
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
From oximes to tertiary alcohols in water, at room temperature and under air: a hybrid one-pot tandem assembly of enzymatic deoximation and RLi/RMgX reagents†
Davide
Arnodo
 ab,
Marina
Ramos-Martín
a,
Luciana
Cicco
ac,
Vito
Capriati
c,
Nicolás
Ríos-Lombardía
ad,
Javier
González-Sabín
*d,
Alejandro
Presa Soto
*a and
Joaquín
García-Álvarez
*a
ab,
Marina
Ramos-Martín
a,
Luciana
Cicco
ac,
Vito
Capriati
c,
Nicolás
Ríos-Lombardía
ad,
Javier
González-Sabín
*d,
Alejandro
Presa Soto
*a and
Joaquín
García-Álvarez
*a
aLaboratorio de Química Sintética Sostenible (QuimSinSos), Departamento de Química Orgánica e Inorgánica, (IUQOEM), Centro de Innovación en Química Avanzada (ORFEO-CINQA), Facultad de Química, Universidad de Oviedo, E33071 Oviedo, Spain. E-mail: garciajoaquin@uniovi.es
bDipartimento di Chimica-Università degli Studi di Torino, Via P. Giuria 7, 10125 Torino, Italy
cDipartimento di Farmacia-Scienze del Farmaco, Università di Bari “Aldo Moro”, Consorzio C.I.N.M.P.I.S., Via E. Orabona 4, I-70125 Bari, Italy
dEntreChem SL, Vivero Ciencias de la Salud. Santo Domingo de Guzmán, Oviedo, 33011, Spain
First published on 28th March 2023
Abstract
The highly efficient biodeoximation of aromatic ketoximes, promoted by the enzymatic oxidative system laccase/TEMPO/O2, has been successfully assembled with the fast and chemoselective addition of highly-polar s-block organometallic reagents (RLi/RMgX) en route to highly-substituted tertiary alcohols. By using this hybrid one-pot tandem protocol, tertiary alcohols have been selectively synthesized in good yields and under mild and bench-type reaction conditions (room temperature, the absence of a protecting atmosphere and aqueous media, which are non-typical conditions for polar organometallic reagents). The overall hybrid one-pot tandem transformation amalgamates two distant organic synthetic tools (RLi/RMgX reagents and enzymes) without the need for any tedious and energy/time-consuming intermediate isolation/purification steps.
Introduction
In recent years, the design of multistep one-pot synthetic protocols has attracted great attention in the synthetic organic community (for the substitution of traditional and tedious stepwise processes), since the implementation of these tandem protocols allows us to avoid the employment of purification steps, usually needed for the isolation of reaction intermediates (with the concomitant reduction of chemical waste and energy/time costs), thus making simpler the practical aspects of the desired synthetic methodology.1 Moreover, these one-pot tandem protocols are also the synthetic tools of choice when it is not possible to isolate highly reactive or transitory-formed species as intermediates.2 Although these protocols have been successfully exploited to create a wide variety of natural products and other complex molecular architectures, there are currently relatively few examples of their hybrid versions3 in which the combination of enzymatic, organocatalytic, and transition-metal or main-group-catalysed processes has been successfully assembled within the different reaction steps.4 In this field, hybrid chemoenzymatic one-pot tandem protocols in which enzymes are successfully combined with transition-metal-based catalysts or organocatalysis are currently considered a hot topic of research. However, and to the best of our knowledge, the combination of enzymatic synthetic protocols (which intrinsically use water as the natural reaction medium, under aerobic conditions and at room temperature)5 and the chemistry of highly reactive and polar organolithium (RLi) or Grignard (RMgX) reagents,6,7 which usually requires the use of dry, often toxic and volatile organic solvents, under an inert atmosphere (N2 or Ar) and at low temperature (ranging from 0 to −78 °C), is still in its infancy.8 The design of these hybrid protocols, which rely on the combination of enzymes and highly-polar main-group organometallic reagents, is based on previous related studies reported by other research groups (for example, C.-J. Li9a or Lipshutz9b) or some of us,10,11 which demonstrated the possibility of promoting organolithium- or organomagnesium-mediated organic reactions in protic solvents [water, glycerol or deep eutectic solvents (DESs)], at room temperature and without the need for a protecting atmosphere (i.e., under air).12–14 To the best of our knowledge, to date, only two protocols have reported the combination of enzymes and polar organometallic chemistry: (i) a lithium carbenoid homologation reaction combined with an enzymatic reduction8a and (ii) the catalytic oxidation of secondary alcohols by the laccase/TEMPO system followed by the addition of organolithium reagents to the in situ formed ketones.8b The latter methodology, in particular, presents several shortcomings such as (i) the limited scope of the reaction as the catalytic biooxidation tolerates only a few alcohols; (ii) low to moderate isolated yields of the resulting tertiary alcohols; and (iii) the ineffectiveness of Grignard reagents in this hybrid tandem protocol.On the other hand, laccase from Trametes versicolor15 is considered one of the most reliable and versatile enzymatic partners available in the synthetic biocatalytic toolbox for the design of chemoenzymatic one-pot tandem protocols.8b,16 Moreover, some of us have reported the first biocatalytic deoximations, which enabled us to recover ketones from ketoximes by means of a laccase/TEMPO system.17,18 This is important also in the perspective of late-stage functionalization as the oxime function is ubiquitous in nature and can be found in the structure of innumerable natural and synthetic bioactive compounds as well as in several metabolic pathways.19 Remarkably, the biodeoximation reaction took place in an aqueous medium under mild reaction conditions, and the products were isolated in excellent yields without the need for chromatographic purification. Herein we report a new hybrid one-pot tandem methodology capable of transforming ketoximes into non-symmetric tertiary alcohols by combining the biodeoximation reaction (by means of a laccase/TEMPO/O2 system) with the subsequent fast, chemoselective and air/moisture/ambient temperature compatible addition of highly polar RLi/RMgX organometallic reagents (see Scheme 1).
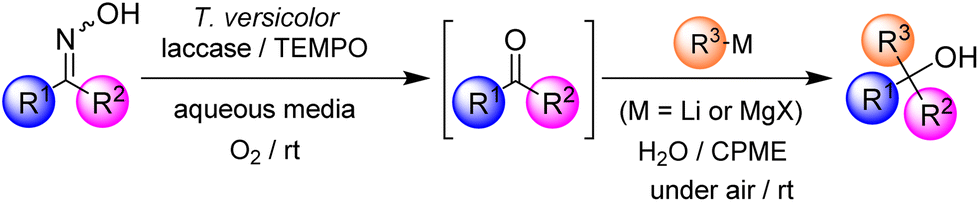 | ||
| Scheme 1 Design of a hybrid one-pot tandem protocol by combining enzymes (laccase from Trametes versicolor) with highly polar s-block organometallic reagents (RLi/RMgX) in an aqueous medium, at room temperature and under air. | ||
The following features of our hybrid one-pot tandem protocol are noteworthy: (i) an aqueous medium is used as the solvent for the combination of both biocatalysed and main-group-promoted organic transformations; (ii) the global one-pot tandem protocol proceeds at room temperature and under aerobic conditions; (iii) no isolation of any reaction intermediate (ketones in this case) is needed, thus reducing the chemical waste and energy/time costs; and (iv) it is an effective and chemoselective methodology for the synthesis of highly-substituted tertiary alcohols (conversions up to 91%), which are often considered components of active pharmaceutical ingredients (APIs), natural products, agrochemicals and synthetic materials.20 This new methodology overcomes the aforementioned drawbacks of our previous chemoenzymatic methodology based on the catalytic oxidation of secondary alcohols by the laccase/TEMPO system followed by the addition of organolithium reagents to the in situ formed ketones,8b as: (i) it tolerates a large variety of starting oximes; (ii) better isolated yields (up to 82%) have been obtained for tertiary alcohols; and (iii) for the first time, Grignard reagents have been successfully employed, thereby expanding the scope of main-group reagents which can be amalgamated with enzymes.
Results and discussion
We started our investigation focusing our attention on the deprotection of aromatic ketoximes under mild reaction conditions promoted by the laccase/TEMPO/O2 system (see Table 1). We selected as the model reaction the biodeoximation of (Z/E)-1-phenylpropan-1-one oxime (1a) in pure water as the solvent, promoted by the laccase/TEMPO system and using aerial O2 as a co-oxidant during 24 h. First, we observed an important effect of the stirring speed on the reaction, that is, a dramatic increase in the conversion of 1a into the corresponding propiophenone (2a) when moving from 800 rpm to 1800 rpm (47% to 84%; entries 1 and 2, Table 1) was found. This experimental observation has been previously reported in biooxidation processes promoted with the laccase/TEMPO/O2 catalytic system and is related to the increase in solubility of the required O2 in the reaction medium.16–18 Once the stirring speed was fixed at 1800 rpm, we decided to parametrize the amount of the co-catalyst (TEMPO in this case), observing an important increase in activity when moving from 10 to 33 mol% (compare entries 2 and 3 in Table 1). Trying to achieve quantitative conversion into the desired propiophenone (2a), we carried out the biocatalytic oxidation reaction in an oxygen atmosphere using an external oxygen balloon (1 atm). Working under these conditions, we achieved the complete and chemoselective conversion of ketoxime 1a into the desired ketone 2a (no by-products were detected, entry 4, Table 1), at room temperature and after 24 h. The effectiveness of a different commercially available laccase (Rhus vernicifera) was also studied (50 U mg−1), but no activity in the deoximation process was observed (entry 5, Table 1). Interestingly, an archetypical transition-metal based catalytic oxidation system (i.e., CuCl2·2H2O/TMEDA; TMEDA = N,N,N′,N′-tetramethylethylenediamine) is not capable of replicating the activity observed when using our biocatalytic system (entry 6, Table 1).21 At this point, we should mention that the biooxidation system under study tolerates the use of other co-catalysts such as AZADO (2-azaadamantane N-oxyl, entry 7, Table 1) or water-miscible co-solvents such as acetonitrile (100 μL; entry 8, Table 1). On the other hand, no biocatalytic reaction was observed in the absence of catalytic amounts of laccase (entry 9) or a co-catalyst (TEMPO in this case, entry 10, Table 1). These experimental findings testify that the biocatalytic system laccase/TEMPO/O2 is responsible for the catalytic activity observed in the deoximation reaction of 1a.|
|
||||
|---|---|---|---|---|
| Entry | Laccaseb,c | Co-catalyst | Oxidant | Conv.d (%) |
| a General conditions: 24 h of reaction at room temperature and at 1800 rpm, using 0.73 mmol of 1a in 1 mL of water. b 280 mg of T. versicolor (0.5 U mg−1) and 2.8 mg of Rhus vernicifera (50 U mg−1) were employed. c U mg−1 = units of activity per mg of enzyme. d Determined by GC; no significant amount of by-products was detected. e Stirring speed: 800 rpm. f 100 μL of CH3CN were added as the co-solvent. | ||||
| 1 | T. versicolor | TEMPO (33 mol%) | Air | 47e |
| 2 | T. versicolor | TEMPO (33 mol%) | Air | 84 |
| 3 | T. versicolor | TEMPO (10 mol%) | Air | 40 |
| 4 | T. versicolor | TEMPO (33 mol%) | O2 | >99 |
| 5 | Rhus vernicifera | TEMPO (33 mol%) | O2 | 1 |
| 6 | CuCl2·2H2O/TMEDA | TEMPO (33 mol%) | O2 | 2 |
| 7 | T. versicolor | AZADO (33 mol%) | O2 | >99 |
| 8 | T. versicolor | TEMPO (33 mol%) | O2 | >99f |
| 9 | — | TEMPO (33 mol%) | O2 | 0 |
| 10 | T. versicolor | — | O2 | 2 |

After setting up the best conditions for the biodeoximation process of 1a into the desired ketone 2a (water as the solvent, room temperature and 24 h of reaction, 1800 rpm), we investigated its combination with the chemoselective addition of RLi/RMgX reagents to the intermediate ketone 2a without any intermediate purification/isolation step, that is in the presence of the biocatalytic system laccase/TEMPO/O2, and working at room temperature, under air and in an aqueous medium, a trio of reaction conditions not usually employed in traditional polar organometallic chemistry.6,7 This hybrid methodology could be considered as another proof-of-concept on the scarcely studied combination of polar organometallic chemistry and biocatalysts, thus being a new strategy for the design of RLi/RMgX-mediated organic transformations in the presence of enzymes and co-factors in aqueous media. The laccase/TEMPO/O2 deoximation of 1a into the corresponding ketone 2a was followed by the direct addition of a variety of RLi/RMgX reagents to the resulting aqueous-based reaction mixture (see Table 2).
|
|
|||||
|---|---|---|---|---|---|
| Entry | R1Li | Equiv. | Solvent | Product | Conv.b (%) |
| a General conditions: 24 h of reaction at room temperature and at 1800 rpm; laccase from T. versicolor (0.5 U mg−1, 280 mg) per 0.73 mmol of 1a and 0.33 eq. of TEMPO in 1 mL of water were used. Then 1 mL of the co-solvent and the RLi reagent [R = Ph (1.9 M in n-Bu2O) or n-Bu (2.5 M in hexanes)] were added without any isolation/purification. b Determined by GC; no significant amount of by-products was detected. | |||||
| 1 | PhLi | 3 | H2O | 3a | 67 |
| 2 | PhLi | 3 | H2O/CPME | 3a | 79 |
| 3 | PhLi | 2 | H2O/CPME | 3a | 60 |
| 4 | PhLi | 3 | H2O/2-MeTHF | 3a | 65 |
| 5 | n-BuLi | 2 | H2O/CPME | 3b | 28 |
| 6 | n-BuLi | 3 | H2O/CPME | 3b | 62 |
| 7 | n-BuLi | 3 | H2O/2-MeTHF | 3b | 37 |
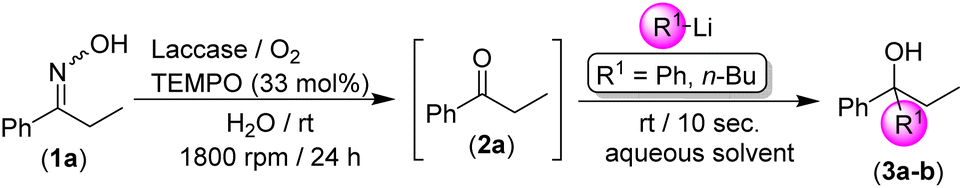
Initially, the reaction mixture formed by ketoxime 1a and the biooxidation system (laccase/TEMPO) was allowed to react in bulk water, at room temperature and under O2 to trigger the desired deoximation reaction. As soon as the complete conversion of 1a into propiophenone (2a) was achieved (24 h, GC analysis), PhLi was directly added to the reaction mixture in the absence of any protecting atmosphere and at room temperature (entry 1, Table 2). Under these bench-type reaction conditions, we observed that PhLi added almost immediately (10 s) to the in situ formed propiophenone (2a), leading to the desired tertiary alcohol 3a with total chemoselectivity, as no by-products were detected even in the presence of the enzyme (laccase) and the co-catalyst (TEMPO). It is worth mentioning that the possible side reaction between the organolithium reagent and TEMPO was not detected,22 thus disclosing a new example in which the addition reaction of the RLi reagent to a carbonyl compound is faster than any other side reactions or hydrolysis.9–13 With the aim to increase the yield of the final product 3a, and taking into account the positive effect associated with the use of biphasic mixtures of water and ethereal solvents [such as cyclopentyl methyl ether (CPME)]23 in the addition of RLi reagents to organic electrophiles in protic solvents, we assayed the use of this mixture in our tandem protocol. An important increase in the final yield of the tertiary alcohol 3a was observed in this case (79%, entry 2, Table 2). On the other hand, when the equivalents of PhLi were decreased from 3 to 2, a lower yield (60%) of the final aromatic alcohol 3a was detected (entry 3, Table 2). Along the same lines and by replacing CPME with other ethereal solvents such as 2-MeTHF,24 the yield of 3a could not be increased either (entry 4, Table 2). Finally, we decided to study the scope of the reaction by investigating: (i) the nature of the starting organolithium reagent (comparing aromatic PhLi with aliphatic n-BuLi); and (ii) the co-solvent in which these commercially available solutions of RLi reagents are accessible [that is an ethereal donor solvent for PhLi (n-Bu2O) and an aliphatic and non-coordinating solvent for n-BuLi (hexanes)]. Thus, a comparison between entries 2–4 (for PhLi, synthesis of alcohol 3a) and 5–7 (for n-BuLi, synthesis of alcohol 3b) in Table 2 clearly indicates that higher yields are always observed when an ethereal-based solution of RLi is employed. These experimental observations could be related to the capability of the ethereal solvent (n-Bu2O) present in the commercial solution of PhLi to break down less reactive aggregates (oligomers) usually present in RLi solutions.25
Given the best conditions for the formation of tertiary alcohols 3a and 3b upon addition of the polar organolithium reagent at room temperature, under air, in the biphasic H2O/CPME mixture and under stirring of the reaction media for 10 s, we extended our studies to other highly polar organometallic reagents (RLi/RMgX, see Table 3). First, we observed almost quantitative conversion into tertiary alcohol 3c (91%, entry 1, Table 3) when using MeLi (commercial solution in Et2O) as the alkylating reagent. As expected, both the gradual increase of steric hindrance (thus decreasing their nucleophile character) from EtLi to s-BuLi and t-BuLi and the use of all these commercially available solutions of RLi reagents in hydrocarbon solvents produced a concomitant reduction of the yield (from 73 to 53%) in the alcohols 3d–f (entries 2–4, Table 3). However, it is worth highlighting the total chemoselectivity of our hybrid protocol, as no side products (aside from the unreacted ketone 2a and the desired alcohols 3d–f) were observed. These results are especially remarkable when considering highly reactive RLi compounds (i.e., s-BuLi and t-BuLi, which are usually stored and employed at low temperatures), as these reagents are known to be prone to undergo a fast β-hydride elimination.6 Moreover, we should mention that not only aliphatic and primary RLi reagents but also heteroaromatic [such as 2-thienyl lithium (thienylLi); 3g; entries 5, Table 3] organolithium reagents can be employed in our hybrid one-pot tandem protocol, working at room temperature and under air. Finally, we also studied the usefulness of Grignard reagents, such as allylMgBr (entry 6, Table 3) and benzylMgCl (entry 7, Table 3), under the previously optimized reaction conditions (room temperature, under air) and in the presence of the enzyme (laccase) and the co-factor (TEMPO). Satisfactorily, we found that magnesium-based polar organometallic reagents can promote the Grignard reaction even under biocatalytic conditions, giving rise to the desired tertiary alcohols 3h and 3i in moderate to good conversions (46–62%; entries 6 and 7, Table 3). Again, both reactions proved to be totally chemoselective giving rise to the expected alcohols after only 10 seconds of reaction as the sole products of the reaction. These results are particularly remarkable taking into account other previous studies which reported complete protonation of the Grignard reagents in competition with carbonyl addition.26
|
|
||||
|---|---|---|---|---|
| Entry | R–M (eq.) | Product | Conv.b (%) | Yieldc (%) |
| a General conditions: 24 h of reaction at room temperature and at 1800 rpm; laccase from T. versicolor (0.5 U mg−1, 280 mg) per 0.73 mmol of 1a and 0.33 eq. of TEMPO in 1 mL of water were used. Then 1 mL of the co-solvent and the RLi [R = Me (1.6 M in Et2O); Et (0.5 M in benzene/cyclohexane); s-Bu (1.4 M in cyclohexane); t-Bu (1.7 M in pentane); 2-thienyl (1.0 M in THF/hexanes)] or RMgX [R = allyl (1.0 M in Et2O); benzyl (2.0 M in THF)] reagents were added without any isolation/purification. b Determined by GC; no significant amount of by-products was detected. c Isolated yield. | ||||
| 1 | MeLi (3) | 3c | 91 | 82 |
| 2 | EtLi (3) | 3d | 73 | 66 |
| 3 | s-BuLi (3) | 3e | 69 | 64 |
| 4 | t-BuLi (3) | 3f | 53 | 40 |
| 5 | ThienylLi (3) | 3g | 55 | 46 |
| 6 | AllylMgBr | 3h | 62 | 50 |
| 7 | BenzylMgCl | 3i | 46 | 33 |

Highlighting the exciting potential of using highly polar s-block organometallic reagents under biocatalytic conditions (air, aqueous media, room temperature) and in the presence of enzymes/co-factors, we decided to run control experiments aimed at corroborating the observed ability of RLi/RMgX reagents to survive under the aforementioned biocatalytic conditions. When the addition order of the reagents was reversed and PhLi or allylMgBr was first introduced into the mixture containing only water/CPME, TEMPO and laccase, followed by the addition of 2a, after 3 s stirring under air and at room temperature, the products 3a and 3h were still isolated in remarkable yields of 66% and 59%, respectively (Scheme 2). Conversely, by prolonging the stirring time to 6 s before adding 2a, the formation of 3a and 3h was totally suppressed. These results are in agreement with our previous studies on the addition of RLi reagents to imines10b,g and nitriles10c,g in protic solvents (e.g., deep eutectic solvents, glycerol, water), thereby reinforcing the concept of kinetic stability of s-block organometallic reagents towards an expected fast hydrolysis when coming into contact with aqueous solutions.
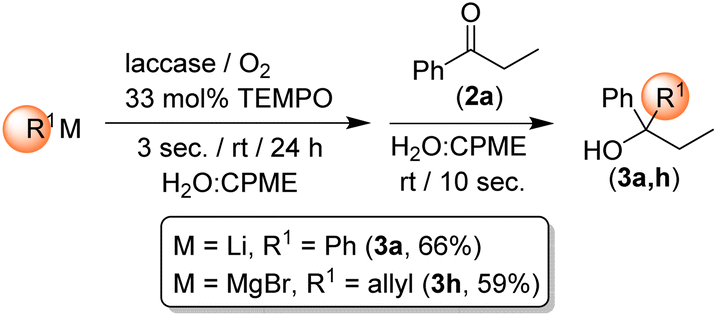 | ||
| Scheme 2 Control experiments for assessing the capability of RLi/RMgX reagents to work under biocatalytic conditions and in the presence of enzymes/co-factors. | ||
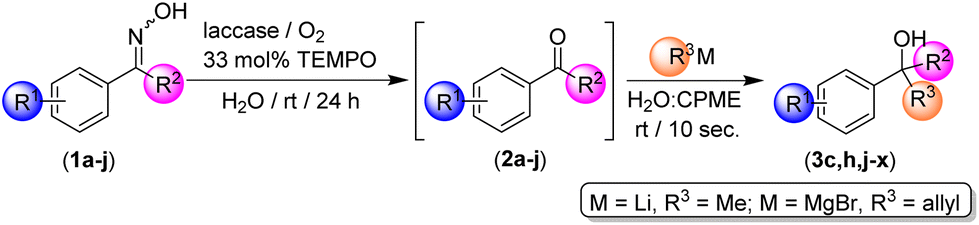
To explore the limitations of our methodology, we decided to test a series of ketoximes (1a–j, Table 4) by employing either MeLi or allylmagnesium bromide as the alkylation/allylation reagent (best conversions were observed when using these polar organometallic reagents; see entries 1 and 6, Table 3) working at room temperature, in an aqueous medium and under air. In all cases studied, the biooxidative system formed by laccase/TEMPO/O2 was able to convert quantitatively all the starting ketoximes 1a–j into the corresponding ketones 2a–j (99% conversions, GC analysis) after 24 h reaction time, independently from: (i) the nature [electron-withdrawing (such as Cl in 1b and 1f–h) or electron-donating (MeO or Me in 1c–d, i–j)] or (ii) the position of the substituent in the aromatic ring (o-, m- and p-positions are tolerated). In addition, it was possible to add directly the desired polar organometallic reagent (MeLi or allylMgBr) to the biocatalytic reaction medium (in the presence of the enzyme and TEMPO), working under bench-type conditions and in aqueous media, thus yielding the desired tertiary alcohols 3c, 3h, and 3j–x in only 10 s reaction time (see Table 4). The conclusion is that our hybrid one-pot tandem protocol tolerates a variety of functional groups in the aromatic ring, being compatible with either electron-withdrawing (Cl, entries 3 and 4, Table 4) or electron-donating groups (Me or OMe, entries 5–8) at the para-position. Higher yields were always observed when MeLi was used as the organometallic reagent in place of allylMgBr. At this point, it is also worth mentioning the high chemoselectivity of our hybrid protocol, as the following side reactions were not observed: (i) Li- or Mg-chloride exchange reaction in the ketoxime 1b; (ii) ortho-metalation in ketoxime 1c; or (iii) metalation of benzylic positions in substrate 1d. Moreover, we have proved that not only propiophenone-type ketoximes (1a–d) but also acetophenone-based ketoximes 1e–j can be used in our hybrid one-pot tandem protocol. In this case, we explored the effect (steric hindrance) of the position of the same substituent (Cl) in the three different positions of the aromatic ring, finding a higher yield for the para-derivative 3r (78%, entry 11, Table 4) and a lower yield in the case of the hindered ortho-derivative 3t (11%, entry 13, Table 4). As expected, the meta-derivative 3s was formed in an intermediate yield of 29% (entry 12, Table 4). These experimental observations support a strong influence of the steric effects in the addition reaction of RLi/RMgX reagents in our hybrid one-pot tandem protocol. Finally, it is also noteworthy that our new hybrid one-pot tandem system is compatible with acetophenone-type ketones 2e–j (Table 4), whereas previously reported addition reactions of RLi/RMgX towards such water-soluble substrates were totally ineffective in aqueous media.13a,27 We correlate this experimental observation with the presence of an immiscible ethereal solvent (CPME) in the aqueous mixture which could decrease the solubility of the acetophenone-type substrates in water.
|
|
|||||
|---|---|---|---|---|---|
| Entry | R1 | R2 | R3M | Product | Yieldb (%) |
| a General conditions: 24 h of reaction at room temperature and at 1800 rpm; laccase from T. versicolor (0.5 U mg−1, 280 mg) per 0.73 mmol of 1a–i and 0.33 eq. of TEMPO in 1 mL of water were used. Then 1 mL of the co-solvent and 3 eq. of MeLi (1.6 M in Et2O) or allylMgBr (1.0 M in Et2O) reagents were added without any isolation/purification. b Isolated yield. | |||||
| 1 | H (1a) | Et | MeLi | 3c | 82 |
| 2 | H (1a) | Et | AllylMgBr | 3h | 50 |
| 3 | p-Cl (1b) | Et | MeLi | 3j | 83 |
| 4 | p-Cl (1b) | Et | AllylMgBr | 3k | 58 |
| 5 | p-OMe (1c) | Et | MeLi | 3l | 72 |
| 6 | p-OMe (1c) | Et | AllylMgBr | 3m | 52 |
| 7 | p-Me (1d) | Et | MeLi | 3n | 54 |
| 8 | p-Me (1d) | Et | AllylMgBr | 3o | 36 |
| 9 | H (1e) | Me | MeLi | 3p | 74 |
| 10 | H (1e) | Me | AllylMgBr | 3q | 43 |
| 11 | p-Cl (1f) | Me | MeLi | 3r | 78 |
| 12 | m-Cl (1g) | Me | MeLi | 3s | 29 |
| 13 | o-Cl (1h) | Me | MeLi | 3t | 11 |
| 14 | p-Cl (1f) | Me | AllylMgBr | 3u | 16 |
| 13 | m-OMe (1i) | Me | MeLi | 3v | 40 |
| 14 | m-OMe (1i) | Me | AllylMgBr | 3w | 28 |
| 15 | p-OMe (1j) | Me | MeLi | 3x | 88 |
Conclusions
In this work, we have extended the possibility of combining enzyme-promoted organic transformation with the chemistry of highly polar organometallic compounds (RLi/RMgX), working in aqueous media and under bench-type reaction conditions (room temperature and the absence of any protecting atmosphere), which are the conditions typically employed in biocatalytic chemistry but non-ideal for polar organometallic reagents (RLi/RMgX). Thus, we have presented the chemoselective and efficient synthesis of highly substituted tertiary alcohols by an efficient one-pot tandem protocol which combines a biocatalytic deoximation step (mediated by the laccase/TEMPO/O2 system) and the chemoselective and fast addition of RLi/RMgX reagents on transiently formed ketones. This two-step protocol led to a variety of functionalized tertiary alcohols in up to 82% yield with a good substrate scope in terms of both oximes and organometallics (organolithium and Grignard reagents). In addition, it is worth mentioning that all reactions were found to proceed under bench-type conditions.The described results represent another successful example of a hybrid synthetic methodology that combines, in this case, a biocatalytic first step with a chemoselective addition of RLi/RMgX reagents. These results help to expand this emerging field of chemistry, not only by demonstrating the possibility of consecutively using two synthetic tools as distant as enzymes and organometallic reagents, but also by carrying out this hybrid combination in protic reaction media and in the absence of a protective atmosphere.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
D. A., M. R.-M., L. C., A. P. S. and J. G. A. thank MCIN/AEI/10.13039/501100011033 (project numbers CTQ2016-75986-P, RED2018-102387-T and PID2020-113473GB-I00) for the financial support. M. R.-M. acknowledges a predoctoral award from “Programa Severo Ochoa para la formación en investigación y docencia del Principado de Asturias” (PA-21-PF-BP20-093). D. A. would also like to acknowledge the Italian Ministry of Research for funding, Prof. Cristina Prandi, Prof. Marco Blangetti and Prof. Salvatore Baldino for PhD supervision and helpful support. L. C. also thanks the Erasmus + Staff Mobility for a Training (SST). V. C. wishes to thank the MUR for supporting this research within the framework of the national PRIN project “Unlocking Sustainable Technologies Through Nature-Inspired Solvents” (NATUREChem) (grant number: 2017A5HXFC_002).References
- (a) Y. Hayashi, Chem. Sci., 2016, 7, 866 RSC; (b) Y. Hayashi, Acc. Chem. Res., 2021, 54, 1385 CrossRef CAS PubMed.
- (a) K. C. Nicolaou, D. J. Edmonds and P. G. Bulger, Angew. Chem., Int. Ed., 2006, 45, 7134 CrossRef CAS PubMed; (b) W. Zhao and F.-Er Chen, Curr. Org. Synth., 2012, 9, 873 CrossRef CAS.
- R. Ye, J. Zhao, B. B. Wickemeyer, F. D. Toste and G. A. Somorjai, Nat. Catal., 2018, 1, 318 CrossRef.
- For recent reviews in this field, see: (a) M. Hönig, P. Sondermann, N. J. Turner and E. M. Carreira, Angew. Chem., Int. Ed., 2017, 56, 8942 CrossRef PubMed; (b) F. Rudroff, M. D. Mihovilovich, H. Gröger, R. Snajdrova, H. Iding and U. T. Bornscheuer, Nat. Catal., 2018, 1, 12 CrossRef; (c) N. Ríos-Lombardía, J. García-Álvarez and J. González-Sabín, Catalysts, 2018, 8, 75 CrossRef; (d) J. Lia, A. Amatunia and H. Renata, Curr. Opin. Chem. Biol., 2020, 55, 111 CrossRef PubMed; (e) C. Ascaso-Alegre and J. Mangas, Eur. J. Org. Chem., 2022, e202200093 CAS; (f) H. Groger, F. Gallou and B. H. Lipshutz, Chem. Rev., 2023, 123 DOI:10.1021/acs.chemrev.2c00416; (g) S. González-Granda, J. Albarrán-Velo, I. Lavandera and V. Gotor-Fernández, Chem. Rev., 2023, 123 DOI:10.1021/acs.chemrev.2c00454; (h) S. González-Granda, L. Escot, I. Lavandera and V. Gotor-Fernández, Angew. Chem., Int. Ed., 2023, 62, e202217713 Search PubMed; (i) L. Cicco, G. Dilauro, M. Pulpito and V. Capriati, Curr. Opin. Green Sustainable Chem, 2023, 41, 100799 CrossRef.
- (a) E. L. Bell, W. Finnigan, S. P. France, A. P. Green, M. A. Hayes, L. J. Hepworth, S. L. Lovelock, H. Niikura, S. Osuna, E. Romero, K. S. Ryan, N. J. Turner and S. L. Flitsch, Nat. Rev. Methods Primers, 2021, 1, 46 CrossRef CAS; (b) J. Whittall and P. W. Sutton, Applied Biocatalysis: The Chemist's Enzyme Toolbox, John Wiley & Sons Ltd., Hoboken, 2021 Search PubMed.
- (a) J. Clayden, Organolithiums: Selectivity for Synthesis, Pergamon, Elsevier, Science Ltd., Oxford, 2002 Search PubMed; (b) H. J. Reich, Chem. Rev., 2013, 113, 7130 CrossRef CAS PubMed; (c) V. Capriati, F. M. Perna and A. Salomone, Dalton Trans., 2014, 43, 14204 RSC; (d) E. Carl and D. Stalke, Lithium Compounds in Organic Synthesis-From Fundamentals to Applications, ed. R. Luisi and V. Capriati, Wiley-VCH, Weinheim, 2014 Search PubMed; (e) U. Wietelmann and J. Klett, Z. Anorg. Allg. Chem., 2018, 644, 194 CrossRef CAS PubMed.
- (a) J. F. Garst and M. P. Soriaga, Chem. Soc. Rev., 2004, 248, 623 CAS; (b) The Chemistry of Organomagnesium Compounds, ed. Z. Rappoport and I. Marek, Patai Series, Wiley, Chichester, 2008 Search PubMed; (c) D. Seyferth, Organometallics, 2009, 28, 1598 CrossRef CAS.
- (a) L. Castoldi, L. Ielo, P. Hoyos, M. J. Hernaiz, L. De Luca, A. R. Alcántara, W. Holzer and V. Pace, Tetrahedron, 2018, 74, 2211 CrossRef CAS; (b) M. Ramos-Martín, R. Lecuna, L. Cicco, P. Vitale, V. Capriati, N. Ríos-Lombardía, J. González-Sabín, A. Presa-Soto and J. García-Álvarez, Chem. Commun., 2021, 57, 13534 RSC.
- For pioneering studies in this field, see: (a) F. Zhou and C.-J. Li, Nat. Commun., 2014, 5, 4254 CrossRef CAS PubMed; (b) A. Bhattacharjya, P. Klumphu and B. H. Lipshutz, Nat. Commun., 2015, 6, 7401 CrossRef PubMed.
- For previous studies in this field, see: (a) C. Vidal, J. García-Álvarez, A. Hernán-Gómez, A. R. Kennedy and E. Hevia, Angew. Chem., Int. Ed., 2014, 53, 5969 CrossRef CAS PubMed; (b) C. Vidal, J. García-Álvarez, A. Hernán-Gómez, A. R. Kennedy and E. Hevia, Angew. Chem., Int. Ed., 2016, 55, 16145 CrossRef CAS PubMed; (c) M. J. Rodríguez-Álvarez, J. García-Álvarez, M. Uzelac, M. Fairley, C. T. O'Hara and E. Hevia, Chem. – Eur. J., 2018, 24, 1720 CrossRef PubMed; (d) A. Sánchez-Condado, G. A. Carriedo, A. Presa Soto, M. J. Rodríguez-Álvarez, J. García-Álvarez and E. Hevia, ChemSusChem, 2019, 12, 3134 CrossRef PubMed; (e) L. Cicco, A. Fombona-Pascual, A. Sánchez-Condado, G. A. Carriedo, F. M. Perna, V. Capriati, A. Presa Soto and J. García-Álvarez, ChemSusChem, 2020, 13, 4967 CrossRef CAS PubMed; (f) L. Cicco, A. Salomone, P. Vitale, N. Ríos-Lombardía, J. González-Sabín, J. García-Álvarez, F. M. Perna and V. Capriati, ChemSusChem, 2020, 13, 3583 CrossRef CAS PubMed; (g) F. F. Mulks, L. J. Bole, L. Davin, A. Hernán-Gómez, A. Kennedy, J. García-Alvarez and E. Hevia, Angew. Chem., Int. Ed., 2020, 59, 19021 CrossRef CAS PubMed; (h) M. Fairley, L. J. Bole, F. F. Mulks, L. Main, A. R. Kennedy, C. T. O'Hara, J. García-Álvarez and E. Hevia, Chem. Sci., 2020, 11, 6500 RSC; (i) D. Arnodo, S. Ghinato, S. Nejrotti, M. Blangetti and C. Prandi, Chem. Commun., 2020, 56, 2391 RSC; (j) A. F. Quivelli, G. D'Addato, P. Vitale, J. García-Álvarez, F. M. Perna and V. Capriati, Tetrahedron, 2021, 81, 131898 CrossRef CAS; (k) S. Ghinato, D. Territo, A. Maranzana, V. Capriati, M. Blangetti and C. Prandi, Chem. – Eur. J., 2021, 27, 2868 CrossRef CAS PubMed; (l) D. Elorriaga, B. Parra-Cadenas, A. Antiñolo, F. Carrillo-Hermosilla and J. García-Álvarez, Green Chem., 2022, 24, 800 RSC; (m) D. Elorriaga, B. Parra-Cadenas, A. Antiñolo, F. Carrillo-Hermosilla and J. García-Álvarez, ChemSusChem, 2022, 15, e202201348 CrossRef CAS PubMed; (n) F. F. Mulks, B. Pinho, A. W. J. Plattern, M. R. Andalibi, A. J. Expósito, K. J. Edler, E. Hevia and L. Torrente-Murciano, Chem, 2022, 8, 3382 CrossRef CAS; (o) P. Slavík, B. R. Trowse, P. O'Brien and D. K. Smith, Nat. Chem., 2023, 15, 319 CrossRef PubMed.
- For other examples of the use of polar organometallic reagents in protic (bio-based) reaction media, see: (a) V. Mallardo, R. Rizzi, F. C. Sassone, R. Mansueto, F. M. Perna, A. Salomone and V. Capriati, Chem. Commun., 2014, 50, 8655 RSC; (b) F. C. Sassone, F. M. Perna, A. Salomone, S. Florio and V. Capriati, Chem. Commun., 2015, 51, 9459 RSC; (c) L. Cicco, S. Sblendorio, R. Mansueto, F. M. Perna, A. Salomone, S. Florio and V. Capriati, Chem. Sci., 2016, 7, 1192 RSC; (d) G. Dilauro, A. F. Quivelli, P. Vitale, V. Capriati and F. M. Perna, Angew. Chem., Int. Ed., 2019, 58, 1799 CrossRef CAS PubMed; (e) G. Dilauro, C. S. Azzollini, P. Vitale, A. Salomone, F. M. Perna and V. Capriati, Angew. Chem., Int. Ed., 2021, 60, 10632 CrossRef CAS PubMed; (f) S. Ghinato, F. De Nardi, P. Bolzoni, A. Antenucci, M. Blangetti and C. Prandi, Chem. – Eur. J., 2022, 28, e202201154 CrossRef CAS PubMed; (g) G. Dilauro, M. Dell'Aera, P. Vitale and F. M. Perna, Angew. Chem., Int. Ed., 2017, 56, 10200 CrossRef CAS PubMed.
- For recent reviews of this topic, see: (a) J. García-Álvarez, Eur. J. Inorg. Chem., 2015, 5147 CrossRef; (b) J. García-Álvarez, E. Hevia and V. Capriati, Eur. J. Org. Chem., 2015, 6779 CrossRef; (c) J. García-Álvarez, E. Hevia and V. Capriati, Chem. – Eur. J., 2018, 24, 14854 CrossRef PubMed; (d) E. Hevia, Chimia, 2020, 74, 681 CrossRef CAS PubMed; (e) T. X. Gentner and R. E. Mulvey, Angew. Chem., Int. Ed., 2021, 60, 9247 CrossRef CAS PubMed; (f) F. M. Perna, P. Vitale and V. Capriati, Curr. Opin. Green Sustainable Chem., 2021, 30, 100487 CrossRef CAS; (g) S. E. García-Garrido, A. Presa Soto, E. Hevia and J. García-Álvarez, Eur. J. Inorg. Chem., 2021, 3116 CrossRef.
- For recent examples of hybrid tandem protocols which merge transition-metal-catalysis or organocatalysis with highly-polar main-group reagents (RLi/RMgX) working at room temperature and in the absence of a protecting atmosphere, see: (a) L. Cicco, M. J. Rodríguez-Álvarez, F. M. Perna, J. García-Álvarez and V. Capriati, Green Chem., 2017, 19, 3069 RSC; (b) S. Ghinato, G. Dilauro, F. M. Perna, V. Capriati, M. Blangetti and C. Prandi, Chem. Commun., 2019, 55, 7741 RSC; (c) D. Elorriaga, M. J. Rodríguez-Álvarez, N. Ríos-Lombardía, F. Morís, A. Presa Soto, J. González-Sabín, E. Hevia and J. García-Álvarez, Chem. Commun., 2020, 56, 8932 RSC; (d) D. Elorriaga, F. de la Cruz-Martínez, M. J. Rodríguez-Álvarez, A. Lara-Sánchez, J. A. Castro-Osma and J. García-Álvarez, ChemSusChem, 2021, 14, 2084 CrossRef CAS PubMed.
- For examples related to the use of RMgX reagents in non-protic ionic liquids, see: (a) T. Ramnial, D. D. Ino and J. A. C. Clyburne, Chem. Commun., 2005, 325 RSC; (b) V. Jurcik and R. Wilhem, Green Chem., 2005, 7, 844 RSC; (c) S. T. Handy, J. Org. Chem., 2006, 71, 4659 CrossRef CAS PubMed; (d) M. C. Law, K.-Y. Wong and T. H. Chan, Chem. Commun., 2006, 2457 RSC; (e) T. Itoh, K. Kude, S. Hayase and M. Kawatsura, Tetrahedron Lett., 2007, 48, 7774 CrossRef CAS; (f) T. Ramnial, S. A. Taylor, J. A. C. Clyburne and C. J. Walsby, Chem. Commun., 2007, 2066 RSC.
- Laccases are copper-containing enzymes belonging to the group of oxidoreductases. In nature, laccases are involved in biological processes such as lignification in plants or lignin degradation in fungi, and catalyse the oxidation of substrates such as phenols, polyphenols and anilines by means of four-electron transfer processes while reducing O2 to water: (a) P. Giardina, V. Faraco, C. Pezzella, A. Piscitelli, S. Vanhulle and G. Sannia, Cell. Mol. Life Sci., 2010, 67, 369 CrossRef CAS PubMed; (b) J.-R. Jeon and Y.-S. Chang, Trends Biotechnol., 2013, 31, 335 CrossRef CAS PubMed; (c) K. W. Wellington, Application of Laccases in Organic Synthesis: A Review, in Green Chemistry, ed. R. Luque, Nova Science Publishers Inc, New York, 2014 Search PubMed; (d) A. C. Sousa, L. O. Martins and M. P. Robalo, Molecules, 2021, 26, 3719 CrossRef CAS PubMed; (e) I. Bassanini, E. E. Ferrandi, S. Riva and D. Monti, Catalysts, 2021, 11, 26 CrossRef CAS; (f) N. Cardullo, V. Muccilli and C. Tringali, RSC Chem. Biol., 2022, 3, 614 RSC.
- For examples of catalytic networks in which laccases have been combined with other enzymes such as lipases, oxidoreductases, ω-transaminases or enoate reductases, see: (a) P. Gavezzotti, C. Navarra, S. Caufin, B. Danieli, P. Magrone, D. Monti and S. Riva, Adv. Synth. Catal., 2011, 353, 2421 CrossRef CAS; (b) A. Díaz-Rodríguez, N. Ríos-Lombardía, J. H. Sattler, I. Lavandera, V. Gotor-Fernández, W. Kroutil and V. Gotor, Catal. Sci. Technol., 2015, 5, 1443 RSC; (c) L. Montero, V. Gotor, V. Gotor-Fernández and I. Lavandera, Green Chem., 2017, 19, 474 RSC; (d) M. J. Rodríguez-Álvarez, N. Ríos-Lombardía, S. Schumacher, D. Pérez-Iglesias, F. Morís, V. Cadierno, J. García-Álvarez and J. González-Sabín, ACS Catal., 2017, 7, 7753 CrossRef; (e) E. Brenna, M. Crotti, M. De Pieri, F. G. Gatti, G. Manenti and D. Monti, Adv. Synth. Catal., 2018, 360, 3677 CrossRef CAS; (f) J. Albarrán-Velo, V. Gotor-Fernández and I. Lavandera, ChemBioChem, 2020, 21, 200 CrossRef PubMed; (g) J. Albarrán-Velo, V. Gotor-Fernández and I. Lavandera, Mol. Catal., 2020, 493, 111087 CrossRef; (h) S. González-Granda, D. Méndez-Sánchez, I. Lavandera and V. Gotor-Fernández, ChemCatChem, 2020, 12, 520 CrossRef.
- (a) J. González-Sabín, N. Ríos-Lombardía, I. García, N. M. Vior, A. F. Braña, C. Méndez, J. A. Salas and F. Morís, Green Chem., 2016, 18, 989 RSC; (b) R. S. Correia Cordeiro, N. Ríos-Lombardía, F. Morís, R. Kourist and J. González-Sabín, ChemCatChem, 2019, 11, 1272 CrossRef CAS.
- For an outstanding example of enzyme-catalysed oxime dehydration, see: T. Betke, P. Rommelmann, K. Oike, Y. Asano and H. Gröger, Angew. Chem., Int. Ed., 2017, 56, 12361 CrossRef CAS PubMed.
- For some examples of natural products containing the oxime moiety, see: (a) H. P. Chen, Z. Z. Zhao, Z. H. Li, Z. J. Dong, K. Wei, X. Bai, L. Zhang, C. N. Wen, T. Feng and J. K. Liu, ChemistryOpen, 2016, 5, 142 CrossRef CAS PubMed; (b) M. Sørensen, E. H. J. Neilson and B. L. Møller, Mol. Plant, 2018, 11, 95 CrossRef PubMed. For some examples of biologically active oximes, see: (c) K. C. Fylaktakidou, D. J. Hadjipavlou-Litina, K. E. Litinas, E. A. N. Varella and D. N. Nicolaides, Curr. Pharm. Des., 2008, 14, 1001 CrossRef CAS PubMed; (d) A. Nikitjuka and A. Jirgensons, Heterocycl. Compd., 2014, 49, 1544 CrossRef CAS; (e) B. Bednarczyk-Cwynar and L. Zaprutko, Phytochem. Rev., 2015, 14, 203 CrossRef CAS PubMed.
- (a) J. G. de Vries, Important Pharmaceuticals and Intermediates, in Quaternary Stereocenters: Challenges and Solutions for Organic Synthesis, ed. J. Christoffers and A. Baro, Wiley-VCH, Weinheim, 2005 Search PubMed; (b) K. C. Nicolaou and T. Montagnon, Molecules that Changed the World, Wiley-VCH, Weinheim, 2008 Search PubMed.
- Some of us have previously reported the use of the CuCl2·2H2O/TMEDA/TEMPO system for the selective oxidation of terminal alcohols into aldehydes: L. Cicco, M. Roggio, M. López-Aguilar, M. Ramos-Martín, F. M. Perna, J. García-Álvarez, P. Vitale and V. Capriati, ChemistryOpen, 2022, 11, e202200160 CrossRef CAS PubMed.
- G. M. Whitesides and T. L. Newirth, J. Org. Chem., 1975, 40, 3449 Search PubMed.
- Cyclopentyl methyl ether (CPME) has been already used as a solvent in combination with RLi/RMgX reagents, working at room temperature and in the presence of air. For recent examples, see ref. 8b, 10i, k and 11a, b, f.
- 2-MeTHF has been employed as the solvent for s-block polar organometallic chemistry at room temperature and in the presence of air/moisture. For recent examples, see ref. 10g, h and 13d.
- The formation of more reactive tetrameric or dimeric species in solutions of PhLi in different ethereal solvents has been previously reported. H. J. Reich, D. P. Green, M. A. Medina, W. S. Goldenberg, B. Ö. Gudmundsson, R. R. Dykstra and N. H. Phillips, J. Am. Chem. Soc., 1998, 120, 7201 CrossRef CAS.
- G. Osztrovszky, T. Holm and R. Madsen, Org. Biomol. Chem., 2010, 8, 3402 RSC.
- D. Elorriaga, M. J. Rodríguez-Álvarez, N. Ríos-Lombardía, F. Morís, A. Presa Soto, J. González-Sabín, E. Hevia and J. García-Álvarez, Chem. Commun., 2020, 56, 8932 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ob00285c |
| This journal is © The Royal Society of Chemistry 2023 |