD and A3A19 prompted us to investigate the possibility of the synthesis and incorporation of more powerful CDA inhibitors for the creation of more powerful A3 inhibitors.Out of all the reported potent CDA inhibitors, 1,3-diazepin-2-one riboside31–35 is notably different structurally as it has no functionality at C5 to coordinate with Zn2+ or to undergo hydration in the active site of CDA. In the crystal structure of 1,3-diazepin-2-one riboside with human CDA (hCDA, PDB ID: 1MQ0),35 the protein self-associates to form a dimer of dimers (tetramer) (Fig. S2 in the ESI†) which is known to be active.19 Each subunit is bound to one molecule of diazepinone riboside but each active site is made up of three of the four hCDA monomers and all of them contribute to the recognition of the inhibitor. It is interesting that diazepinone is not coordinated to Zn2+. Instead, a water molecule is bound to Zn2+. The nucleobase N3–H is engaged in interactions with Glu67 of hCDA and the oxygen of the carbonyl C2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O is hydrogen bonded to the main chain NH of Ala66 of the first subunit. The seven-membered nucleobase is puckered and the C5–C6 double bond of the molecule is involved in a canonical π–π interaction with Phe137 residue of the third subunit of hCDA. Reduction of the double bond to the corresponding saturated nucleoside led to significant loss in binding affinity to CDA which highlights the importance of the double bond for the inhibition of hCDA.31 Interestingly, aromatic amino acids Tyr130 and Tyr313 are present at the same position in the active site of monomeric A3A and A3BCTD, respectively, instead of Phe137 in hCDA (Fig. 2). We hypothesised that a π–π interaction between the double bonds of diazepinone and Tyr130(A3A)/Tyr313(A3Bctd) will be preserved for A3A and A3B and potentially result in potent inhibition of these enzymes by diazepinone. These observations make this compound interesting to be synthesised as a 2′-deoxy analogue (dDiAzep, Fig. 1B) for testing as an A3 inhibitor when incorporated into ssDNA.
O is hydrogen bonded to the main chain NH of Ala66 of the first subunit. The seven-membered nucleobase is puckered and the C5–C6 double bond of the molecule is involved in a canonical π–π interaction with Phe137 residue of the third subunit of hCDA. Reduction of the double bond to the corresponding saturated nucleoside led to significant loss in binding affinity to CDA which highlights the importance of the double bond for the inhibition of hCDA.31 Interestingly, aromatic amino acids Tyr130 and Tyr313 are present at the same position in the active site of monomeric A3A and A3BCTD, respectively, instead of Phe137 in hCDA (Fig. 2). We hypothesised that a π–π interaction between the double bonds of diazepinone and Tyr130(A3A)/Tyr313(A3Bctd) will be preserved for A3A and A3B and potentially result in potent inhibition of these enzymes by diazepinone. These observations make this compound interesting to be synthesised as a 2′-deoxy analogue (dDiAzep, Fig. 1B) for testing as an A3 inhibitor when incorporated into ssDNA.
 | ||
| Fig. 2 Active site residues (grey) and diazepinone riboside (green) of hCDA (1MQ0) overlaid with the active site of A3A (5KEG) (cyan). Tyr 130 (cyan) of A3A is on the same place as Phe 137 (orange) of hCDA interacting with diazepinone; zinc2+ atoms are shown as spheres. Structures were overlaid in pymol. The CDA tetramer is shown in ESI Fig. S2.† | ||
To mimic the respective tetrahedral substrate intermediate in the context of 7-membered ring nucleobase, diazepinone derivatives carrying an OH-group on carbon C5 have been prepared as R and S isomers in the past (rDiAzep-OH, Fig. 1B).31 One of the isomers was identified as a slow-binding inhibitor of CDA, as potent as THU. Unfortunately, the authors were unable to determine which isomer was more potent. The lack of proper stereochemical information prompted us to attempt the synthesis, isolation, and characterisation of the stereochemistry of both dDiAzep-OH isomers before their incorporation into ssDNA as A3 inhibitors.
2.2. Synthesis of modified 2′-deoxynucleosides, their DMT-protected phosphoramidites and derived oligodeoxynucleotides
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :α ratio of 9:1. The β isomer as the major product was confirmed by a NOESY NMR experiment. The spectrum shows cross-peaks of the 1′ and 4′-protons of the major isomer of compound 3 through space, confirming it as a β-nucleoside, whereas the minor α-anomer had a cross-peak between the 1′ and 3′-protons (see Fig. S3 in the ESI†).
:α ratio of 9:1. The β isomer as the major product was confirmed by a NOESY NMR experiment. The spectrum shows cross-peaks of the 1′ and 4′-protons of the major isomer of compound 3 through space, confirming it as a β-nucleoside, whereas the minor α-anomer had a cross-peak between the 1′ and 3′-protons (see Fig. S3 in the ESI†).
 | ||
| Scheme 1 Synthesis of 2′-deoxy diazepine phosphoramidite. Reagents and conditions: (i) N,N’-disuccinimidyl carbonate, THF, r.t., 2 h; (ii) TMS chloride, Et3N, benzene, r.t., overnight; (iii) Hoffer's chlorosugar, SnCl4, 1,2-dichloroethane, −35 °C, 1 h; (iv) benzoyl chloride, pyridine, 0 °C → r.t., overnight; (v) GreenCat™, dry DCM, reflux, 2 h; (vi) 30% aq. NH3, MeOH (1:10, v/v), r.t., 3 days; (vii) 4,4′-dimethoxytrityl chloride (DMTCl), pyridine, 0 °C → r.t., overnight; (viii) N,N-diisopropylamino-2-cyanoethoxychlorophosphine (CEP-Cl), Et3N, dry DCM, 0 °C, 20 min; and (ix) 30% aq. NH3, r.t., 15 min. | ||
As reported in the literature, acylation of the amide 3 was required to lock the allyl groups in a cis orientation for the successful RCM.47 The amide 3 was protected with benzoyl chloride to provide compound 4. The subsequent RCM with the GreenCat™ catalyst provided the required seven-membered ring 5 in 41% isolated yield. Resuspending and washing the precipitate 5 in methanol gave a >99% pure β anomer. The toluoyl groups on compound 5 were selectively deprotected with a solution of aqueous ammonia in methanol followed by selective protection of the 5′-hydroxyl group using standard DMT protection conditions to provide compound 6 in 82% yield. Phosphoramidite 7 was prepared using standard phosphitylation conditions with an isolated yield of 82%.
The fully deprotected nucleoside, dDiAzep (8), was obtained by the deprotection of compound 5 in concentrated aqueous ammonia and isolated by preparative thin-layer chromatography (TLC) in 58% yield.
 | ||
| Scheme 2 Attempted hydroboration/oxidation of 1,3-diazepin-2-one nucleoside and nucleobase. Reagents and conditions: (i) 1 M BH3·THF, 0 °C → r.t., 2.5 h, H2O, 1 N NaOH, 30% H2O2, r.t., 1 h; (ii) benzoyl chloride, pyridine, 0 °C → r.t., overnight; (iii) Boc-anhydride, DMAP, Et3N, THF, reflux, overnight; (iv) GreenCat™, dry DCM, reflux, 2 h; (v) 30% aq. ammonia in MeOH (1:10, v/v), 15 min then TFA, DCM, 15 min. | ||
The above failed reaction on substrate 5 prompted us to investigate the possibility of hydroboration/oxidation on a protected nucleobase 12. We started the synthesis with the previously obtained bis-allyl urea 2. Benzoyl protection of the compound 2 followed by tert-butyloxy carbonyl (boc) protection allowed the facile RCM of compound 11 producing the protected nucleobase 12 in 50% overall yield over three steps. The attempted hydroboration/oxidation protocol resulted only in benzoyl deprotection. Next, we tried the same hydroboration/oxidation protocol on a free nucleobase 14 which was reported previously for the synthesis of compound 15.32 Unfortunately, the reported procedure did not work in our study and the starting material was isolated (Scheme 2). In the past this protocol was not chosen for the synthesis of the target rDiAzep-OH. Instead, the formation of a diastereomeric mixture of ribosides of R- and S-5-hydroxy-diazepin-2-ones (Fig. 3A) relied on the use of toxic mercury salts (HgO/HgBr2) and perhydro-1,3-diazepine-2,5-dione that has to be synthesised.31
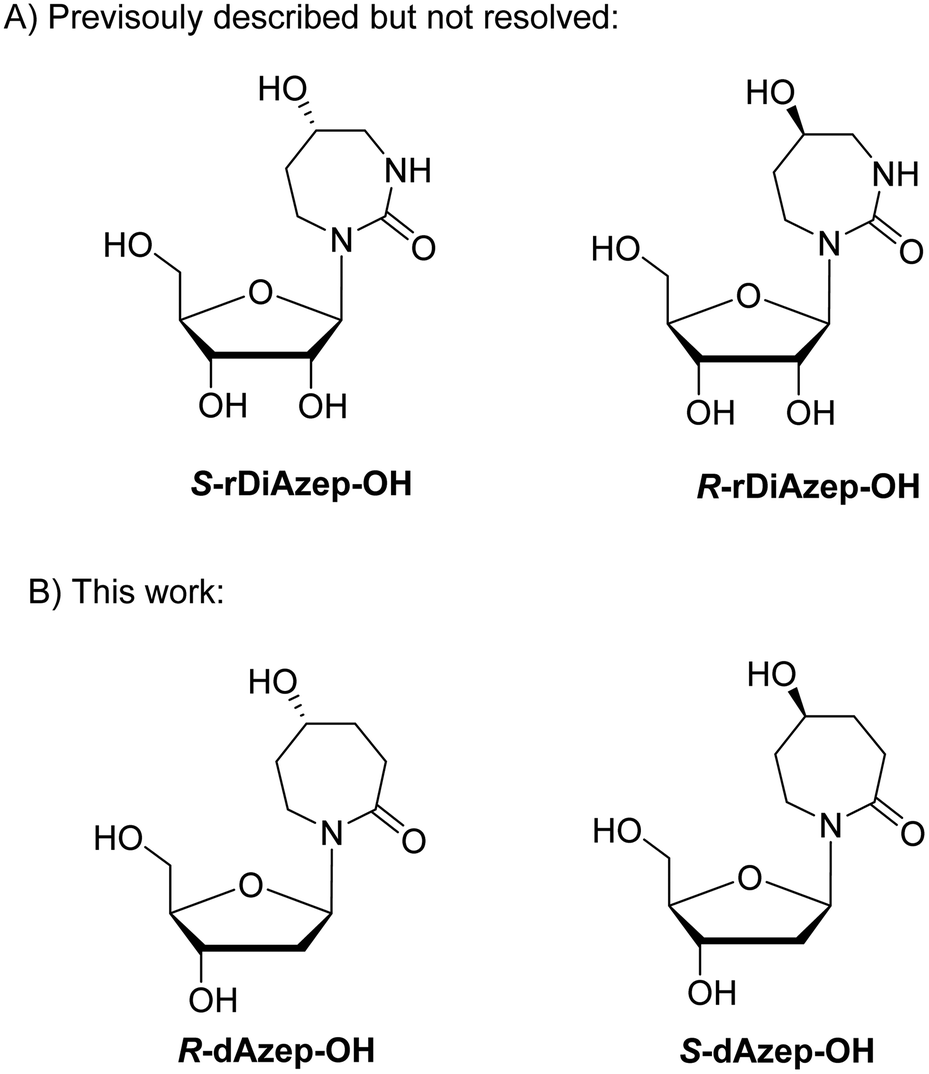 | ||
| Fig. 3 (A) Previously described ribosides of R- and S-5-hydroxy-1,3-diazepin-2-ones. (B) Proposed structure of 2′-deoxyribosides of 5-hydroxy-azepin-2-ones. | ||
We envisioned that a potent CDA and A3 inhibitor can be based on the nucleoside carrying 1,3-diazepine-2-one in which the N3 atom is replaced with CH2. One should note that the IUPAC nomenclature assignment of R and S isomers is opposite for 1,3-diazepin-2-one and azepin-2-one (Fig. 3).
The synthesis started from commercially available 1,4-cyclohexanedione cyclic ethylene monoketal (16) that was converted to oxime and then, using a Beckmann rearrangement, provided the required protected nucleobase as a 7-membered cyclic amide (17).50 This compound was coupled with Hoffer's chlorosugar51 in the presence of K-tBuO in 1,4-dioxane at room temperature to provide the required nucleoside 18 with a β:α ratio of 99:1 as determined by NOESY NMR. However, deprotection of the ketal in compound 18 under various acidic conditions failed. The difficulties of deprotecting the cyclic ketal in 7-membered ring compounds have been reported in the past.32 To circumvent these difficulties, we decided to change the synthetic route (Scheme 3) and rely on the benzyl-protecting group on the alcohol at C5 of the hexahydro azepin-2-one (21).
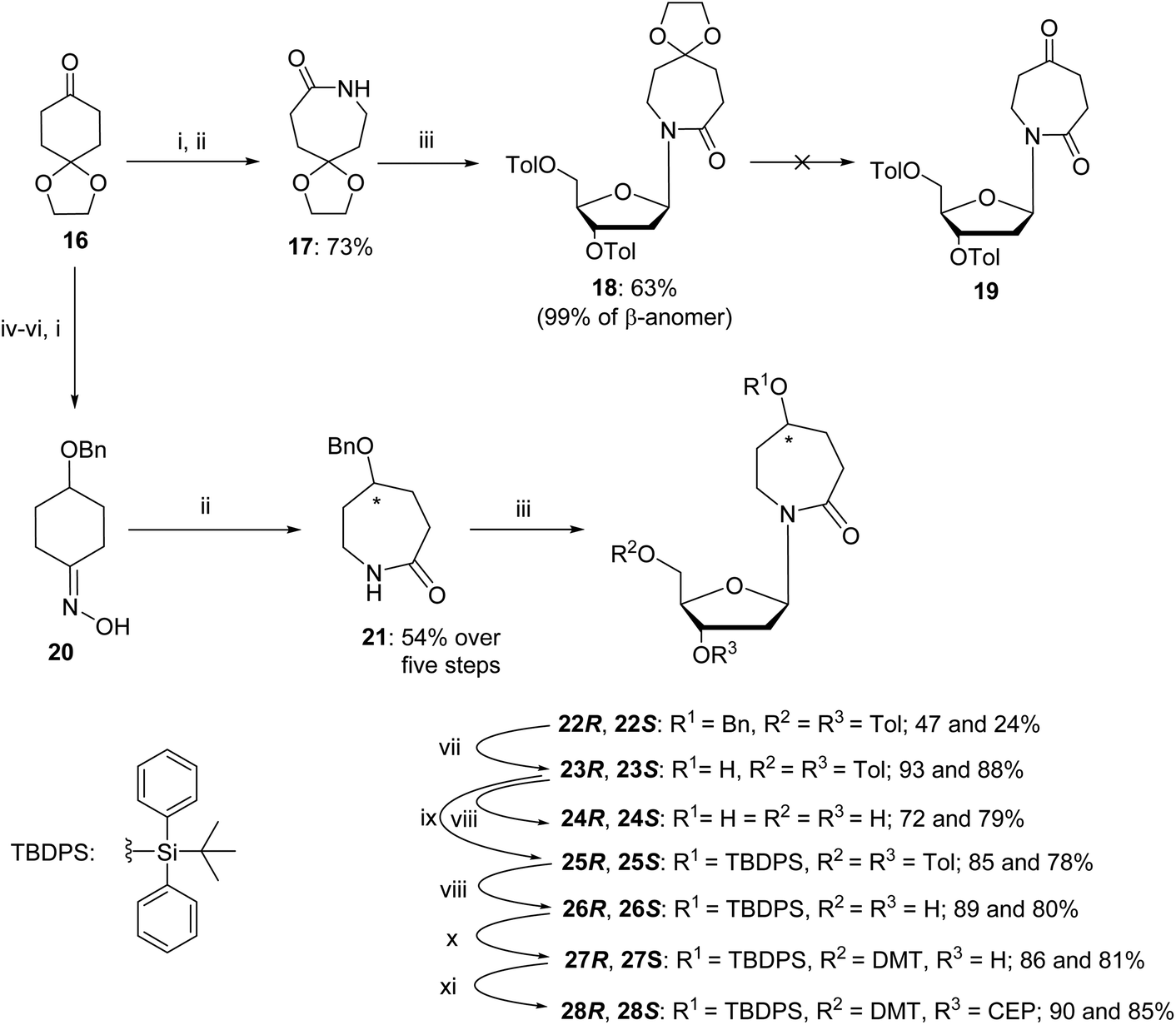 | ||
| Scheme 3 Synthesis of R-and S-dAzep nucleosides. Reagents and conditions: (i) NH2OH·HCl, NaOH, H2O, MeOH, r.t., 10 min; (ii) tosyl chloride, 4N NaOH, acetone/H2O r.t., 2 h then 4 N HCl, r.t., overnight; (iii) Hoffer's chlorosugar, K-tBuO, 1,4-dioxane, r.t., 2 h; (iv) NaBH4, MeOH, 0 °C → r.t., overnight; (v) benzyl chloride, Et3N, DMF, 0 °C → r.t., overnight; (vi) 1 N HCl, THF, r.t., overnight; (vii) 10% Pd/C, H2, EtOH, r.t., 6 h; (viii) 30% aq. NH3, MeOH, r.t., 3 days; (ix) TBDPS chloride, imidazole, DCM, 0 °C → r.t., overnight; (x) 4,4′-dimethoxytrityl chloride (DMTCl), pyridine, 0 °C → r.t., overnight; and (xi) N,N-diisopropylamino-2-cyanoethoxychlorophosphine, Et3N, DCM, r.t., 20 min. | ||
A pathway reported in the current paper includes the synthesis of the protected azepan nucleobase 21 from the cyclic ketal 16 using the Beckmann rearrangement on oxime 20 (Scheme 3).50 The cyclic amide 21 and Hoffer's chlorosugar were coupled in the presence of K-tBuO in 1,4-dioxane at room temperature to provide a pair of diastereomers 22R and 22S in moderate yields (47 and 24%, respectively) and a β:α ratio of 99:1. These diastereomers were purified using silica gel column chromatography and crystallised. The stereochemistry of each compound was assigned based on X-ray crystal data (Fig. 4). Here onwards, these diastereomers were subjected to the same sequence of reactions separately. Benzyl deprotection of 22 was performed by catalytic hydrogenation using 10% Pd/C at room temperature to give 23R and 23S with 93 and 88%, respectively. One part of the benzyl-deprotected compounds was treated with 30% aq. NH3 in MeOH at r.t. for 3 days to provide free nucleosides 24R and 24S with 72% and 79% yield, respectively, for evaluation as inhibitors of hCDA. The rest of each compound was protected with tert-butyl diphenylsilyl (TBDPS). After the removal of toluoyl groups, 5′-O-DMT-protected 3′-O-phosphoramidites 28R and 28S were prepared employing standard protocols.
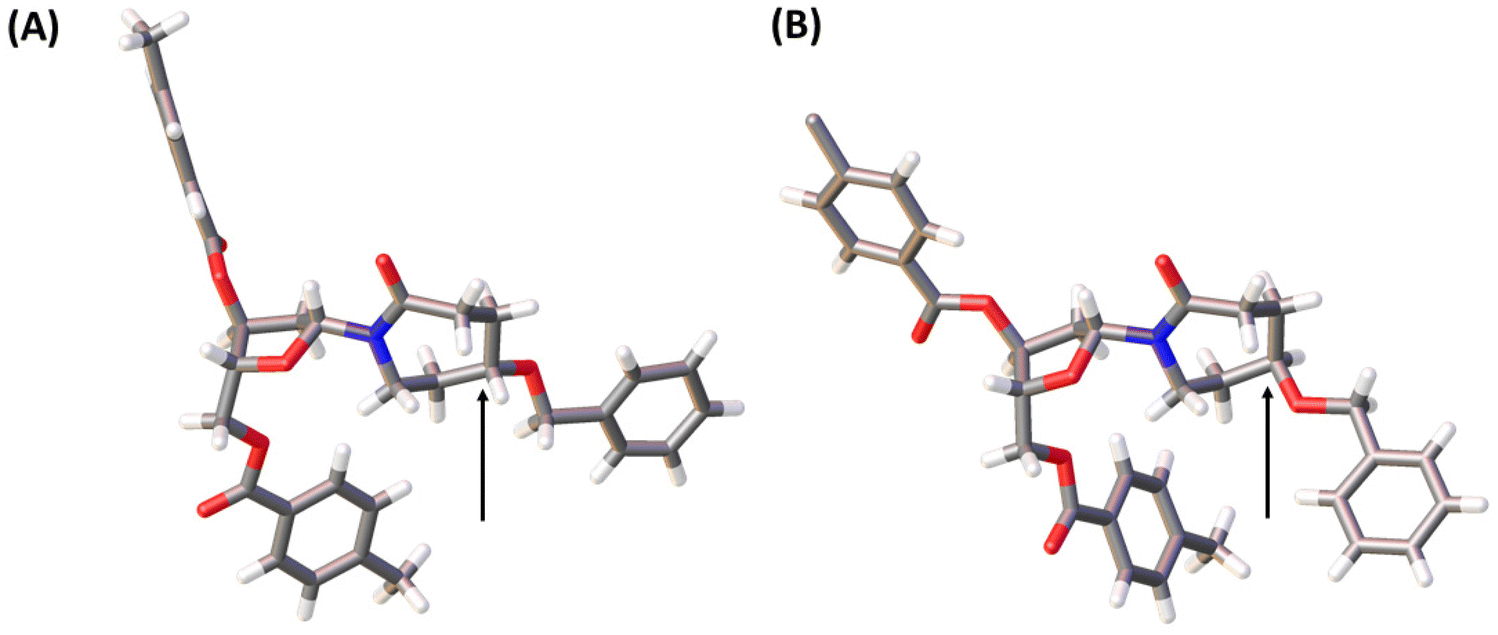 | ||
| Fig. 4 X-ray structures of 22S (A) and 22R (B). Arrows indicate the stereocenters of these compounds where they differ. The structures have been deposited with the CCDC with deposition numbers 2207174 (22S) and 2207173 (22R).† | ||
We incorporated the modified nucleosides at the location of dC in the preferred A3 substrate motifs. Detailed procedures for oligodeoxynucleotide synthesis are provided in the ESI.† All oligodeoxynucleotides were purified by reverse-phase HPLC with detection at 260 nm. ESI-MS (Table 1) confirmed their compositions.
| Name | DNA sequence, 5′ → 3′ | Retention timea (min) | ESI-MS [Da], found/calculated |
|---|---|---|---|
| a Reverse-phase HPLC on a 250/4.6 mm, 5 μm, 300 Å C18 column (Thermo Fisher Scientific) using a gradient of CH3CN (0 → 20% for 20 min, 1.3 mL min−1) in 0.1 M TEAA buffer (pH 7.0) with detection at 260 nm. | |||
| 7-mer dZ-linear | TTTT dZ AT | 14.2 | 2045.37/2045.37 |
| 9-mer dDiAzep | ATTT dDiAzep ATTT | 15.9 | 2679.48/2678.50 |
| 7-mer R-dAzep-OH | TTTT R-dAzep-OH AT | 14.9 | 2078.39/2078.41 |
| 7-mer S-dAzep-OH | TTTT S-dAzep-OH AT | 14.8 | 2078.38/2078.41 |
| FdZ-hairpin | (GC)2TT FdZ (GC)2 | 14.3 | 3311.50/3310.60 |
| dDiAzep-hairpin | T(GC)2TT dDiAzep (GC)2T | 14.3 | 3916.65/3916.69 |
| S-dAzep-OH-hairpin | T(GC)2TT S-dAzep-OH (GC)2T | 14.0 | 3934.65/3933.71 |
2.3. Evaluation of modified diazepinone derivatives as inhibitors of human CDA, engineered APOBEC3B and wild-type APOBEC3A
 | ||
| Fig. 5 Initial rate of hCDA-catalysed deamination of dC with and without inhibitors. Conditions: 100 μM dC as a substrate at specified concentrations of inhibitors, 27 nM hCDA, pH 6.0 at 25 °C. Measurements were taken at 286 nm. The suffix “pre” means that these samples were preincubated with hCDA for 5 min. | ||
By analysing the initial rate of deamination, we observed that the S-isomer of 24 was more potent than the R-isomer at all inhibitor concentrations with and without preincubation with hCDA. However, both analogues were not as powerful hCDA inhibitors as the previously described dZ. Therefore, no detailed analysis was performed. Instead, we focused our attention on the 1,3-diazepin-2-one 2′-deoxyriboside 8.
The 1,3-diazepin-2-one riboside was previously reported as a powerful inhibitor of CDA. In order to accurately compare the 2′-deoxy form of 1,3-diazepin-2-one riboside and dZ we monitored the deamination of dC at 286 nm and analysed the kinetic profiles at various inhibitor concentrations using a global regression analysis of the kinetic data using Lambert's W function.52 This method provides better estimates for Km and Vmax than non-linear regression analysis of the initial rate or any of the known linearised transformations of the Michaelis–Menten equation, such as Lineweaver–Burk, Hanes–Woolf and Eadie–Hofstee transformations.52 Then the Michaelis–Menten constant (KM) for the substrate and the inhibition constant (Ki) for each inhibitor were calculated (Table 2) assuming a competitive nature of inhibitors.
Indeed, dDiAzep nucleoside is an at least 20 times more powerful inhibitor of hCDA than dZ if Km/Ki values are compared. This warrants evaluation of dDiAzep incorporated in ssDNA as an inhibitor of A3 enzymes.
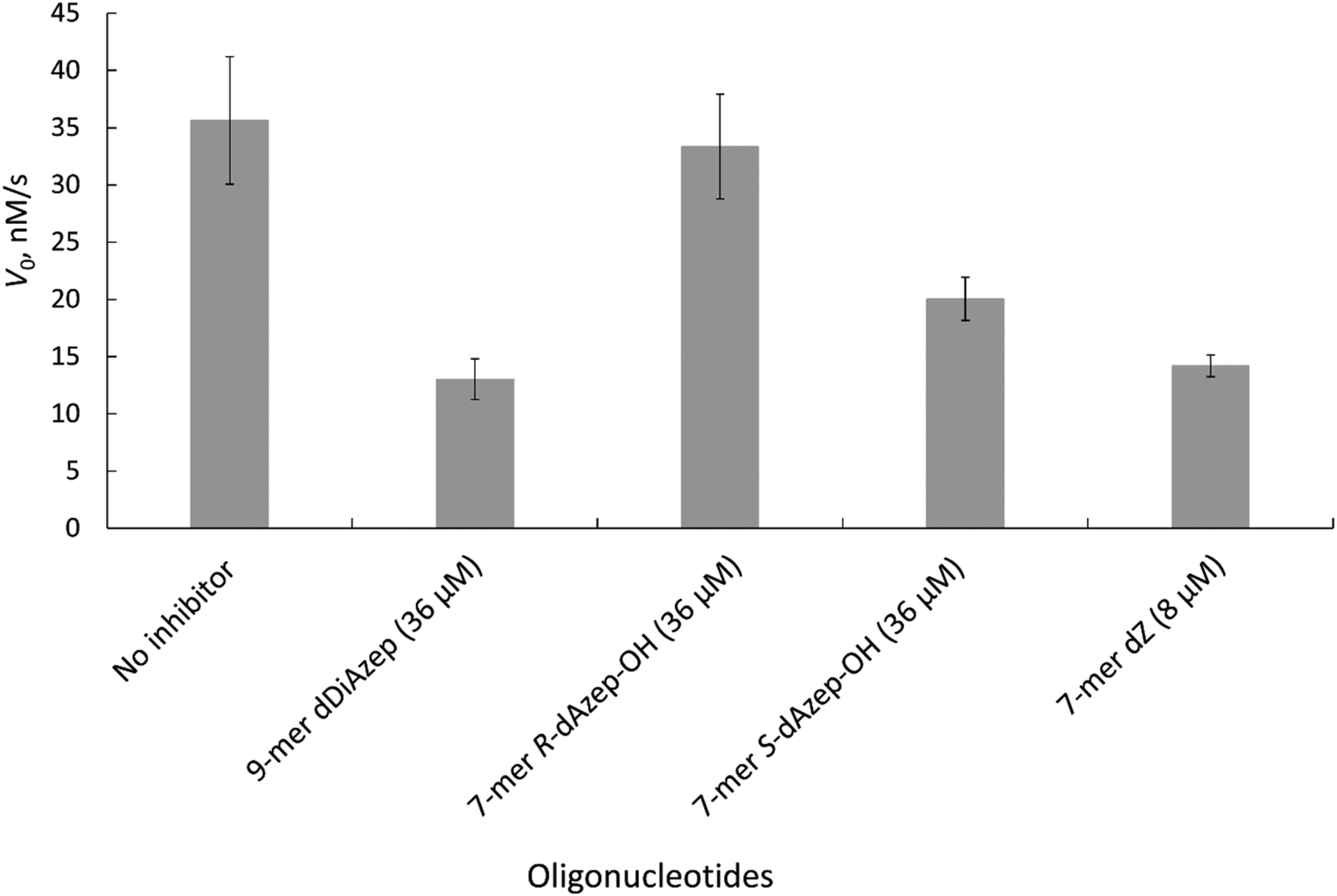 | ||
| Fig. 6 Initial rate of A3BCTD-QM-ΔL3-AL1swap-catalysed deamination of 5′-T4CAT in the absence (no inhibitor) and presence of inhibitors at the concentrations indicated. Conditions: 400 μM 5′-T4CAT, 36 μM seven-membered ring containing oligodeoxynucleotides and 8 μM dZ-containing oligodeoxynucleotide, 300 nM A3BCTD-QM-ΔL3-AL1swap in a 50 mM sodium phosphate buffer (pH 6.0) containing 100 mM NaCl, 2.5 mM β-mercaptoethanol, 50 μM 3-(trimethylsilyl)-2,2,3,3-tetradeuteropropionic acid (TSP) and 20% D2O at 25 °C. Error bars are estimated standard deviations from triplicate measurements. | ||
The results revealed that to achieve a similar level of inhibition induced by 7-mer-dZ we had to increase the concentration of oligodeoxynucleotides containing azepinone nucleotides from 8 to 36 μM. Despite the fact that dDiAzep was a more powerful inhibitor of hCDA in comparison with dZ, it did not exhibit a similar effect on engineered A3BCTD. Interestingly, the inhibition by R-dAzep-OH and S-dAzep-OH containing oligodeoxynucleotides was poor compared to the standard inhibitor (7-mer-dZ), but the difference in the inhibitory potential observed between R and S isomers was similar to that seen for hCDA (Fig. 5). This suggests the orientation of –OH on the nucleobase which mimics the intermediate formed after the attack of H2O at C4 of cytosine by A3BCTD-QM-ΔL3-AL1swap.
| Inhibitor | K i (nM) |
|---|---|
| Conditions: 50 mM Na+/K+ phosphate buffer, pH 7.4 supplemented with 100 mM NaCl, 1 mM TCEP, 100 μM DSS and 10% D2O at 20 °C; enzyme concentration: 140 nM; substrate concentration (dC-hairpin): 500 μM. Km for the dC-hairpin against A3A is 21 ± 10 μM (ref. 43) (33 ± 7 μM in experiments with the S-dazep-OH-hairpin). Full structural characterisation of the FdZ-hairpin in the complex with A3A and the activity of the FdZ-hairpin in cells will be published elsewhere.43 | |
| FdZ-linear | 2400 ± 940 |
| FdZ-hairpin43 | 117 ± 15 |
| dDiAzep-hairpin | 290 ± 40 |
| S-dAzep-OH-hairpin | 6000 ± 1000 |
The Ki values show that FdZ and dDiAzep-containing hairpins are superior inhibitors of the wild-type A3A in comparison with the best linear oligodeoxynucleotide (FdZ-linear). It is also evident from these data that FdZ and dDiAzep are more potent inhibitors of A3 when used instead of dC in the structure of the hairpin than in the linear 9-mer sequence (Fig. 6 and Table 3). A hairpin carrying S-dAzep-OH instead of dC has a Ki of 6 μM which is worse than the Ki for FdZ-linear. These data support our assertion that nucleoside-like inhibitors of CDA can be converted into inhibitors of A3 when used instead of dC in a DNA hairpin.
3. Discussion
A3 enzymes catalyse the deamination of cytosine to uracil in ssDNA substrates, thus playing a key role in innate antiviral immunity. However, the A3 family has also been linked to many mutational signatures in cancers, which sparked our interest in developing inhibitors of A3's catalytic activity as potential therapeutics and tools to study the biochemistry, structure and cellular function of these enzymes.Recently, we have shown that the incorporation of cytosine-like 2′-deoxyzebularine45,57 and 5-fluoro-2′-deoxyzebularine (dZ and FdZ on Fig. 1B) into short, linear ssDNA led to the first selective A3 inhibitors17–19 with low micromolar inhibition constants (Ki). We have proposed that the inhibitory potential of ssDNAs can be further improved through the incorporation of more potent inhibitors of CDA into ssDNA.19 Here we have considered several diazepin derivatives known to inhibit CDA and incorporated them into ssDNA as possible inhibitors of A3 enzymes. It was pleasing to note that the RCM methodology allowed us to obtain and evaluate diazepinone 2′-deoxyriboside (dDiAzep, Fig. 1B) as an inhibitor of hCDA, engineered A3BCTD and A3A. In comparison with dZ, dDiAzep with a Ki of 430 ± 80 nM was a 20 times more potent inhibitor of recombinantly expressed hCDA but it did not reach the Ki of 25 nM against human liver CDA reported forty years ago for the diazepinone riboside.36 It is plausible that different sugar puckering of ribose and 2′-deoxyribose had an effect here, as well as a different source of hCDA enzyme. The use of dDiAzep instead of dC in the loop of a DNA hairpin led to an A3A inhibitor with nM potency, exhibiting at least 70 times tighter binding to A3A than a dC-hairpin if their Ki and Km values are compared. However, the inhibitory effect of this nucleoside against hCDA was substantially higher (Km/Ki = 512, Table 2). This may be due to the differences in the π–π interactions of dDiAzep with Phe and Tyr because the binding energy of π–π interactions depends on interacting pairs.58
As a control for the evaluation of dDiAzep in the structure of a DNA hairpin, we synthesised and tested an FdZ-hairpin that showed powerful inhibition of wild-type A3A with a Ki of 117 nM.43 Full characterisation of this compound and its crystal structure with wild-type A3A will be published elsewhere.43
Unfortunately, due to chemical difficulties we had to abandon the synthesis of the 2′-deoxy form of dDiAzep-OH (Fig. 1B). Instead, we synthesised the R and S isomers of 1,3-diazepine-2-one 2′-deoxyriboside in which the N3 atom is replaced with CH2. The replacement of the nitrogen atom by CH2 might be a reason for a considerable decrease in the inhibitory potential of nucleosides against hCDA and engineered A3BCTD. However, the S-isomer was more potent than the R-isomer against both hCDA and engineered A3BCTD. Very recently, using X-ray crystallography it was revealed that FdZ and dZ form tetrahedral intermediates (4-(R)-hydroxy-3,4-dihydro-2′-deoxy-5-fluorozebularine and 4-(R)-hydroxy-3,4-dihydro-2′-deoxyzebularine) in the enzyme's active site with wild-type A3A42,43 and catalytically active C-terminal domain of A3G.59 The position of OH in hydrated FdZ and dZ is the same in the complex with CDA and A3 with carbon C4 having R-stereochemistry. Considering that the nomenclature changes when we replace N3 in 1,3-diazepin-2-one to CH2 in azepin-2-one (Fig. 3), our observation that the S-isomer is more active than the R-isomer for 5-hydroxy-azepin-2-ones is consistent with the same conformation seen for the S-isomer in the model (Fig. 7A) and for the hydrated FdZ in the crystal structure (Fig. 7B).43 This observation also implies that the 5-hydroxy group of S-dAzep-OH coordinates to Zn2+ and possibly displaces a water molecule that is usually coordinated to Zn2+ in A3's active site (Fig. 7).
 | ||
| Fig. 7 (A) Chemical structure of S-dAzep-OH in ssDNA; (B) hydration of FdZ and dZ by A3 results in 4-(R)-hydroxy-3,4-dihydro-2′-deoxyzebularines. | ||
In the past, high-throughput screening efforts led to the identification of covalent small molecule A3G inhibitors with low micromolar potencies.60,61 During our study two articles have been published on drug discovery efforts to obtain small molecule inhibitors of A3.62,63 None approaches the nM potency exhibited by our FdZ43 and dDiAzep-containing hairpins. This highlights the interest and need for powerful A3 inhibitors. It is gratifying that the 7-membered ring nucleobase upon incorporation into DNA inhibits A3A, which provides another useful scaffold for further development of A3 inhibitors.
4. Conclusion
In this study we developed a synthetic protocol for the synthesis of the 2′-deoxy derivatives of a known CDA inhibitor and a pair of new CDA inhibitors and their facile incorporation into the previously described ssDNA sequence as inhibitors of A3 enzymes. The inhibitor with the double bond (dDiAzep) was a powerful inhibitor of hCDA and showed inhibition of wild-type A3A in the nM range when used instead of dC in the hairpin with a short TTC loop. However, its inhibition of A3A was not as powerful as that exhibited by the FdZ-containing hairpin.43 The differences in the inhibitory potential of the R and S isomers of 2′-deoxyribosides of hexahydro 5-hydroxy-azepin-2-one shed light on the relative stereochemistry of the hydroxyl substituent which is important in the inhibition of CDA and A3. This study therefore provides useful information for future development of mimics of an intermediate and of transition states of cytidine deamination as powerful inhibitors of A3 enzymes.Author contributions
M. V. K., G. B. J., E. H., and V. V. F. designed the research, M. V. K. and H. M. K. performed the synthesis, and S. H. and H. M. K. performed enzymatic assays. All authors analysed the data, wrote the article, and have read and agreed to the published version of the manuscript.Conflicts of interest
The authors have filed a patent on single-stranded DNA enzyme inhibitors describing inhibitors created in this work.42Acknowledgements
The NMR, mass spectrometry and X-ray facilities at Massey University and the assistance of Dr Patrick J. B. Edwards, Mr David Lun and Dr Subo Lee are gratefully acknowledged. We thank Prof. Reuben S. Harris (HHMI and University of Texas Health, San Antonio, TX, USA) and members of his cancer research program for many helpful discussions. We are grateful for the financial support provided by the Worldwide Cancer Research (grant 16-1197), Health Research Council of New Zealand – breast cancer partnership (grant 20/1355), Palmerston North Medical Research Foundation, Maurice Wilkins Centre for Molecular Biodiscovery, Massey University Research Fund (MURF 2015, 7003 and RM20734), Kiwi Innovation Network (KiwiNet) and Massey Ventures Ltd (RM22772) and the School of Natural Sciences, Massey University.References
- R. S. Harris and J. P. Dudley, Virology, 2015, 479–480, 131–145 CrossRef CAS PubMed.
- T. Izumi, K. Shirakawa and A. Takaori-Kondo, Mini-Rev. Med. Chem., 2008, 8, 231–238 CrossRef CAS PubMed.
- R. Suspene, M.-M. Aynaud, D. Guetard, M. Henry, G. Eckhoff, A. Marchio, P. Pineau, A. Dejean, J.-P. Vartanian and S. Wain-Hobson, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 4858–4863 CrossRef CAS PubMed.
- C. Swanton, N. McGranahan, G. J. Starrett and R. S. Harris, Cancer Discovery, 2015, 5, 704–712 CrossRef CAS PubMed.
- E.-Y. Kim, R. Lorenzo-Redondo, S. J. Little, Y.-S. Chung, P. K. Phalora, I. Maljkovic Berry, J. Archer, S. Penugonda, W. Fischer, D. D. Richman, T. Bhattacharya, M. H. Malim and S. M. Wolinsky, PLoS Pathog., 2014, 10, e1004281 CrossRef PubMed.
- S. Venkatesan, R. Rosenthal, N. Kanu, N. McGranahan, J. Bartek, S. A. Quezada, J. Hare, R. S. Harris and C. Swanton, Ann. Oncol., 2018, 29, 563–572 CrossRef CAS PubMed.
- E. K. Law, A. M. Sieuwerts, K. LaPara, B. Leonard, G. J. Starrett, A. M. Molan, N. A. Temiz, R. I. Vogel, M. E. Meijer-van Gelder, F. C. G. J. Sweep, P. N. Span, J. A. Foekens, J. W. M. Martens, D. Yee and R. S. Harris, Sci. Adv., 2016, 2, e1601737 CrossRef PubMed.
- J. Zou, C. Wang, X. Ma, E. Wang and G. Peng, Cell Biosci., 2017, 7, 29 CrossRef PubMed.
- E. K. Law, R. Levin-Klein, M. C. Jarvis, H. Kim, P. P. Argyris, M. A. Carpenter, G. J. Starrett, N. A. Temiz, L. K. Larson, C. Durfee, M. B. Burns, R. I. Vogel, S. Stavrou, A. N. Aguilera, S. Wagner, D. A. Largaespada, T. K. Starr, S. R. Ross and R. S. Harris, J. Exp. Med., 2020, 217, e20200261 CrossRef CAS PubMed.
- C. Durfee, N. A. Temiz, R. Levin-Klein, P. P. Argyris, L. Alsøe, S. Carracedo, A. A. de la Vega, J. Proehl, A. M. Holzhauer, Z. J. Seeman, Y.-H. T. Lin, R. I. Vogel, R. Sotillo, H. Nilsen and R. S. Harris, bioRxiv, 2023, DOI:10.1101/2023.02.24.529970.
- A. M. Sieuwerts, S. Willis, M. B. Burns, M. P. Look, M. E. M.-V. Gelder, A. Schlicker, M. R. Heideman, H. Jacobs, L. Wessels, B. Leyland-Jones, K. P. Gray, J. A. Foekens, R. S. Harris and J. W. M. Martens, Horm. Cancer, 2014, 5, 405–413 CrossRef CAS PubMed.
- L. M. Cortez, A. L. Brown, M. A. Dennis, C. D. Collins, A. J. Brown, D. Mitchell, T. M. Mertz and S. A. Roberts, PLoS Genet., 2019, 15, e1008545 CrossRef CAS PubMed.
- M. Petljak, A. Dananberg, K. Chu, E. N. Bergstrom, J. Striepen, P. von Morgen, Y. Chen, H. Shah, J. E. Sale, L. B. Alexandrov, M. R. Stratton and J. Maciejowski, Nature, 2022, 607, 799–807 CrossRef CAS PubMed.
- P. Jalili, D. Bowen, A. Langenbucher, S. Park, K. Aguirre, R. B. Corcoran, A. G. Fleischman, M. S. Lawrence, L. Zou and R. Buisson, Nat. Commun., 2020, 11, 2971 CrossRef CAS PubMed.
- J. M. Kidd, T. L. Newman, E. Tuzun, R. Kaul and E. E. Eichler, PLoS Genet., 2007, 3, 584–592 CAS.
- M. E. Olson, R. S. Harris and D. A. Harki, Cell Chem. Biol., 2018, 25, 36–49 CrossRef CAS PubMed.
- M. V. Kvach, F. M. Barzak, S. Harjes, H. A. M. Schares, G. B. Jameson, A. M. Ayoub, R. Moorthy, H. Aihara, R. S. Harris, V. V. Filichev, D. A. Harki and E. Harjes, Biochemistry, 2019, 58, 391–400 CrossRef CAS PubMed.
- F. M. Barzak, S. Harjes, M. V. Kvach, H. M. Kurup, G. B. Jameson, V. V. Filichev and E. Harjes, Org. Biomol. Chem., 2019, 17, 9435–9441 RSC.
- M. V. Kvach, F. M. Barzak, S. Harjes, H. A. M. Schares, H. M. Kurup, K. F. Jones, L. Sutton, J. Donahue, R. T. D'Aquila, G. B. Jameson, D. A. Harki, K. L. Krause, E. Harjes and V. V. Filichev, ChemBioChem, 2020, 21, 1028–1035 CrossRef CAS PubMed.
- M. Liu, A. Mallinger, M. Tortorici, Y. Newbatt, M. Richards, A. Mirza, R. L. M. van Montfort, R. Burke, J. Blagg and T. Kaserer, ACS Chem. Biol., 2018, 13, 2427–2432 CrossRef CAS PubMed.
- T. Cacciamani, A. Vita, G. Cristalli, S. Vincenzetti, P. Natalini, S. Ruggieri, A. Amici and G. Magni, Arch. Biochem. Biophys., 1991, 290, 285–292 CrossRef CAS PubMed.
- T. P. Ko, J. J. Lin, C. Y. Hu, Y. H. Hsu, A. H. Wang and S. H. Liaw, J. Biol. Chem., 2003, 278, 19111–19117 CrossRef CAS PubMed.
- G. K. Schroeder and R. Wolfenden, Biochemistry, 2007, 46, 13638–13647 CrossRef CAS PubMed.
- M. Chen, M. Herde and C. P. Witte, Plant Physiol., 2016, 171, 799–809 Search PubMed.
- A. Frances and P. Cordelier, Mol. Ther., 2020, 28, 357–366 CrossRef CAS PubMed.
- R. Z. Mahfouz, A. Jankowska, Q. Ebrahem, X. Gu, V. Visconte, A. Tabarroki, P. Terse, J. Covey, K. Chan, Y. Ling, K. J. Engelke, M. A. Sekeres, R. Tiu, J. Maciejewski, T. Radivoyevitch and Y. Saunthararajah, Clin. Cancer Res., 2013, 19, 938–948 CrossRef CAS PubMed.
- T. K. Bjånes, L. P. Jordheim, J. Schjøtt, T. Kamceva, E. Cros-Perrial, A. Langer, G. R. de Garibay, S. Kotopoulis, E. McCormack and B. Riedel, Drug Metab. Dispos., 2020, 48, 153–158 CrossRef PubMed.
- D. Lavelle, K. Vaitkus, Y. Ling, M. A. Ruiz, R. Mahfouz, K. P. Ng, S. Negrotto, N. Smith, P. Terse, K. J. Engelke, J. Covey, K. K. Chan, J. Desimone and Y. Saunthararajah, Blood, 2012, 119, 1240–1247 CrossRef CAS PubMed.
- D. Ferraris, B. Duvall, G. Delahanty, B. Mistry, J. Alt, C. Rojas, C. Rowbottom, K. Sanders, E. Schuck, K.-C. Huang, S. Redkar, B. B. Slusher and T. Tsukamoto, J. Med. Chem., 2014, 57, 2582–2588 CrossRef CAS PubMed.
- A. A. Patel, K. Cahill, C. Saygin and O. Odenike, Blood Adv., 2021, 5, 2264–2271 CrossRef CAS PubMed.
- V. E. Marquez, P. S. Liu, J. A. Kelley, J. S. Driscoll and J. J. McCormack, J. Med. Chem., 1980, 23, 713–715 CrossRef CAS PubMed.
- V. E. Marquez, P. S. Liu, J. A. Kelley and J. S. Driscoll, J. Org. Chem., 1980, 45, 485–489 CrossRef CAS.
- V. E. Marquez, K. V. B. Rao, J. V. Silverton and J. A. Kelley, J. Org. Chem., 1984, 49, 912–919 CrossRef CAS.
- M. Kim, K. Gajulapati, C. Kim, H. Y. Jung, J. Goo, K. Lee, N. Kaur, H. J. Kang, S. J. Chung and Y. Choi, ChemComm, 2012, 48, 11443–11445 RSC.
- S. J. Chung, J. C. Fromme and G. L. Verdine, J. Med. Chem., 2005, 48, 658–660 CrossRef CAS PubMed.
- P. S. Liu, V. E. Marquez, J. S. Driscoll, R. W. Fuller and J. J. McCormack, J. Med. Chem., 1981, 24, 662–666 CrossRef CAS PubMed.
- G. W. Ashley and P. A. Bartlett, J. Biol. Chem., 1984, 259, 13621–13627 CrossRef CAS PubMed.
- R. M. Cohen and R. Wolfenden, J. Biol. Chem., 1971, 246, 7561–7565 CrossRef CAS PubMed.
- C. H. Kim, V. E. Marquez, D. T. Mao, D. R. Haines and J. J. McCormack, J. Med. Chem., 1986, 29, 1374–1380 CrossRef CAS PubMed.
- L. Frick, C. Yang, V. E. Marquez and R. V. Wolfenden, Biochemistry, 1989, 28, 9423–9430 CrossRef CAS PubMed.
- H. M. Kurup, M. V. Kvach, S. Harjes, F. M. Barzak, G. B. Jameson, E. Harjes and V. V. Filichev, Biochemistry, 2022, 61, 2568–2578 CrossRef CAS PubMed.
- F. M. Y. Barzak, V. V. Filichev, E. Harjes, S. Harjes, G. B. Jameson, H. M. Kurup, M. V. Kvach and Y. Su, WO2022162536, 2022.
- S. Harjes, H. M. Kurup, A. E. Rieffer, M. Bayarjagal, J. Filitcheva, Y. Su, T. K. Hale, V. V. Filichev, E. Harjes, R. S. Harris and G. B. Jameson, bioRxiv, 2023, DOI:10.1101/2023.02.17.528918.
- J. C. Serrano, D. von Trentini, K. N. Berrios, A. Barka, I. J. Dmochowski and R. M. Kohli, ACS Chem. Biol., 2022, 17, 3379–3388 CrossRef CAS PubMed.
- J. J. Barchi, A. Haces, V. E. Marquez and J. J. McCormack, Nucleosides Nucleotides, 1992, 11, 1781–1793 CrossRef CAS.
- P. S. Liu, V. E. Marquez, J. A. Kelley and J. S. Driscoll, J. Org. Chem., 1980, 45, 5225–5227 CrossRef CAS.
- O. R. Ludek, G. K. Schroeder, C. Liao, P. L. Russ, R. Wolfenden and V. E. Marquez, J. Org. Chem., 2009, 74, 6212–6223 CrossRef CAS PubMed.
- S. Belyakov, B. Duvall, D. Ferraris, G. Hamilton and M. Vaal, WO2010118010, 2010.
- M. P. Kotick, C. Szantay and T. J. Bardos, J. Org. Chem., 1969, 34, 3806–3813 CrossRef CAS PubMed.
- E. Tarkin-Tas and L. J. Mathias, Macromolecules, 2010, 43, 968–974 CrossRef CAS.
- V. Rolland, M. Kotera and J. Lhomme, Synth. Commun., 1997, 27, 3505–3511 CrossRef CAS.
- M. Paar, W. Schrabmair, M. Mairold, K. Oettl and G. Reibnegger, ChemistrySelect, 2019, 4, 1903–1908 CrossRef CAS.
- M. J. Grillo, K. F. M. Jones, M. A. Carpenter, R. S. Harris and D. A. Harki, Trends Pharmacol. Sci., 2022, 43, 362–377 CrossRef CAS PubMed.
- T. V. Silvas, S. Hou, W. Myint, E. Nalivaika, M. Somasundaran, B. A. Kelch, H. Matsuo, N. K. Yilmaz and C. A. Schiffer, Sci. Rep., 2018, 8, 7511 CrossRef PubMed.
- S. Hou, T. V. Silvas, F. Leidner, E. A. Nalivaika, H. Matsuo, N. K. Yilmaz and C. A. Schiffer, J. Chem. Theory Comput., 2019, 15, 637–647 CrossRef CAS PubMed.
- R. Buisson, A. Langenbucher, D. Bowen, E. E. Kwan, C. H. Benes, L. Zou and M. S. Lawrence, Science, 2019, 364, eaaw2872 CrossRef CAS PubMed.
- V. E. Marquez, J. J. Barchi Jr., J. A. Kelley, K. V. Rao, R. Agbaria, T. Ben-Kasus, J. C. Cheng, C. B. Yoo and P. A. Jones, Nucleosides, Nucleotides Nucleic Acids, 2005, 24, 305–318 CrossRef CAS PubMed.
- K. A. Wilson, J. L. Kellie and S. D. Wetmore, Nucleic Acids Res., 2014, 42, 6726–6741 CrossRef CAS PubMed.
- A. Maiti, A. K. Hedger, W. Myint, V. Balachandran, J. K. Watts, C. A. Schiffer and H. Matsuo, Nat. Commun., 2022, 13, 7117 CrossRef PubMed.
- M. Li, S. M. D. Shandilya, M. A. Carpenter, A. Rathore, W. L. Brown, A. L. Perkins, D. A. Harki, J. Solberg, D. J. Hook, K. K. Pandey, M. A. Parniak, J. R. Johnson, N. J. Krogan, M. Somasundaran, A. Ali, C. A. Schiffer and R. S. Harris, ACS Chem. Biol., 2012, 7, 506–517 CrossRef CAS PubMed.
- M. E. Olson, M. Li, R. S. Harris and D. A. Harki, ChemMedChem, 2013, 8, 112–117 CrossRef CAS PubMed.
- J. J. King, F. Borzooee, J. Im, M. Asgharpour, A. Ghorbani, C. P. Diamond, H. Fifield, L. Berghuis and M. Larijani, ACS Pharmacol. Transl. Sci., 2021, 4, 1390–1407 CrossRef CAS PubMed.
- Y.-H. Zhang, X.-C. Guo, J.-B. Zhong, D.-X. Zhong, X.-H. Huang, Z.-Y. Fang, C. Zhang and Y.-J. Lu, ChemistrySelect, 2022, 7, e202201456 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details of the synthesis of nucleosides and modified oligodeoxynucleotides and enzymatic assays; and the sequence alignment of proteins used in this study, 1H, 13C, 31P NMR, IR and HRMS (ESI) spectra of new compounds synthesised, RP-HPLC profiles and HRMS (ESI) spectra of oligodeoxynucleotides. CCDC 2207173 and 2207174. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3ob00392b |
| This journal is © The Royal Society of Chemistry 2023 |