Synthesis of SARS-CoV-2 Mpro inhibitors bearing a cinnamic ester warhead with in vitro activity against human coronaviruses†
Received
10th March 2023
, Accepted 12th April 2023
First published on 13th April 2023
Abstract
COVID-19 now ranks among the most devastating global pandemics in history. The causative virus, SARS-CoV-2, is a new human coronavirus (hCoV) that spreads among humans and animals. Great efforts have been made to develop therapeutic agents to treat COVID-19, and among the available viral molecular targets, the cysteine protease SARS-CoV-2 Mpro is considered the most appealing one due to its essential role in viral replication. However, the inhibition of Mpro activity is an interesting challenge and several small molecules and peptidomimetics have been synthesized for this purpose. In this work, the Michael acceptor cinnamic ester was employed as an electrophilic warhead for the covalent inhibition of Mpro by endowing some peptidomimetic derivatives with such a functionality. Among the synthesized compounds, the indole-based inhibitors 17 and 18 efficiently impaired the in vitro replication of beta hCoV-OC-43 in the low micromolar range (EC50 = 9.14 μM and 10.1 μM, respectively). Moreover, the carbamate derivative 12 showed an antiviral activity of note (EC50 = 5.27 μM) against another hCoV, namely hCoV-229E, thus suggesting the potential applicability of such cinnamic pseudopeptides also against human alpha CoVs. Taken together, these results support the feasibility of considering the cinnamic framework for the development of new Mpro inhibitors endowed with antiviral activity against human coronaviruses.
Introduction
Coronaviruses (CoVs) belong to a family of RNA viruses responsible for human Respiratory Tract Infections (RTIs) ranging from the common cold (hCoV-229E, hCoV-OC43, hCoV-NL63, and hCoV-HKU1) to severe pneumonia (SARS-CoV, MERS-CoV and SARS-CoV-2).1–5 The ongoing COVID-19 pandemic, caused by SARS-CoV-2, is to date responsible for more than 6.7 million deaths worldwide and despite the currently available vaccines showing high efficacy in preventing serious respiratory complications in infected patients, the incessant mutations of the corresponding viral S antigen may reduce their effectivness.6 In this regard, the development of secure and efficacious drugs to suppress viral replication and treat acute forms of COVID-19 remains a valid therapeutic option. Among the limited SARS-CoV-2 molecular targets, Main Protease (called Mpro or 3CLpro) turned out to be the most appealing and “druggable” one, even for the development of new pan-CoV Direct-Acting Antivirals (DAA), because of its high degree of conservation among Coronaviridae family members. The Mpro binding site is highly different from that of known human cysteine proteases, a feature that allows specific enzymatic inhibitions with reduced toxicity.7 Mpro plays an indispensable role in the replication of hCoVs by orchestrating the proteolytic processing of 11 cleavage sites within the non-structural polyproteins pp1a and pp1ab to produce mature functional proteins.8 The homodimeric Mpro is a chymotrypsin-like cysteine protease characterized by three distinct domains (I, II and III) in each monomer. In a cleft between domain I and II, a catalytic dyad formed by Cys145 and His41 performs proteolytic cleavage, with Cys acting as a nucleophile in the course of an acyl transfer enzymatic reaction with the help of a buried water molecule.9 Mpro is so far the most characterized SARS-CoV-2 druggable target for the development of small-molecule DAA for oral therapies, and to date, a significant number of studies have been conducted to identify potent and selective Mpro inhibitors.10 In contrast, the specific inhibition of papain-like protease (PLpro), the other SARS-CoV-2 protease involved in polyprotein processing, is more challenging, as this enzyme possesses a deubiquitinating activity. Both PLpro and human DUBs bind ubiquitin at the extended C-terminus with the consensus sequence Leu-X-Gly-Gly, thus raising potential concerns about the aspecific effects of PLpro inhibitors against human DUBs.11 Therefore, one of the main issues in the development of anti-CoV Mpro inhibitors lies in their selectivity in comparison with PLpro inhibitors. Although many small molecules have been designed and synthesized to selectively inhibit the Mpro activity, peptidomimetics, which mimic natural peptide substrates, are the most investigated chemotypes, as demonstrated by Pfizer's marketing of Paxlovid (combination of the Mpro inhibitor nirmatrelvir and the antiretroviral drug ritonavir) as the first oral anti-COVID-19 treatment.12 The most rational approach in the design of peptidomimetic inhibitors is based on the insertion of a C-terminal electrophilic “warhead” capable of covalently binding to the thiol group of the catalytic Cys145, providing the peptide backbone with suitable recognition elements involved in the interaction with the enzymatic pocket. Since 2020, a large number of peptidomimetic inhibitors have been discovered or repurposed in an attempt to target Mpro (Fig. 1), bearing various electrophilic warheads, such as epoxy- (1), hydroxymethyl- (2), fluoromethyl- (3), acyloxymethyl ketone (4), keto amide (5), aldehyde (6), nitrile (as in nirmatrelvir) and Michael acceptor vinyl ester (7).7
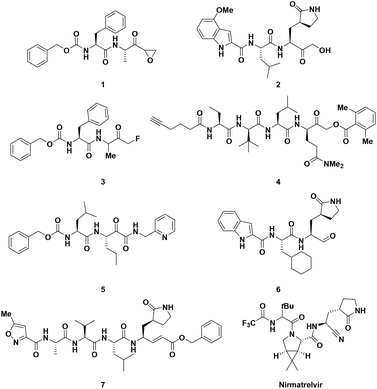 |
| | Fig. 1 Examples of peptidomimetic inhibitors of SARS-CoV-2 Mpro bearing different electrophilic warheads. | |
α,β-Unsaturated carbonyl compounds have long been studied and used as electrophilic “warheads” to covalently bind the thiol groups of cysteine proteases.13 However, very little interest has been directed to the use of the cinnamic ester functional group, although cinnamic derivatives have found various other applications, mostly as anticancer, antioxidant, and antimicrobial agents, inter alia.14–16 Taking into account the relevant reactivity of conjugated double bonds towards nucleophiles, we decided to exploit the cinnamic ester moiety for the design of novel peptide-based SARS-CoV-2 Mpro inhibitors, in the context of our ongoing research activity aimed at the identification of protease inhibitors.17–19 In this study we present a panel of 11 unprecedented p-aminocinnamic ethyl esters derivatives, which are joined to a L-Phe residue variously decorated at the N-terminus with carbamate, urea and indole-bearing amide recognition functionalities. The enzymatic assays against viral proteases revealed that these compounds are selective inhibitors of SARS-CoV-2 Mpro with IC50 values in the lower micromolar range, in comparison to that of PLpro (no inhibition at 20 μM). The best inhibitor turned out to be the carbamate derivative 11 that exhibited a single digit micromolar value (IC50 = 1.9 μM) and was selected for in silico docking studies inside the Mpro binding pocket to clarify the most relevant interactions. All cinnamic derivatives were also tested against two representative human coronaviruses, hCoV-229E and hCoV-OC43, to evaluate their potential in vitro antiviral activity. The obtained results indicate that the indole-based amide inhibitors 17 and 18 efficiently reduce the replication of hCoV-OC43 in the low micromolar range (EC50 = 9.14 μM and 10.1 μM, respectively), while the carbamate derivative 12 was found to be a potent and selective inhibitor of hCoV-229E replication (EC50 = 5.27 μM).
Results and discussion
Preliminary studies
The design of the lead compound started from the selection of the irreversible inhibitor of Mpro epoxy ketone 1, already investigated in previous literature studies.17,20 The chemical structure of 1 consists of a pseudo-dipeptide backbone made of an N-terminal carbobenzyloxy-L-phenylalanine residue, selected as the recognition motif for Mpro, linked through an amide bond to a modified alanine. A C-terminal electrophilic epoxy ketone warhead was introduced to trap the thiol group of Cys145 (Fig. 2). Replacement of the epoxy ketone moiety could represent a suitable approach to avoid the side effect due to non-specific reactivity. Elagawany et al. performed a ligand-based design supplanting the electrophile warhead of 1 with a 5-nitrothiazole moiety, extracted from nitazoxanide, a micromolar inhibitor of SARS-CoV-2 replication in vitro, producing a panel of inhibitors that showed modest inhibitory activity against SARS-CoV-2 Mpro.20 Recently, we discovered MPD112 (Fig. 2), a trifluoromethyl diazirine inhibitor, that showed potent inhibitory activity against SARS-CoV-2 Mpro (IC50 = 4.1 μM) and an optimal safety profile.17 Inspired by the aforementioned rationale, we decided to combine the N-terminal carbobenzyloxy-L-phenylalanine recognition motif with a cinnamic ester electrophile warhead, which is able to react with the thiol group of Cys145. Therefore, compound 9 was synthesized (Scheme 1) and evaluated against SARS-CoV-2 Mpro in a FRET-based assay, leading to 40% inhibition of the enzymatic activity at 20 μM concentration (screening test). Such a preliminary result prompted us to maintain the cinnamic ester moiety as the steady electrophilic warhead in the final structure of all designed derivatives and to expand the chemical diversity by modifying the N-terminal region. The L-phenylalanine (Phe) core was then connected to different decorated (hetero)aromatic rings by replacing the carbamate linkage of 9 with a urea bond (to enhance chemical stability) or an indole-bearing amide (according to the recurring presence of the indole scaffold among Mpro inhibitors).21
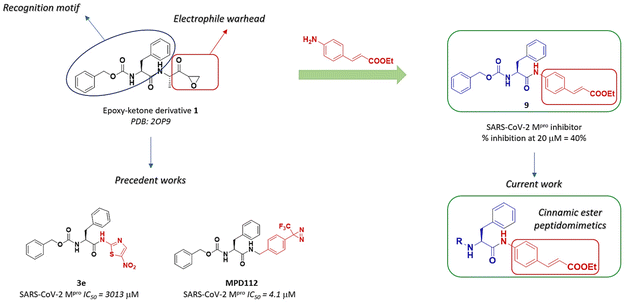 |
| | Fig. 2 Design of target p-aminocinnamic ethyl ester derivatives. | |
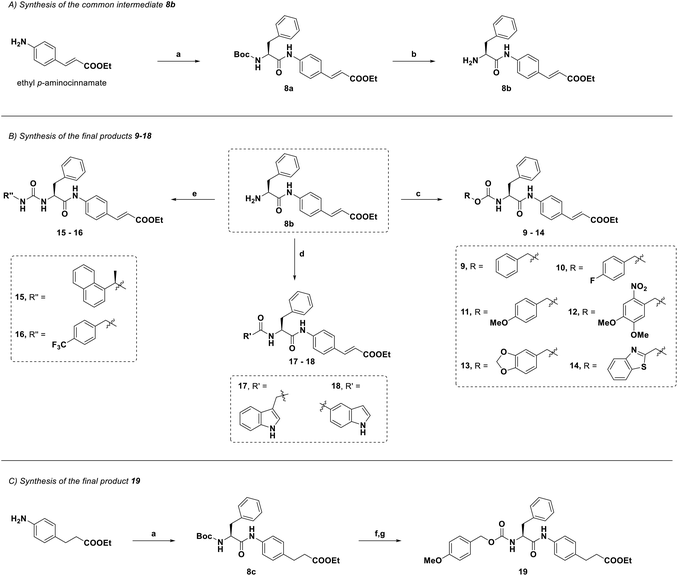 |
| | Scheme 1 Synthesis of 9–19. Reagents and conditions: (a) Boc-Phe-OH, EDCI, HOBt, dry DMF, rt, overnight, 84–49%; (b) TFA, dry DCM, rt, 4 h, then K2CO3, 91%; (c) 4-NO2PhOCO2R, DIPEA, dry THF, 0 °C to rt, overnight, 32–68%; (d) R'CO2H, EDCI, HOBt, dry DMF, rt, overnight, 31–45%; (e) R′′NCO, NEt3, dry THF, 0 °C to rt, overnight, 60–88%. (f) TFA, dry DCM, rt, 4 h, quant. yield; (g) 4-methoxybenzyl (4-nitrophenyl) carbonate, DIPEA, dry THF, rt, overnight, 38%. | |
Chemistry
The synthetic strategy is depicted in Scheme 1. The common intermediate 8b was obtained by EDCI/HOBt-mediated amide coupling between ethyl p-aminocinnamate and Boc-Phe-OH (a) and subsequent Boc-deprotection by TFA (b). Then, by reacting with the appropriate carbonate/isocyanate in the presence of a base, 8b afforded the corresponding carbamates 9–14 and ureas 15 and 16 in good yields (c and e). By means of a coupling reaction with an indole-bearing carboxylic acid, 8b was then converted to amides 17 and 18 (d). To afford 19, a two-step method was performed starting from the coupling of the saturated analogue of the ethyl p-aminocinnamate starting material with Boc-Phe-OH. Intermediate 8c was then deprotected and reacted with the appropriate carbonate derivative to afford 19. The purity of all final compounds was confirmed as >95% by HPLC analysis.
Enzyme inhibition assays
Compounds 9–18 were tested for their inhibitory activity against the recombinant SARS-CoV-2 Mpro and the results are shown in Table 1. Among the carbamate derivatives, 11, bearing the p-OMe substituent on the phenyl ring, turned out to be the most active compound, with an IC50 of 1.9 μM. The replacement of the methoxy group with a fluorine atom (10, 26% at 20 μM) led to a significant loss of activity. The presence of an additional –OMe and a –NO2 group on the phenyl ring (12, IC50 = 14.0 μM) or the substitution of the benzyl group with the piperonyl one (13, IC50 = 18.72 μM) afforded compounds with moderate activity. The two urea derivatives 15 and 16 showed contrasting outcomes in the assay on the basis of the chemical difference of the substituents. The derivative bearing the bulkier 1-(2-naphthyl)ethyl substituent displayed effective inhibition (15, IC50 = 6.8 μM), whereas the other (i.e. the p-CF3-Bn-substituted) showed a very modest inhibition (16, 17% at 20 μM). The indole-bearing amides 17 and 18 also showed good inhibition profiles (IC50 = 12.4 μM and 14.5 μM, respectively). All compounds were also evaluated against SARS-CoV-2 PLpro and they showed no significant inhibition percentage at 20 μM, therefore demonstrating to be extremely selective towards SARS-CoV-2 Mpro (Table 1).
Table 1
In vitro inhibitory activity (% inh. at 20 μM and IC50, μM) of compounds 9–19 against SARS-CoV-2 Mpro and PLpro
| Compound |
SARS-CoV-2 Mpro |
SARS-CoV-2 Mpro |
SARS-CoV-2 PLpro |
| % inh. |
IC50 (μM) |
% inh. |
| n.d. = not determined; n.i. = no inhibition. IC50 values are the means ± SD of data derived from two experiments performed in triplicate |
|
9
|
40% |
n.d. |
n.i. |
|
10
|
26% |
n.d. |
n.i. |
|
11
|
∼100% |
1.9 ± 0.8 |
n.i. |
|
12
|
59% |
14.0 ± 0.2 |
n.i. |
|
13
|
53% |
18.7 ± 0.02 |
n.i. |
|
14
|
31% |
n.d. |
n.i. |
|
15
|
66% |
6.8 ± 0.6 |
n.i. |
|
16
|
17% |
n.d. |
n.i. |
|
17
|
59% |
12.4 ± 0.7 |
n.i. |
|
18
|
58% |
14.5 ± 0.5 |
n.i. |
|
19
|
n.i. |
— |
n.i. |
|
Nirmatrelvir
|
|
0.03 ± 0.0004 |
|
Biomimetic HPLC-MS and ESI-MS experiments with Mpro
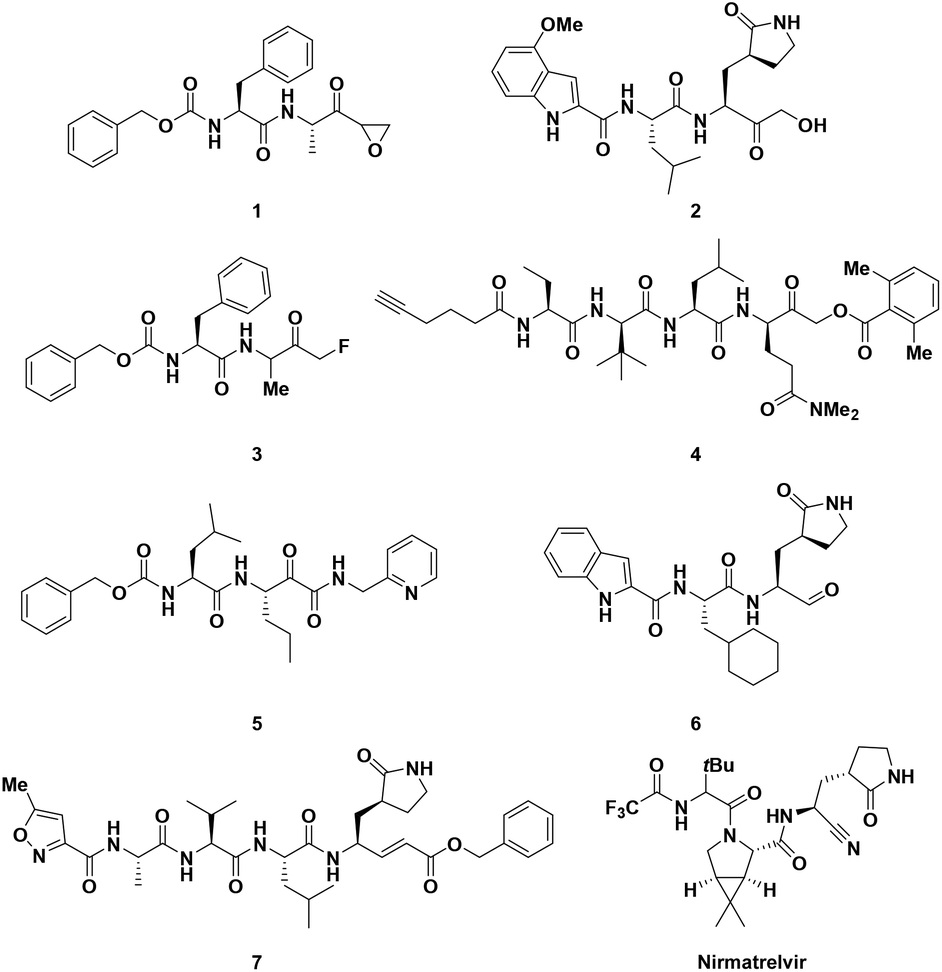
Biomimetic experiments were performed to shed light on the interaction of 11 with the target cysteine protease.2211 was incubated with N-acetyl cysteine in buffer (pH = 7.4) containing 10% ACN at 37 °C and the outcome was analyzed via HPLC-MS to measure the consumption of the cinnamic derivative (Fig. 3). The analysis revealed the appearance of a new peak (m/z = 666.4) corresponding to the Michael adduct of N-acetyl cysteine with 11. After 24 h, a conversion of about 70% was observed. Aiming to confirm the supposed mechanism of action, we then synthesized 19 (Scheme 1), the analogue of 11, which does not have the double bond. Compound 19 proved to have no inhibitory activity and it also did not undergo Michael addition with the thiol group of N-acetyl cysteine in the prementioned test, supporting our hypothesis of covalent inhibition.
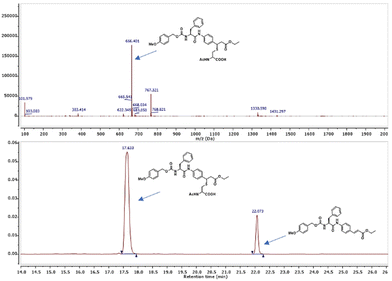 |
| | Fig. 3 LC-MS biomimetic experiment to confirm the reactivity of 11 with the thiol group of N-acetyl cysteine. | |
Mass analyses were also used to further investigate the reactivity of Cys145 with the cinnamic compounds and thereby confirm the mechanism of action.23 Mpro was incubated with 11 (compound![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :protein ratio 50:1) and the experiment was followed by ESI-MS analyses (Fig. S1†). After 1 h of incubation, in addition to the signals corresponding to multiple charge states of the non-reacted protein, the appearance of a new distribution of signals corresponding to the Mpro covalently modified by 11 was observed.
:protein ratio 50:1) and the experiment was followed by ESI-MS analyses (Fig. S1†). After 1 h of incubation, in addition to the signals corresponding to multiple charge states of the non-reacted protein, the appearance of a new distribution of signals corresponding to the Mpro covalently modified by 11 was observed.
Docking studies
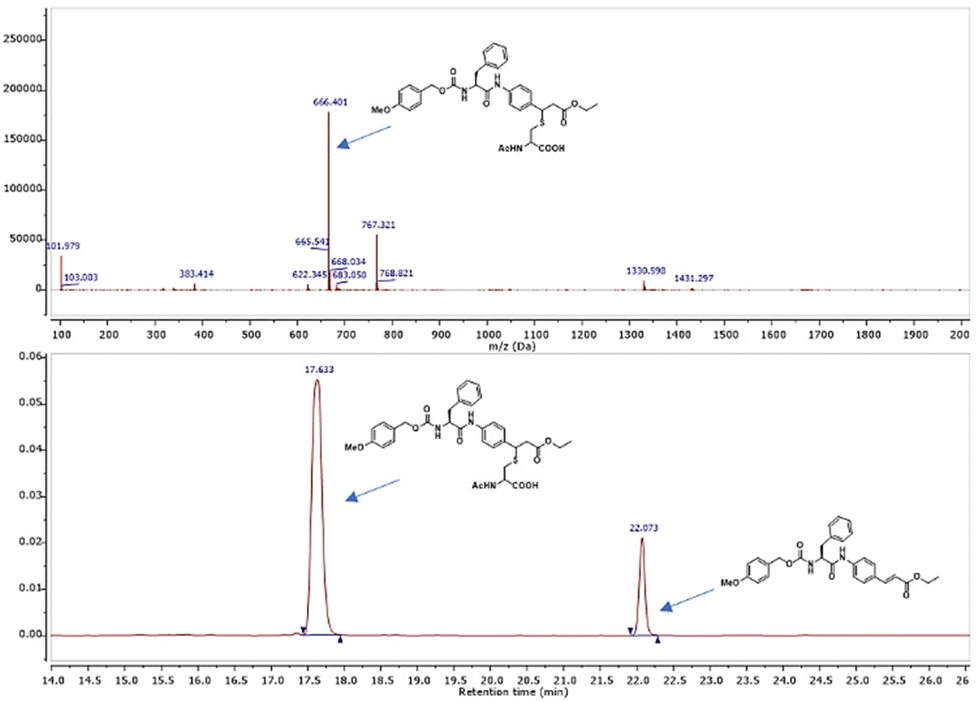
Compounds 9–18 were computationally evaluated using a combination of non-covalent and covalent docking techniques. Non-covalent docking was performed to identify the best-fit conformation of each molecule within the Mpro binding site in an unbiased manner and to predict the likeness of the warhead to be located in close proximity to the thiol group of Cys145. The non-covalent docking calculations positioned the warhead in a location that would allow for a covalent reaction to occur, taking into account the dynamicity of the protein–ligand binding and the interactions established between the chemical groups present in the molecule and amino acids in the binding site. Additionally, covalent docking was also performed to hypothesize the conformation that the molecules would adopt if a covalent bond was formed between the warhead and the thiol group of Cys145. This approach provided valuable insights into the potential binding interactions established between the molecules and the amino acids present in the Mpro binding site and can be useful for guiding the design of new derivatives with improved binding properties. In Fig. 4 the main interactions predicted by the covalent docking experiment between 11 and the active site of Mpro are described. A covalent bond between the thiol group of Cys145 and the β-carbon of the cinnamic ester was established, highlighting the presence of a hydrogen bond between the oxygen of the cinnamic ester and the NH of Gly 143, and of two hydrophobic interactions occurring between the ethyl group and the aliphatic side chains of Thr25 and Leu27. The cinnamic phenyl ring is able to have π–π stacking interactions with His141, while Phe was found to be in close proximity with the side chain of Glu166. The p-OMe substituent orients itself so as to establish a hydrophobic interaction with His143.
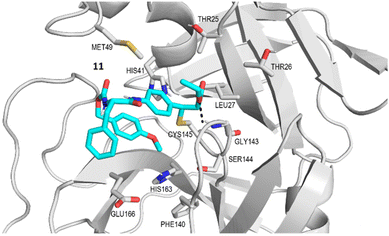 |
| | Fig. 4 Best conformer of 11 (cyan) obtained by covalent docking inside the SARS-CoV-2 Mpro active site (gray). The hydrogen bond interaction is represented as a black dotted line connecting the atoms implicated in the interaction. | |
Antiviral studies
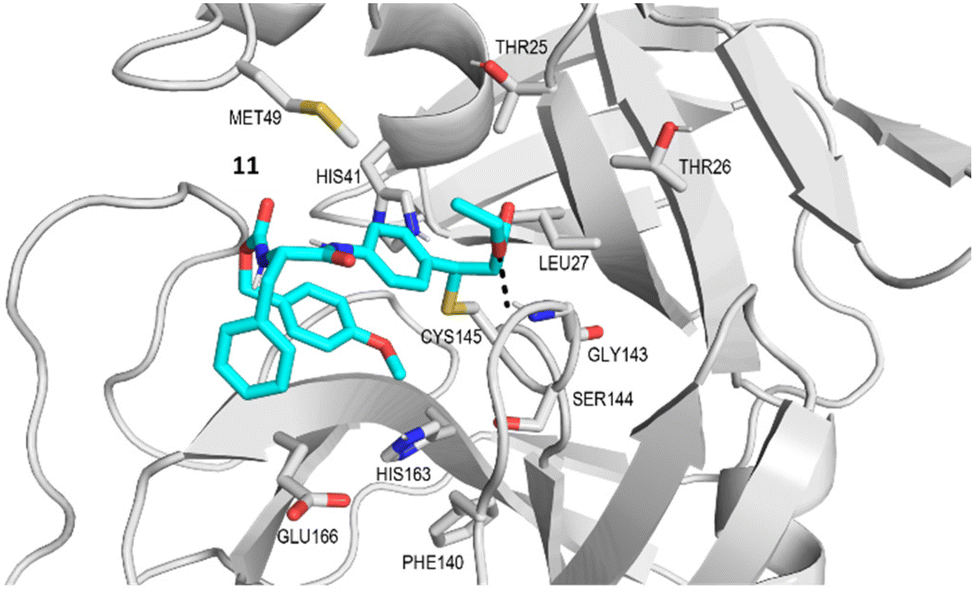
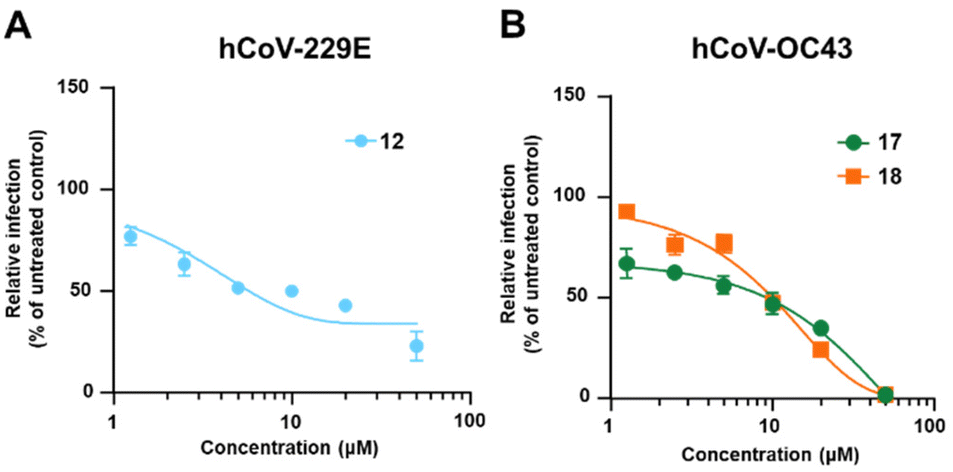
The potential anti-hCoVs activity of the cinnamic esters was then evaluated in vitro against the prototypical human α-CoV, hCoV-229E, and β-CoV, hCoV-OC43.24 Manipulation of these hCoVs, while it does not require Biosafety laboratory level 3 (BSL3) facilities, can provide results supporting further development of both new SARS-CoV-2 and pan-CoV antiviral agents. A panel of seven newly designed cinnamic esters, chosen based on their promising enzyme activity inhibition values, were therefore evaluated for antiviral activity against the two human endemic coronaviruses. While four compounds (11, 13, 15 and 19) were ineffective against both viruses when tested at up to 50 μM (Table S1†), a valuable concentration-dependent inhibition of hCoV-229 replication was observed for the carbamate derivative compound 12 (Fig. 5 – panel A), with a measured EC50 value in the low micromolar range (EC50 = 5.27 ± 0.25 μM). However, no significant antiviral effect was observed for 12 against hCoV-OC43 (Table 2). In contrast, the amides derivatives 17 and 18, which did not significantly inhibit the replication of hCoV-229E, showed a noteworthy inhibitory activity against hCoV-OC43 in the micromolar range (EC50 = 9.14 ± 0.70 μM and 10.1 ± 0.17 μM, respectively) (Fig. 5 – panel B and Table 2). The anti-hCoVs activity of 12, 17 and 18 was not due to the toxicity of target cells, given that their cytotoxic concentrations (CC50), as determined in uninfected cells, were >400 μM, 44.83 ± 0.76 μM and 64.3 ± 4.19 μM, respectively (Table 2).
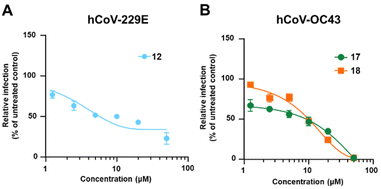 |
| | Fig. 5 Dose–response curves of 12 against hCoV-229E and 17 and 18 against hCoV-OC43. | |
Table 2 Antiviral activity of 12, 17 and 18 against different human coronaviruses
| Compound |
hCoV-229E EC50a (μM) |
hCoV-OC43 EC50a (μM) |
CC50 MRC5b (μM) |
CC50 HCT8b (μM) |
SIc |
|
EC50 was measured by evaluating the MRC-5 cell viability as a surrogate of viral CPE against hCoV-229E, or by focus forming reduction assay (FFRA) against hCoV-OC43 in HCT-8 cells.
CC50 was determined by cell viability assay (CellTiter-Glo Luminescence assay) in uninfected MRC5 or HTC-8 cells. EC50 and CC50 values are the means ± SD of data derived from two experiments performed in triplicate.
SI, selectivity index is determined as the ratio between CC50 and EC50.
|
|
12
|
5.27 ± 0.25 |
>50 |
>400 |
— |
>75.9 |
|
17
|
>50 |
9.14 ± 0.70 |
— |
44.83 ± 0.76 |
4.9 |
|
18
|
>50 |
10.1 ± 0.17 |
— |
64.3 ± 4.19 |
6.4 |
In silico homology modelling studies
Additional covalent docking calculations of 11, 12, 17, and 18 were performed to study their binding to SARS-CoV-2 Mpro, hCoV-229E Mpro and hCoV-OC43 Mpro. For the performance of the docking experiments, we retrieved the three three-dimensional (3D) structures of amino acids 1–200, which encompass the binding site of interest, for the prementioned coronavirus Mpros: (1) the crystal structure of the SARS-CoV-2 Mpro covalently bound to the inhibitor MG-132, resolved at 1.80 Å (PDB 7NG3); (2) the crystal structure of hCoV-229E Mpro, resolved at 2.37 Å (PDB 1P9U); and (3) the homology model of the free enzyme of hCoV-OC43 Mpro. The three structures were superimposed for a comparison of the conformation acquired by their substrate binding sites (Fig. 6). Visual analysis shows that the three protein structures are nicely superimposed and present a conserved 3D configuration.
 |
| | Fig. 6 Superimposition of the section of the X-ray structures of SARS-CoV-2 Mpro (pink) and hCoV-229E Mpro (cyan) and the homology model of hCoV-OC43 Mpro (gray) in which the targeted binding site is present. The structures are displayed in a ribbon representation. | |
In addition, a quantitative analysis was conducted to compare the three protein structures. The evaluation involved computing the values of the root mean square deviation (RMSD) between them, and the results are presented in Table 3. The findings demonstrated that the RMSD values were consistently below 0.65 Å, signifying a high level of structural similarity. This suggests that the conformation of these regions is consistently preserved across the three main coronavirus proteases. Consequently, it is plausible that the development of compounds targeting SARS-CoV-2 Mpro may have the potential to target hCoV-229E Mpro and hCoV-OC43 Mpro, as they share similar structural features at the binding site of interest.
Table 3 RMSD values calculated between pairs of protein structures of the studied proteases
| |
SARS-CoV-2 Mprovs. hCoV-229E Mpro |
SARS-CoV-2 Mprovs. hCoV-OC43 Mpro |
hCoV-229E Mprovs. hCoV-OC43 Mpro |
| RMSD |
0.623 Å over 162 aligned Cα positions |
0.519 Å over 170 aligned Cα positions |
0.604 Å over 156 aligned Cα positions |
The results of our docking simulations showed that all four compounds were predicted to fit well within the binding sites of the three proteases. Our analysis revealed that the binding modes of the ligands could vary across the three studied binding sites, depending on the nature of the interacting residues present in each site and their spatial orientation, since during the docking experiments, the binding site residues were kept fixed. An analysis of the sequence alignment of the first 200 amino acids in the three proteases under study demonstrated variations in amino acid residues, resulting in a sequence similarity of 36% (Fig. S2†). However, we also identified several key residues that were conserved in the three proteins, including His41, Cys145, Leu27 and Gly143. These amino acids play a key role in substrate recognition, binding, and catalytic activity. Cys145 and His41 are the most important amino acids in the binding site of the three Mpro, since they are the catalytic dyad of the proteases and are the ones targeted by the covalent inhibitors.25,26 Cys145 is the nucleophile in the catalytic dyad. His41 is involved in the stabilization of the negatively charged tetrahedral intermediate during substrate cleavage. Additionally, His41 is capable of forming hydrogen bonds with other residues within the binding site, providing additional stability both to the substrate and to the intermediate state. Moreover, residues Arg40 and Asp187, also present in the catalytic site of Mpro from SARS-CoV-2, are conserved in the three proteases.27 Then, the presented covalent inhibitors contain a Michael acceptor group that reacts with the thiol group of Cys145 and a hydrophobic group that interacts with the S1 pocket through hydrophobic interactions with Leu27 placed in close proximity to Cys145 in the 3D structure of the binding sites. Gly143 is also close in spatial distance to Cys145 and its relatively small lateral chain enables it to accommodate a diverse range of inhibitor groups (Fig. S2†).
ADME and drug-likeness studies
The analogues of the selected cinnamic esters 11, 12, 17 and 18 display good pharmacokinetic properties as shown by the Swiss ADME (Absorption, Distribution, Metabolism, Excretion) predictions and show favourable GI absorption (Table 4). Concerning their physico-chemical properties, all of the synthesised compounds are moderately soluble in water according to ESOL solubility and thus much fewer problems may be encountered during drug formulation. The lipophilicity (log P) of all the synthesized molecules is predicted to be <5, indicating good permeability of these compounds to the target tissue. However, the tested compounds showed no blood–brain barrier penetration and, except for 12, are predicted to follow the Lipinski rule of 5 and have a comparable bioavailability score of 0.55.
Table 4 ADME and drug-likeness of cinnamic esters 12, 17 and 18
| |
11
|
12
|
17
|
18
|
| Consensus logP |
4.33 |
3.51 |
4.21 |
4.28 |
| ESOL solubility |
Mod. soluble |
Mod. soluble |
Mod. soluble |
Mod. soluble |
| GI absorption |
High |
Low |
High |
High |
| BBB permeant |
No |
No |
No |
No |
| Bioavailability |
0.55 |
0.17 |
0.55 |
0.55 |
Conclusions
To conclude, overall, our results support a further in-depth exploration of the ethyl cinnamate functional group as a new electrophile warhead for the Cys145 of Mpro of both SARS-CoV-2 and other hCoVs. From the experimental evidence collected, the synthesized compounds are able to inhibit Mpro within the micromolar range with a covalent mechanism. Importantly, three of these compounds, namely the carbamate derivative 12 and the indole-bearing amides 17 and 18, show in vitro antiviral activity against representative hCoVs within the low micromolar range, thus enabling further development of cinnamate-based Mpro inhibitors as drug candidates for the treatment of hCoV infections.
Experimental section
Materials and methods
Chemistry.
Unless otherwise stated, reagents and solvents were purchased from Merck (Milan, Italy), Fluorochem (Hadfield, United Kingdom) or TCI (Zwijndrecht, Belgium) and used without further purification. All reactions were carried out in oven-dried glassware, using dry solvents under a nitrogen atmosphere, and monitored by TLC on silica gel (Merck precoated 60F254 plates), with detection by UV light (254 nm) or by permanganate or by HPLC. HPLC was performed on an Agilent 1100 Series System using a Gemini 5 μM C18 110 Å LC column 150 × 3 mm and with a gradient of H2O/ACN ranging from 5% ACN up to 100% ACN in 40 min (flux of 1.0 mL min−1 and sample injection of 20 μL), choosing 220 nm as the wavelength for the detection of compounds. Products were purified by flash column chromatography, using silica gel Merck 60 (230–400 mesh) as the stationary phase. The purity of the final tested compounds was confirmed to be >95%, as assessed by NMR and HPLC analysis. 1H NMR and 13C NMR spectra were recorded at 298 K on a Brüker Avance Spectrometer (400 MHz), using commercially available deuterated solvents (dichloromethane-d2, chloroform-d, DMSO-d6). Chemical shifts are reported in parts per million (δppm), compared to TMS as the internal standard. Coupling constants (J) are given in hertz (Hz) and are quoted to the nearest 0.5 Hz. Peak multiplicities are described as follows: s, singlet; bs, broad singlet; d, doublet; m, multiplet; and br, broad. High-resolution mass spectra (HRMS) were recorded using the Q-ToF Synapt G2-Si HDMS Acquity UPLC I-Class Photodiode Detector Array (PDA) (Waters).
Biomimetic experiments.
To a solution of 11 (15 mg, 0.03 mmol, 1 equiv.) in 5 mL of PBS (pH = 7.4) with 10% of ACN, N-acetyl cysteine (5 mg, 0.033 mmol, 1.1 equiv.) was added and the mixture was left stirring for 24 h at 37 °C. The reaction was checked via HPLC-MS and the adduct of 11 with N-acetyl cysteine was observed, as reported in Fig. 3. 11 (3.34 μL, 20 μM) was incubated with purified Mpro (2 μL, 0.4 μM) for 1 h at 4 °C, in the dark, in a mixture of water:MeOH 1:1 with 0.1% HCOOH (final volume 600 μL). The sample was concentrated and filtered through Amicon Centrifugal Filter Devices with a 3 kDa cut-off (Merck Millipore, Milan, Italy) for 30 min at 3030g. Then, the eluent solution (5 μL) was analyzed for protein-free ligands using LC-MS/MS. The supernatant solution (ligand bound to Mpro) was analysed by LC-MS. Mass spectra were recorded on a Thermo Fisher LCQ Fleet Ion Trap Mass Spectrometer with an ESI-probe for electrospray ionization and equipped with an UltiMate™ 3000 HPLC system using a Gemini 5 μM C18 110 Å LC column 150 × 3 mm and a gradient of H2O(+0.1% HCOOH)/ACN(+0.1% HCOOH) from 5% ACN to 100% ACN in 40 min, flux of 1.0 mL min−1 and sample injection of 20 μL. HRMS spectra were obtained using a Synapt G2-Si QTof mass spectrometer (Waters) with a ZsprayTM ESI-probe for electrospray ionization (Waters).
Enzyme inhibition assays.
The inhibitory activity of the compounds was evaluated by means of a Förster resonance energy transfer (FRET)-based enzymatic cleavage assay on a TECAN Infinite F2000 PRO plate reader (Agilent Technologies, Santa Clara, USA) using white flat-bottom 96-well microtiter plates (Greiner bio-one, Kremsmünster, Austria). Nirmatrelvir was purchased from AOBIUS (Gloucester, Massachusetts, USA) and used as the positive control; the IC50 of nirmatrelvir was determined as previously described by us.28 Recombinant SARS-CoV-2 Mpro was expressed and purified as previously described,29 whereas the peptidic substrate Dabcyl-KTSAVLQ↓SGFRKME-Edans (TFA salt) was obtained from a commercial source (Genescript, New Jersey, USA). The arrow indicates the cleavage position. The proteolytic activity of the SARS-CoV-2 Mpro was measured by monitoring the increasing fluorescence of SGFRKME-Edans upon hydrolytic shedding of the quencher Dabcyl-KTSAVLQ at 25 °C with a 335 nm excitation filter and a 493 nm emission filter. Each well contained 200 μL solution, composed of 185 μL of reaction buffer (20 mM Tris pH = 7.5, 0.1 mM EDTA, 1 mM DTT and 200 mM NaCl), 5 μL of SARS-CoV-2 Mpro in enzyme buffer at a final concentration of 50 nM together with 5 μL of the fluorogenic substrate (final concentration 25 μM) and 10 μL of the compounds present at a final concentration of 20 μM (screening assay) or at variable concentrations (IC50 assay). The proteolytic activity of the SARS-CoV-2 PLpro was measured in an Infinite 200 PRO microplate reader (Tecan, Männedorf, Switzerland), the reaction buffer used was 20 mM Tris-HCl (pH 7.5), 200 mM NaCl, 1 mM DTT, 0.1 mM EDTA, at 25 °C with a 360 nm excitation filter and a 460 nm emission filter. The concentrations for all compounds: 0–0.5–1–2.5–5–10–20–40 μM. DMSO was used as a negative control. The inhibitors and the substrate were dissolved and diluted in DMSO, leading to a final DMSO concentration of 7.5% (v/v). The compounds and enzyme were incubated for 10 min at 25 °C prior to substrate addition. The product released from the substrate hydrolysis was monitored in 30 s increments over a period of 10 min. The related KM value was determined in a separate experiment (33 μM). The IC50 value was calculated with GraFit (Version 6.0.12; Erithacus Software Limited, East Grinstead, West Sussex, UK) by fitting the relative enzymatic activities plotted against the respective inhibitor concentration to the four-parameter equation.
In silico studies.
The crystal structure of the main protease (Mpro) from SARS-CoV-2, used for the docking experiments, was obtained from the Protein Data Bank (PDB), ID 7NG3. The first random three-dimensional structure of the compounds was generated using the program MarvinSketch from ChemAxon. Docking input files for the target protein Mpro and the ligand structure were prepared with UCSF Chimera 1.142. The calculations were performed using AutoDock Vina 3, which was run in UCSF Chimera 1.14 for the analysis and visualization of the results. AutoDock Vina considered all the atoms of the target site included in a cubic grid box with a grid spacing of 0.375 Å and a grid size of 20 Å for the geometry search within the docking process. The origin of the grid was positioned at the center of the Mpro binding site where Cys145 is located. Finally, the resulting docking models were classified using the value of the binding-free energy ΔG0 (kcal mol−1), and the best solution with the lowest energy was selected. The images were rendered using PyMol 2.5.44. The covalent docking experiments were performed using the software Sampler for Multiple Protein–Ligand Entities (S4MPLE), which is a docking program developed at the University of Strasbourg.30 S4MPLE cannot rigorously simulate covalent docking by FF-based approaches, because FF simulations are not typically equipped to handle covalent bond breaking and formation and instead rely on a predetermined table of bonded and non-bonded interactions. Moreover, since the FF parametrization strategies differ for the protein (AMBER) and the ligand (GAFF), it makes it technically harder to program a simulation in which the two moieties (protein and ligand) are covalently bound to each other. Therefore, a covalent docking trick was employed by S4MPLE. This is because S4MPLE was originally designed to virtually screen for optimal linkers or growing moieties of “anchor” fragments prepositioned in the protein site as a result of fragment soaking in fragment-based drug design.31 This ability can be exploited to emulate the binding of warheads to protein residues by (1) mutating the target amino acid with the reactive side chain (Cys, Lys, Glu, Asp, etc.) to Gly and (2) inserting the organic fragment representing the side chain as an “anchor” fragment, with the coordinates of the original sidechain atoms. As is customary in the S4MPLE growth/linking strategy, this anchor fragment will be covalently connected to the linker or growth moiety, but in the resulting ligand, the atoms of the anchor fragment are kept fixed. Hence, the algorithm will search for linker atom positions that are both compatible with the introduced covalent bond to the fixed anchor and establishing favorable contacts with the surrounding protein site. Eventually, the best poses found above are energy-minimized after unfixing the anchor atoms but expected not to undergo any significant geometric rearrangement (proving that they correspond to “natural” poses in the site, not to constrained geometries which only result from fixing the anchor fragment). Those drifting away by more than a specified RMS deviation higher than 1.5 Å are discarded. The remaining poses are (indirect) proof that it is possible to connect the considered linker to the amino acid side chain, all while maintaining favorable interactions with the rest of the protein. The sequence alignment of SARS-CoV-2 Mpro, hCoV-229E Mpro, and hCoV-OC43 Mpro was performed on the BLASTp web server32 using the Needleman–Wüncsh algorithm with the default substitution matrix (BLOSUM62).33 To give a clearer visual representation of the results, the sequence alignment was downloaded as a text file from BLASTp and converted to an ALI format file to visualize and produce the sequence alignment images in Molsoft Browser 3.9.34 The homology modeling process for the prediction of the structure of hCoV-OC43 Mpro was performed using the SWISS-MODEL server.35 The amino acid sequence was extracted from the NCBI database [GenBank: AEN19363], and the homology model was constructed using the structure with PDB ID 6JIJ as the template, which corresponds to the crystal structure of the MPro from mouse hepatitis virus A59 in complex with an inhibitor. The superimposition and calculation of the RMSD values of the protein structures of SARS-CoV-2 Mpro, hCoV229E Mpro, and hCoV-OC43 Mpro were performed using the structure comparison tool align implemented in the software PyMOL Molecular Graphics System, Version 2.0 Schrödinger, LLC.
Cell studies.
The human colorectal carcinoma HCT-8 (ATCC CCL-244) and the human lung fibroblast MRC5 (ATCC CCL-171) cell lines were obtained from the American Type Culture Collection (ATCC) and cultured in Dulbecco's Modified Eagle Medium (DMEM; Euroclone) supplemented with 10% fetal bovine serum (FBS, Euroclone), 2 mM glutamine, 1 mM sodium pyruvate, 100 U mL−1 penicillin, and 100 g mL−1 streptomycin sulfate (P/S, both from Euroclone). hCoV-OC43 (ATCC VR-1558) and hCoV-229E (ATCC VR-740) were purchased from ATCC, propagated and titrated in MRC5 and HCT-8 cells, respectively. For antiviral assays, MRC5 or HCT-8 cell monolayers were treated with increasing concentrations of compounds 1 h before infection with hCoV-229E (100 PFU per well) or hCoV-OC43 (150 PFU per well). The compounds remained in the culture medium throughout the assay. hCoV-229E replication was measured by assessing the residual MRC5 cell viability at 72 h p.i. as a surrogate of viral CPE using the CellTiter-Glo luminescence assay (Promega, Madison, WI, USA) according to the specifications of the manufacturer. hCoV-OC43 replication was quantified at 72 h p.i. by focus forming reduction assay (FFRA) based on indirect immunoperoxidase staining with a mAb against the hCoV-OC43 N protein (clone.542-D7; Millipore, Burlington, MA, USA). Viral foci were microscopically counted and the mean counts for each drug concentration were expressed as a percentage of the mean plaque counts of the control virus (DMSO). The compound concentration producing 50% reduction of the viral replication (EC50) was determined by GraphPad Prism.
General procedure for the synthesis of the carbonate intermediates.
Carbonate intermediates were prepared by modifying a previously reported procedure.36 The appropriate alcohol (1.0 equiv.) was dissolved in dry DCM and cooled to 0 °C, then DIPEA (2.0 equiv.) was added, followed by 4-nitrophenyl chloroformate (1.15 equiv.) and the mixture was left stirring overnight. The reaction mixture was concentrated under reduced pressure, then diluted with ethyl acetate and washed with a saturated aq. solution of NaHCO3 and brine. The organic layer was dried with anhydrous Na2SO4, filtered and the solvent was removed under reduced pressure. The resulting crude product was treated with diethyl ether/n-hexane 1:1 (v/v) and the white precipitate obtained was filtered and dried under vacuum to afford the corresponding crude carbonate that was used in the next step without any further purification.
Synthesis of intermediates 8a, b, and c
(S,E)-Ethyl 3-(4-(2-((tert-butoxycarbonyl)amino)-3-phenylpropanamido)phenyl)acrylate, 8a.
Boc-Phe-OH (1.66 g, 6.27 mmol, 1.2 equiv.), HOBt (989 mg, 7.32 mmol, 1.4 equiv.), EDCI (1.4 g, 7.32 mmol, 1.4 equiv.) and trans-ethyl 4-aminocinnamate (1.0 g, 5.23 mmol, 1.0 equiv.) were dissolved in dry DMF (20 mL) and the solution was left stirring at room temperature overnight. The reaction mixture was then diluted with ethyl acetate (30 mL) and washed with a saturated aq. solution of NH4Cl (3 × 20 mL), saturated aq. solution of NaHCO3 (3 × 20 mL) and brine (3 × 20 mL). The organic layer was dried with anhydrous Na2SO4, filtered and concentrated under reduced pressure. The crude product was purified by column chromatography using n-hexane/ethyl acetate (4:6, v/v) as the eluent to afford 8a as a white amorphous solid (49% yield, 1.13 g). 1H NMR (400 MHz, CDCl3): δ 8.07 (bs, 1H, –NH), 7.61 (d, J = 16.0 Hz, 1H, Ar–CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH–), 7.43 (d, J = 8.9 Hz, 2H, Ar–H), 7.40 (d, J = 8.9 Hz, 2H, Ar–H), 7.33–7.23 (m, 5H, Ar–H), 6.34 (d, J = 16.0 Hz, 1H, Ar–CHCH–), 5.14 (bs, 1H, –NH), 4.48 (bs, 1H, a-CH), 4.25 (q, J = 7.1 Hz, 2H, –CH2CH3), 3.15 (m, 2H, Ar–CH2CH), 1.42 (s, 9H, –C(CH3)3), 1.33 (t, J = 7.1 Hz, 3H, –CH2CH3). 13C NMR (100 MHz, CDCl3): δ 170.1, 167.2, 156.1, 144.0, 139.4, 136.6, 130.6, 129.4, 128.9, 127.2, 123.7, 120.0, 117.3, 81.0, 60.6, 56.8, 38.4, 28.4, 14.4. MS (ESI), m/z [M + H]+: 439.4.
CH–), 7.43 (d, J = 8.9 Hz, 2H, Ar–H), 7.40 (d, J = 8.9 Hz, 2H, Ar–H), 7.33–7.23 (m, 5H, Ar–H), 6.34 (d, J = 16.0 Hz, 1H, Ar–CHCH–), 5.14 (bs, 1H, –NH), 4.48 (bs, 1H, a-CH), 4.25 (q, J = 7.1 Hz, 2H, –CH2CH3), 3.15 (m, 2H, Ar–CH2CH), 1.42 (s, 9H, –C(CH3)3), 1.33 (t, J = 7.1 Hz, 3H, –CH2CH3). 13C NMR (100 MHz, CDCl3): δ 170.1, 167.2, 156.1, 144.0, 139.4, 136.6, 130.6, 129.4, 128.9, 127.2, 123.7, 120.0, 117.3, 81.0, 60.6, 56.8, 38.4, 28.4, 14.4. MS (ESI), m/z [M + H]+: 439.4.
(S,E)-Ethyl 3-(4-(2-amino-3-phenylpropanamido)phenyl)acrylate, 8b.
To a solution of 8a (1.13 g, 2.6 mmol, 1.0 equiv.) in dry DCM (20 mL), TFA (7 mL) was added dropwise and the reaction mixture was left stirring for 4 h at room temperature. The solvent was evaporated under reduced pressure and the crude oil was treated with a saturated aq. solution of K2CO3. The precipitate was collected, filtered and washed with water to afford 8b as a pure white amorphous solid (800 mg, 91%). 1H NMR (400 MHz, CDCl3): δ 9.57 (s, 1H, –NH), 7.64 (m, 3H, Ar–H), 7.50 (m, 2H, Ar–H), 7.35–7.24 (m, 4H, Ar–H), 6.37 (d, J = 16.3 Hz, 1H, Ar–CHCH–), 4.26 (q, J = 6.7 Hz, 2H, –CH2CH3), 3.77 (m, 1H, a-CH), 3.37 (m, 1H, Ar–CH2CH), 2.82 (m, 1H, Ar–CH2CH), 1.33 (t, J = 7.1 Hz, 3H, –CH2CH3). 13C NMR (100 MHz, CDCl3): δ 172.7, 167.3, 144.1, 139.6, 137.7, 130.4, 129.4, 129.2, 129.0, 127.2, 119.6, 117.2, 60.6, 56.9, 40.8, 14.5. MS (ESI), m/z [M + H]+: 339.9.
(S)-Ethyl 3-(4-(2-((tert-butoxycarbonyl)amino)-3-phenylpropanamido)phenyl)propanoate, 8c.
Boc-Phe-OH (1.62 g, 6.12 mmol, 1.2 equiv.), HOBt (965 mg, 7.14 mmol, 1.4 equiv.), EDCI (1.4 g, 7.14 mmol, 1.4 equiv.) and ethyl 3-(4-aminophenyl)propanoate (1.0 g, 5.1 mmol, 1.0 equiv.) were dissolved in dry DMF (20 mL) and the solution was left stirring at room temperature overnight. The reaction mixture was then diluted with ethyl acetate (30 mL) and washed with a saturated aq. solution of NH4Cl (3 × 20 mL), a saturated aq. solution of NaHCO3 (3 × 20 mL) and brine (3 × 20 mL). The organic layer was dried with anhydrous Na2SO4, filtered and concentrated under reduced pressure to give the crude product that was purified by column chromatography using n-hexane/ethyl acetate (4:6, v/v) as the eluent to afford 8c as a white solid (84% yield, 1.9 g). 1H NMR (400 MHz, CDCl3): δ 7.83 (bs, 1H, –NH), 7.36–7.22 (m, 7H, Ar–H), 7.13 (d, J = 8.3 Hz, 2H, Ar–H), 5.22 (bs, 1H, –NH), 4.49 (bs, 1H, a-CH), 4.14 (q, J = 7.1 Hz, 2H, –CH2CH3), 3.16 (d, J = 6.9 Hz, 2H, Ar–CH2CH–), 2.92 (t, J = 7.8 Hz, 2H, Ar–CH2CH2–), 2.60 (t, J = 7.8 Hz, 2H, Ar–CH2CH2–), 1.44 (s, 9H, –C(CH3)3), 1.26 (t, J = 7.1 Hz, 3H, –CH2CH3). 13C NMR (100 MHz, CDCl3): δ 173.0, 169.7, 155.9, 136.9, 136.8, 135.6, 129.4, 128.9, 128.9, 127.1, 120.4, 80.6, 60.5, 56.8, 38.6, 36.0, 30.5, 28.4, 14.3. MS (ESI), m/z [M + H]+: 441.3.
Synthesis of the final products 9–19
(S,E)-Ethyl 3-(4-(2-(((benzyloxy)carbonyl)amino)-3-phenylpropanamido)phenyl)acrylate, 9.
To a solution of 8b (250 mg, 0.55 mmol, 1.0 equiv.) and DIPEA (0.20 mL, 1.1 mmol, 2.0 equiv.) in dry THF (3 mL) at 0 °C, benzyl (4-nitrophenyl) carbonate (180 mg, 0.66 mmol, 1.2 equiv.) in dry THF (2 mL) was added dropwise and the mixture was left stirring overnight. The reaction mixture was concentrated under reduced pressure, then diluted with ethyl acetate (10 mL) and washed with a saturated aq. solution of NaHCO3 (3 × 8 mL) and brine (3 × 8 mL). The organic layer was dried with anhydrous Na2SO4, filtered and the solvent was removed under reduced pressure. The resulting crude product was treated with diethyl ether and the white precipitate obtained was filtered and dried under vacuum to afford 9 as a white amorphous solid (68% yield, 177 mg). 1H NMR (400 MHz, DMSO-d6): δ 10.32 (s, 1H, –NH), 7.77–7.62 (m, 5H, Ar–H), 7.59 (d, J = 16.0 Hz, 1H, Ar–CHCH–), 7.39–7.17 (m, 10H, Ar–H), 6.52 (d, J = 16.0 Hz, 1H, Ar–CHCH–), 4.97 (s, 2H, CH2–O), 4.43 (td, J = 9.8, 4.9 Hz, 1H, a-CH), 4.18 (q, J = 7.1 Hz, 2H, –CH2CH3), 3.03 (dd, J = 13.7, 4.6 Hz, 1H, Ar–CH2CH), 2.86 (dd, J = 13.6, 10.2 Hz, 1H, Ar–CH2CH), 1.26 (t, J = 7.1 Hz, 3H, –CH2CH3). 13C NMR (100 MHz, DMSO-d6): δ 170.9, 166.3, 156.0, 143.9, 140.8, 137.7, 136.9, 129.2, 129.2, 129.0, 128.3, 128.1, 127.7, 127.5, 126.4, 119.2, 116.4, 65.3, 59.9, 57.0, 37.4, 14.2. MS (ESI), m/z [M + H]+: 473.9. HRMS (ESI), m/z [M + H]+: calculated for C28H29N2O5+ 473.2071; found 473.2074. HPLC rt: 26.0 min.
Ethyl (S,E)-3-(4-(2-((((4-fluorobenzyl)oxy)carbonyl)amino)-3-phenylpropanamido)phenyl)acrylate, 10.
To a solution of 8b (150 mg, 0.44 mmol, 1.0 equiv.) and DIPEA (0.23 mL, 1.32 mmol, 3.0 equiv.) in dry THF (3 mL) at 0 °C, 4-fluorobenzyl (4-nitrophenyl) carbonate (68 mg, 0.53 mmol, 1.2 equiv.) in dry THF (2 mL) was added dropwise and the mixture was left stirring overnight. The reaction mixture was concentrated under reduced pressure, then diluted with ethyl acetate (12 mL) and washed with a saturated aq. solution of NaHCO3 (3 × 10 mL) and brine (3 × 10 mL). The organic layer was dried with anhydrous Na2SO4, filtered and the solvent was removed under reduced pressure. The resulting crude product was purified by column chromatography using n-hexane/ethyl acetate (6:4, v/v) as the eluent to afford 10 as a white amorphous solid (32% yield, 71 mg). 1H NMR (400 MHz, DMSO-d6): δ 10.32 (s, 1H, –NH), 7.74–7.64 (m, 5H, Ar–H), 7.59 (d, J = 15.6 Hz, 1H, Ar–CHCH–), 7.35–7.14 (m, 9H, Ar–H, –NH), 6.53 (d, J = 15.9 Hz, 1H, Ar–CHCH–), 4.95 (s, 2H, Ar–CH2–O), 4.42 (m, 1H, α-CH), 4.18 (q, J = 7.0 Hz, 2H, –CH2CH3), 3.03 (m, 1H, Ar–CH2CH), 2.85 (m, 1H, Ar–CH2CH), 1.25 (t, J = 7.0 Hz, 3H, –CH2CH3). 13C NMR (100 MHz, CDCl3): δ 169.8, 167.2, 164.0, 161.5, 156.5, 143.8, 139.1, 136.3, 131.8, 130.7, 130.05 (J = 7.9 Hz), 129.4, 128.9, 127.3, 120.1, 117.5, 115.7, 115.5, 66.7, 60.6, 57.3, 38.7, 14.4. MS (ESI), m/z [M + H]+: 491.20. HRMS (ESI), m/z [M + H]+: calculated for C28H28FN2O5+ 491.1977; found 491.1973. HPLC rt: 22.5 min.
(S,E)-Ethyl 3-(4-(2-((((4-methoxybenzyl)oxy)carbonyl)amino)-3-phenylpropanamido)phenyl)acrylate, 11.
To a solution of 8b (55 mg, 0.12 mmol, 1.0 equiv.) and DIPEA (0.08 mL, 0.48 mmol, 4.0 equiv.) in dry THF (1 mL) at 0 °C, 4-methoxybenzyl (4-nitrophenyl) carbonate (42 mg, 0.14 mmol, 1.2 equiv.) in dry THF (1 mL) was added dropwise and the mixture was left stirring overnight. The reaction mixture was concentrated under reduced pressure, then diluted with ethyl acetate (6 mL) and washed with a saturated aq. solution of NaHCO3 (3 × 6 mL) and brine (3 × 5 mL). The organic layer was dried with anhydrous Na2SO4, filtered and the solvent was removed under reduced pressure. The resulting crude product was purified by column chromatography using n-hexane/ethyl acetate (4:6, v/v) as the eluent to afford 11 as a white amorphous solid (67% yield, 40 mg). 1H NMR (400 MHz, CDCl3): δ 8.35 (s, 1H, –NH), 7.65 (d, J = 16.0 Hz, 1H, Ar–CHCH–), 7.46–7.36 (m, 4H, Ar–H), 7.35–7.19 (m, 8H, Ar–H), 6.89 (d, J = 8.2 Hz, 2H, Ar–H), 6.38 (d, J = 16.0 Hz, 1H, Ar–CHCH–), 5.68 (d, J = 7.9 Hz, 1H, –NH), 5.05 (s, 2H, Ar–CH2–O), 4.73–4.58 (m, 1H, α-CH), 4.31 (q, J = 7.1 Hz, 2H, –CH2CH3), 3.83 (s, 3H, –OCH3), 3.18 (dd, J = 7.1, 3.1 Hz, 2H, Ar–CH2CH), 1.39 (t, J = 7.1 Hz, 3H, CH2CH3). 13C NMR (100 MHz, CDCl3): δ 169.7, 167.2, 159.8, 143.9, 139.2, 136.3, 130.6, 130.0, 129.4, 128.9, 128.0, 127.3, 120.0, 117.4, 114.1, 67.4, 60.6, 55.4, 38.6, 29.8, 14.4. MS (ESI), m/z [M + H]+: 503.5. HRMS (ESI), m/z [M + H]+: calculated for C29H31N2O6+ 503.2177; found 503.2176. HPLC rt: 22.0 min.
Ethyl (S,E)-3-(4-(2-((((4,5-dimethoxy-2-nitrobenzyl)oxy)carbonyl)amino)-3-phenylpropanamido)phenyl)acrylate, 12.
To a solution of 8b (160 mg, 0.55 mmol, 1.0 equiv.) and DIPEA (0.16 mL, 0.94 mmol, 2.0 equiv.) in dry THF (3 mL) at 0 °C, 4,5-dimethoxy-2-nitrobenzyl (4-nitrophenyl) carbonate (211 mg, 0.56 mmol, 1.2 equiv.) in dry THF (2 mL) was added dropwise and the mixture was left stirring overnight. The reaction mixture was concentrated under reduced pressure, then diluted with ethyl acetate (10 mL) and washed with a saturated aq. solution of NaHCO3 (3 × 8 mL) and brine (3 × 8 mL). The organic layer was dried with anhydrous Na2SO4, filtered and the solvent was removed under reduced pressure. The resulting crude product was treated with diethyl ether and the white precipitate obtained was filtered and dried under vacuum to afford 12 as a white amorphous solid (45% yield, 143 mg). 1H NMR (400 MHz, CDCl3): δ 7.77 (bs, 1H, –NH), 7.62 (s, 1H, Ar–H), 7.53 (d, J = 16.2 Hz, 1H, Ar–CHCH–), 7.34 (d, J = 8.3 Hz, 1H, Ar–H), 7.28–7.15 (m, 8H, Ar–H), 6.86 (s, 1H, Ar–H), 6.27 (d, J = 16.2 Hz, 1H, Ar–CHCH–), 5.60 (bs, 1H, -NH), 5.44 (q, J = 15.0 Hz, 2H, Ar–CH2–O), 4.49 (m, 1H, α-CH), 4.19 (q, J = 7.4 Hz, 2H, –CH2CH3), 3.87 (s, 3H, –OCH3), 3.83 (s, 3H, –OCH3), 3.10 (m, 2H, Ar–CH2CH), 1.27 (t, J = 7.4 Hz, 3H, –CH2CH3). 13C NMR (100 MHz, CDCl3): δ 169.4, 167.2, 156.2, 153.2, 148.1, 143.0, 139.3, 138.2, 136.2, 130.9, 129.4, 129.1, 129.0, 127.5, 120.1, 117.6, 110.0, 108.4, 64.4, 60.6, 57.4, 56.5, 38.6, 14.5. MS (ESI), m/z [M + H]+: 578.1. HRMS (ESI), m/z [M + H]+: calculated for C30H32N3O9+ 578.2133; found 578.2140. HPLC rt: 22.0 min.
(S,E)-Ethyl 3-(4-(2-(((benzo[d][1,3]dioxol-5-ylmethoxy)carbonyl)amino)-3-phenylpropanamido)phenyl)acrylate, 13.
To a solution of 8b (250 mg, 0.55 mmol, 1.0 equiv.) and DIPEA (0.20 mL, 1.1 mmol, 2.0 equiv.) in dry THF (3 mL) at 0 °C, benzo[d][1,3]dioxol-5-ylmethyl (4-nitrophenyl) carbonate (210 mg, 0.66 mmol, 1.2 equiv.) in dry THF (2 mL) was added dropwise and the mixture was left stirring overnight. The reaction mixture was concentrated under reduced pressure, then diluted with ethyl acetate (12 mL) and washed with saturated aq. solution of NaHCO3 (3 × 10 mL) and brine (3 × 10 mL). The organic layer was dried with anhydrous Na2SO4, filtered and the solvent was removed under reduced pressure. The resulting crude product was treated with t-butyl methyl ether and the white precipitate obtained was filtered and dried under vacuum to afford 13 as a white amorphous solid (48% yield, 137 mg). 1H NMR (400 MHz, DMSO-d6): δ 10.30 (s, 1H, –NH), 7.70–7.62 (m, 4H, Ar–H), 7.59 (d, J = 16.0 Hz, 1H, Ar–CHCH–), 7.35–7.24 (m, 4H, Ar–H), 7.23–7.17 (m, 1H, Ar–H), 6.86 (m, 2H, Ar–H), 6.78 (m, 1H, Ar–H), 6.52 (d, J = 16.0 Hz, 1H, Ar–CHCH–), 6.00 (s, 2H, O–CH2–O), 4.86 (s, 2H, Ar–CH2–O), 4.41 (td, J = 9.0, 4.7 Hz, 1H, α-CH), 4.18 (q, J = 7.1 Hz, 2H, –CH2CH3), 3.02 (m, 1H, Ar–CH2CH), 2.85 (m, 1H, Ar–CH2CH), 1.26 (t, J = 7.1 Hz, 3H, –CH2CH3). 13C NMR (100 MHz, DMSO-d6): δ 171.4, 166.8, 156.5, 147.7, 147.4, 144.4, 141.3, 138.2, 131.1, 129.7, 129.6, 129.5, 128.6, 126.9, 122.1, 119.7, 116.9, 109.0, 108.5, 101.5, 65.8, 60.4, 57.5, 37.9, 14.7. MS (ESI), m/z [M + H]+: 517.20. HRMS (ESI), m/z [M + H]+: calculated for C29H29N2O7+ 517.1970; found 517.1972. HPLC rt: 21.8 min.
Ethyl(S,E)-3-(4-(2-(((benzo[d]thiazol-2-ylmethoxy)carbonyl)amino)-3-phenylpropanamido)phenyl)acrylate, 14.
To a solution of 8b (300 mg, 0.89 mmol, 1.0 equiv.) and DIPEA (0.31 mL, 1.78 mmol, 2.0 equiv.) in dry THF (4 mL) at 0 °C, benzo[d]thiazol-2-ylmethyl (4-nitrophenyl) carbonate (353 mg, 1.07 mmol, 1.2 equiv.) in dry THF (2 mL) was added dropwise and the mixture was left stirring overnight. The reaction mixture was concentrated under reduced pressure, then diluted with ethyl acetate (12 mL) and washed with a saturated aq. solution of NaHCO3 (3 × 10 mL) and brine (3 × 10 mL). The organic layer was dried with anhydrous Na2SO4, filtered and the solvent was removed under reduced pressure. The resulting crude product was treated with diethyl ether and the white precipitate obtained was filtered and dried under vacuum to afford 14 as a white amorphous solid (57% yield, 271 mg). 1H NMR (400 MHz, CD2Cl2): δ 8.00 (d, J = 8.4 Hz, 1H, Ar–H), 7.59 (d, J = 15.8 Hz, 1H, Ar–CHCH–), 7.89 (m, 2H, Ar–H), 7.50 (t, J = 8.0 Hz, 1H, Ar–H), 7.43 (m, 5H, Ar–H), 7.27 (m, 5H, Ar–H), 6.35 (d, J = 15.8 Hz, 1H, Ar–CHCH–), 5.80 (bs, 1H, –NH), 5.47 (m, 2H, Ar–CH2–O), 4.57 (m, 1H, α-CH), 4.22 (q, J = 7.0 Hz, 2H, –CH2CH3), 3.19 (m, 2H, Ar–CH2CH), 1.31 (t, J = 7.0 Hz, 3H, –CH2CH3). 13C NMR (100 MHz, CD2Cl2): δ 169.5, 167.3, 167.2, 155.9, 152.9, 144.0, 139.6, 136.9, 131.3, 129.9, 129.4, 129.4, 127.8, 127.0, 126.2, 123.6, 122.4, 120.5, 118.0, 64.5, 60.9, 57.9, 38.8, 14.7. MS (ESI), m/z [M + H]+: 530.92. HRMS (ESI), m/z [M + H]+: calculated for C29H28N3O5S+ 530.1744; found 530.1750. HPLC rt: 21.9 min.
Ethyl (E)-3-(4-((S)-2-(3-((R)-1-(naphthalen-1-yl)ethyl)ureido)-3-phenylpropanamido)phenyl)acrylate, 15.
To a solution of 8b (150 mg, 0.44 mmol, 1.0 equiv.) and TEA (0.12 mL, 0.88 mmol, 2.0 equiv.) in dry THF (3 mL) at 0 °C, (R)-(−)-1-(1-naphthyl)ethyl isocyanate (104 mg, 0.53 mmol, 1.2 equiv.) in dry THF (2 mL) was added dropwise and the mixture was left stirring overnight. The reaction mixture was concentrated under reduced pressure, then diluted with ethyl acetate (12 mL) and washed with brine (3 × 10 mL). The organic layer was dried with anhydrous Na2SO4, filtered and the solvent was removed under reduced pressure. The resulting crude product was treated with diethyl ether and the white precipitate obtained was filtered and dried under vacuum to afford 15 as a white amorphous solid (60% yield, 141 mg). 1H NMR (400 MHz, DMSO-d6): δ 10.27 (s, 1H, –NH), 8.08 (m, 1H, Ar–H), 7.91 (m, 1H, Ar–H), 7.79 (d, J = 8.4 Hz, 1H, Ar–H), 7.66 (m, 2H, Ar–H), 7.59 (m, 2H, Ar–H), 7.57 (m, 1H, Ar–H), 7.49 (m, 4H, Ar–H), 7.24 (m, 5H, Ar–H), 6.77 (d, J = 8.4 Hz, 1H, Ar–H), 6.51 (d, J = 16.7 Hz, 1H, –CHCH–), 6.24 (d, J = 8.4 Hz, 1H, Ar–H), 5.51 (q, J = 7.7 Hz, 1H, Ar–CH), 4.58 (q, 1H, J = 6.9 Hz, 1H, α-CH), 4.18 (q, J = 7.1 Hz, 2H, –CH2CH3), 3.03 (dd, J = 13.0 Hz, 1H, Ar–CH2CH), 2.86 (dd, J = 13.0 Hz, 1H, Ar–CH2CH), 1.42 (t, J = 7.7 Hz, 3H, –CHCH3), 1.25 (t, J = 7.1 Hz, 3H, –CH2CH3). 13C NMR (100 MHz, DMSO-d6): δ 171.3, 166.3, 156.6, 143.9, 141.0, 140.8, 137.4, 133.4, 130.3, 129.3, 129.1, 128.9, 128.5, 128.1, 127.1, 126.3, 126.0, 125.5, 125.4, 123.2, 122.0, 119.2, 116.3, 59.8, 55.2, 44.7, 22.5, 14.2. MS (ESI), m/z [M + H]+: 536.70. HRMS (ESI), m/z [M + H]+: calculated for C33H34N3O4+ 536.2544; found 536.2541. HPLC rt: 22.8 min.
(S,E)-Ethyl 3-(4-(3-phenyl-2-(3-(4-(trifluoromethyl)benzyl)ureido)propanamido)phenyl)acrylate, 16.
To a solution of 8b (155 mg, 0.34 mmol, 1.0 equiv.) and TEA (0.12 mL, 0.86 mmol, 2.0 equiv.) in dry THF (3 mL) at 0 °C, 4-(trifluoromethyl)benzyl isocyanate (0.07 mL, 0.41 mmol, 1.2 equiv.) in dry THF (2 mL) was added dropwise and the mixture was left stirring overnight. The reaction mixture was concentrated under reduced pressure, then diluted with ethyl acetate (12 mL) and washed with brine (3 × 10 mL). The organic layer was dried with anhydrous Na2SO4, filtered and the solvent was removed under reduced pressure. The resulting crude product was treated with t-butyl methyl ether and the white precipitate obtained was filtered and dried under vacuum to afford 16 as a white amorphous solid (88% yield, 162 mg). 1H NMR (400 MHz, DMSO-d6): δ 10.30 (s, 1H, –NH), 7.72–7.61 (m, 6H, Ar–H), 7.59 (d, J = 15.9 Hz, 1H, Ar–CHCH–), 7.38 (d, J = 7.9 Hz, 2H, Ar–H), 7.32–7.11 (m, 5H, Ar–H), 6.68 (t, J = 6.1 Hz, 1H, –NH), 6.52 (d, J = 15.9 Hz, 1H, Ar–CHCH–), 6.44 (d, J = 8.4 Hz, 1H, –NH), 4.61 (q, J = 7.7 Hz, 1H, α-CH), 4.27 (d, J = 6.0 Hz, 2H, –CH2–NH), 4.18 (q, J = 7.1 Hz, 2H, –CH2CH3), 3.04 (m, 1H, Ar–CH2CH), 2.86 (m, 1H, Ar–CH2CH), 1.26 (t, J = 7.3 Hz, 3H, –CH2CH3). 13C NMR (100 MHz, DMSO-d6): δ 171.4, 166.3, 157.5, 145.7, 144.0, 140.8, 137.4, 129.3, 129.2, 129.0, 128.1, 127.4, 127.1, 126.3, 125.7, 125.0, 119.2, 116.4, 59.9, 55.3, 42.4, 38.6, 14.2. MS (ESI), m/z [M + H]+: 540.41. HRMS (ESI), m/z [M + H]+: calculated for C29H29F3N3O4+ 540.2105; found 540.2103. HPLC rt: 21.8 min.
(S,E)-Ethyl 3-(4-(2-(2-(1H-indol-3-yl)acetamido)-3-phenylpropanamido)phenyl)acrylate, 17.
2-(1H-indol-3-yl)acetic acid (197 mg, 1.12 mmol, 1.2 equiv.), HOBt (173 mg, 1.31 mmol, 1.4 equiv.), EDCI (250 mg, 1.31 mmol, 1.4 equiv.), DIPEA (0.20 mL, 1.12 mmol, 1.2 equiv.) and 8b (423 mg, 0.83 mmol, 1.0 equiv.) were dissolved in dry DMF (10 mL) and the solution was left stirring at room temperature overnight. The reaction mixture was then diluted with ethyl acetate (12 mL) and washed with a saturated aq. solution of NH4Cl (3 × 12 mL), a saturated aq. solution of NaHCO3 (3 × 12 mL) and brine (3 × 12 mL). The organic layer was dried with anhydrous Na2SO4, filtered and concentrated under reduced pressure. The resulting crude product was treated with t-butyl methyl ether and the precipitate obtained was filtered and dried under vacuum to afford 17 as a white amorphous solid (31% yield, 156 mg). 1H NMR (400 MHz, DMSO-d6): δ 10.82 (s, 1H, –NH), 10.34 (s, 1H, –NH), 8.37 (d, J = 7.9 Hz, 1H, Ar–H), 7.75–7.53 (m, 5H, Ar–H), 7.45–7.14 (m, 7H, Ar–H), 7.05 (dd, J = 14.8, 7.2 Hz, 2H, Ar–H), 6.91 (t, J = 7.4 Hz, 1H, Ar–H), 6.53 (d, J = 16.0 Hz, 1H, Ar–CHCH–), 4.71 (q, J = 8.3 Hz, 1H, a-CH), 4.19 (q, J = 7.0 Hz, 2H, –CH2CH3), 3.54 (s, 2H, –CH2–Indole), 3.06 (m, J = 13.3, 9.5 Hz, 1H, Ar–CH2CH), 2.91 (dd, J = 13.3, 9.5 Hz, 1H, Ar–CH2CH), 1.26 (t, J = 7.0 Hz, 3H, –CH2CH3). 13C NMR (100 MHz, DMSO-d6): δ 171.2, 171.1, 166.8, 144.4, 141.3, 137.9, 136.5, 129.7, 129.6, 129.5, 128.5, 127.7, 126.8, 124.2, 121.3, 119.7, 119.2, 118.7, 116.9, 111.7, 109.1, 60.3, 55.4, 38.2, 32.8, 14.7. MS (ESI), m/z [M + H]+: 496.29. HRMS (ESI), m/z [M + H]+: calculated for C30H30N3O4+ 496.2231; found 496.2228. HPLC rt: 20.0 min.
(S,E)-Ethyl 3-(4-(2-(1H-indole-5-carboxamido)-3-phenylpropanamido)phenyl)acrylate, 18.
1H-indole-5-carboxylic acid (105 mg, 0.66 mmol, 1.2 equiv.), HOBt (102 mg, 0.77 mmol, 1.4 equiv.), EDCI (148 mg, 0.77 mmol, 1.4 equiv.), DIPEA (0.19 mL, 1.1 mmol, 2.0 equiv.) and 8b (250 mg, 0.55 mmol, 1.0 equiv.) were dissolved in dry DMF (10 mL) and the solution was left stirring at room temperature overnight. The reaction mixture was then diluted with ethyl acetate (12 mL) and washed with a saturated aq. solution of NH4Cl (3 × 10 mL), a saturated aq. solution of NaHCO3 (3 × 10 mL) and brine (3 × 10 mL). The organic layer was dried with anhydrous Na2SO4, filtered and concentrated under reduced pressure. The resulting crude product was treated with diethyl ether and the precipitate obtained was filtered and dried under vacuum to afford 18 as a pink amorphous solid (45% yield, 138 mg). 1H NMR (400 MHz, DMSO-d6): δ 11.3 (s, 1H, –NH), 10.41 (s, 1H, –NH), 8.56 (d, J = 7.8 Hz, 1H, Ar–H), 8.16 (s, 1H, Ar–H), 7.69–7.57 (m, 6H, Ar–H), 7.42 (m, 4H, Ar–H), 7.28 (t, J = 8.0 Hz, 2H, Ar–H), 7.18 (t, J = 7.4 Hz, 1H, Ar–H), 6.52 (d, J = 16.3 Hz, 1H, Ar–CHCH–), 6.52 (bs, 1H, –NH), 4.88 (q, J = 7.0 Hz, 1H, a-CH), 4.18 (q, J = 6.7 Hz, 2H, –CH2CH3), 3.15 (d, J = 7.0 Hz, 2H, Ar–CH2CH), 1.25 (t, J = 6.7 Hz, 3H, –CH2CH3). 13C NMR (100 MHz, DMSO-d6): δ 171.2, 167.6, 166.3, 143.9, 141.0, 138.2, 137.5, 129.2, 129.2, 128.9, 128.1, 126.9, 126.6, 126.3, 124.8, 120.7, 120.3, 119.2, 116.4, 110.8, 102.1, 59.8, 55.9, 37.1, 14.2. MS (ESI), m/z [M + H]+: 482.30. HRMS (ESI), m/z [M + H]+: calculated for C29H28N3O4+ 482.2075; found 482.2077. HPLC rt: 19.9 min.
Ethyl (S)-3-(4-(2-((((4-methoxybenzyl)oxy)carbonyl)amino)-3-phenylpropanamido)phenyl)propanoate, 19.
Step 1: to a solution of 8c (500 mg, 1.13 mmol, 1.0 equiv.) in dry DCM (10 mL) was added TFA (5 mL) and the reaction was left stirring for 4 h at room temperature. After the consumption of the starting material, the solvent was evaporated under reduced pressure and the crude oil was used for the next step without purification. Step 2: a solution of the precedent TFA salt was dissolved in dry THF (10 mL) and DIPEA (0.8 mL, 4.52 mmol, 4.0 equiv.) was added dropwise. 4-Methoxybenzyl (4-nitrophenyl) carbonate (412 mg, 1.36 mmol, 1.2 equiv.) in dry THF (4 mL) was added dropwise and the mixture was left stirring overnight. The reaction mixture was concentrated under reduced pressure, then diluted with ethyl acetate (10 mL) and washed with a saturated aq. solution of NaHCO3 (3 × 12 mL) and brine (3 × 12 mL). The organic layer was dried with anhydrous Na2SO4, filtered and the solvent was removed under reduced pressure. The resulting crude product was purified by column chromatography using n-hexane/ethyl acetate (4:6, v/v) as the eluent to afford 19 as a white amorphous solid (38% yield, 215 mg). 1H NMR (400 MHz, DMSO-d6): δ 10.03 (s, 1H, –NH), 7.59 (d, J = 8.3 Hz, 1H, –NH), 7.48 (d, J = 8.2 Hz, 2H, Ar–H), 7.30 (m, 4H, Ar–H), 7.24–7.12 (m, 5H, Ar–H), 6.88 (d, J = 8.2 Hz, 2H, Ar–H), 4.88 (s, 2H, Ar–CH2–O), 4.37 (m, 1H, α-CH), 4.04 (q, J = 7.1 Hz, 2H, –CH2CH3), 3.74 (s, 3H, O–CH3), 3.00 (m, 1H, Ar–CH2CH), 2.84 (m, 1H, Ar–CH2CH), 2.80 (t, J = 8.2 Hz, 2H, CH2-CH2), 2.58 (t, J = 8,2 Hz, 2H, –CH2–CH2), 1.15 (t, J = 7.1 Hz, 3H, –CH2CH3). 13C NMR (100 MHz, DMSO-d6): δ 172.1, 170.4, 158.9, 156.0, 137.8, 136.9, 135.5, 129.6, 129.2, 128.8, 128.4, 128.0, 126.3, 119.3, 113.7, 65.2, 59.8, 56.9, 55.1, 37.5, 35.2, 29.7, 14.1. MS (ESI), m/z [M + H]+: 505.81. HRMS (ESI), m/z [M + H]+: calculated for C29H33N2O6+ 505.2334; found 505.2336. HPLC rt: 21.4 min.
Conflicts of interest
There are no conflicts of interest to declare.
Acknowledgements
This research was supported by EU funding within the MUR PNRR Extended Partnership initiative on Emerging Infectious Diseases (Project no. PE00000007, INF-ACT) and by Regione Piemonte (PAR FSC INFRA-P2B) to G. G.
References
- S. Adachi, T. Koma, N. Doi, M. Nomaguchi and A. Adachi, Commentary: Origin and evolution of pathogenic coronaviruses, Front. Immunol., 2020, 11, 811 CrossRef CAS.
- J. F. Chan, K. S. Li, K. K. To, V. C. Cheng, H. Chen and K. Y. Yuen, Is the discovery of the novel human betacoronavirus 2c EMC/2012 (HCoV-EMC) the beginning of another SARS-like pandemic?, J. Infect., 2012, 65(6), 477–489 CrossRef PubMed.
- C. Drosten, S. Günther, W. Preiser, S. van der Werf, H. R. Brodt, S. Becker, H. Rabenau, M. Panning, L. Kolesnikova, R. A. Fouchier, A. Berger, A. M. Burguière, J. Cinatl, M. Eickmann, N. Escriou, K. Grywna, S. Kramme, J. C. Manuguerra, S. Müller, V. Rickerts, M. Stürmer, S. Vieth, H. D. Klenk, A. D. Osterhaus, H. Schmitz and H. W. Doerr, Identification of a novel coronavirus in patients with severe acute respiratory syndrome, N. Engl. J. Med., 2003, 348(20), 1967–1976 CrossRef CAS PubMed.
- C. Huang, Y. Wang, X. Li, L. Ren, J. Zhao, Y. Hu, L. Zhang, G. Fan, J. Xu, X. Gu, Z. Cheng, T. Yu, J. Xia, Y. Wei, W. Wu, X. Xie, W. Yin, H. Li, M. Liu, Y. Xiao, H. Gao, L. Guo, J. Xie, G. Wang, R. Jiang, Z. Gao, Q. Jin, J. Wang and B. Cao, Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China, Lancet, 2020, 395(10223), 497–506 CrossRef CAS PubMed.
- P. Zhou, X. L. Yang, X. G. Wang, B. Hu, L. Zhang, W. Zhang, H. R. Si, Y. Zhu, B. Li, C. L. Huang, H. D. Chen, J. Chen, Y. Luo, H. Guo, R. D. Jiang, M. Q. Liu, Y. Chen, X. R. Shen, X. Wang, X. S. Zheng, K. Zhao, Q. J. Chen, F. Deng, L. L. Liu, B. Yan, F. X. Zhan, Y. Y. Wang, G. F. Xiao and Z. L. Shi, A pneumonia outbreak associated with a new coronavirus of probable bat origin, Nature, 2020, 579(7798), 270–273 CrossRef CAS PubMed.
- C. Zheng, W. Shao, X. Chen, B. Zhang, G. Wang and W. Zhang, Real-world effectiveness of COVID-19 vaccines: a literature review and meta-analysis, Int. J. Infect. Dis., 2022, 114, 252–260 CrossRef CAS PubMed.
- A. Citarella, A. Scala, A. Piperno and N. Micale, SARS-CoV-2 M(pro): A Potential Target for Peptidomimetics and Small-Molecule Inhibitors, Biomolecules, 2021, 11(4), 607 CrossRef CAS PubMed.
- A. E. Gorbalenya, E. V. Koonin, A. P. Donchenko and V. M. Blinov, Coronavirus genome: prediction of putative functional domains in the non-structural polyprotein by comparative amino acid sequence analysis, Nucleic Acids Res., 1989, 17(12), 4847–4861 CrossRef CAS PubMed.
- V. M. Balaramnavar, K. Ahmad, M. Saeed, I. Ahmad, M. Kamal and T. Jawed, Pharmacophore-based approaches in the rational repurposing technique for FDA approved drugs targeting SARS-CoV-2 M(pro), RSC Adv., 2020, 10(66), 40264–40275 RSC.
- Z. Jin, X. Du, Y. Xu, Y. Deng, M. Liu, Y. Zhao, B. Zhang, X. Li, L. Zhang, C. Peng, Y. Duan, J. Yu, L. Wang, K. Yang, F. Liu, R. Jiang, X. Yang, T. You, X. Liu, X. Yang, F. Bai, H. Liu, X. Liu, L. W. Guddat, W. Xu, G. Xiao, C. Qin, Z. Shi, H. Jiang, Z. Rao and H. Yang, Structure of M(pro) from SARS-CoV-2 and discovery of its inhibitors, Nature, 2020, 582(7811), 289–293 CrossRef CAS PubMed.
- Z. Shen, K. Ratia, L. Cooper, D. Kong, H. Lee, Y. Kwon, Y. Li, S. Alqarni, F. Huang, O. Dubrovskyi, L. Rong, G. R. J. Thatcher and R. Xiong, Design of SARS-CoV-2 PLpro Inhibitors for COVID-19 Antiviral Therapy Leveraging Binding Cooperativity, J. Med. Chem., 2022, 65(4), 2940–2955 CrossRef CAS PubMed.
- E. Mahase, Covid-19: Pfizer's paxlovid is 89% effective in patients at risk of serious illness, company reports, BMJ, 2021, 375, n2713 CrossRef PubMed.
- H. H. Otto and T. Schirmeister, Cysteine Proteases and Their Inhibitors, Chem. Rev., 1997, 97(1), 133–172 CrossRef CAS.
- P. De, M. Baltas and F. Bedos-Belval, Cinnamic acid derivatives as anticancer agents-a review, Curr. Med. Chem., 2011, 18(11), 1672–1703 CrossRef CAS PubMed.
- N. Ruwizhi and B. A. Aderibigbe, Cinnamic acid derivatives and their biological efficacy, Int. J. Mol. Sci., 2020, 21(16), 5712 CrossRef CAS PubMed.
- M. Sova, Antioxidant and antimicrobial activities of cinnamic acid derivatives, Mini-Rev. Med. Chem., 2012, 12(8), 749–767 CrossRef CAS PubMed.
- A. Citarella, D. Moi, M. Pedrini, H. Pérez-Peña, S. Pieraccini, C. Stagno, N. Micale, T. Schirmeister, G. Sibille, G. Gribaudo, A. Silvani, D. Passarella and C. Giannini, Discovery of a Novel Trifluoromethyl Diazirine Inhibitor of SARS-CoV-2 Mpro, Molecules, 2023, 28(2), 514 CrossRef CAS PubMed.
- A. Citarella, D. Gentile, A. Rescifina, A. Piperno, B. Mognetti, G. Gribaudo, M. T. Sciortino, W. Holzer, V. Pace and N. Micale, Pseudo-Dipeptide Bearing α,α-Difluoromethyl Ketone Moiety as Electrophilic Warhead with Activity against Coronaviruses, Int. J. Mol. Sci., 2021, 22(3), 1398 CrossRef CAS PubMed.
- L. Ielo, V. Patamia, A. Citarella, T. Efferth, N. Shahhamzehei, T. Schirmeister, C. Stagno, T. Langer, A. Rescifina, N. Micale and V. Pace, Novel Class of Proteasome Inhibitors: In Silico and In Vitro Evaluation of Diverse Chloro(trifluoromethyl)aziridines, Int. J. Mol. Sci., 2022, 23(20), 12363 CrossRef CAS.
- M. Elagawany, A. A. Elmaaty, A. Mostafa, N. M. Abo Shama, E. Y. Santali, B. Elgendy and A. A. Al-Karmalawy, Ligand-based design, synthesis, computational insights, and in vitro studies of novel N-(5-Nitrothiazol-2-yl)-carboxamido derivatives as potent inhibitors of SARS-CoV-2 main protease, J. Enzyme Inhib. Med. Chem., 2022, 37(1), 2112–2132 CrossRef CAS.
- S. I. Hattori, N. Higashi-Kuwata, H. Hayashi, S. R. Allu, J. Raghavaiah, H. Bulut, D. Das, B. J. Anson, E. K. Lendy, Y. Takamatsu, N. Takamune, N. Kishimoto, K. Murayama, K. Hasegawa, M. Li, D. A. Davis, E. N. Kodama, R. Yarchoan, A. Wlodawer, S. Misumi, A. D. Mesecar, A. K. Ghosh and H. Mitsuya, A small molecule compound with an indole moiety inhibits the main protease of SARS-CoV-2 and blocks virus replication, Nat. Commun., 2021, 12(1), 668 CrossRef CAS.
- P. A. Jackson, J. C. Widen, D. A. Harki and K. M. Brummond, Covalent Modifiers: A Chemical Perspective on the Reactivity of α,β-Unsaturated Carbonyls with Thiols via Hetero-Michael Addition Reactions, J. Med. Chem., 2017, 60(3), 839–885 CrossRef CAS PubMed.
- L. El Khoury, Z. Jing, A. Cuzzolin, A. Deplano, D. Loco, B. Sattarov, F. Hédin, S. Wendeborn, C. Ho and D. El Ahdab, Computationally driven discovery of SARS-CoV-2 M pro inhibitors: from design to experimental validation, Chem. Sci., 2022, 13(13), 3674–3687 RSC.
- A. Calistri, A. Luganini, B. Mognetti, E. Elder, G. Sibille, V. Conciatori, C. Del Vecchio, S. Sainas, D. Boschi, N. Montserrat, A. Mirazimi, M. L. Lolli, G. Gribaudo and C. Parolin, The New Generation hDHODH Inhibitor MEDS433 Hinders the In Vitro Replication of SARS-CoV-2 and Other Human Coronaviruses, Microorganisms, 2021, 9(8), 1731 CrossRef CAS PubMed.
- J. C. Ferreira, S. Fadl, A. J. Villanueva and W. M. Rabeh, Catalytic dyad residues His41 and Cys145 impact the catalytic activity and overall conformational fold of the main SARS-CoV-2 protease 3-chymotrypsin-like protease, Front. Chem., 2021, 9, 692168 CrossRef CAS PubMed.
- A. Narayanan, M. Narwal, S. A. Majowicz, C. Varricchio, S. A. Toner, C. Ballatore, A. Brancale, K. S. Murakami and J. Jose, Identification of SARS-CoV-2 inhibitors targeting Mpro and PLpro using in-cell-protease assay, Commun. Biol., 2022, 5(1), 169 CrossRef CAS PubMed.
- D. W. Kneller, G. Phillips, H. M. O'Neill, R. Jedrzejczak, L. Stols, P. Langan, A. Joachimiak, L. Coates and A. Kovalevsky, Structural plasticity of SARS-CoV-2 3CL Mpro active site cavity revealed by room temperature X-ray crystallography, Nat. Commun., 2020, 11(1), 3202 CrossRef CAS PubMed.
- E. Fassi, M. Manenti, A. Citarella, M. D. Cas, S. Casati, N. Micale, T. Schirmeister, G. Roda, A. Silvani and G. Grazioso, Computational Design, Synthesis, and Biophysical Evaluation of β-Amido Boronic Acids as SARS-CoV-2 Mpro Inhibitors, Molecules, 2023, 28(5), 2356 CrossRef CAS PubMed.
- G. Amendola, R. Ettari, S. Previti, C. Di Chio, A. Messere, S. Di Maro, S. J. Hammerschmidt, C. Zimmer, R. A. Zimmermann, T. Schirmeister, M. Zappalà and S. Cosconati, Lead Discovery of SARS-CoV-2 Main Protease Inhibitors through Covalent Docking-Based Virtual Screening, J. Chem. Inf. Model., 2021, 61(4), 2062–2073 CrossRef CAS PubMed.
- L. Hoffer, C. Chira, G. Marcou, A. Varnek and D. Horvath, S4MPLE–Sampler for Multiple Protein-Ligand Entities: Methodology and Rigid-Site Docking Benchmarking, Molecules, 2015, 20(5), 8997–9028 CrossRef CAS PubMed.
- L. Hoffer, J. P. Renaud and D. Horvath, In silico fragment-based drug discovery: setup and validation of a fragment-to-lead computational protocol using S4MPLE, J. Chem. Inf. Model., 2013, 53(4), 836–851 CrossRef CAS PubMed.
- S. F. Altschul, W. Gish, W. Miller, E. W. Myers and D. J. Lipman, Basic local alignment search tool, J. Mol. Biol., 1990, 215(3), 403–410 CrossRef CAS PubMed.
- S. Henikoff and J. G. Henikoff, Amino acid substitution matrices from protein blocks, Proc. Natl. Acad. Sci. U. S. A., 1992, 89(22), 10915–10919 CrossRef CAS PubMed.
- R. A. Abagyan and S. Batalov, Do aligned sequences share the same fold?, J. Mol. Biol., 1997, 273(1), 355–368 CrossRef CAS PubMed.
- A. Waterhouse, M. Bertoni, S. Bienert, G. Studer, G. Tauriello, R. Gumienny, F. T. Heer, T. A. P. de Beer, C. Rempfer, L. Bordoli, R. Lepore and T. Schwede, SWISS-MODEL: homology modelling of protein structures and complexes, Nucleic Acids Res., 2018, 46(W1), W296–W303 CrossRef CAS PubMed.
- Q. Fu, H. Li, D. Duan, C. Wang, S. Shen, H. Ma and Z. Liu, External-Radiation-Induced Local Hydroxylation Enables Remote Release of Functional Molecules in Tumors, Angew. Chem., Int. Ed. Engl., 2020, 59(48), 21546–21552 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *a,
Davide
Moi
*a,
Davide
Moi