
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Tetramate derivatives by chemoselective Dieckmann ring closure of allo-phenylserines, and their antibacterial activity†
Liban
Saney
a,
Kirsten E.
Christensen
a,
Miroslav
Genov
 c,
Alexander
Pretsch
c,
Dagmar
Pretsch
c and
Mark G.
Moloney
*ab
c,
Alexander
Pretsch
c,
Dagmar
Pretsch
c and
Mark G.
Moloney
*ab
aThe Department of Chemistry, Chemistry Research Laboratory, University of Oxford, 12 Mansfield Road, Oxford, OX1 3TA, UK. E-mail: mark.moloney@chem.ox.ac.uk
bOxford Suzhou Centre for Advanced Research, Building A, 388 Ruo Shui Road, Suzhou Industrial Park, Jiangsu, 215123, P.R. China
cOxford Antibiotic Group, The Oxford Science Park, Magdalen Centre, Oxford OX4 4GA, UK
First published on 14th April 2023
Abstract
A general route which provides direct access to substituted bicyclic tetramates, making use of Dieckmann cyclisation of oxazolidine derivatives derived from allo-phenylserines, is reported. Of interest is the high level of diastereoselectivity observed for the N-acylation reaction of oxazolidines and the complete chemoselectivity of their ring closure in the Dieckmann cyclisation. Significantly, the sense of the chemoselectivity is different to earlier reported threo-phenylserine systems, showing the importance of steric bulk around the bicyclic ring system. The derived C7-carboxamidotetramates, but not C7-acyl systems, exhibited potent antibacterial activity against MRSA, with the most active compounds exhibiting well-defined physicochemical and structure–activity properties. This work clearly demonstrates that densely functionalised tetramates are both readily available and may exhibit high levels of antibacterial activity.
Introduction
The tetramate system occurs as a core skeleton in natural products which exhibit a wide range of bioactivity,1–3 and we have shown that bicyclic tetramates, which are accessible by chemoselective and stereoselective Dieckmann ring closures of oxazolidine or thiazolidine templates 1 (Fig. 1),4,5 may also exhibit potent antibacterial activity, mostly against Gram-positive bacteria. The chemoselectivity of this cyclisation depends upon the identity of R1, R2, X, along with the 2,5-relative stereochemistry of the N-acyloxazolidine or thiazolidine starting material 1. Normally, path A mode of cyclisation is preferred and predominates for 2,5-cis-malonyloxazolidines (X = O, R1 = tBu, R2 = H/Me) derived from serine5,6 and threonines7,8 leading to tetramates 2, although 2,5-trans-malonyloxazolidines (X = O) if formed, usually close by path B leading to tetramates 3. This intrinsic chemoselectivity may be controlled in some cases by adjusting the conditions for the Dieckmann cyclisation, thus tetramates (X = O, R2 = H) arising by path A were most readily accessed in good yield using the reaction conditions (KOtBu (1.1 eq.), dry tBuOH, reflux), while those from path B were available by the use of alternate conditions (KOtBu (2.2 eq.), wet tBuOH, reflux).9 On the other hand, N-acylthiazolidines derived from cysteine that had been condensed with aromatic aldehydes (X = S, R1 = Ar, R2 = H) gave tetramate products by path B, that is, in a different direction to the t-butyl series (X = S, R1 = tBu, R2 = H) which close by path A, and this arises due to differing stabilities of the cis- and trans-oxa(or thia)zolidine starting materials.4,10,11 However, by contrast to threonines (X = O, R1 = tBu, R2 = Me) which close by path A, threo-β-arylserines ((2S*,3R*)-2-amino-3-hydroxy-3-arylpropanoic acids, X = O, R1 = tBu, R2 = Ar) close by path B as a result of the steric influence of the C-4 aryl group.12 Of interest was the chemoselectivity which would be observed for allo-β-arylserines ((2S*,3S*)-2-amino-3-hydroxy-3-arylpropanoic acid), which are epimers at C-4 of threo-β-arylserines; we report here the results of that investigation.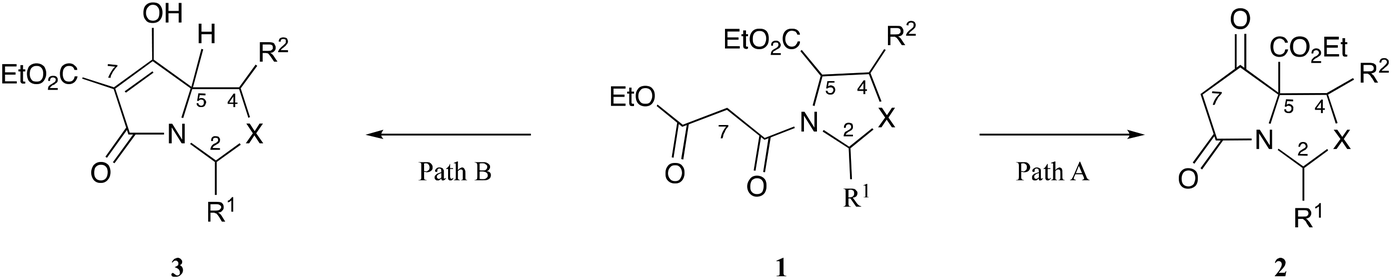 | ||
| Fig. 1 Chemoselectivity of Dieckmann cyclisation leading to tetramates. | ||
Results and discussion
The initial requirement was access to allo-β-hydroxy-β-aryl-α-amino acids, and such systems have previously been reported by Chang et al.13 along with Inoue et al.14 who exploited fully diastereoselective catalytic reductions of oximes using palladium on carbon, controlled by formation of a substrate–catalyst complex where the palladium coordinates with both the oxygen and nitrogen atoms of the substrate.13,14 The required ethyl aroylacetate starting materials were either commercially available, or could be readily prepared using the methodology of Clay et al.15 Compound 5a is commercially available, but for the others, ethyl potassium malonate 4 was dissolved in acetonitrile and was reacted with acid chlorides using a MgCl2–Et3N base system to obtain β-oxoesters 5b–g (Scheme 1), typically in quantitative yields and with a very high level of purity, not requiring chromatography; more than 3 eq. of Et3N was required to suppress the formation of side products.15 NMR analysis indicated that these compounds existed as a mixture of keto–enol tautomers (Table S1, ESI†). These were readily converted to their oxime derivatives 6a–g by reaction with sodium nitrite in glacial acetic acid using the conditions of Fell et al. (Scheme 1 and Table S2, ESI†), which required the temperature to be kept below 10 °C, giving the products in yields of between 71–93% in excellent purity, again not requiring chromatography.16 The major isomer for 6a was assumed to be E, based upon the work of Fell et al.,16 although this assignment was not material for subsequent work. Oximes 6a–g were dissolved in ethanol and conc. HCl and hydrogenated at ambient temperature and pressure, using palladium on carbon as a catalyst, for 1–3 days, and this gave allo-arylserines 7a–f fully diastereoselectively, in variable yields but again of high purity, not requiring chromatography (Scheme 1 and Table S3, ESI†).13,14 A sizeable amount of catalyst was required to produce the desired product in acceptable yields, and as a result, oxime 6g gave compound 7a instead of the desired bromo 7g by concomitant hydrogenolysis. Confirmation that the phenylserine derivatives were indeed the allo-isomer was achieved by conversion of phenylserine 7a to cis-oxazolidinone 8 and comparison to literature data (Scheme 2 and Table S4, ESI†).17 Furthermore, allo-phenylserine 7a was converted to crystalline Boc-protected allo-phenylserine 9a (Scheme 2), and a single-crystal X-ray structure confirmed the allo-assignment (Fig. S1, ESI†).18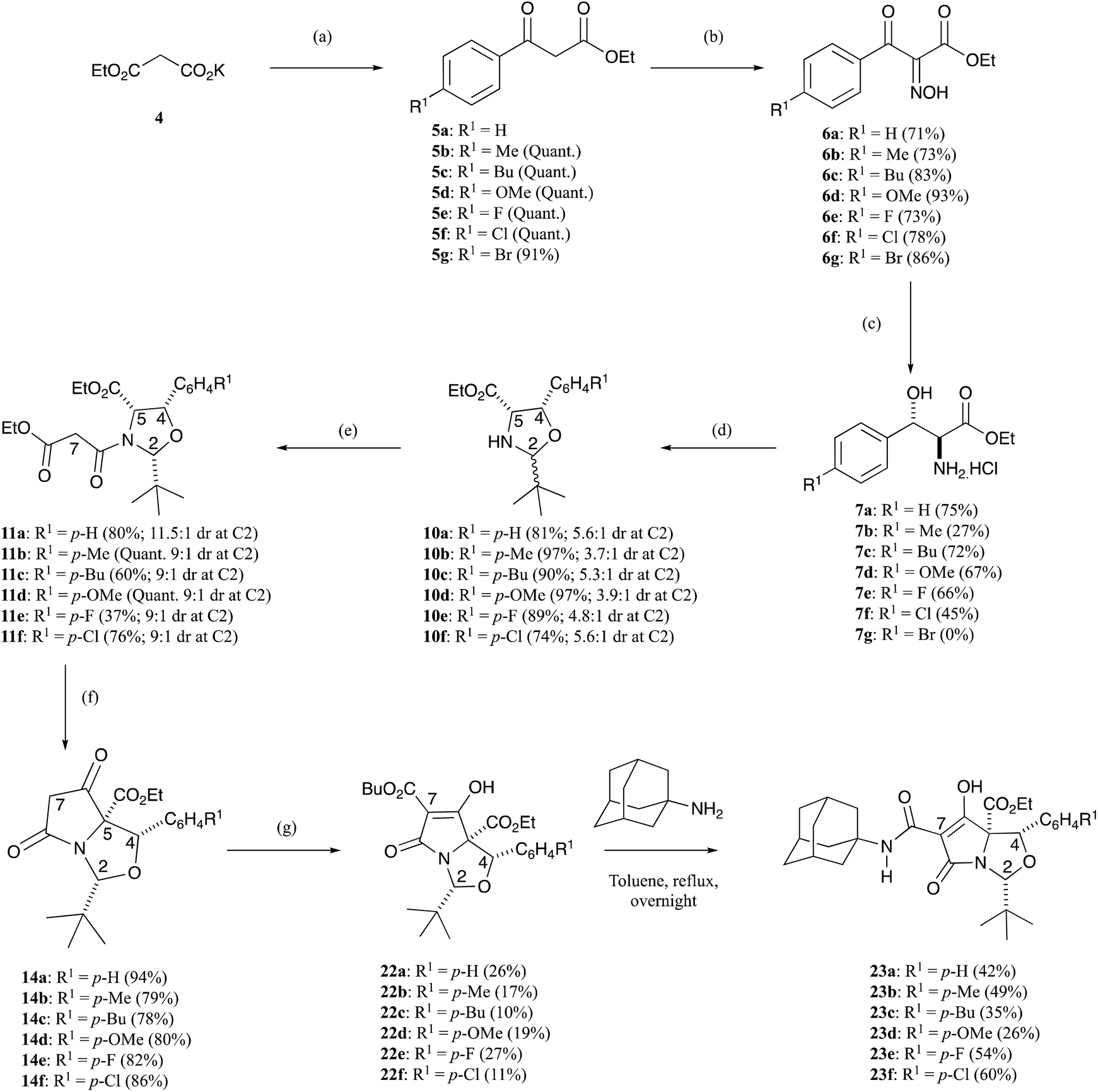 | ||
Scheme 1 Synthetic route to tetramates. Synthesis of tetramates 14, 22 and 23 from allo-phenylserine derivatives 7a–f; (a) MeCN, MgCl2, Et3N, 10–25 °C, 2.5 h then R1C6H4C(O)Cl, Et3N, rt, overnight; (b) NaNO2 in H2O, AcOH, rt, 30 min-overnight; (c) EtOH, conc. HCl, H2, Pd–C, rt, 1–3 d; (d) pivaldehyde, petroleum ether 40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :60, Et3N, Dean–Stark, >100 °C, overnight; (e) ethyl malonyl chloride, pyridine, DCM, reflux, overnight; (f) DBU, THF, rt, overnight; (g) butyl chloroformate, DCM, DMAP, reflux, overnight. :60, Et3N, Dean–Stark, >100 °C, overnight; (e) ethyl malonyl chloride, pyridine, DCM, reflux, overnight; (f) DBU, THF, rt, overnight; (g) butyl chloroformate, DCM, DMAP, reflux, overnight. | ||
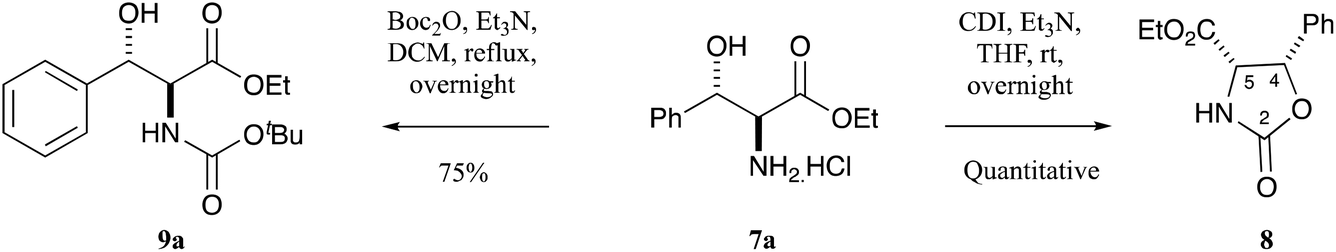 | ||
| Scheme 2 Confirmation of relative stereochemistry of phenylserine. | ||
With amino esters 7a–f in hand, reaction with pivaldehyde in the presence of Et3N and petroleum ether 40:60 with heating to over 100 °C under a Dean–Stark trap for 16–24 h formed oxazolidines 10a–f in good yields – typically between 74–97% – as roughly a 5:1 mixture of diastereomers at the C2-centre, as determined from NMR analysis (Scheme 1 and Table S5, ESI†).5,7 Confirmation of their relative stereochemistry was achieved by nOe analysis. Since we had earlier found that oxazolidines derived from threo-phenylserines were not very stable to chromatographic purification,12 these products were taken directly into the next reaction sequence. Oxazolidines 10a–f were readily N-acylated by reaction with ethyl malonyl chloride under basic conditions to form malonamides 11a–f in 37% up to quantitative yields (Scheme 1). Once again, the crude products were pure by NMR analysis but could easily be chromatographically purified if required to obtain malonamides 11a–f with high levels of purity (Table S6, ESI†), being typically a 9:1 mixture of diastereomers at the C2-position. nOe studies clearly demonstrated the cis-relationship between the H2, H4 and H5-protons for malonamide 11a and this was also confirmed by a single-crystal X-ray structure (Fig. S1, ESI†).18 This stereochemical outcome was also assumed for the compound series as a whole on the basis of the similarity of their NMR spectra.
Interestingly, the N-acylation of oxazolidines 10a–f was highly diastereoselective in favour of the all cis product (Scheme 1 and Fig. 2), and much more so than the related threo-phenylserine derivatives.12 Therefore, it was apparent that the cis-C4–C5 stereochemistry had a significant effect on the diastereoselectivity of both the oxazolidine cyclisation and N-acylation reactions. While the all cis oxazolidine diastereomer 10a–f is the major one, favoured ca. 5:1 over the minor one, it is also the most reactive in the N-acylation reaction, since there is an exposed face for reactions which is away from all three ring substituents, and this gives the major N-acyl derivative 11a–f. The minor oxazolidine is not so reactive to N-acylation, since both sides of attack on the oxazolidine experience some steric hindrance from ring substituents, allowing for equilibration to the major oxazolidine and onward reaction to the major N-acyl product, leading to an overall 9:1 mixture of malonamide diastereomers 11a–f, starting from roughly a 5:1 mixture of oxazolidine diastereomers 10a–f.
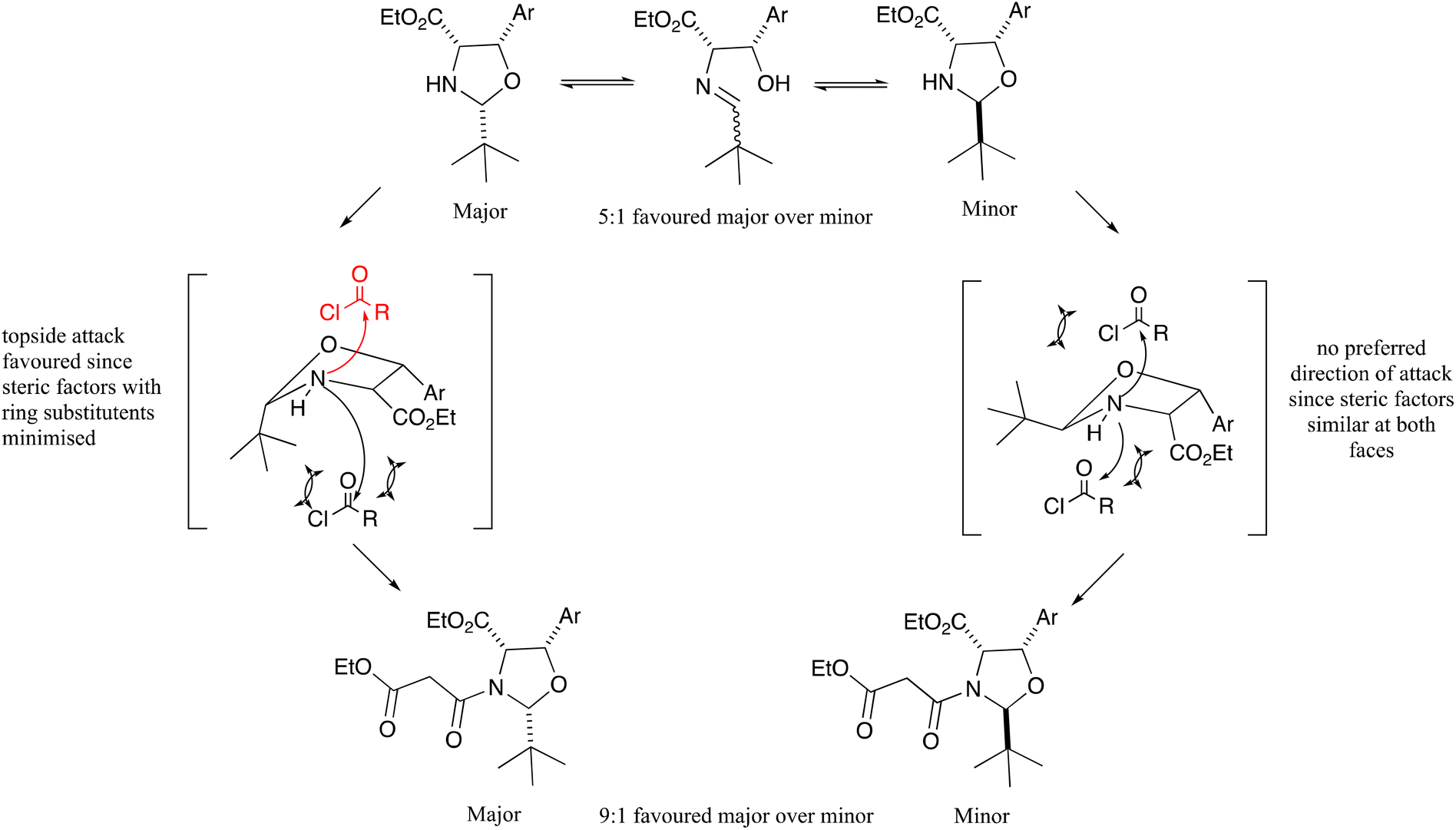 | ||
| Fig. 2 Diastereoselectivity in oxazolidine ring closure and N-acylation. | ||
When oxazolidine 10a was reacted with ethyl methylmalonyl chloride 12c (Scheme 3),19 malonamide 13a was obtained as a 1.8:1 mixture of diastereomers at the C7-position, which could be partially separated by careful chromatographic purification, and whose ring relative stereochemistry was confirmed by nOe and NOESY analysis with full assignment achievable by a single-crystal X-ray analysis (Fig. S1, ESI†).18 Similarly, oxazolidine 10a was reacted with ethyl phenylmalonyl chloride 12d (Scheme 3) and malonamide 13b was obtained as a 1.7:1 mixture of diastereomers at the C7-position in good yield, but unlike methyl derivatives 13a, were inseparable by chromatographic purification.19 The relative stereochemistry of the ring protons of malonamides 13b was confirmed as being all cis by a single-crystal X-ray structure of one of the diastereomers (Fig. S1, ESI†).18 Equilibration of malonamide diastereomers 13a and 13b in CDCl3 solution over several weeks was observed.
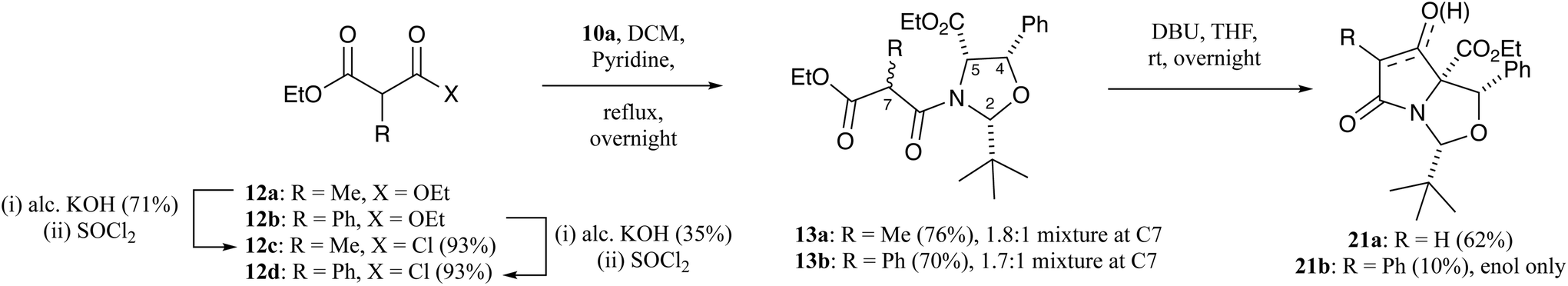 | ||
| Scheme 3 Synthetic route to C-7 alkyl and aryl tetramates. | ||
Malonamides 11a–f were subjected to the Dieckmann cyclisation to form the desired bicyclic tetramates 14a–f (Scheme 1). Initially, malonamide 11a was reacted with KOtBu in THF at reflux overnight, but this led to a complex mixture of unidentifiable products. However, changing the base to DBU, following the procedure of Josa-Culleré et al.,20 effectively gave tetramates 14a–f (Table S7, ESI†) in excellent yields as single diastereomers (Scheme 1) and with full chemoselectivity, following path A for ring closure (Fig. 1). This corresponds to a switch in the chemoselectivity observed for threo-phenylserines12 and this is discussed in more detail below. These tetramates did not require chromatographic purification, being pure by NMR analysis, since aqueous extraction of the highly acidic tetramate salts proved to be effective. Overall, pure tetramates 14a–f could be synthesised from β-oxoesters 5a–g in 5 synthetic steps without the need for purification by chromatography at any stage of the reaction sequence. Additionally, tetramates 14a–f were stable in CDCl3 solution and as an oil for many months. Of interest is that long-range coupling between the H2 and H7-protons (coupling constant of 0.8 Hz) was observed for tetramates 14 (Fig. S2, ESI†). The C5-ethyl ester stereochemistry was later confirmed from a single-crystal X-ray structure of C7-carboxamides 23a and 23d (Fig. S1, ESI†).18 Substituted tetramates 21a and 21b were formed via the Dieckmann cyclisation of malonamides 13a and 13b respectively (Scheme 3). Tetramate 21a was obtained in 62% yield as a mixture of keto/enol tautomers, but in the case of tetramate 21b, a yield of only 10% was obtained, and the product was observed exclusively in the enolic form in CDCl3 solvent.
Of interest was both the high level of, and the switch in, chemoselectivity in the Dieckmann cyclisation of malonamides 11a–f derived from allo-phenylserines (Fig. 3) when compared with those derived from threo-phenylserine derivatives.12 In the formation of enolates 15a–f and then 16a–f from malonamides 11a–f which ultimately led to the formation of the observed bicyclic tetramates 14a–f, both the tBu and aryl groups are on the exo-face in pseudo-diequatorial locations, thus minimising steric interactions. Tetramates 17a–f which might have formed via the ring closure of enolates 18a–f would require C-5 epimerisation in the original substrate, in which both the tBu and aryl groups would be found on the exo-face of the bicyclic lactam, but this material was not observed experimentally. Of interest is that tetramates 19a–f and 20a–f were also not observed experimentally and this is most likely due to the fact that the tBu and aryl groups are on the endo-face of the bicyclic lactam in both cases, causing unfavourable steric congestion – consistent with that observed for the threo-phenylserine-derived tetramic acids, for which detailed theoretical calculations were made.12 Overall, this work demonstrates that allo-phenylserines behave in a similar manner to the reactions of L-serine, L-threonine, L-allo-threonine and L-cysteine-derived products that were condensed with pivaldehyde,5,7 reacting by path A (Fig. 1) but very differently to threo-phenylserine derivatives in Dieckmann ring closures,12 reacting by path B (Fig. 1).
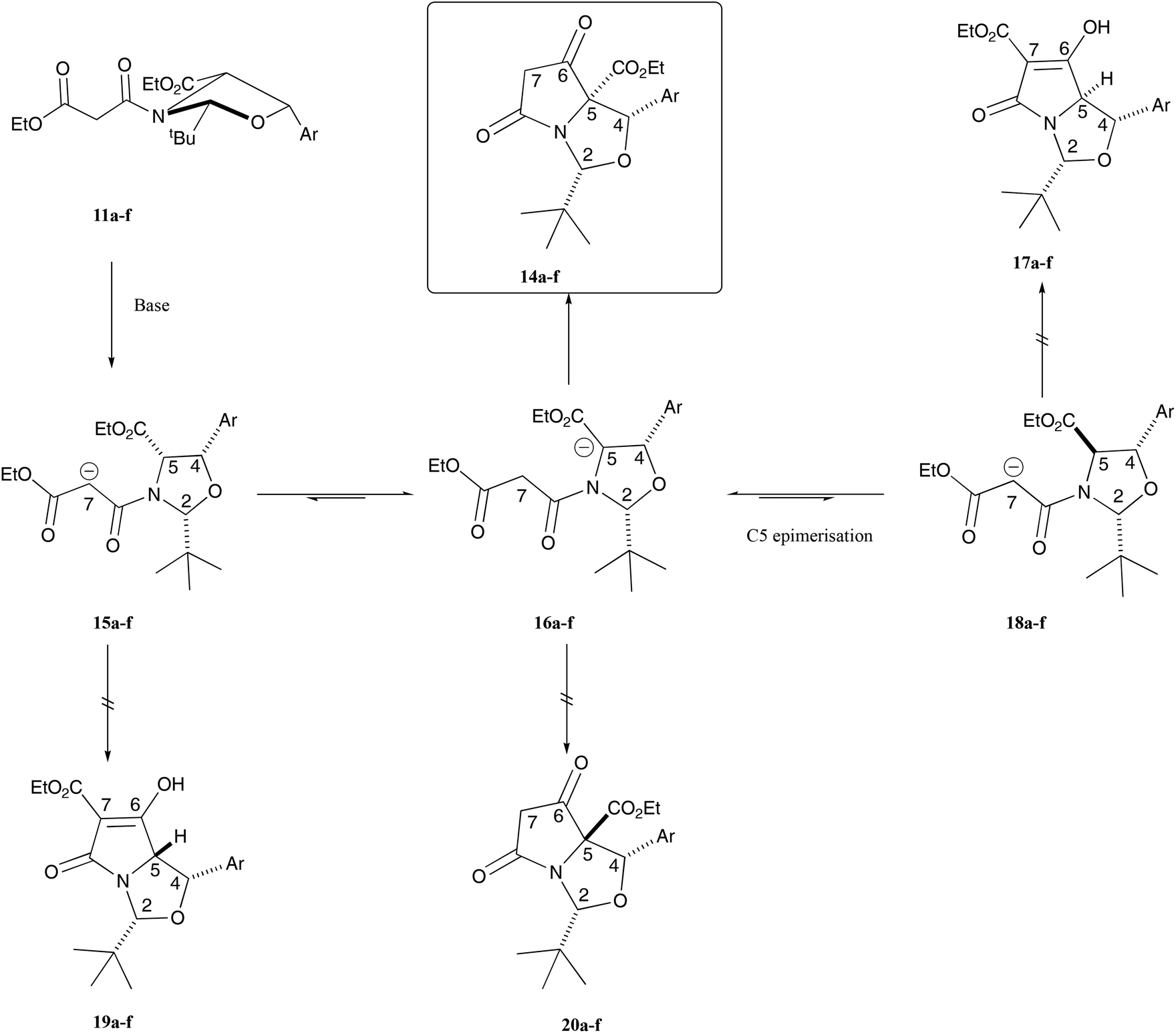 | ||
| Fig. 3 Dieckmann ring closure leading to tetramates. | ||
The chemoselective formation of tetramates 14a–f from malonamides 11a–f led to unfunctionalised C7-tetramates. In order to incorporate functionality at this position, tetramates 14a–f were reacted with butyl chloroformate under basic conditions, following the reaction conditions of Jeong et al.,21 to form C7-butyl ester tetramates, to allow differentiation of C5-ethyl esters 22a–f (Scheme 1 and Table S8, ESI†). The yields for these conversions were poor – typically between 10–27%, with a considerable amount of starting material being present in the reaction mixture after 24 h, probably reflecting the low reactivity of the intermediate tetramate enolate. Moreover, the C7-butyl ester tetramates 22a–f were extremely unstable in CDCl3 solution – analogous to C7-ethyl ester tetramates of type 3 (Fig. 1) reported earlier,12 needing to be both chromatographically purified and reacted on very quickly; this instability could also explain the presence of starting material in the initial reaction mixture. These compounds also picked up trace metal ions during chromatographic purification, a phenomenon we had observed earlier, but which could be readily removed with a 10% citric acid (aq.) workup. The importance of metal chelation in these systems has recently become increasingly apparent and has been reviewed.22,23
These compounds could be converted to C7-carboxamidotetramic acids 23a–f by reaction with adamantylamine in yields that ranged from 26–60% (Scheme 1 and Table S9, ESI†) and which were suitable for chromatographic purification as well as being stable in both the pure form and in CDCl3 solution for many months. The aminolysis reaction was fully chemoselective for the C7-butyl ester, with no exchange at the more hindered C5-ethyl ester. Single-crystal X-ray structures of compounds 23a and 23d confirmed their relative stereochemistry, and that no epimerisation had occurred at any of the ring stereogenic centres during the aminolysis reactions (Fig. S1, ESI†).18 Compounds 23a–f, after chromatographic purification on silica gel, picked up trace metal ions to form metal-chelates across the tri-carbonyl unit; this was immediately evident in the 1H NMR spectrum, where broad peaks were observed after chromatographic purification, a phenomenon which has been seen previously.4,21 In the case of compound 23a, the H2 and H4-signals were relatively sharp – indicating these protons were relatively far away from the metal-chelation site – whereas the adamantyl and aromatic peaks were relatively broad – indicating these groups were relatively close to the metal-chelation site (Fig. S3a, ESI†). The acid-washed tetramic acids 23a–f existed as approximately a 50:50 mixture of AB:CD tautomers in CDCl3 solution (Fig. 4), as has been observed in related systems,21 but this differed from C7-carboxamides 24 that lacked a C5-ethyl ester (i.e. those derived from threo-phenylserine), whose ratio was typically an 80:20 AB:CD tautomeric mixture in CDCl3 solution (Fig. 4).12 It appears that the allo-derived 23a binds metal ions with less affinity than threo-derived carboxamidotetramate 24a,12 since the NMR peaks were far sharper for tetramate 23a (Fig. S3, ESI†) than for tetramate 24a. Since the only difference between these two compounds was a missing C5-ethyl ester in the case of tetramate 24a, it seems that the C5-ethyl esters might play an important (steric) role in the reduction of metal-chelation across the proximal tricarbonyl core. The NMR peaks of tetramates 23a–f could be further sharpened by washing metal-chelated tetramic acids with 10% citric acid solution (aq.) (Fig. S3b, ESI†). Interestingly, the peaks for both the adamantyl and aromatic groups were sharpened by a simple acid wash, and changes in the chemical shift values for the tBu, adamantyl and H4-groups before and after acid wash (Fig. S3b, ESI†) were observed.
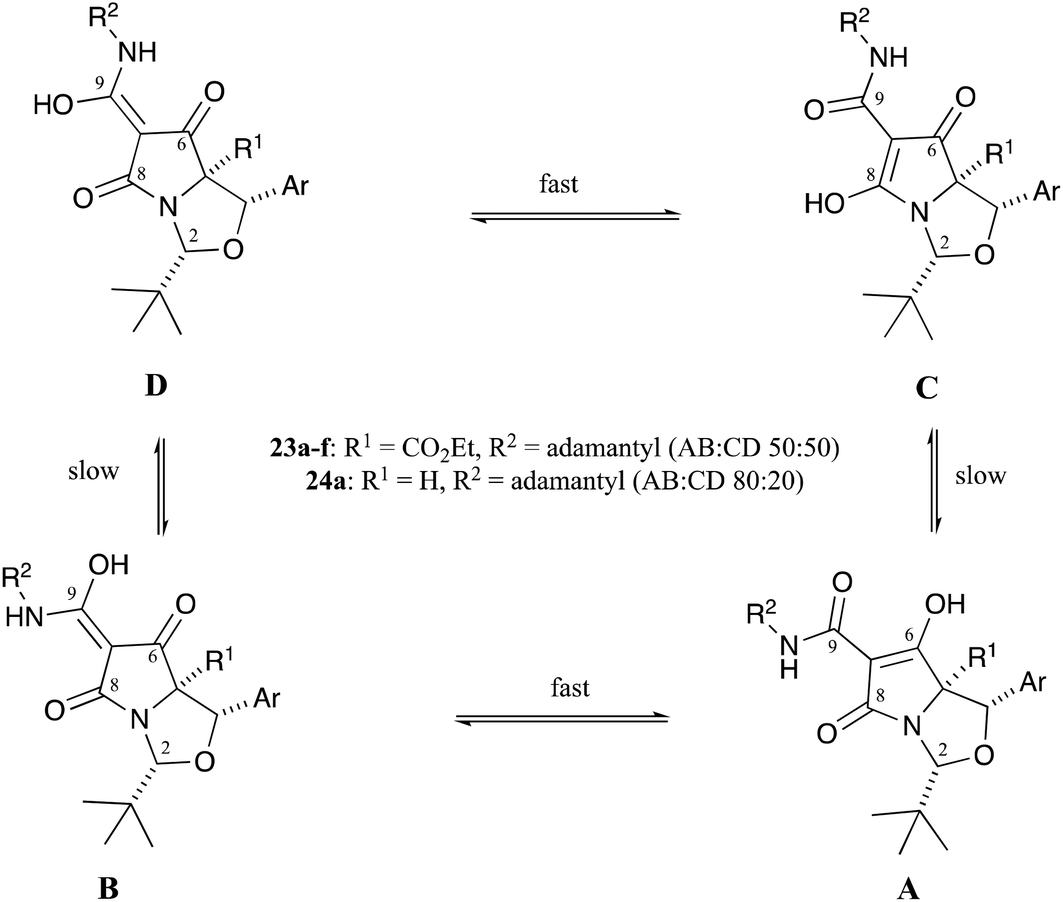 | ||
| Fig. 4 Tautomeric equilibria in carboxamidotetramates. | ||
Inspired by an earlier report of the direct acylation of tetramates using carboxylic acids,24 tetramate 14a was coupled with aromatic and aliphatic carboxylic acids using DCC and stoichiometric amounts of DMAP (1.3 eq.) to form C7-acyltetramates 25a–s in acceptable yields (Scheme 4 and Table S10, ESI†). In this process, substituents were tolerated at the para-position of the aromatic ring system, but unsurprisingly less so at the ortho-position. Moreover, more hindered acids such as 1-naphthoic acid, 2,6-dichlorobenzoic acid, 2-chlorobenzoic acid and 9-anthracenecarboxylic acid gave no products. While the 2-thienyl substituent was obtained in poor yield, aliphatic acids were obtained in moderate to good yields. NOESY studies suggested no epimerisation had occurred at any of the stereogenic centres after DCC/DMAP coupling. Once again, purification of these tetramic acids on silica gel led to the introduction of trace metal ions, evidenced by very broad resonances in their 1H NMR spectra (Fig. S4, ESI†), and which were much broader than metal-chelated C7-carboxamides 23a described above (Fig. S3, ESI†). For example, in the case of carboxamidotetramate 23a, while discernible peaks are visible, for acyltetramate 25d, no assignable peaks were initially visible but sharp peaks arose after a simple acid wash with 10% citric acid solution (aq.).
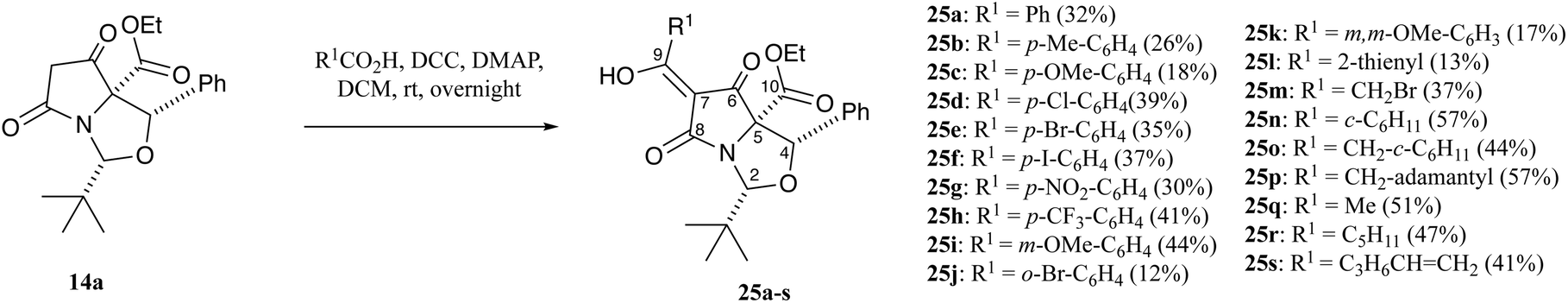 | ||
| Scheme 4 Synthetic route to C-7 acyl tetramates. | ||
Additionally, the products in all cases were obtained as the CD tautomers, with the exception of 25o–q, which were observed as a mixture of tautomers but in which the CD tautomers were the major ones. Tan et al. had earlier demonstrated that the major tautomers for C7-acyltetramates 26 was the CD pair, in which the C6-carbonyl peaks from the AB tautomers were more downfield due to deshielding from hydrogen bond effects (Fig. 5),21,24–26 and the spectra for 25a–s were assigned by analogy.
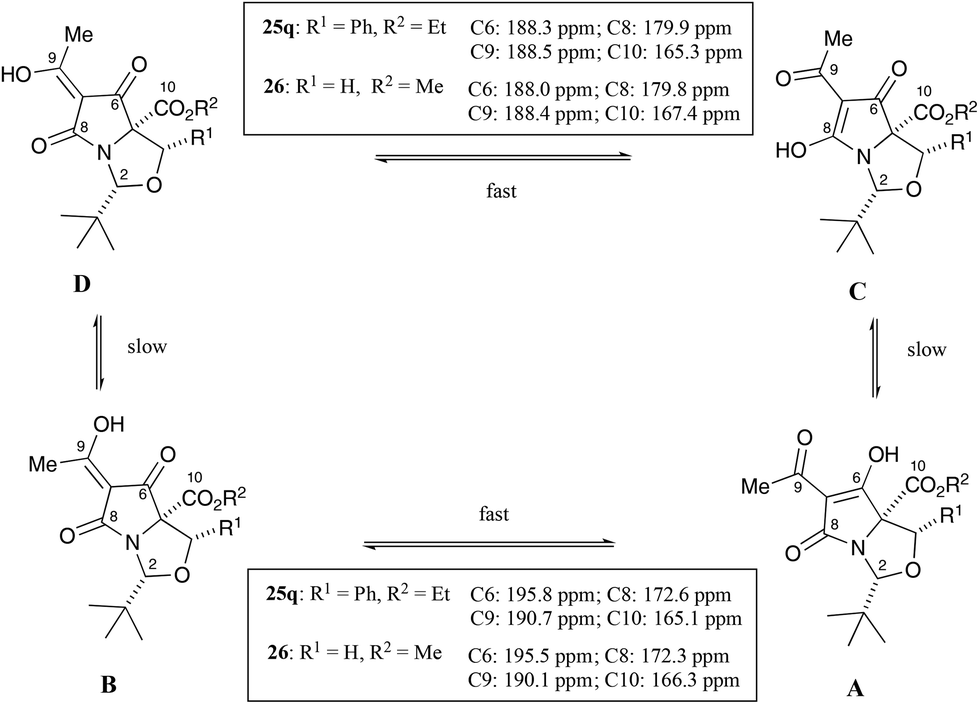 | ||
| Fig. 5 Tautomeric equilibria in acyltetramates. | ||
Although the above described synthesis of allo-phenylserines 7a–f from β-oxoesters 5a–fvia Pd–C catalysed hydrogenation was very diastereoselective, it suffered from four major drawbacks. Firstly, all the β-oxoesters 5b–f and oximes 6a–f – with the exception of 5a – had to be synthesised. Secondly, a sizeable amount of Pd–C was required to convert the oximes 6a–f to allo-phenylserines 7a–f, and thirdly, the yields were modest. Finally, chloro- and bromo-substituted allo-phenylserine derivatives further reacted by hydrogenolysis under the reduction conditions to give dehalogenated side-products. An alternative, based upon a report by Chaumont et al.27 of the erythro-diasteroselective synthesis of Boc-protected allo-phenylserine derivatives via a decarboxylative aldol reaction between the hemi-malonate derivative of diethyl (Boc-amino)malonate 27c and various aldehydes (Scheme 5),27 was therefore examined; starting 27c could be readily synthesised by reacting inexpensive diethyl aminomalonate hydrochloride 27a with Boc2O. Boc-protected allo-phenylserine 9a synthesised this way was fully identical to that both prepared by Chaumont et al.27 and the material prepared earlier (Scheme 1). This approach proved to be applicable to a wide variety of substituted allo-phenylserine derivatives 9a–m (Scheme 5 and Table S11, ESI†).
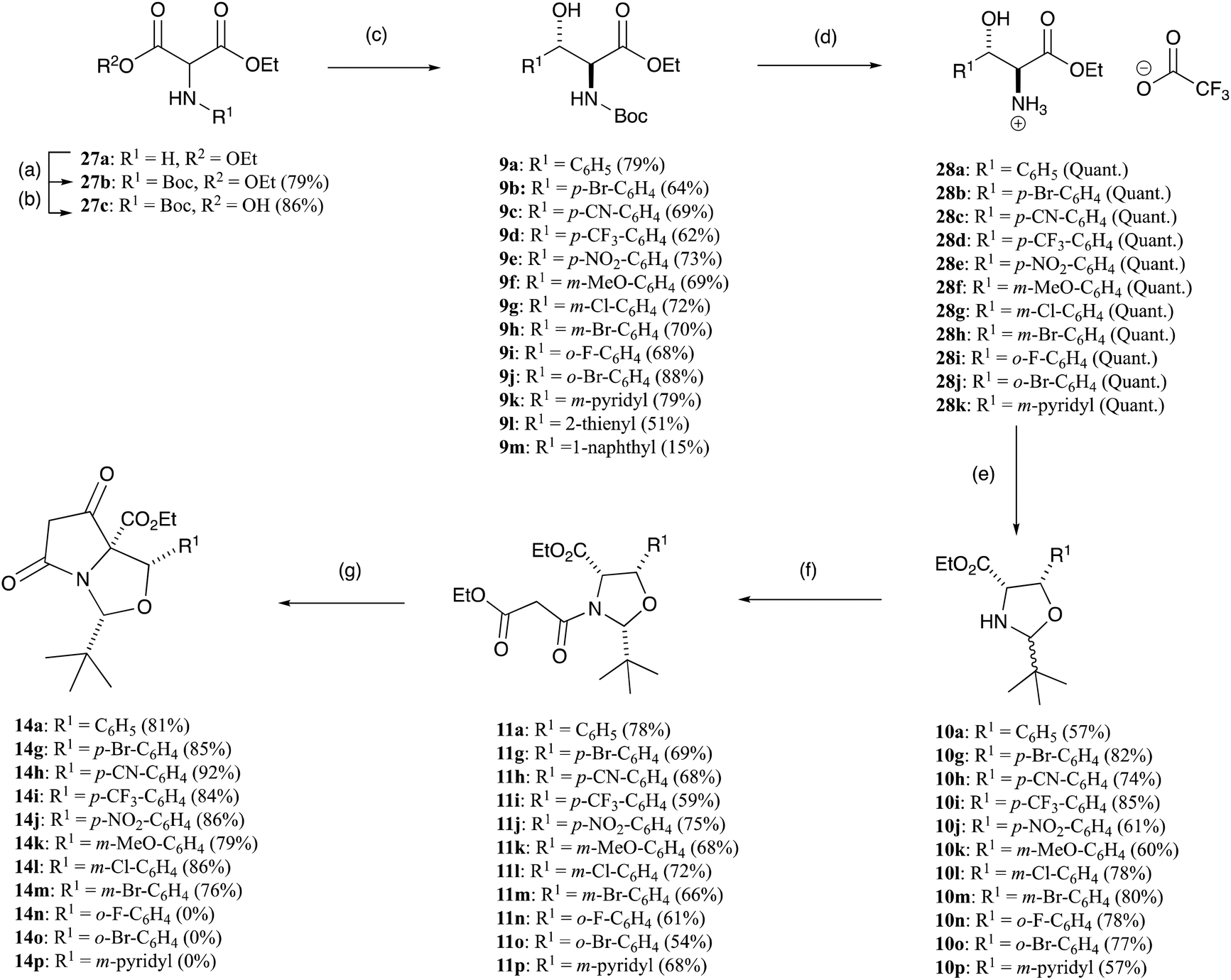 | ||
| Scheme 5 Expanded functionalisation of C4-aromatic tetramates 14a,g–mvia the decarboxylative aldol reactions of hemimalonate 27c; (a) Boc2O, 1 M NaOH (aq.), 1,4-dioxane, rt, overnight; (b) KOH, EtOH, rt, overnight; (c) R1CHO, Et3N, 50 °C, 2 d; (d) TFA, DCM, rt, 2 h; (e) petroleum ether 40:60, pivaldehyde, Et3N, >100 °C, Dean–Stark, overnight; (f) ethyl malonyl chloride, pyridine, DCM, reflux, overnight; (g) DBU, THF, rt, overnight. | ||
Boc-protected-allo-phenylserine derivatives 9a–l were deprotected using TFA and were stirred at room temperature for 2 h to furnish allo-phenylserines 28a–k as the trifluoroacetate ammonium salts in quantitative yields (Scheme 5 and Table S12 ESI†). The exception was 9l, which gave a complex mixture of side-products, and was thus not used further, and 9m was discontinued due to its low yield. The allo-phenylserine derivatives 28a–k were then reacted with pivaldehyde using the established protocol outlined above, to obtain oxazolidines 10a,g–p in good yields as mixtures of diastereomers (Scheme 5 and Table S13, ESI†). The spectroscopic consistencies of oxazolidines 10a,g–p with those of 10a–f derived from β-oxoesters 5a–f prepared above confirmed their cis-relationship (see Tables S5 and S13, ESI†). The major and minor diastereomers of oxazolidines 10g–p were readily assigned by comparison to the independently prepared and characterised materials described above. These compounds were then N-acylated with ethyl malonyl chloride using the established protocol to obtain malonamides 11a,g–p in good yields as predominantly one diastereomer (Scheme 5 and Table S14, ESI†), again consistent with that of malonamides 11a–f. Comparison to earlier compounds and NOESY studies confirmed their relative stereochemistry. These malonamides 11a,g–p were then reacted with DBU in THF overnight (Scheme 5) to form tetramates 14a,g–m in good yields (Table S15, ESI†). However, surprisingly, tetramate 14n was formed as a very minor product, and was not used further, and tetramate 14o did not form at all, presumably due to hindrance at the ortho-position. Additionally, the Dieckmann cyclisation of the 3-pyridyl substituent 11p was not successful.
Evaluation of antibacterial activity
While C7-unfunctionalised tetramates have no intrinsic antibacterial activity against Gram (+) and Gram (−) bacteria,28 compounds 14a–f showed appreciable activity against MRSA, with MIC values ranging between 7.8–15.6 μg mL−1, whereas 14g and 14k was more weakly acting with MIC of 31 μg mL−1 (Table 1 and Table S16, ESI†). However, tetramates 14h–j were much less active and had activity ranging between 125–250 μg mL−1, clearly indicating that substitution around the C4-aromatic ring system imparts antibacterial activity. However, no activity was observed against E. coli, a feature commonly observed with such tetramates, and which is likely to arise due to poor bacterial cell membrane permeability and/or efficient efflux. Surprisingly, C7-acyltetramates 25 – which were more analogous to natural product antibacterial tetramates – were relatively inactive against both MRSA and E. coli (Table S16, ESI†).| Compound | MIC(S. aureus) (μg mL−1) | Cell lines EC50 (μg mL−1) | Therapeutic ratioa | |||
|---|---|---|---|---|---|---|
| HeLa | heK-293 | CaCo | MDCK | |||
| a Calculated from MIC against S. aureus using the HeLa cell line (EC50 HeLa cell line/MIC(S. aureus)). | ||||||
| 14a | 15.6 | 15.6 | 31.3 | 62.5 | 31.3 | 1 |
| 14b | 7.8 | 15.6 | 15.6 | 31.3 | 31.3 | 2 |
| 14c | 15.6 | 7.8 | 3.9 | 31.3 | 15.6 | 0.5 |
| 14d | 15.6 | 15.6 | 31.3 | 62.5 | 31.3 | 1 |
| 14e | 15.6 | 15.6 | 31.3 | 62.5 | 31.3 | 1 |
| 14f | 15.6 | 15.6 | 15.6 | 31.3 | 31.3 | 1 |
| 23a | 0.25 | 15.6 | 15.6 | 31.3 | 31.3 | 62 |
| 23b | 0.25 | 15.6 | 15.6 | 31.3 | 31.3 | 62 |
| 23c | 3.9 | 15.6 | 31.3 | 31.3 | 62.5 | 4 |
| 23d | 0.25 | 7.8 | 15.6 | 7.8 | 31.3 | 31 |
| 23e | 0.25 | 7.8 | 15.6 | 15.6 | 31.3 | 31 |
| 23f | 0.25 | 7.8 | 7.8 | 15.6 | 15.6 | 31 |
However, C7-carboxamides 23a–f displayed potent antibacterial activities against MRSA, similar to previously reported C7-carboxamides,12,21 and of interest was that a C5-ethyl ester group was well tolerated, particularly when compared with threo-derived C7-carboxamide 24a,12 although none exhibited antibacterial activity against E. coli. The antibacterial activity against MRSA for C7-carboxamides 23a,b,d–f was extremely potent – MIC values of 0.25 μg mL−1 – with the exception of tetramate 23c, which had a MIC value of 3.9 μg mL−1 (Table 1), suggesting that hydrophobic groups at the para-position worsen bioactivity. The physicochemical property space of the most potent compounds (MIC < 4 μg mL−1) was confined to a small area of chemical structure space (523 < Mw < 579 g mol−1; 3.9 < clogP < 5.9; 105 < PSA < 114 Å2; 777 < MSA < 898 Å2; HBD = 2; HBA = 5, see Fig. 6 and Table S16, ESI†), with many compliant with the Ro5 – even though the initial design of these compounds did not explicitly seek to obey Lipinski's parameters. The high clogP values of many of these may also explain, at least in part, the lack of activity observed against Gram (−) E. coli bacteria. These bicyclic tetramates were significantly more lipophilic and less polar than other antibiotic classes (Fig. S5, ESI†) and the structure–activity profile indicates distinct regions of the bicyclic nucleus responsible for antibacterial activity and/or metal chelation (Fig. 7).
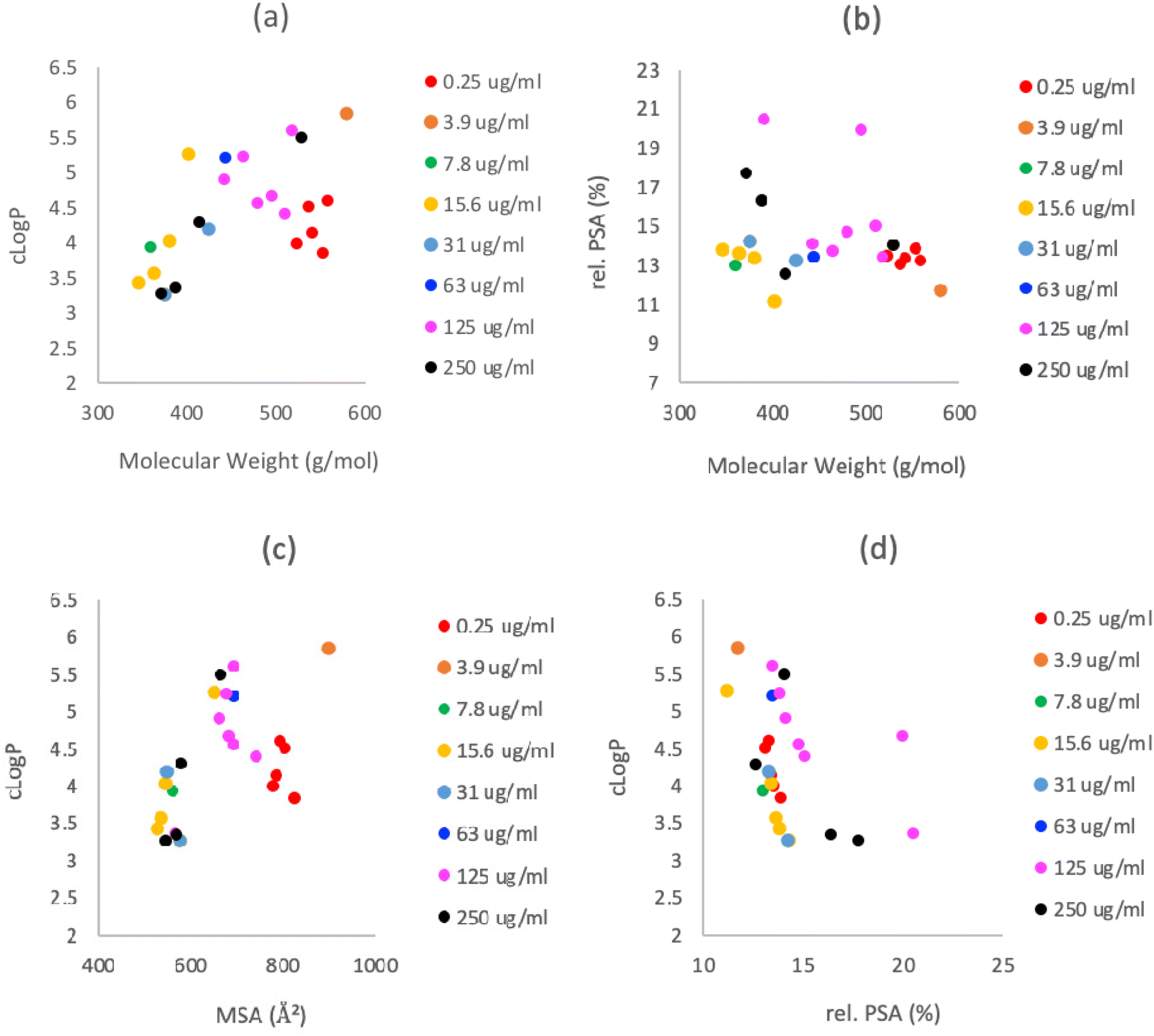 | ||
| Fig. 6 Physicochemical property space of tetramates; (a) clogP plotted against Mw; (b) rel. PSA plotted against Mw; (c) clogP plotted against MSA and (d) clogP plotted against rel. PSA. clogP, MSA and PSA were calculated using Marvin (19.9.0), 2019, ChemAxon. | ||
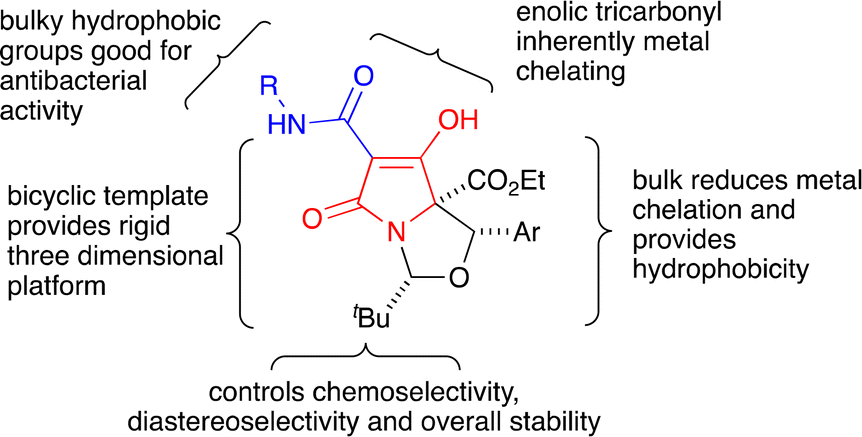 | ||
| Fig. 7 Overview of the chemical components necessary for potent antibacterial activity against MRSA, derived from an in-depth SAR analysis of the N,O-bicyclic tetramates. | ||
This compound series showed some cytotoxicity against HeLa, heK-293, CaCo and MDCK human cell lines, and the therapeutic ratio (calculated from MIC against S. aureus using the HeLa cell line (EC50 HeLa cell line/MIC(S. aureus)) (Table 1) shows that C5-ethyl ester tetramates 14a–f possessed low selectivity, although this was mainly due to the lack of potency observed against MRSA. However, carboxamides 23a,b,d–f showed high selectivity for bacteria, with a therapeutic ratio of either 31 or 62. Panduwawala et al.4 and Jeong et al.21 had earlier demonstrated that carboxamidotetramic acids typically lost their antibacterial activity in the presence of human serum albumin (HSA). When bicyclic tetramates 23a–f were tested against MRSA in the presence of 1/3 the serum albumin concentration as that found in human blood, for carboxamides 23a,b, 23d–f the activity dropped 16-fold, and in the case of 23e, 33-fold. However, compound 23c only showed a 4-fold decrease in antibacterial activity and noteworthy is that it is the most lipophilic in the carboxamide series 23a–f. It was clear from these results that MIC shifted to higher values in the presence of HSA, consistent with the known behaviour of plasma protein binding small molecules; it has earlier been shown that PPB strongly correlates with increased hydrophobicity.29 However, good bioactivity (MIC < 4 μg mL−1) is still observed in the presence of 1/3 the HSA concentration as that found in blood, and this result is important, indicating that the aromatic substitution placed at the C4-position of the bicyclic tetramate could not only be tolerated, but could also be crucial to adjusting the physicochemical properties of these bicyclic tetramates to lend itself towards potential in vivo studies and therapeutic applications.
Conclusion
It has been demonstrated that oxazolidines derived from allo-phenylserines efficiently close by Dieckmann ring closure giving tetramates with full diastereocontrol; the sense of this closure follows analogous reactions of serine, threonine and cysteine but is different to threo-phenylserines. Importantly, the oxazolidine ring substituents have been shown to reduce metal cation chelation by steric interaction. C7-unfunctionalised tetramates 14a–f had appreciable activity against MRSA, and conversion to C7-carboxamides 23 led to much improved antibacterial activity. A large, bulky, hydrophobic carboxamide pendant group at the C7-position led to more potent antibacterial activity. All the compounds screened showed no antibacterial activity against E. coli. C7-adamantylcarboxamidotetramic acids 23a–f were very selective for bacteria over mammalian cells. Many of the compounds synthesised were within the constraints of Ro5. These compounds occupied a distinct chemical property space different from that occupied by other known antibiotics, with the most potent compounds occupying 523 < Mw < 579 g mol−1; 3.9 < clogP < 5.9; 11.7 < rel-PSA < 13.9%. Unfortunately, these compounds suffered from PPB, leading to the MIC values being shifted to higher concentrations when tested in the presence of HSA. From a consideration of physicochemical properties, potency and toxicity of these compounds, the most promising candidates for further optimisation were 23a,b,d–f.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
L. S. gratefully acknowledges DTA funding from EPSRC.References
- M. Petermichl and R. Schobert, 3-Acyltetramic acids: A decades-long approach to a fascinating natural product family, Synlett, 2017, 28, 654–663 CrossRef CAS.
- R. Schobert and A. Schlenk, Tetramic and tetronic acids: An update on new derivatives and biological aspects, Bioorg. Med. Chem., 2008, 16, 4203–4221 CrossRef CAS PubMed.
- B. J. L. Royles, Naturally-occurring tetramic acids: Structure, isolation, and synthesis, Chem. Rev., 1995, 95, 1981–2001 CrossRef CAS.
- T. D. Panduwawala, S. Iqbal, A. L. Thompson, M. Genov, A. Pretsch, D. Pretsch, S. Liu, R. H. Ebright, A. Howells, A. Maxwell and M. G. Moloney, Functionalised bicyclic tetramates derived from cysteine as antibacterial agents, Org. Biomol. Chem., 2019, 17, 5615–5632 RSC.
- M. D. Andrews, A. G. Brewster, K. M. Crapnell, A. J. Ibbett, T. Jones, M. G. Moloney, K. Prout and D. Watkin, Regioselective Dieckmann cyclisations leading to enantiopure highly functionalised tetramic acid derivatives, J. Chem. Soc., Perkin Trans. 1, 1998, 223–235 RSC.
- H. Bagum, B. R. Shire, K. E. Christensen, M. Genov, A. Pretsch, D. Pretsch and M. G. Moloney, Bicyclic Lactams Derived from Serine or Cysteine and 2-Methylpropanal, Synlett, 2020, 31, 378–382 CrossRef CAS.
- H. Bagum, K. E. Christensen, M. Genov, A. Pretsch, D. Pretsch and M. G. Moloney, Synthetic Access to 3-Substituted Pyroglutamic acids from Tetramate Derivatives of Serine, Threonine, allo-Threonine and Cysteine, J. Org. Chem., 2019, 84, 10257–10279 CrossRef CAS PubMed.
- M. Anwar and M. G. Moloney, Efficient enantioselective synthesis of tetramic acids and lactams from threonine, Tetrahedron Lett., 2007, 48, 7259–7262 CrossRef CAS.
- Y.-C. Jeong, M. Anwar, T. M. Nguyen, B. S. W. Tan, C. L. L. Chai and M. G. Moloney, Control of chemoselectivity in Dieckmann ring closures leading to tetramic acids, Org. Biomol. Chem., 2011, 9, 6663–6669 RSC.
- R. Zhang, X. Li, M. Genov, A. Pretsch, D. Pretsch and M. G. Moloney, Synthetic Access to Hydrophilic Tetramate Derivatives of Cysteine, J. Org. Chem., 2020, 85, 12393–12407 CrossRef CAS PubMed.
- T. D. Panduwawala, S. Iqbal, R. Tirfoin and M. G. Moloney, Chemoselectivity and stereoselectivity of cyclisation pathways leading to bicyclic tetramates controlled by ring-chain tautomerisation in thiazolidines, Org. Biomol. Chem., 2016, 14, 4464–4478 RSC.
- L. Saney, K. E. Christensen, X. Li, M. Genov, A. Pretsch, D. Pretsch and M. G. Moloney, Tetramate Derivatives by Chemoselective Dieckmann Ring Closure of threo-Phenylserines and Their Antibacterial Activity, J. Org. Chem., 2022, 87, 12240–12249 CrossRef CAS PubMed.
- Y.-T. Chang and W. H. Hartung, Palladium Catalysis. V. The Hydrogenation of alpha-Oximino Ketones., J. Am. Chem. Soc., 1953, 75, 89–91 CrossRef CAS.
- H. Inoue, K. Matsuki and T. Oh-ishi, A New Enantioselective Synthesis of (2R, 3S)-3-(4-Methoxyphenyl)glycidic Ester via the Enzymetic Hydrolysis of erythro-N-acetyl-beta-(4-methoxyphenyl)serine, Chem. Pharm. Bull., 1993, 41, 1521–1523 CrossRef CAS.
- R. J. Clay, T. A. Collom, G. L. Karrick and J. Wemple, A Safe, Economical Method for the Preparation of beta-Oxo Esters, Synthesis, 1993, 290–292 CrossRef CAS.
- S. C. M. Fell, M. J. Pearson, G. Burton and J. S. Elder, Synthesis and biological activity of new C-6 and C-7 substituted vinyloxyimino-penicillins and -cephalosporins, J. Chem. Soc., Perkin Trans. 1, 1995, 1483–1493 RSC.
- Y. Z. Z. Lu and W. D. Wulff, Direct Access to N-H Aziridines from the Asymmetric Catalytic Aziridination with Borate Catalysts Derived from the VANOL and VAPOL Ligands, J. Am. Chem. Soc., 2007, 129, 7185–7194 CrossRef PubMed.
- Low temperature single crystal X-ray diffraction data for 9a, 11a, 13a, 13b, 23a, and 23d were collected using a Rigaku Oxford SuperNova diffractometer and at Diamond Light Source, Beamline I19-1. Raw frame data were reduced using CrysAlisPro and the structures were solved using ‘Superflip’ before refinement with CRYSTALS as per the CIF. Full refinement details are given in the ESI† (CIF); Crystallographic data have been deposited with the Cambridge Crystallographic Data Centre (CCDC 2160046–56†).30–32.
- H. Bagum, K. E. Christensen, A. Pretsch, M. Genov, D. Pretsch and M. G. Moloney, Synthetic access to 3,4-disubstituted pyroglutamic acids from tetramate derivatives of serine, threonine, allo-threonine and cysteine, Tetrahedron, 2019, 75, 130561–130583 CrossRef CAS.
- L. Josa-Culleré, A. Pretsch, D. Pretsch and M. G. Moloney, Antibacterial Mimics of Natural Products by Side-Chain Functionalization of Bicyclic Tetramic Acids, J. Org. Chem., 2018, 83, 10303–10317 CrossRef.
- Y.-C. Jeong, M. Anwar, M. G. Moloney, Z. Bikadi and E. Hazai, Natural product inspired antibacterial tetramic acid libraries with dual enzyme inhibition, Chem. Sci., 2013, 4, 1008–1015 RSC.
- D. Repac Antić, M. Parčina, I. Gobin and M. Petković Didović, Chelation in Antibacterial Drugs: From Nitroxoline to Cefiderocol and Beyond, Antibiotics, 2022, 11, 1105 CrossRef.
- F. Albini, S. Bormann, P. Gerschel, V. A. Ludwig and W. Neumann, Dithiolopyrrolones are Prochelators that Are Activated by Glutathione, Chem. – Eur. J., 2023, 29, e202202567 CrossRef CAS PubMed.
- Y.-C. Jeong and M. G. Moloney, Synthesis of and Tautomerism in 3-Acyltetramic Acids, J. Org. Chem., 2011, 76, 1342–1354 CrossRef CAS PubMed.
- Y.-C. Jeong, M. G. Moloney, Z. Bikadi and E. Hazai, A Detailed Study of Antibacterial 3-Acyltetramic Acids and 3-Acylpiperidine-2,4-diones, ChemMedChem, 2014, 9, 1826–1837 CAS.
- S. W. B. Tan, C. Chai and M. G. Moloney, Synthesis of 3-Acyltetramates by Side Chain Manipulation and their Antibacterial Activity, Org. Biomol. Chem., 2014, 12, 1711–1716 RSC.
- P. Chaumont, J. Baudoux, J. Maddaluno, J. Rouden and A. Harrison-Marchand, Access to Anti or Syn 2-Amino-1,3-diol Scaffolds from a Common Decarboxylative Aldol Adduct, J. Org. Chem., 2018, 83, 8081–8091 CrossRef CAS PubMed.
- Y.-C. Jeong and M. G. Moloney, Tetramic acids as bioactive templates: synthesis, tautomeric and antibacterial behaviour, Synlett, 2009, 2487–2491 CAS.
- D. G. Brown, T. L. May-Dracka, M. M. Gagnon and R. Tommasi, Trends and Exceptions of Physical Properties on Antibacterial Activity for Gram-positive and Gram-negative Pathogens, J. Med. Chem., 2014, 57, 10144–10161 CrossRef CAS PubMed.
- P. Parois, R. I. Cooper and A. L. Thompson, Crystal structures of increasingly large molecules: meeting the challenges with CRYSTALS software, Chem. Cent. J., 2015, 9, 30 CrossRef PubMed.
- R. I. Cooper, A. L. Thompson and D. J. Watkin, CRYSTALS enhancements: dealing with hydrogen atoms in refinement, J. Appl. Crystallogr., 2010, 43, 1100–1107 CrossRef CAS.
- L. Palatinus and G. Chapuis, SUPERFLIP—A computer program for the solution of crystal structures by charge flipping in arbitrary dimensions, J. Appl. Crystallogr., 2007, 40, 786–790 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details; 1H and 13C NMR spectra; X-ray crystallographic data (CIF); bioassay and cheminformatic data. 2235613–2235618. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3ob00376k |
| This journal is © The Royal Society of Chemistry 2023 |