
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of a fully protected long-chain polyamine subunit of aculeine B using the photoremovable NPEC group†
Masayoshi
Miyahara
,
Ryoya
Wakabayashi
,
Raku
Irie
 and
Masato
Oikawa
*
and
Masato
Oikawa
*
Graduate School of Nanobioscience, Yokohama City University, Seto 22-2, Kanazawa-ku, Yokohama 236-0027, Japan. E-mail: moikawa@yokohama-cu.ac.jp
First published on 10th April 2023
Abstract
Polyamines are ubiquitously found in nature. In this paper, we disclose our iterative coupling strategy for the synthesis of a structurally defined polymer of 1,3-propanediamine, and the polymer can be used for the synthesis of both the initially proposed structure and the revised structure of protoaculeine B isolated from a marine sponge. We first attempted the synthesis of polyamines using “the Ns strategy” but found that a polyamine with eleven Ns groups has solubility problems. We then examined the versatility of the photoremovable NPEC protecting group in polyamine synthesis. Finally, the synthesis of a suitably protected 12-mer polyamine was achieved employing the NPEC group for the temporary protection of a terminal amino group.
Introduction
Aculeine B (1a in Fig. 1A, ACU-B) is a natural product isolated by Sakai et al. in 20111 from a marine sponge Axinyssa aculeata collected inshore around Iriomote island, Japan. The structure of ACU-B consists of a 44-amino acid residue peptide, designated AcuPep, bound to a 12-, 13-, 14-, or 15-mer of 1,3-propanediamine (long-chain polyamine, LCPA)2via a modified tryptophan (W′, shown in red in Fig. 1A). The Trp-AcuPep peptide is thought to be post-translationally modified by structurally heterogeneous LCPAs by an enzymatic Pictet–Spengler reaction, in the biosynthesis of ACU-B.3 The sponge contains analogs of ACU-B with various amino acids/peptides conjugated to 12- to 15-mer LCPAs via W′.1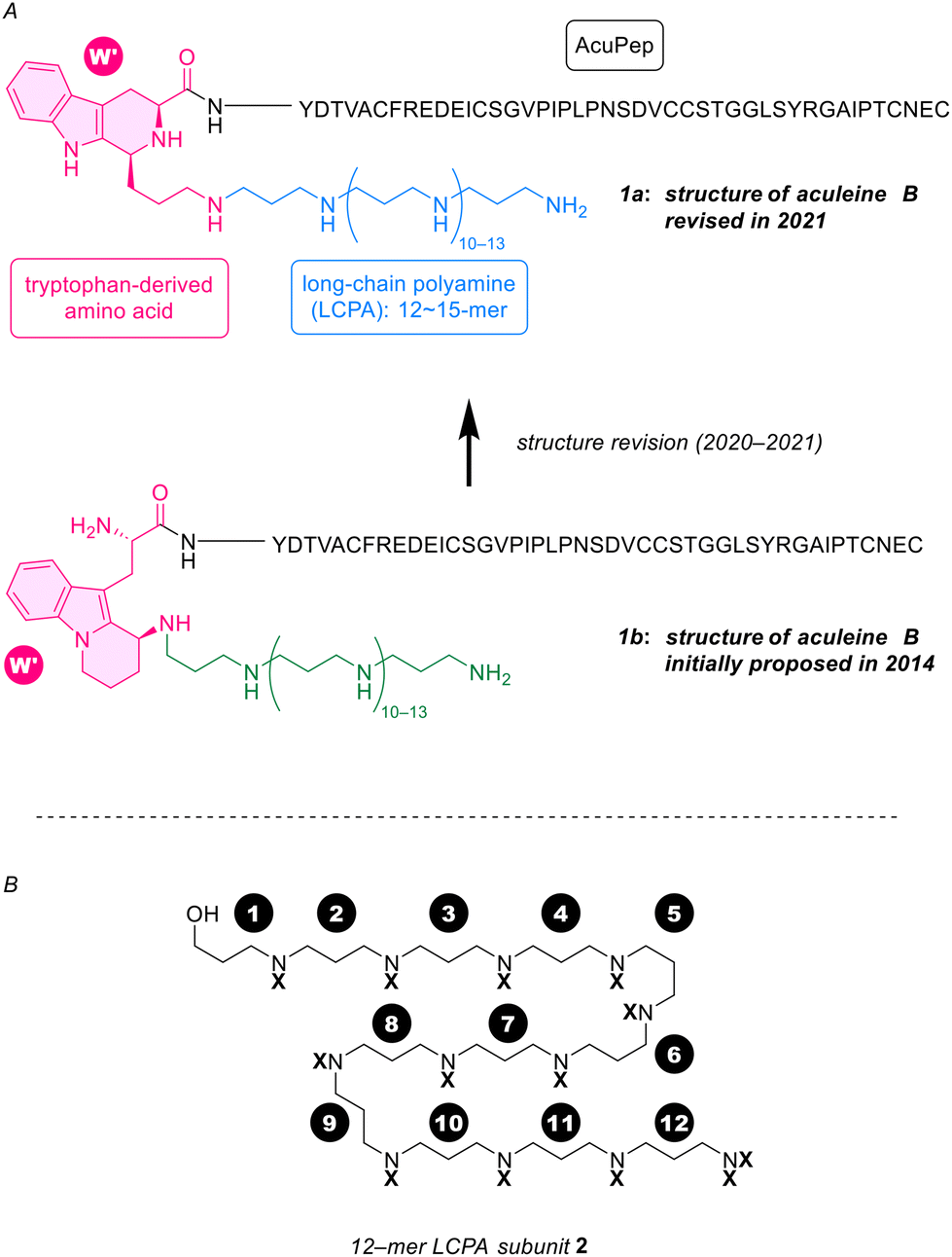 | ||
Fig. 1 (A) The structure of aculeine B (ACU-B) 1a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 1 isolated from A. aculeata and its initially proposed misassigned structure 1b.9 (B) Suitably protected 12-mer LCPA subunit 2 for chemical syntheses of 1a and 1b. 1 isolated from A. aculeata and its initially proposed misassigned structure 1b.9 (B) Suitably protected 12-mer LCPA subunit 2 for chemical syntheses of 1a and 1b. | ||
The biological functions of ACUs were investigated mainly on aculeine A (ACU-A, structure not shown),1 which has an additional LCPA chain conjugated to ACU-B. ACU-A was found to be neurotoxic and hemolytically active in erythrocytes. ACU-A showed membrane permeabilizing activity when exposed to 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) liposomes with 10% cholesterol.1 Furthermore, ACU-A was recently shown to be a naturally occurring cell penetrating peptide (CPP) that can internalize in the cells in an energy dependent manner.4
We then planned to study the biological properties of ACUs quantitatively using structurally less complex ACU-B. However, as noted above, the LCPA in the natural ACUs is a mixture of 12-mer to 15-mer of 1,3-propanediamine and hence is structurally heterogeneous. Furthermore, ACU-B is less abundant among the series of ACUs isolated from the sponge, and was obtained in only a limited amount. It was, therefore, necessary to establish a chemical synthesis method for obtaining large amounts of homogeneous ACUs (1) to quantitatively evaluate the biological properties and (2) to investigate the membrane permeabilizing and cell penetrating activities.
Here we report our practical synthesis of suitably protected, homogeneous polyamines.5 Although many attempts to chemically synthesize polyamines have been reported,6 synthesis of the LCPAs of ACUs is challenging because of their large molecular size. In this study, therefore, we de novo investigated the synthesis of a suitably protected 12-mer of 1,3-propanediamine. Our synthetic plan includes (1) employment of “the Ns strategy”7 and (2) the use of a 3-mer as the smallest structural unit and elaboration of a larger 6-mer and 12-mer by its iterative coupling.
As shown in Fig. 1A, the chemical structure of the modified tryptophan (W′, red part) of ACUs underwent a revision in 2021.3 The 12-mer LCPA subunit 2 (Fig. 1B) targeted in this study was first designed as an alcohol to directly serve the synthesis of the misassigned structure 1b (see Fig. 1A, the green part)8 initially proposed in 2014.8,9 However, the alcohol 2 was expected to also be utilized in the chemical synthesis of the revised structure 1a, the part shown in blue. Indeed, we have recently succeeded in the total synthesis of the revised LCPA-W′ part3 named protoaculeine B using 2 (see Scheme 11 for the structure).10 Furthermore, our preliminary studies using homogeneous synthetic LCPA prepared in this study have recently shown that the above activities characteristic of ACU-A require the LCPA moiety in addition to the AcuPep motif (unpublished). This paper reports our efforts to develop and synthesize the LCPA subunit 2 of ACUs in the following order.
(1) Our unfruitful attempt at the synthesis of the first-generation 12-mer LCPA subunit 2a (see Scheme 3) with eleven Ns groups by the Ns strategy.
(2) Revisiting synthesis strategies, and the evaluation of the suitability of the photoremovable NPEC protecting group in the modified Ns strategy.5
(3) Achievement of the synthesis of a suitably protected, second-generation 12-mer LCPA subunit 2b (see Scheme 10), with favourable physical properties owing to fewer Ns groups (four).9b
Results and discussion
Synthesis of the first-generation 12-mer LCPA subunit 2a protected with eleven Ns groups
For the synthesis of the 12-mer LCPA subunit 2, we decided to adopt a strategy starting from the smallest 3-mer structural unit and first doubling the size to construct a 6-mer polyamine. A similar process was planned to further extend the 6-mer polyamine to a 12-mer polyamine. In such an iterative strategy, the choice of the protecting group seemed important, and we decided to use the Boc group and the Ns group as persistent protecting groups for amino groups.We first attempted the synthesis of a suitably protected 12-mer polyamine 2a (see Scheme 3 for the structure) by a well-established method called the Ns strategy.7Scheme 1 shows the synthesis of a 3-mer triamine 9 which is the smallest unit for iterative synthesis of the 12-mer LCPA 2a. The synthesis was performed according to the procedure reported by Fukuyama and Kan.7d Selective mono-N-Ns-protection of 1,3-propanediamine (3) gave 4 quantitatively (99% yield). N-Boc protection of 4, followed by coupling with 1,3-dibromopropane (6), provided 2-mer diamine 7 in 80% in 2 steps. After treatment of the bromide 7 with N-Ns-3-aminopropanol (8) prepared from 3-aminopropanol by N-Ns protection,7d the key 3-mer alcohol 9 was obtained in good yield (83%).
 | ||
| Scheme 1 Preparation of the smallest 3-mer triamine unit 9.7d | ||
From the 3-mer triamine unit 9, a 6-mer polyamine 14 was synthesized as shown in Scheme 2. First, the hydroxy group was converted to iodide 11via mesylate 10 in 93% yield over 2 steps. NMR data of 11 were consistent with those reported.7d On the other hand, a two-step protecting group manipulation (SOCl2, MeOH; NsCl, Et3N) was performed on 9 to obtain 3-mer alcohol 13 with three Ns groups via free amine 12 in 94% yield.
 | ||
| Scheme 2 Synthesis of 6-mer polyamine 14 with five Ns protecting groups. | ||
With two 3-mer triamines 11 and 13 in hand, we then attempted to construct a 6-mer polyamine bearing five Ns groups by coupling them. Gratifyingly, it was found that the coupling induced by Cs2CO3 proceeded smoothly in CH3CN to give 14 in excellent yield (94%).
From the 6-mer polyamine 14, we further attempted to synthesize a 12-mer polyamine. Here, polyamine 2a (Scheme 3) of twice the size of 14 was synthesized following the procedure for synthesizing the 6-mer polyamine 14 from the 3-mer triamine 9 shown in Scheme 2. That is, as shown in Scheme 3, the hydroxy group of 14 was converted to iodide 16 in 83% yield (2 steps) by mesylation followed by iodination. On the other hand, the N-Boc group was replaced with an N-Ns group over two-step manipulation to furnish hexa-N-Ns-polyamine 18 in moderate yield (62%, 2 steps). The two 6-mer polyamines (16 and 18) were then subjected to a coupling reaction induced by Cs2CO3 to give 12-mer polyamine 2a. Unfortunately, 2a was found to be insoluble in common organic solvents except DMF and pyridine and could not be purified. The poor solubility of 2a may be due to the many (eleven) Ns groups that cause hydrophobic and/or π–π stacking interactions. Therefore, we decided to consider another 12-mer LCPA subunit with a different pattern of N-protecting groups.
 | ||
| Scheme 3 Synthesis of the first-generation 12-mer LCPA subunit 2a with eleven Ns protecting groups. | ||
Revisiting synthesis strategies
Since the insolubility of the initially synthesized 12-mer LCPA subunit 2a was thought to be due to the interactions between N-Ns groups, we decided to synthesize a 12-mer LCPA subunit with fewer Ns groups. To make this possible, the number of N-Boc and N-Ns groups in the 3-mer triamine 9 must be changed. We planned to increase the number of N-Boc groups to “two”, and reduce the number of N-Ns groups to “one” in the newly designed 3-mer triamine unit (see 36 in Scheme 8).To achieve the synthesis, it was necessary to employ another temporary group orthogonal to the Boc and Ns groups, for protection of a terminal amino group. Although N-Alloc and N-Cbz groups had been used orthogonally,7c in the present study we decided to examine the applicability of a photoremovable group in polyamine synthesis. Among the photoremovable protecting groups for amines, we focused on the 1-(2-nitrophenyl)ethoxy carbonyl (NPEC) group11 (Scheme 4) used for photocaged compounds. The NPEC group belongs to the nitrophenylethyl (NPE)-type caging groups12 and was developed by Walker et al. in 198913 as a more reactive group than the popular o-nitrobenzyl group.14 The NPE-type caging groups had been used in biochemistry to control in vitro biological events.15 However, there were only a few instances in which the NPEC group was used in organic synthesis.16 One reason for this may be that the NPEC group contains a stereogenic center. We expected that the issue of the NPEC group will not be a major problem in the synthesis of achiral polyamines; rather, the photoremovable nature was expected to bring efficiency to polyamine synthesis.
 | ||
| Scheme 4 Mode of photodegradation of N-NPEC amine.12 | ||
Scheme 5 shows one cycle of the iterative process planned for the synthesis of polyamines, wherein the NPEC group is used to temporarily protect a terminal amino group. That is, starting with an oligomer of 1,3-propanediamine A bearing NPEC-NNs- and TrO- groups at both ends, the Tr group would be selectively removed under acidic conditions to give alcohol B. On the other hand, N-Ns amine C is expected to be synthesized by photoirradiating A to remove the NPEC group. A Mitsunobu reaction17 of B and C is expected to give polyamine D twice the size of A. Since the product D has NPEC-NNs- and TrO- groups at both ends, a polyamine twice the size of D would be further synthesized by repeating the same operation as A → D for D. Thus, the number of Ns groups was expected to be reduced in this synthetic strategy.
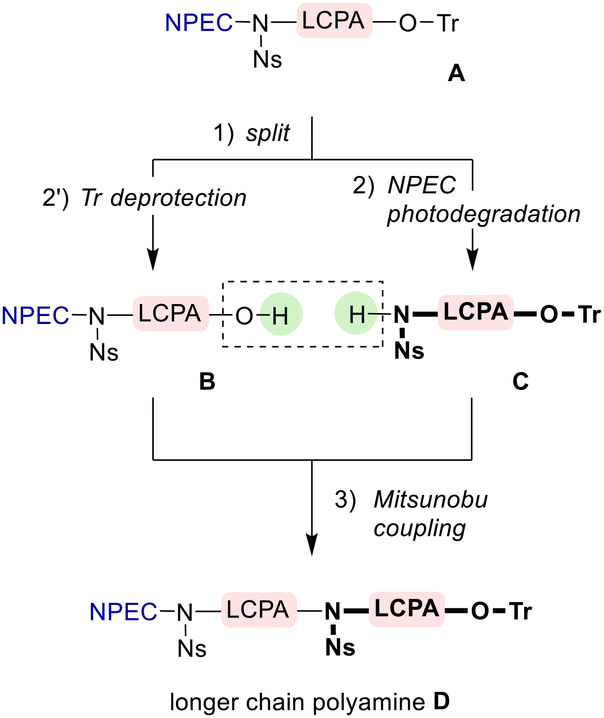 | ||
| Scheme 5 Our plan for polyamine synthesis using the modified Ns strategy. “LCPA” denotes the protected oligomer of 1,3-propanediamine. | ||
Evaluation of the photoremovable NPEC protecting group
Preparation of three NPEC reagents, NPEC-OSu 21, NPEC-Cl 22, and NPEC-OBt 23, is shown in Scheme 6. The syntheses were carried out starting from commercially available 2′-nitroacetophenone (19).18 Reduction of 19 afforded alcohol 20 in 99% yield. Treatment of 20 with (SuO)2C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and Et3N gave the chemically stable NPEC-OSu 21 in good yield (94%) after silica-gel column chromatography. Alcohol 20 was also converted to chemically rather unstable NPEC-Cl 22 upon exposure to triphosgene and Na2CO3. From NPEC-Cl 22, NPEC-OBt 23 (HOBt, Et3N)19 was elaborated as another stable NPEC reagent in 63% yield.
O and Et3N gave the chemically stable NPEC-OSu 21 in good yield (94%) after silica-gel column chromatography. Alcohol 20 was also converted to chemically rather unstable NPEC-Cl 22 upon exposure to triphosgene and Na2CO3. From NPEC-Cl 22, NPEC-OBt 23 (HOBt, Et3N)19 was elaborated as another stable NPEC reagent in 63% yield.
 | ||
| Scheme 6 Preparation of three NPEC reagents NPEC-OSu 21, NPEC-Cl 22, and NPEC-OBt 23. | ||
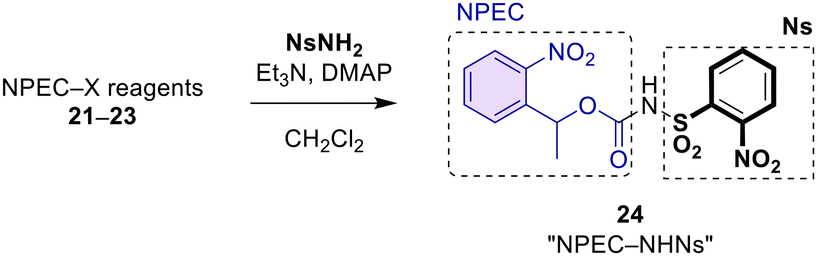
With three NPEC reagents thus obtained, we synthesized NPEC-NHNs 24. As shown in Table 1, succinimidyl ester 21 was treated with NsNH2, Et3N, and DMAP to furnish NPEC-NHNs 24 in 69% yield (entry 1). When chloroformate 22 was used, the reaction gave a better yield (85%, run 2). With NPEC-OBt 23, the reaction was sluggish and the yield for 24 was only 13% (entry 3). Therefore, NPEC-Cl 22 was decided to be employed for the preparation of NPEC-NHNs reagent 24 that is used for the introduction of the NPEC-NNs- group (see below) in the present study.
|
|
||
|---|---|---|
| Entry | NPEC-X | Yield of 24 |
| 1 | NPEC-OSu (21) | 69% |
| 2 | NPEC-Cl (22) | 85% |
| 3 | NPEC-OBt (23) | 13% |
Reactivity of NPEC-NHNs 24 was then evaluated in the (A) coupling reaction with an alkyl bromide or (B) Mitsunobu reaction with an alcohol (Table 2).7c The former coupling reaction (procedure A) was examined with two bromides using K2CO3 and Bu4NI in DMF at 80 °C. As shown in entry 1, heptylamine 25 was readily synthesized from NPEC-NHNs 24 and heptyl bromide in 77% yield. With more reactive benzyl bromide, benzylamine 26 was obtained in higher yield (98%, entry 2).



Amine synthesis by the Mitsunobu reaction of NPEC-NHNs 24 with alcohols using DEAD and PPh3 (procedure B in Table 2) was next examined in benzene at rt. From 1-octanol, N-NPEC-N-Ns-octylamine (27) was provided in good yield (92%) (entry 3). The product 27 was later used as a model compound for the photodegradation reaction (see Table 3 and the text). Naphthalenylethylamine 28 was smoothly provided in 95% yield by the Mitsunobu reaction of 24 with naphthalene-2-ethanol (entry 4). Even a secondary alcohol can react in the Mitsunobu reaction; alanine ester 29 was constructed from ethyl lactate in good yield (85%, entry 5).
|
|
|||||
|---|---|---|---|---|---|
| Entrya | Irradiation source | Wavelength | Solvent | Time | Yieldb (NMR/isolation) |
| a Milligram quantities of N-NPEC amine 27 were used for the photodegradation. b N.D.: not determined. | |||||
| 1 | UV lamp (3 W) | 365 nm | MeOH | 1.5 h | 36%/N.D. |
| 2 | UV lamp (3 W) | 365 nm | MeOH | 9 h | 85%/N.D. |
| 3 | UV lamp (3 W) | 365 nm | TFE | 9 h | 100%/92% |
| 4 | UV lamp (3 W) | 365 nm | HFIP | 9 h | 100%/94% |
| 5 | LED lamp (3 W) | 365 nm | MeOH | 20 min | 100%/95% |
| 6 | LED lamp (1.2 W) | 395 nm | MeOH | 9 h | 3.7%/N.D. |
| 7 | LED lamp (1.2 W) | 395 nm | TFE | 9 h | 6.1%/N.D. |
| 8 | LED lamp (1.2 W) | 395 nm | HFIP | 9 h | 8.2%/N.D. |
| 9 | Blue LED (0.5 W) | ca. 460 nm | MeOH | 9 h | 6.5%/N.D. |
| 10 | High-pressure Hg lamp (435 W) | — | MeOH | 20 min | 100%/91% |
| 11 | High-pressure Hg lamp (435 W) | — | MeOH | 60 min | 100%/94% |
| 12 | High-pressure Hg lamp (435 W) | — | TFE | 20 min | 100%/N.D. |
| 13 | High-pressure Hg lamp (435 W) | — | HFIP | 20 min | 100%/N.D. |

For efficient synthesis of primary and secondary amines, Boc-NHNs7c,20 and Cbz-NHNs7c,21 have been generally used in the Ns strategy.7 From the good reactivity of NPEC-NHNs 24 shown in Table 2, 24 is expected to serve as a new entry for this purpose.
With N-NPEC-N-Ns amines 25–29, we studied the orthogonal reactivity of NPEC and Ns protecting groups under their deprotection conditions. Selective deprotection of the N-Ns group was first examined (Scheme 7, see the ESI for further details†). Treatment of 25–29 with PhSH and Cs2CO3 in CH3CN, the standard reagents developed by Fukuyama and Kan,7c afforded des-N-Ns amines 25a–29a in good yields (88–91%). No side reaction was observed, and all reactions were completed at rt within 120 min.
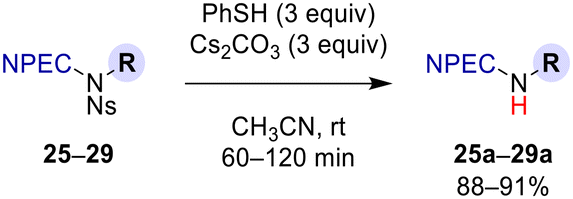 | ||
| Scheme 7 Selective deprotection of the Ns group in N-NPEC-N-Ns amines 25–29. For structures of R groups, see Table 2. For further details, see the ESI†. | ||
We next screened selective deprotection conditions for the NPEC group using N-NPEC-N-Ns-octylamine (27, Table 3) as a representative substrate. The photodegradations were performed using various irradiation sources at rt in alcohol solvents. As shown in entry 1, irradiation of 27 in MeOH, with a UV lamp (365 nm), induced selective NPEC deprotection in 36% conversion (NMR yield) after 1.5 h. When the reaction time was extended to 9 h, the conversion yield was 85% (entry 2). When the solvent for the reaction was changed to 2,2,2-trifluoroethanol (TFE, entry 3) or 1,1,1,3,3,3-hexafluoro-2-propanol (HFIP, entry 4), the reaction proceeded to completion and the isolated yield was satisfactory (92% for TFE, 94% for HFIP). Thus, TFE and HFIP were found to be the solvents that increase the conversion efficiency in the NPEC photodegradation. Interestingly, irradiation with an LED at the same wavelength (365 nm) completed the deprotection in a much shorter reaction time (20 min, entry 5). As shown in entries 6–8, however, irradiation with a longer wavelength LED (395 nm) was less effective in the three solvents (MeOH, TFE, and HFIP). A blue LED did not induce deprotection as well (entry 9).
Then irradiation with a high-pressure Hg lamp was examined. As shown in entry 10, the reaction smoothly proceeded in MeOH in 20 min and the product 27b was obtained in 91% isolated yield. Even under prolonged irradiation conditions (60 min), the reaction was reproducible, and furthermore, no decomposition of the N-Ns amine product 27b was observed (entry 11). With TFE (entry 12) or HFIP (entry 13) as a solvent, photoremoval of the NPEC group proceeded also smoothly in 20 min as expected from entries 3 and 4.
From these irradiation studies, we concluded that reactions under an LED (365 nm) or a high-pressure Hg lamp are sufficient to remove the NPEC group.
To examine the versatility of NPEC deprotection by LED (365 nm) irradiation, further reactions with other substrates (25, 26, 28, 29) were also conducted (Table 4). Here, irradiation was carried out in MeOH at rt. Under these conditions, N-NPEC-N-Ns-heptylamine (25, entry 1) and two other substrates 26 (entry 2) and 29 (entry 4) wherein R = benzyl and R = ethyl lactate, respectively, were smoothly photodegraded in 20 min as in the case for 27 (Table 3, entry 5). The only exception was N-NPEC-N-Ns-naphthylamine (28, Table 4, entry 3), which took 40 min to complete the reaction. The substrate 28 has a light-absorbing naphthyl group, which is assumed to reduce irradiation efficiency.
|
|
|||
|---|---|---|---|
| Entry | Substrate | Time | Yield (NMR) |
| a For structures of R groups, see Table 2. b Milligram quantities of N-NPEC amines 25, 26, 28, and 29 were used for the photodegradation. | |||
| 1 | 25 (R = heptyl) | 20 min | 100% (25b) |
| 2 | 26 (R = benzyl) | 20 min | 100% (26b) |
| 3 | 28 (R = naphthylethyl) | 2 × 20 min | 63% (20 min), 100% (40 min) (28b) |
| 4 |
29 (R = ethyl lactate, dr 57:43) |
20 min | 100% (29b) |
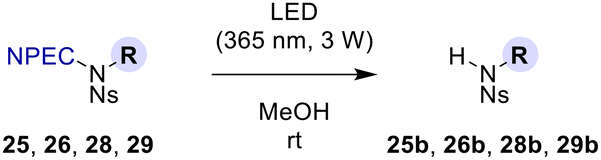
Achievement of the synthesis of a suitably protected, second-generation 12-mer LCPA subunit 2b
With the N-NPEC method in hand, synthesis of the suitably protected 12-mer LCPA subunit 2 (see Fig. 1B) was retried. Our attempt to prepare the second-generation key 3-mer unit 36 is shown in Scheme 8. Di-N-Ns protection of 1,3-propanediamine (3) yielded 30 in 84% yield. Mono-N-alkylation of 30 with bromide 31, prepared from commercially available 3-bromopropanol by tritylation,22 provided 2-mer 32 in good yield (94%) which, in turn, was subjected to the second N-alkylation with 3-bromopropanol (67% yield). Two Ns groups of 33 were then replaced with Boc groups, using the Fukuyama protocol (PhSH, Cs2CO3, CH3CN)7c followed by N-Boc protection (Boc2O, Et3N, CH2Cl2). However, the yields of these two steps were quite low (18%) due to the hydrophilic properties of intermediate 34. Synthesis of 36 by this pathway was deemed impractical. | ||
| Scheme 8 An attempt to prepare the second-generation 3-mer triamine unit 36 protected with the NPEC group. | ||
Therefore, we decided to adopt the route of introducing the Boc groups in the beginning of the synthesis, not by replacing Ns groups. Ultimately, we were able to synthesize the 12-mer LCPA subunit 2b by the new route (see below). Scheme 9 shows our successful synthesis of the fully protected key 3-mer structural unit 36. First, 1,3-propanediamine (3) was reacted with 2.1 equiv. of methyl acrylate to give 37,6a which was then treated with Boc2O to furnish bis-N-Boc amine 38 in 80% yield in 2 steps. After reduction of two ester groups with LAH (88%), the symmetric diol 39 was monoprotected as Tr ether to provide 35 (51%) and the unreacted 39 (38%) was recovered. Finally, NPEC-NHNs 24 was introduced into alcohol 35 by a Mitsunobu reaction (see Table 2) to obtain the suitably protected key 3-mer unit 36 in moderate yield (78%).
 | ||
| Scheme 9 Successful preparation of the second-generation 3-mer triamine unit 36 protected with the NPEC group. | ||
Repeated splitting and coupling operations converted the key 3-mer triamine unit 36 to the 12-mer LCPA subunit 2b (Scheme 10). Removal of the Tr group of 36 by TFE under reflux conditions gave alcohol 40 (88%).9b On the other hand, the NPEC group of 36 was removed by photoirradiation with LED (365 nm) quantitatively. It should be noted that the photodegradation can also be performed with a high-pressure Hg lamp in 94% yield. Mitsunobu coupling of alcohol 40 and the N-Ns amine 41 thus obtained proceeded smoothly, yielding 6-mer polyamine 42 in reasonable yield (82%). From 42, the same deprotections provided alcohol 43 and N-Ns amine 44, which were then coupled by a Mitsunobu reaction (DEAD, PPh3, benzene) to furnish 12-mer polyamine 45 (78% yield). Finally, the Tr group of 45 was removed under mild conditions (TFE, rt) to accomplish the synthesis of the suitably protected 12-mer LCPA subunit alcohol 2b in 40% yield. Unreacted Tr ether 45 (56%) was recovered intact. From 1,3-propanediamine (3), 2b was thus synthesized in 6.9% total yield in 10 steps. The subunit 2b contained only four Ns groups, fewer than 2a (eleven Ns groups). As expected, 12-mer LCPA 2b was found to be soluble in most common organic solvents.
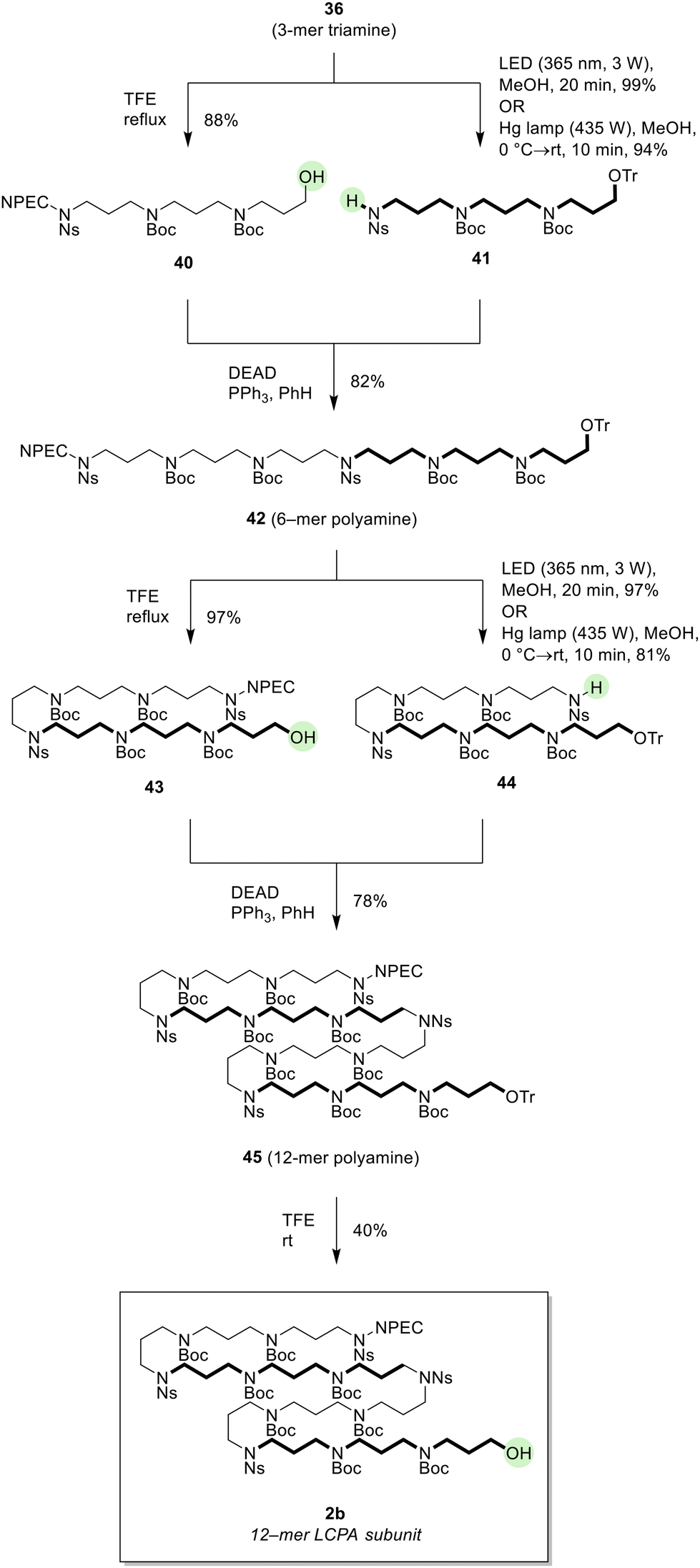 | ||
| Scheme 10 Iterative synthesis of the second-generation 12-mer LCPA subunit 2b. | ||
The 12-mer LCPA subunit 2b was later converted to the structure 46b initially proposed as protoaculeine B (pACU-B, LCPA-W′) corresponding also to a terminal substructure of ACU-B (Scheme 11).8,9b However, a comparison of synthetic 46b with the natural product in terms of chemical reactivity and spectroscopic profiles unfortunately revealed that the initially proposed structure of pACU-B needed revision.8 Then 2b was further used in the synthetic study of the revised structure 46a of pACU-B. Recently, we have successfully completed the synthesis of 46a, and determined the true structure of pACU-B as 46a (manuscript in preparation).10 These studies were made possible by the establishment of the synthesis of the protected polyamines reported here. In the future, we plan to study the structure–activity relationships of ACUs with structurally homogeneous LCPAs. Such studies are expected to reveal how ACUs, with their unique polyamine–peptide conjugate structures, show diverse activities such as membrane permeabilizing1 and cell penetrating activities.4
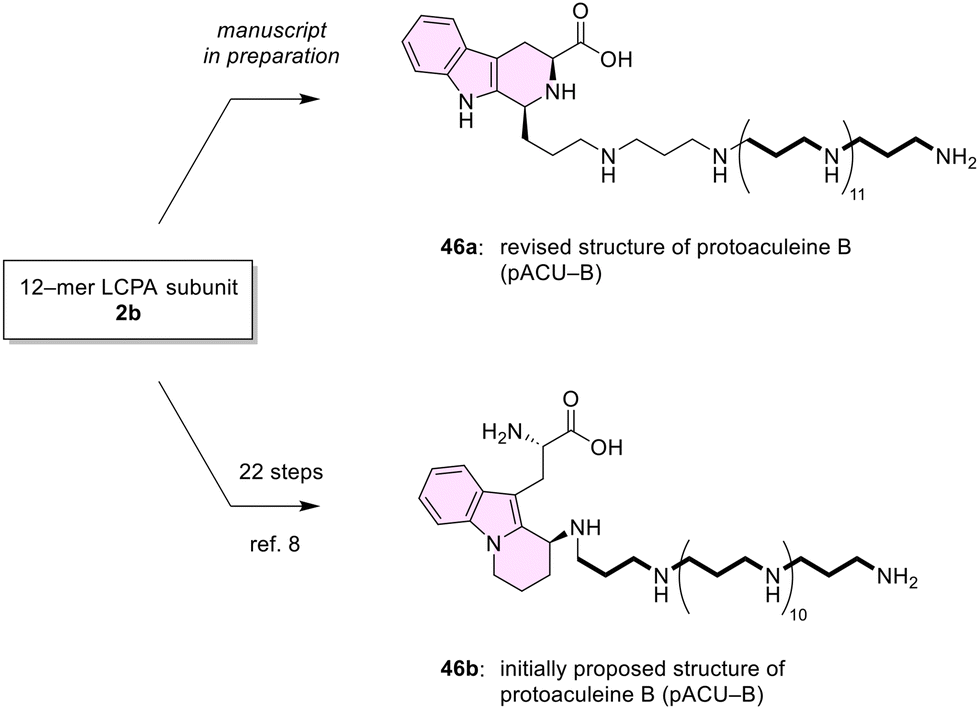 | ||
| Scheme 11 Syntheses of the revised structure 46a10 and the initially proposed structure 46b8 of protoaculeine B (pACU-B) from the LCPA subunit 2b. The work established the true structure of pACU-B from Axinyssa aculeata as 46a.10 Bold lines denote the part derived from the 12-mer LCPA subunit 2b. | ||
Conclusions
In this study, we investigated the use of Boc and Ns groups for persistent protection of amino groups to efficiently synthesize suitably protected and structurally defined polymers of 1,3-propanediamine. To achieve this goal, we studied the use of an additional photoremovable NPEC group as a temporary protecting group, whose reactivity is orthogonal to that of Boc and Ns groups. Finally, a method was successfully developed to synthesize 6-mer LCPA 42 by dimerization and 12-mer LCPA 45 by tetramerization, using the 3-mer triamine 36 as the smallest structural unit. Polyamines are important structural components used in a variety of applications.23 We believe that the method developed in this study will be useful for supplying structurally defined, diverse, and homogeneous polyamines for a variety of biochemical studies.Author contributions
M. O. conceptualized the research. M. M. and R. W. performed the experiments and collected the data. R. I. and M. O. analysed the data and wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Mr Hiroki Shiozaki for some experiments in the early stage of this study. This work was supported by a JSPS grant in aid for scientific research (22H02211).References
- S. Matsunaga, M. Jimbo, M. B. Gill, L. L. Lash-Van Wyhe, M. Murata, K. i. Nonomura, G. T. Swanson and R. Sakai, ChemBioChem, 2011, 12, 2191–2200 CrossRef CAS.
- S. Matsunaga, R. Sakai, M. Jimbo and H. Kamiya, ChemBioChem, 2007, 8, 1729–1735 CrossRef CAS PubMed.
- R. Irie, K. Miyako, S. Matsunaga, R. Sakai and M. Oikawa, J. Nat. Prod., 2021, 84, 1203–1209 CrossRef CAS PubMed.
- H. Watari, R. Kishi, S. Matsunaga, T. Nishikawa, Y. Sawada, A. Honda, M. J. Fujita and R. Sakai, Chem. Lett., 2023, 52, 185–189 CrossRef CAS.
- For preliminary studies on this work, see; M. Miyahara, H. Shiozaki, H. Tukada, Y. Ishikawa and M. Oikawa, Tetrahedron Lett., 2018, 59, 4259–4262 CrossRef CAS.
- (a) M. Matsushita, T. Kanemura, S. Hatakeyama, H. Irie, T. Toki and M. Miyashita, Tetrahedron, 1995, 51, 10687–10698 CrossRef CAS; (b) K.-i. Nihei, M. J. Kato, T. Yamane and K. Konno, Tetrahedron, 2006, 62, 8335–8350 CrossRef CAS; (c) A. H. Alkhzem, M. Laabei, T. J. Woodman and I. S. Blagbrough, ChemistryOpen, 2022, 11, e202200147 CrossRef CAS PubMed.
- (a) T. Fukuyama, C. K. Jow and M. Cheung, Tetrahedron Lett., 1995, 36, 6373–6374 CrossRef CAS; (b) T. Fukuyama, M. Cheung, C.-K. Jow, Y. Hidai and T. Kan, Tetrahedron Lett., 1997, 38, 5831–5834 CrossRef CAS; (c) T. Fukuyama, M. Cheung and T. Kan, Synlett, 1999, 1301–1303 CrossRef CAS; (d) Y. Hidai, T. Kan and T. Fukuyama, Chem. Pharm. Bull., 2000, 48, 1570–1576 CrossRef CAS PubMed; (e) T. Kan and T. Fukuyama, Chem. Commun., 2004, 353–359 RSC.
- R. Irie, M. Miyahara, S. Nakamura, A. Honda, R. Sakai and M. Oikawa, J. Nat. Prod., 2020, 83, 2769–2775 CrossRef CAS PubMed.
- (a) S. Matsunaga, R. Kishi, K. Otsuka, M. Fujita, M. Oikawa and R. Sakai, Org. Lett., 2014, 16, 3090–3093 CrossRef CAS PubMed; (b) H. Shiozaki, M. Miyahara, K. Otsuka, K. Miyako, A. Honda, Y. Takasaki, S. Takamizawa, H. Tukada, Y. Ishikawa, R. Sakai and M. Oikawa, Org. Lett., 2018, 20, 3403–3407 CrossRef CAS PubMed; (c) K. Otsuka, M. Miyahara, S. Takaki, R. Wakabayashi, K. Miyako, R. Irie, S. Takamizawa, R. Sakai and M. Oikawa, Eur. J. Org. Chem., 2022, e202200669 CAS.
- R. Irie, R. Wakabayashi, S. Takaki and M. Oikawa, manuscript in preparation.
- J. E. Corrie, A. DeSantis, Y. Katayama, K. Khodakhah, J. B. Messenger, D. C. Ogden and D. R. Trentham, J. Physiol., 1993, 465, 1–8 CrossRef CAS.
- G. Ciamician and P. Silber, Ber. Dtsch. Chem. Ges., 1901, 34, 2040–2046 CrossRef CAS.
- (a) J. W. Walker, G. P. Reid, J. A. McCray and D. R. Trentham, J. Am. Chem. Soc., 1988, 110, 7170–7177 CrossRef CAS; (b) J. W. Walker, G. P. Reid and D. R. Trentham, in Methods in Enzymology, Academic Press, 1989, vol. 172, pp. 288–301 Search PubMed.
- A. Patchornik, B. Amit and R. B. Woodward, J. Am. Chem. Soc., 1970, 92, 6333–6335 CrossRef CAS.
- F. Palma-Cerda, C. Auger, D. J. Crawford, A. C. Hodgson, S. J. Reynolds, J. K. Cowell, K. A. Swift, O. Cais, L. Vyklicky, J. E. Corrie and D. Ogden, Neuropharmacology, 2012, 63, 624–634 CrossRef CAS PubMed.
- M. Su, J. Wang and X. Tang, Chem. – Eur. J., 2012, 18, 9628–9637 CrossRef CAS PubMed.
- O. Mitsunobu, Synthesis, 1981, 1–28 CrossRef CAS.
- S. Tang, Z. Wan, Y. Gao, J.-S. Zheng, J. Wang, Y.-Y. Si, X. Chen, H. Qi, L. Liu and W. Liu, Chem. Sci., 2016, 7, 1891–1895 RSC.
- Y. Hayakawa, S. Wakabayashi, H. Kato and R. Noyori, J. Am. Chem. Soc., 1990, 112, 1691–1696 CrossRef CAS.
- H. Aoki and T. Mukaiyama, Chem. Lett., 2006, 35, 456–457 CrossRef CAS.
- J. Taguchi, T. Ikeda, R. Takahashi, I. Sasaki, Y. Ogasawara, T. Dairi, N. Kato, Y. Yamamoto, J. W. Bode and H. Ito, Angew. Chem., Int. Ed., 2017, 56, 13847–13851 CrossRef CAS PubMed.
- T. Niittymäki, U. Kaukinen, P. Virta, S. Mikkola and H. Lönnberg, Bioconjugate Chem., 2004, 15, 174–184 CrossRef PubMed.
- (a) A. Bernecker, R. Wieneke, R. Riedel, M. Seibt, A. Geyer and C. Steinem, J. Am. Chem. Soc., 2010, 132, 1023–1031 CrossRef CAS PubMed; (b) R. P. Dewangan, D. P. Verma, N. K. Verma, A. Gupta, G. Pant, K. Mitra, S. Habib and J. K. Ghosh, J. Med. Chem., 2022, 5433–5448 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Complete experimental procedures, analytical and spectroscopic data, and copies of NMR spectra. See DOI: https://doi.org/10.1039/d3ob00369h |
| This journal is © The Royal Society of Chemistry 2023 |