
Catalytic I2-moist DMSO-mediated synthesis of valuable α-amidohydroxyketones and unsymmetrical gem-bisamides from benzimidates†
Shobhon
Aich
a,
Rajesh
Nandi
a,
Nirbhik
Chatterjee
 b,
Krishnanka S.
Gayen
c,
Subhasis
Pal
a and
Dilip K.
Maiti
*a
b,
Krishnanka S.
Gayen
c,
Subhasis
Pal
a and
Dilip K.
Maiti
*a
aDepartment of Chemistry, University of Calcutta, 92 A. P. C. Road, Kolkata-700009, India. E-mail: dkmchem@caluniv.ac.in; Fax: +91-33-2351 9755; Tel: +91-33-2350 1014
bDepartment of Chemistry, Kanchrapara College, North 24 parganas-743145, India
cDepartment of Chemistry, Raja Peary Mohan College, West Bengal, India
First published on 14th February 2023
Abstract
We developed an efficient and straightforward I2-catalyzed strategy for the synthesis of functionalized α-amidohydroxyketones and symmetrical and unsymmetrical bisamides using incipient benzimidate scaffolds as starting materials and moist-DMSO as a reagent and solvent. The developed method proceeds through chemoselective intermolecular N–C-bond formation of benzimidates and the α-C(sp3)–H bond of acetophenone moieties. The key advantages of these design approaches include broad substrate scope and moderate yields. High-resolution mass spectrometry of the reaction progress and labeling experiments provided suitable evidence regarding the possible mechanism. 1H nuclear magnetic resonance titration revealed notable interaction between the synthesized α-amidohydroxyketones and some anions as well as biologically important molecules, which revealed a promising recognition property of these valuable motifs.
Introduction
Decoration of valuable structural motifs from easily available starting materials is a challenging task in synthetic organic chemistry. α-Amidoketone1 is used proficiently as a synthetic equivalent for the development of N-heterocyclic scaffolds such as oxazoles,2a imidazoles,2b and thiazoles.2c Likewise, symmetrical and unsymmetrical N,N′-alkylidenebisamide scaffolds are important fragments of peptidomimetics and pharmaceutically active drug molecules, including the anticancer and antidepressant agents leuconoxine (B) and scholarisine G (A), antioxidant and antimicrobial agent JBIR-94, and drugs to counteract high blood pressure.3,4 Several methods have been developed for the construction of α-amidoketone derivatives. The major concerns arising from these methodologies are the utilization of expensive catalysts, harsh reaction conditions, and low atom economy. Notably, Wu et al. developed the synthesis of α-ketoimides through the reaction between aryl methyl ketones and benzamidine hydrochloride in the presence of low-cost molecular iodine and DMSO.5 Few reports of methylene bisamide6–9 derivatives include: the reaction of nitriles and formaldehyde in the presence of strong sulfuric acid solution;6 construction of switched N,N′-oxydimethylenebisalkanamide derivatives from the reaction of simple alkanamide and polymeric formaldehyde;7a an operationally modest method to provide methylene bisamides under heating conditions7b and ammonium persulfate-activated DMSO as a source of one carbon synthon.8 In recent times, one of our major research foci has been imidate chemistry.10i Imidate (or imino-ether) frameworks have garnered considerable interest in synthetic organic chemistry. In general, imidate scaffolds are quite distinctive compared with stereotypical functional groups in that they can be electrophiles and nucleophiles. Hence, they can cover a wide range of strategies to provide homo atomic and hetero atomic couplings.10–12 Based on this fundamental science, recently synthetic chemists have reported interesting breakthroughs regarding some biologically important molecules.13,14 Herein, we describe a highly proficient synthetic protocol for the simultaneous two bond-forming cross-coupling reaction of benzimidate skeletons (1) with acetophenone analogues to provide α-amidohydroxyketones (3, eqn (2), Scheme 1). Wu and colleagues5 stated that the product was an imide moiety where the –NH group was flanked between two carbonyl cores (eqn (1), Scheme 1). However, in our work, by merely using benzimidate rather than benzamidine hydrochloride and moist DMSO, we joined an –OH group adjacent to an –NH group to provide an α-amidohydroxyketone. Stringing of this acidic –OH group in the product made it a suitable candidate for recognition. In addition, we highlighted the unique insertion of a methylene moiety, emerging from the methyl group of DMSO, between two benzimidate scaffolds to furnish valuable unsymmetrical and symmetrical bisamide analogues (4, eqn (4), Scheme 1). However, the synthesis of symmetrical bisamides has been described in a Chinese patent9 utilizing 120–500 mol% of I2 and benzamides (eqn (3), Scheme 1), thereby making the method hazardous. We needed only a catalytic amount of I2 (10 mol%). | ||
| Scheme 1 Developed hydroxyamidation and gem-diamidation reaction. | ||
“Recognition” is one of the “hotspots” of supramolecular chemistry15–18 and ensures wide applicability in various fields. Numerous modes of noncovalent interactions between a host and guest are predicted as major reasons for their origination. In most cases, the receptor parts are cage-like motifs or acyclic compounds capable of providing suitable orientation that are responsible for appropriate interaction with the guest. Hydroxyl groups bearing α-ketoamide have a unique architecture so we explored their recognition properties using 1H nuclear magnetic resonance (1HNMR) titration.
Our initial attempts for the construction of α-amidohydroxyketone (3aa) employing ethyl benzimidate (1a) and acetophenone (2a) covered several prospective catalysts (10 mol%), such as Cu(OTf)2, CuCl2, CuI, AgOTf, AgOAc and Zn(OTf)2 (Table 1, entry 1), in DMSO/H2O at 120 °C, but none of them provided a suitable outcome. Lewis acids such as ZnCl2, Ni(OTf)2, NiCl2·4H2O and AlCl3 exhibited promising results, but the yields were low (Table 1, entries 2–5). Only FeCl3·6H2O led to marginal improvement of the yield (30%, Table 1, entry 6). Gratifyingly, when we shifted our focus to molecular iodine (I2, 10 mol%) in DMSO/H2O, the yield was enhanced markedly (80%) with concomitant reduction of the reaction time (12 h, Table 1, entry 7). In the absence of iodine (I2), the reaction did not progress (Table 1, entry 8). This clearly indicated that molecular iodine was essential for a strategy for the construction of a N–C bond. Detailed solvent studies (Table 1, entries 9–16) showed DMSO/H2O to be the best choice (Table 1, entry 7) in comparison with other common polar and nonpolar organic solvents. Variation in catalyst load had a detrimental effect on the yield of the product (Table 1, entries 17 and 18). Hence, the optimum reaction conditions were 10 mol% iodine (I2) in DMSO at 120 °C to deliver product 3 in good yield (Table 1, entry 7). A gram-scale synthesis under standard reaction conditions afforded the anticipated product 3aa in good yield (75%) within 14 h (Table 1, entry 19), which demonstrated the applicability of this approach in academia and industry. We showed that using an exact amount of moisture (Table 1, entries 20–22) was suitable for the new reaction, and a DMSO![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :water ratio of 10:1 (v/v, Table 1, entry 21) was employed.
:water ratio of 10:1 (v/v, Table 1, entry 21) was employed.
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Catalyst | mol% | Solvent | Conditions (°C) | Time (h) | Yieldb (%) |
| a Reactions were carried out using ethyl benzimidate (1a, 1 mmol) and acetophenone (2a, 1 mmol) and I2 catalyst (0.1 mmol). b Yield of the isolated product after column purification. c No reaction. d Unreacted 1a and 2a were recovered. e Gram-scale synthesis. f DMSO (2 ml) and water (0.1 ml). g DMSO (2 ml) and water (0.2 ml). h DMSO (2 ml) and water (0.3 ml). | ||||||
| 1 | Zn(OTf)2 | 10 | DMSO/H2O | 120 | 24 | NRc |
| 2 | ZnCl2 | 10 | DMSO/H2O | 120 | 24 | 20d |
| 3 | Ni(OTf)2 | 10 | DMSO/H2O | 120 | 24 | 25d |
| 4 | NiCl2·4H2O | 10 | DMSO/H2O | 120 | 20 | 24d |
| 5 | AlCl3 | 10 | DMSO/H2O | 120 | 24 | 24 |
| 6 | FeCl3·6H2O | 10 | DMSO/H2O | 120 | 24 | 30d |
| 7 | I 2 | 10 | DMSO/H 2 O | 120 | 12 | 80 |
| 8 | — | — | DMSO/H2O | 120 | 24 | NRc |
| 9 | I2 | 10 | THF/H2O | Reflux | 24 | NRc |
| 10 | I2 | 10 | Dioxane/H2O | Reflux | 24 | 23 d |
| 11 | I2 | 10 | DCE/H2O | Reflux | 20 | NRc |
| 12 | I2 | 10 | DCM/H2O | Reflux | 22 | NRc |
| 13 | I2 | 10 | DMF/H2O | 120 | 20 | 22d |
| 14 | I2 | 10 | EtOH/H2O | Reflux | 12 | NRc |
| 15 | I2 | 10 | Toluene/H2O | Reflux | 24 | NRc |
| 16 | I2 | 10 | Benzene/H2O | Reflux | 24 | NRc |
| 17 | I2 | 7 | DMSO/H2O | 120 | 20 | 55 |
| 18 | I2 | 15 | DMSO/H2O | 120 | 12 | 65 |
| 19 | I2 | 10 | DMSO/H2O | 120 | 14 | 75e |
| 20 | I2 | 10 | DMSO/H2O | 120 | 12 | 45f |
| 21 | I2 | 10 | DMSO/H2O | 120 | 12 | 80g |
| 22 | I2 | 10 | DMSO/H2O | 120 | 12 | 79h |

After this successful survey, we proceeded to scrutinize the substrate scope utilizing benzimidate (1a) and a wide range of acetophenones (2a–j) under the developed reaction conditions. The desired α-amidohydroxyketone derivatives (3aa–aj) were obtained with good-to-high yields (72–85%, Scheme 2). To our delight, the reactions with sterically hindered ortho-substituted acetophenones (2b and 2c) were also well tolerated to furnish the crowded 3ab and 3ac, respectively. Replacement of acetophenone by cyclohexanone or cyclopentanone did not provide the expected product but instead led to complex reaction mixture.
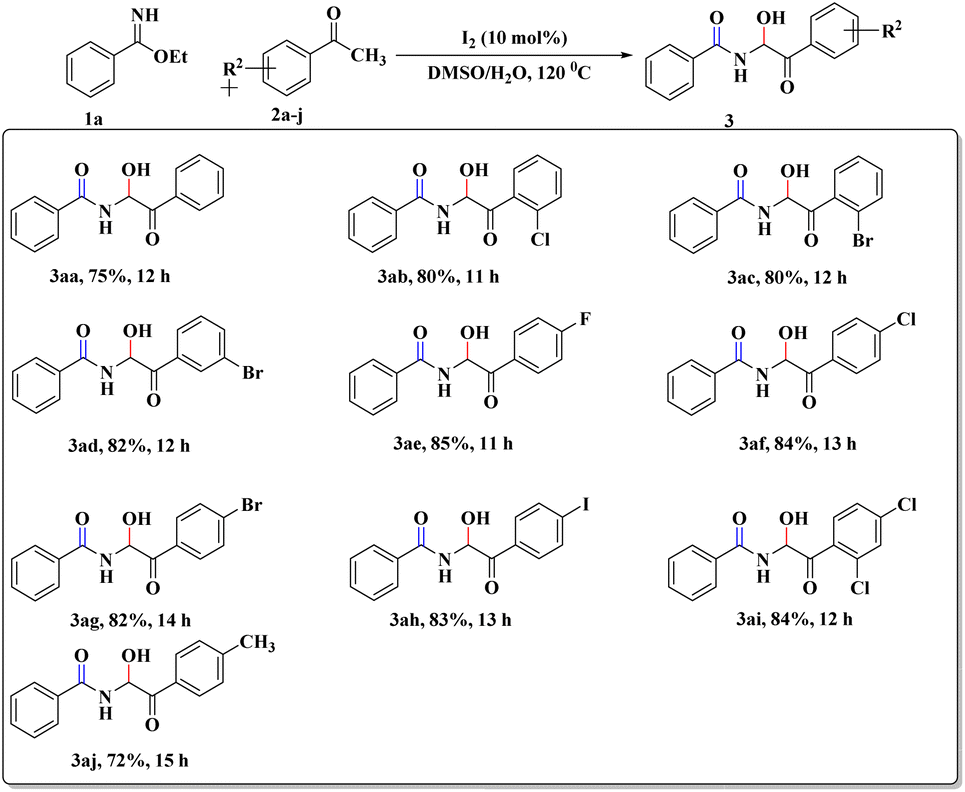 | ||
| Scheme 2 Synthesized α-amidohydroxyketones. | ||
To explore the substrate scope further, reactions between electronically enriched and poor benzimidates and electronically poor, sterically hindered acetophenone analogues were examined to obtain the desired functionalized α-amidohydroxyketones. Gratifyingly, electron-rich p-methylbenzimidate (1b) and p-methoxybenzimidate (1c) were compatible with electronically poor, electronically rich and sterically hindered ketones to provide the desired products 3ba–cj with significant yields. Again, electronically poor benzimidates (1d-p-NO2 and 1g-m-B(OH)2) also provided delightful outcomes (3de, 3gd). Heterocyclic imidates, such as ethyl thiophene-2-carbimidate 1e (3ea–ei, 76–78%) and ethyl furan-2-carbimidate 1f (3fa, 80%) were well tolerated. However, we did not obtain the desired product for pyridine or quinoline. All synthesized compounds were fully characterized by spectroscopic data (ESI) and the structural skeleton of 3ea was confirmed with the help of single-crystal X-ray diffraction analysis (Scheme 3).
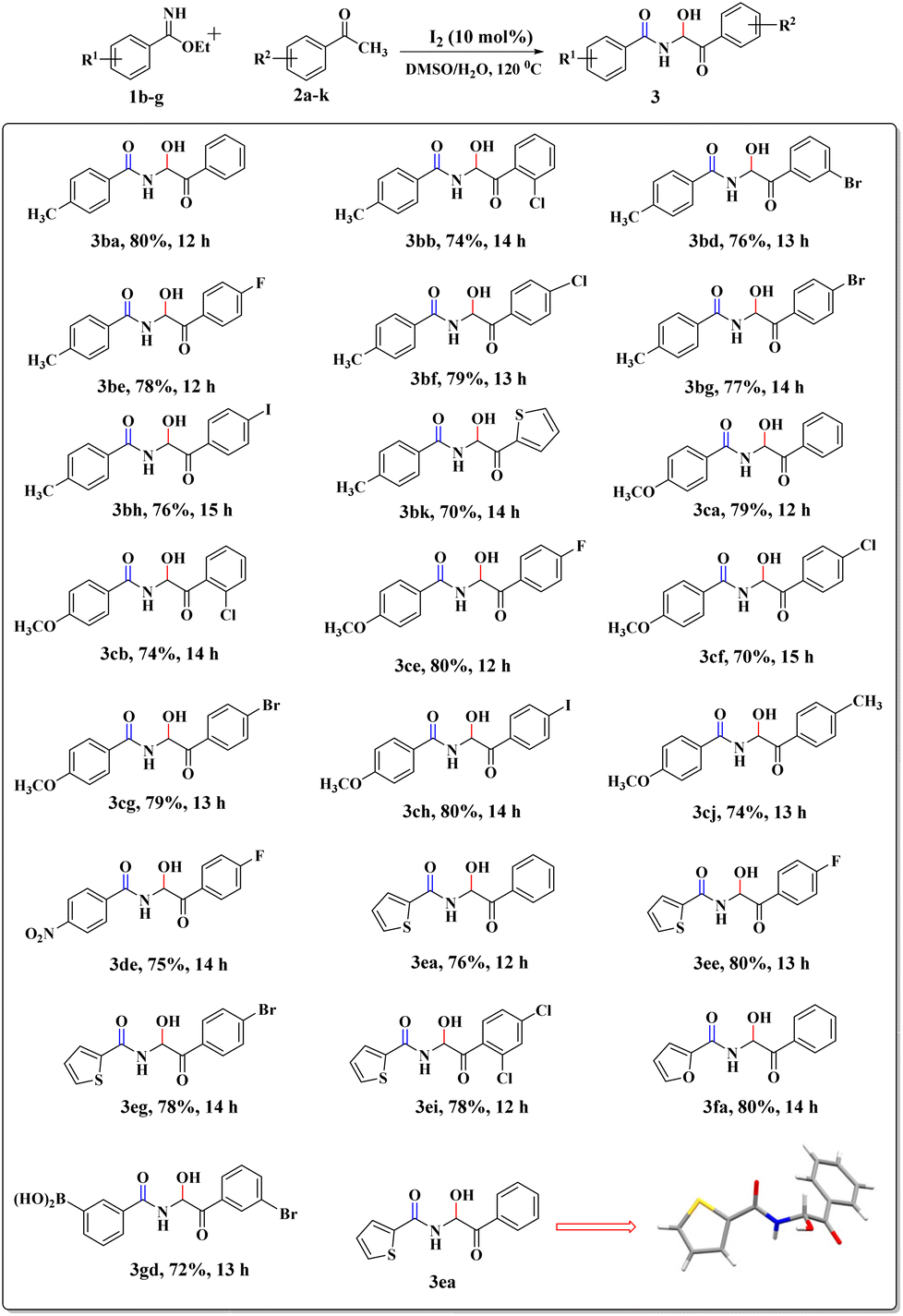 | ||
| Scheme 3 Scope of synthesized α-amidohydroxyketone derivatives. | ||
To acquire a clear-cut vision of the mechanistic pathway, we pursued another important study whereby, under the developed reaction conditions, H2O was replaced by D2O. 1H NMR spectroscopy of the corresponding product (3aa(d), Scheme 4) clarified the absence of an O–H proton (ESI†), which indicated the involvement of nucleophilic attack by H2O (or D2O) during the course of the reaction.
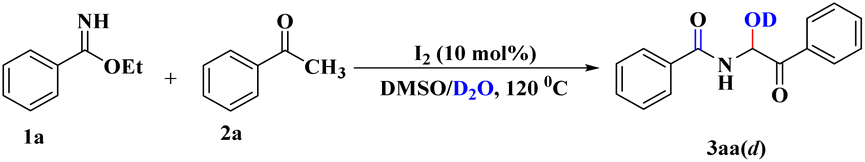 | ||
| Scheme 4 Reaction with D2O instead of H2O. | ||
During our survey, we noticed that the benzimidate moiety itself underwent unique coupling under the developed reaction conditions to provide gem-bisamide (Schemes 5 and 6), and the source of the methylene group was from DMSO. This prediction was justified by the fact that when DMSO-d6 was used instead of DMSO, there was no relevant peak of the –CH2 group in 1H NMR spectroscopy (ESI). It had been replaced by –CD2. Unlike previous reports6–9 our strategy was also capable of providing unsymmetrical gem-bisamides (Scheme 6).
 | ||
| Scheme 5 Synthesis and scope towards symmetrical gem-bisamides. | ||
 | ||
| Scheme 6 Synthesis and scope towards unsymmetrical gem-bisamides. | ||
In addition to the experiment described in Scheme 4, high-resolution MS was undertaken to find suitable evidence of the intermediates formed during the course of the reaction. This allowed justification of the reaction mechanism (calculated and observed values are given in parenthesis, Scheme 7 and ESI†). At the very beginning, acetophenone is converted to intermediate A in the presence of the catalyst (molecular iodine). Then, nucleophilic attack by the benzimidate and concomitant release of HI generates intermediate B. Hydrolysis of B results in formation of the iodo amidoketone C. Finally, release of the iodide ion from C provides the new iminium intermediate D and simultaneously leads to aerial oxidation to regenerate molecular iodine, and the cycle proceeds as usual. Nucleophilic attack upon D by a water molecule furnishes the desired architecture 3aa. A similar mode of investigation was carried out for the synthesis of bisamide 4cc.
 | ||
| Scheme 7 Proposed mechanistic pathway towards the formation of 3aa. | ||
In this regard, the proposal (Scheme 8) was that first DMSO is converted to A′ in the presence of molecular iodine. Now, as usual, the nucleophilic imidate reacts with A′ to provide substituted sulfoxide B′. Oxidation of B′ generates the sulfone C′. Elimination of MeSO2− from C′ results in the active iminium intermediate D′. Finally, a second molecule of the imidate moiety provides condensation with D′ to furnish the desired bisamide 4cc.
 | ||
| Scheme 8 Proposed mechanistic pathway to construct 4cc. | ||
Recognition properties
We carried out traditional 1H NMR titration to reveal the interaction between the substrate 3de and anions, and discovered some promising outcomes. Details of the titration mode are highlighted in Table 2. Initially, addition of 10 μl of the anion solution to the substrate solution indicated that some anions provided a shift in the 1H NMR spectrum of the O–H proton and notable splittings of the O–H and C–H protons (Table 2, entries 1–5). Hence, a gradual increase of the amount of anion solutions (from 10 μl to 40 μl) ultimately led to further change in this shift and disappearance of the O–H proton. However, F− did not disappear completely (Table 2, entry 5, Fig. 1). Interestingly, dramatic observations for the guests OAc− and Cl− were documented (Table 2, entries 1 and 2). Addition of such guests to the receptor had a profound effect in the 1H NMR spectrum of 3de in O–H and C–H regions, but also in aromatic and N–H regions (Fig. 1a). One can postulate that, in the presence of such intimate guests, the receptor acquires a folded pattern (Fig. 1b) to encapsulate these anions. The magnetic environment of all types of protons is affected and ultimately it is responsible for the change in 1H NMR spectrum of the entire framework. However, for other guests, the receptor may be extended slightly (Fig. 1c) so that the interaction is restricted in a confined region between O–H and the guest, and the effect is not as pronounced like in previous cases (Fig. 1). This hypothesis requires more elaborative studies. Addition of other anions (Table 2, entries 6–9) did not provide a notable change, and the specificity of 3de was preserved. Furthermore, we focused on some biologically important molecules, such as adenine, thymine, uracil, and ATP. For adenine (Table 2, entry 10), we did not observe disappearance of the O–H peak after addition of solution (40 μl), though changes in the splitting pattern in the O–H, C–H and aromatic regions were distinctive (ESI†). However, for uracil and ATP (Table 2, entries 12 and 13), the scenario was opposite to that for adenine in terms of disappearance of the O–H peak and aromatic regions (ESI†). | ||
| Fig. 1 (a) 1H NMR titration, (b) and (c) schematic presentations of host–guest interaction. | ||
|
|
||||
|---|---|---|---|---|
| Entry | Aniona | Notable change in O–H and C–H regions in 1H NMR after addition of 40 μl of anion solution | Disappearance of O–H peak in 1H NMR after addition of 40 μl of anion solution | Notable change in aromatic and N–H regions in 1H NMR after addition of 40 μl of anion solution |
|
a Anion solution = 1 mg in 1 ml of distilled water (same for uracil and thymine, for adenine solvent is 1:1 distilled water and DMSO-d6). Substrate solution = 5 mg in 0.5 ml of DMSO-d6. NMR titration was done by gradual addition of the anion solution (from 10 μl to 40 μl) to the substrate solution.
|
||||
| 1 | OAc− | Yes | Yes | Yes |
| 2 | Cl− | Yes | Yes | Yes |
| 3 | CN− | Yes | Yes | No |
| 4 | H2PO4− | Yes | Yes | No |
| 5 | F− | Yes | No | No |
| 6 | NO3− | No | — | — |
| 7 | NO2− | No | — | — |
| 8 | HPF6− | No | — | — |
| 9 | HCO3− | No | — | — |
| 10 | Adenine | Yes | No | Yes |
| 11 | Thymine | Yes | Yes | Yes |
| 12 | Uracil | Yes | Yes | No |
| 13 | ATP | Yes | Yes | No |

Quite interestingly, thymine (Table 2, entry 11) was similar to chloride and acetate, and had a notable appearance in the entire 1H NMR spectrum (ESI†).
The 1H NMR spectrum of 3aa revealed the –OH and –CH peaks to have merged (Fig. 2b) but their appearances were quite distinctive for 3de (Fig. 2a). This scenario is relevant for other products in which the aromatic rings are substituted. Hence, we extended our NMR-titration study to observe the change in the 1H NMR spectrum of 3aa. After addition of Cl(−) solution (30 μl) to the sample solution, the –OH and –CH peaks began to separate (Fig. 2c) and the gap became slightly wider upon addition of Cl(−) (40 μl) (ESI†) but, unlike 3de, the –OH peak did not vanish.
 | ||
| Fig. 2 (a) 1H NMR of 3de, (b) 1H NMR of 3aa and (c) 1H NMR of 3aa after addition of Cl(−) solution (30 μl). | ||
Next, to establish the competence of the recognition property of synthesized products, we conducted a comparative 1H NMR-titration study between 3aa and a similar compound (compound 6), in which a methylene group is adjacent to –NH instead of a hydroxyl-bearing methine. We synthesized 6 according to the literature.19 Even after addition of Cl(−) solution (40 μl), a change in the 1H NMR spectrum of 6 was not observed (Fig. 3). Hence, the presence of the –CH(OH) part in our synthesized α-amidohydroxyketones had a profound effect upon the recognition properties.
 | ||
| Fig. 3 1H NMR titration of compound 6 with Cl(−) solution. | ||
Conclusions
We highlighted promising iodine-catalyzed integration of benzimidates and acetophenones in moist DMSO to provide valuable α-amidohydroxyketones. Self-coupling of benzimidate molecules under identical reaction conditions furnished gem-bisamides. Numerous examples with a wide variety of substituents were well tolerated. Labelling experiments and high-resolution mass spectrometry of the reaction progress provided evidence to justify the predicted mechanism of the new coupling reaction. Among the several synthesized α-amidohydroxyketones, chosen electron-withdrawing groups bearing a candidate displayed excellent recognition properties for some anions and biologically important molecules. Detailed theoretical and kinetic studies regarding the recognition properties and development of new methodologies as well as construction of valuable architectures sufficiently competent to explore new showcases in supramolecular chemistry are in progress.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Funding from the Ministry of Mines (Met4-14/19/2021), research fellowship from UGC (SRF to S. A.) and CSIR (SRF to S. P.), Government of India, are gratefully acknowledged. CAS-UGC, CRNN and CU are acknowledged for use of instrument facilities.References
- (a) C. Gao, Q. Zhou, L. Yang, X. Zhang and X. Fan, J. Org. Chem., 2020, 85, 13710–13720 CrossRef CAS PubMed; (b) A. De, S. Santra, G. V. Zyryanov and A. Majee, Org. Lett., 2020, 22, 3926–3930 CrossRef CAS PubMed; (c) F. Xu, X. J. Si, Y. Y. Song, X. D. Wang, C. S. Liu, P. F. Geng and M. Du, J. Org. Chem., 2019, 84, 2200–2208 CrossRef CAS PubMed; (d) B. H. Oh, I. Nakamura and Y. Yamamoto, J. Org. Chem., 2004, 69, 2856–2858 CrossRef CAS PubMed.
- (a) R.-R. Zhou, Q. Cai, D.-K. Li, S.-Y. Zhuang, Y.-D. Wu and A.-X. Wu, J. Org. Chem., 2017, 82, 6450–6456 CrossRef CAS PubMed; (b) D. E. Frantz, L. Morency, A. Soheili, J. A. Murry, E. J. J. Grabowski and R. D. Tillyer, Org. Lett., 2004, 6, 843–846 CrossRef CAS PubMed; (c) E. Biron, J. Chatterjee and H. Kessler, Org. Lett., 2006, 8, 2417–2420 CrossRef CAS PubMed.
- (a) C. Aleman and J. Puiggali, J. Org. Chem., 1995, 60, 910–924 CrossRef CAS; (b) K. I. Nunami, T. Yamazaki and M. Goodman, Biopolymers, 1991, 31, 1503–1512 CrossRef CAS PubMed; (c) M. Rodriguez, P. Dubreuil, J. P. Bali and J. Martinez, J. Med. Chem., 1987, 30, 758–763 CrossRef CAS PubMed.
- (a) S. Rayavarapu, S. K. Kadiri, M. B. Gajula, M. Nakka, R. Tadikonda, N. S. Yarla and S. Vidavalur, Med. Chem., 2014, 4, 367–372 Search PubMed; (b) Z. Xu, Q. Wang and J. Zhu, J. Am. Chem. Soc., 2013, 135, 19127–19130 CrossRef CAS PubMed; (c) B. M. Verdel, P. C. Souverein, A. C. G. Egberts and H. G. M. Leufkens, Ann. Pharm., 2006, 40, 1040–1046 CAS.
- X. Wu, Q. Gao, S. Liu and A. Wu, Org. Lett., 2014, 16, 2888–2891 CrossRef CAS PubMed.
- E. E. Magat, F. Farisj, E. Reith and L. F. Salisbu, J. Am. Chem. Soc., 1951, 73, 1028–1031 CrossRef CAS.
- (a) N. O. Brace and G. J. Mantell, J. Org. Chem., 1961, 26, 5176–5180 CrossRef CAS; (b) Q. Wang, L. Sun, Y. Jiang and C. Li, Beilstein J. Org. Chem., 2008, 4, 51–56 Search PubMed.
- P. S. Mahajan, S. D. Tanpure, N. A. More, J. M. Gajbhiye and S. B. Mhaske, RSC Adv., 2015, 5, 101641–101646 RSC.
- Ch. Pat., 105237432A, 2016 Search PubMed.
- (a) R. Thakur, Y. Jaiswal and A. Kumar, Org. Biomol. Chem., 2019, 17, 9829–9843 RSC; (b) Y. Kumar, Y. Jaiswal and A. Kumar, Org. Lett., 2018, 20, 4964–4969 CrossRef CAS PubMed; (c) Y. Kumar, Y. Jaiswal and A. Kumar, J. Org. Chem., 2016, 81, 12247–12257 CrossRef CAS PubMed; (d) Y. Kumar, M. Shaw, R. Thakur and A. Kumar, J. Org. Chem., 2016, 81, 6617–6625 CrossRef CAS PubMed; (e) R. Karmakar, P. Pahari and D. Mal, Chem. Rev., 2014, 114, 6213–6284 CrossRef CAS PubMed; (f) B. I. No, Yu. L. Zotov, E. V. Shishkin and D. S. Klimov, Russ. J. Gen. Chem., 2001, 71, 1807–1810 CrossRef CAS; (g) A. R. Katritzky, O. Meth-Cohn and C. W. Rees, Comprehensive Organic Functional Group Transformations, Elsevier Science Ltd, Pergamon, 1995, Vol. 6, p. 70 Search PubMed; (h) R. Roger and D. G. Neilson, Chem. Rev., 1961, 61, 179–211 CrossRef CAS; (i) R. Nandi, P. K. Mandal, A. Kayet, T. Bhattachariya, S. Ghosh and D. K. Maiti, Org. Lett., 2020, 20, 3474–3478 CrossRef PubMed.
- D. G. Nelson, The Chemistry of Amidines and Imidates, Wiley, 1975, pp. 385–489 Search PubMed.
- K. Popov and P. Somfai, J. Org. Chem., 2016, 81, 3470–3472 CrossRef CAS PubMed.
- M. Shaw and A. Kumar, Org. Lett., 2019, 21, 3108–3113 CrossRef CAS PubMed.
- D. G. Yu, M. Suri and F. Glorius, J. Am. Chem. Soc., 2013, 135, 8802–8805 CrossRef CAS PubMed.
- J. L. Sessler, C. M. Lawrence and J. Jayawickramarajah, Chem. Soc. Rev., 2007, 36, 314–325 RSC.
- V. Amendola, L. Fabbrizzi and L. Mosca, Chem. Soc. Rev., 2010, 39, 3889–3915 RSC.
- P. Molina, F. Zapata and A. Caballero, Chem. Rev., 2017, 117, 9907–9972 CrossRef CAS PubMed.
- S. A. Boer, E. M. Foyle, C. M. Thomas and N. G. White, Chem. Soc. Rev., 2019, 48, 2596–2614 RSC.
- Y. Wang, M. Yang, C. Laoa and Z. Jiang, Org. Lett., 2022, 24, 2625–2629 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Details experimental procedures and full spectroscopic data of all the new synthesised compounds are provided. CCDC 2152756. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3ob00165b |
| This journal is © The Royal Society of Chemistry 2023 |