
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Two-step conversion of uridine and cytidine to variously C5-C functionalized analogs†
Karolina
Podskoczyj
 ,
Anna
Klos
,
Szymon
Drewniak
and
Grazyna
Leszczynska
*
,
Anna
Klos
,
Szymon
Drewniak
and
Grazyna
Leszczynska
*
Institute of Organic Chemistry, Faculty of Chemistry, Lodz University of Technology, Zeromskiego 116, Lodz 90-924, Poland. E-mail: grazyna.leszczynska@p.lodz.pl; Tel: +48 426313150
First published on 9th March 2023
Abstract
C5-substituted pyrimidine nucleosides are an important class of molecules that have practical use as biological probes and pharmaceuticals. Herein we report an operationally simple protocol for C5-functionalization of uridine and cytidine via transformation of underexploited 5-trifluoromethyluridine or 5-trifluoromethylcytidine, respectively. The unique reactivity of the CF3 group in the aromatic ring allowed the direct incorporation of several distinct C5-C “carbon substituents”: carboxyl, nitrile, ester, amide, and amidine.
Introduction
Modified ribonucleosides constitute an exceptionally important class of biomolecules that are present in all three domains of life.1 To date, more than 150 modifications have been identified in RNA. About 100 of them have been identified in tRNAs, including C5-substituted cytidines and uridines. Most of them are located at the 34 position of the tRNA anticodon (known as the wobble position) and play a crucial role in the accuracy and efficiency of protein biosynthesis. Wobble nucleosides facilitate the third codon letter recognition, particularly in non-cognate codons by stabilizing the codon–anticodon interactions and modelling the ribosome-acceptable anticodon loop architecture.2–4 Recently, the regulatory and signalling role of RNA ribonucleosides has been discovered,4,5e.g. two epigenetic modifications, 5-methylcytidine (m5C) and 5-hydroxymethylcytidine (hm5C), and products of their oxidation, 5-formylcytidine (f5C) and 5-carboxycytidine (ca5C), were supposed to regulate the translation process.5–8 Notably, their shortage was associated with intellectual disabilities and cancer.6,9,10Due to their ubiquity, naturally modified nucleosides have found widespread applications as biological probes in biochemistry and medicine. For instance, fluorescently labelled m5C-oxidized products were investigated to find a more sensitive method for their identification.7 In addition, 5-substituted uridines were used as models to assess the structure–function relationships.11
Unnatural modified nucleosides are frequently evaluated as potential drugs in the treatment of existing and emerging viral and bacterial diseases and cancer.12–14 Incorporation of the C5-substituent in pyrimidine bases was found to increase drug bioavailability, biological activity, and/or stability under cellular conditions. Among others, 5-fluorine- and 5-CF3-substituted pyrimidine nucleosides are widely used drugs with antitumor and antiviral activities.14 Notably, the 5-CF3-dU nucleoside is an antiviral drug commercially known as trifluridine, approved by the Food and Drug Administration (FDA) for the treatment of epithelial keratitis caused by Herpes Simplex virus 1 and 2 (HSV-1 and HCV-2).
In the light of the foregoing discussion, it is important to develop straightforward and effective methods for the synthesis of natural and artificial modified nucleosides.
According to literature data, the incorporation of a “carbon substituent” at the C5 position of uridine or cytidine involved hydroxymethylation,15 aminomethylation by the Mannich reaction,16 Pd-catalyzed reaction of 5-iodo-nucleosides to introduce alkyl, alkenyl, alkynyl or aryl groups,13,17 reaction of a C5-lithiated nucleoside with an appropriate electrophile,18,19 substitution of 5-halo derivatives with the generated carboanion19,20 and radical malonylation induced by Mn(III) or Ce(IV) compounds.21 All these strategies require, however, the additional ribose protection and final deprotection steps, which are costly and time-consuming.
Herein, we present a facile, two-step approach to introduce several chemically diverse “carbon substituents” (carboxyl, nitrile, ester, amide, or amidine) at the C5 position of uridine (1a, Scheme 1) and cytidine (1b) nucleosides using underexploited 5-trifluoromethyluridine (5-CF3U, 2a) or 5-trifluoromethylcytidine (5-CF3C, 2b) precursors. The employed strategy does not require any protecting groups for regioselective C5-trifluoromethylation of pyrimidine ribonucleosides and the subsequent 5-CF3 conversion.
 | ||
| Scheme 1 Synthesis of 5-trifluoromethyluridine (5-CF3U, 2a) and 5-trifluoromethylcytidine (5-CF3C, 2b). | ||
Results and discussion
Synthesis of 5-trifluoromethyluridine (5-CF3U) and 5-trifluoromethylcytidine (5-CF3C)
Radical trifluoromethylation is a valuable method for the preparation of CF3-tagged aromatic heterocycles, in which the hydrogen atom bonded to the aromatic system is replaced with a trifluoromethyl group without prior substrate prefunctionalization. Pivotal progress in radical trifluoromethylation was made by Baran et al., who developed a transition metal-free approach based on two easy-to-handle, commercially available reactants: sodium trifluoromethanesulfinate (CF3SO2Na, known as the Langlois reagent) in combination with tert-butylhydroperoxide (t-BuOOH) as the radical source.22 This method was successfully applied to monotrifluoromethylation of 2′-deoxyuridine (dU) providing 5-CF3dU as the only regiomer in 57% yield.22Using the CF3SO2Na and t-BuOOH reagent system, several other CF3-containing nucleosides (mostly 2′-deoxy derivatives) have been successfully synthesized and evaluated as potential antiviral or antitumor drugs,23 biochemical probes for 19F NMR studies24 or substrates in the preparation of CF3-functionalized oligonucleotides.25 In case of the 5-CF3U and 5-CF3C ribonucleosides, two general methods based on the peroxide-generated CF3 radicals have been reported in the literature: trifluoromethylation of U/C with gaseous CF3I in the presence of the Fenton oxidation reagent (Fe2+/H2O2/H2SO4; Y = 11–53%)26 or photoinduced reaction with e.g. trifluoromethyl sulfones or the Langlois reagent as the source of CF3 radicals (Y = 38–42%).27
In our studies, 5-CF3-uridine (2a, Scheme 1) and 5-CF3-cytidine (2b) were synthesized under metal-free conditions, using an excess of CF3SO2Na (3 equiv.) and 70% aqueous (aq.) solution of t-BuOOH (5 equiv.). The reaction mixture was left for 3–4 h at room temperature. Products 2a and 2b were isolated by column chromatography with 81% and 72% yields, respectively, and characterized by NMR and MS spectroscopy (ESI, Fig. S1–S6†). Since the starting material remained unreacted, the reaction conditions were optimized by extending the reaction time and/or increasing the excess of the reagents; however, no improvement in the substrate consumption was observed. Notably, when the trifluoromethylation of U/C was carried out using a 4-fold higher amount of the starting material (2 vs. 0.5 g) using the same reactant ratios and conditions as for the small-scale reaction, an ca. 10% decrease in the yields of CF3-nucleosides was observed, with similar recovery yields of the starting nucleosides.
Reactivity of 5-CF3U and 5-CF3C under alkaline conditions
In general, the CF3 group attached to the aromatic ring is stable and chemically inert under neutral conditions. The reactivity of the aromatic CF3 group rises rapidly in alkaline solutions at elevated temperature. The first conversions of the 5-CF3-2′-deoxyuridine nucleoside into 5-carboxy- or 5-cyano derivatives in heated aqueous sodium hydroxide or ammonia, respectively, were presented three decades ago.28Very recently, Ito et al. demonstrated the synthetic scope of 5-CF3dU and 5-CF3dC conversions at the oligonucleotide level.29 Since CF3-modified nucleosides in the ribo series have never been considered as precursors to introduce various C5-functional groups, we evaluated the reactivity of 5-CF3U (2a) and 5-CF3C (2b) with several nucleophiles, involving hydroxide and alkoxide ions, ammonia, and methylamine (Table 1). To select the most suitable conversion conditions, small-scale preliminary experiments were performed with CF3-uridine (2a) (ca. 150 μmol of the starting material). The developed reaction conditions were then used for 5-CF3C (2b) and optimized. To assess whether the method is reproducible and scalable, some of the 5-CF3U/C conversions were performed on a larger scale (1 g of 2a/2bvs. 0.15 g).
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Substrate | –X | Reagent | Time (h) | Product | Yield (%) |
| a Small-scale reaction yield (ca. 150 μmol of 2a/2b). | ||||||
| 1 | 2a |
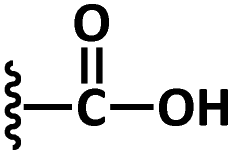
|
0.5 M aq. NaOH | 4 | 3a | 90 |
| 2 | 2b | 0.5 M aq. NaOH | 1 | 3b | 85 | |
| 3 | 2a |

|
(1) 50 mM K2CO3/MeOH | 72 | 4a | 90 |
| (2) Dowex H+ | ||||||
| 4 | 2b |

|
(1) 50 mM K2CO3/MeOH | 72 | 4b | 72 |
| (2) 0.5 M aq. HCl | ||||||
| 5 | 2a |

|
30% aq. NH3/EtOH (3/1 v/v) | 4 | 5a | 70 |
| 6 | 2b | 30% aq. NH3/EtOH (3/1 v/v) | 3 | 5b | 70 | |
| 7 | 2a |

|
4% aq. MeNH2 | 20 | 6a | 65 |
| 8 | 2a |

|
40% aq. MeNH2 | 3 | 7a | 82a |
| 9 | 2b |

|
4% aq. MeNH2 | 4 | 6b | 60 |
| 10 | 2b | 40% aq. MeNH2 | 2 | 6b | 65a | |

First, we investigated the alkaline hydrolysis of 5-CF3U (2a) into 5-carboxyuridine (ca5U, 3a, Table 1, entry 1). NaOH aq. solution at 60 °C was used since hydroxide anions were reported as the most effective reactants to convert CF3-containing aromatic heterocycles to COOH-functionalized derivatives.28–30 To optimize the reaction conditions, we tested the reactivity of 5-CF3U at various NaOH concentrations. When 20 mM aq. NaOH was used, we observed incomplete 5-CF3U conversion even after 20 h. A 5-fold increase in NaOH concentration improved the reaction yield (65% after 12 h), although an unconsumed substrate was still observed. With further increase in aq. NaOH concentration to 0.5 M, 90% yield was achieved and the reaction time was reduced to 4 h (Table 1, entry 1). We found these conditions of 2a hydrolysis to be optimal and used them for 5-CF3-cytidine (2b) conversion (Table 1, entry 2). In this case, the reaction took only 1 h, affording 5-carboxycytidine (3b) in 85% yield. It is worth noting that 5-carboxycytidine (ca5C) is a naturally existing modification identified in mRNA sequences as a product of m5C or hm5C epigenetic nucleoside oxidation.10 The biological function of ca5C is still elusive, although its regulatory role in the translation process has been speculated.7,10
5-CF3U was then exploited for conversion of the CF3 group to the methyl ester residue (–COOMe). Analysis of the literature revealed that sodium methoxide in methanol is a commonly used reagent for CF3-compound methanolysis.31,32 More recently, Markley et al. found that prolonged incubation of CF3dU (2 weeks, 37 °C) in a methanolic solution of K2CO3 leads to the 5-methoxycarbonyl derivative in a quantitative yield.33 We tested both conditions reported in the literature. The reaction of 2a with 50 mM solution of K2CO3 in dry MeOH heated to 60 °C afforded the ortho ester intermediate 5-(trimethoxymethyl)uridine (5-(MeO)3C-uridine) after 72 h (ESI, Fig. S10†). The reaction mixture was diluted with methanol and acidified with Dowex-H+, furnishing 5-methoxycarbonyluridine (4a) in 90% yield (Table 1, entry 3). In turn, the use of 75% MeONa methanolic solution at 60 °C turned out to be less effective, affording 5-methoxycarbonyluridine (4a) in 70% yield after 24 h.
The former conditions of methanolysis were established as optimal and used for 5-CF3-cytidine (2b) conversion. In this case, the reaction of 2b with 50 mM K2CO3 methanolic solution (72 h, 60 °C) resulted in the formation of the 5-(MeO)3C-cytidine orthoester derivative, stable under Dowex-H+ acidification conditions (ESI, Fig. S26†). Thus, in the next experiment, the ortho ester was in situ readily hydrolyzed with mild aq. acid (0.5 M aq. HCl, 12 h, rt), affording 5-methoxycarbonylcytidine (4b) in 72% yield (Table 1, entry 4). Notably, 5-methoxycarbonylcytidine (4b) can be considered as a COOH-protected synthon for the preparation of ca5C-phosphoramidite and then the ca5C-modified RNA fragment.19
Next, we tested the reactivity of 5-CF3U (2a) and 5-CF3C (2b) with ammonia. According to the literature, the ammonolysis of CF3-aromatic systems provided exclusively cyano derivatives; none of the primary amides was formed.28c,29,33 For instance, the incubation of 5-CF3dU with conc. NH3(aq.) for 16 h at 55 °C furnished 5-cyano-dU in 95% yield.33 The high conversion efficiency encouraged us to apply these conditions to 5-CF3U ammonolysis. The reaction was conducted in a sealed vial tube for 5 h at 60 °C, affording 5-cyanouridine (5a) in 70% yield. In the next experiment, the mixture of 30% aq. NH3 and EtOH (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v) was used at 60 °C (Table 1, entry 5). Full conversion of 2a was observed after 4 h (TLC analysis), affording product 5a in 70% yield. Since the latter conditions are commonly used during alkaline deprotection of RNA oligomers, this variant of ammonolysis was employed for the synthesis of 5-CN-cytidine (5b). The reaction of 5-CF3-cytidine 2b with 30% aq. NH3–EtOH (3:1, v/v) proceeded for 3 h at 60 °C to afford 5b in 70% yield (Table 1, entry 6).
:1, v/v) was used at 60 °C (Table 1, entry 5). Full conversion of 2a was observed after 4 h (TLC analysis), affording product 5a in 70% yield. Since the latter conditions are commonly used during alkaline deprotection of RNA oligomers, this variant of ammonolysis was employed for the synthesis of 5-CN-cytidine (5b). The reaction of 5-CF3-cytidine 2b with 30% aq. NH3–EtOH (3:1, v/v) proceeded for 3 h at 60 °C to afford 5b in 70% yield (Table 1, entry 6).
Finally, we performed several trials of 5-CF3U aminolysis with methylamine as a nucleophilic reagent. Ito et al. previously reported the reactivity of 5-CF3dU- and 5-CF3dC-DNA with 2–4% aq. methylamine or butylamine, demonstrating the formation of primary amides or symmetrical amidine compounds.29 It is beneficial that the applied methodology of amide bond formation does not require any coupling reagent. In our studies, 4% and 40% aq. MeNH2 solutions were tested. Incubation of 2a with 4% aq. MeNH2 at 60 °C for 20 h (Table 1, entry 7) resulted in the full conversion of the CF3 group to N-methylcarbamoyl C(O)NHMe (product 6a) in 65% yield. The use of a 10-fold higher MeNH2 concentration for 3 h led to the formation of 7a containing the N,N′-dimethylamidinyl group in 82% yield (Table 1, entry 8). The TLC-controlled analysis of the reaction clearly showed that the amidinyl residue is formed via the amide intermediate. Interestingly, the reaction of 5-CF3-cytidine (2b) with 4% aq. MeNH2 (4 h, 60 °C) or 40% aq. MeNH2 (2 h, 60 °C) afforded exclusively the amide-type compound 6b in ca. 60% yields (Table 1, entries 9 and 10). The extension of the reaction time to 20 h did not change the chemical status of the amide product. In the past, Ito et al. reported a similar divergence between the reactivity of 5-CF3dU and 5-CF3-dC with MeNH2.29 Based on the DFT calculations, the authors showed that the imine carbon in the amidinyl residue of cytosine is more sensitive to water attack leading to amide group formation than in the case of amidinyl-uridine.
Conclusions
Compared to the multi-step and complex procedures reported in the literature for C5-C functionalization of uridine and cytidine,15–21 the approach described herein involves only two reactions and stable, easy to handle commercially available reactants. The regioselectively obtained 5-trifluoromethylated uridines and cytidines (5-CF3U, 2a and 5-CF3C, 2b) proved to be useful substrates for a rapid, effective (Y = 60–90%) and scalable process of the C5-functionality with the carboxyl, nitrile, ester, amide, or amidine group.Experimental section
General methods
All solid compounds were dried under a high vacuum prior to use. The reaction progress was monitored by analytical thin layer chromatography (TLC) on silica gel-coated plates (60F254). Column chromatography was performed on silica gel 60 (230–400 mesh, Fluka). NMR spectra were recorded on a Bruker Avance II Plus 700 spectrometer at 700 (for 1H) and 176 (for 13C) MHz or on a JEOL 400 spectrometer at 376 (for 19F) MHz. Chemical shifts (δ) are reported in ppm relative to TMS (internal standard) for 1H and 13C. Multiplicities are described as s (singlet), d (doublet), dd (doublet of doublets), dt (doublet of triplets), q (quartet) and m (multiplet). Coupling constants (J) are reported in hertz. IR spectra were recorded using an FTIR ALPHA instrument (Bruker) equipped with a Platinum ATR QuickSnap module in the range of 4000–400 cm−1. High-resolution mass spectrometry (HRMS) measurements were performed using a Synapt G2Si mass spectrometer (Waters) equipped with an ESI source and a quadrupole-time-of-flight mass analyzer. The measurement was performed in the negative ion mode. The results of the measurements were processed using the MassLynx 4.1 software (Waters).Synthesis of 5-trifluoromethyluridine (2a)
Uridine 1a (0.50 g, 2.05 mmol) was dissolved in H2O (6.2 mL), and CF3SO2Na (0.96 g, 6.15 mmol) was added. After cooling the reaction mixture to 0 °C, t-BuOOH (70% in H2O, 1.34 mL, 10.25 mmol) was added dropwise; then the reaction mixture was warmed to rt and stirred for 3 h. The reaction mixture was concentrated under reduced pressure, and co-evaporated with anhydrous toluene (2 × 5 mL). The crude product was purified by column chromatography (silica gel, 0–12% MeOH in CHCl3) to give 5-trifluoromethyluridine 2a (0.52 g, 81%). TLC: Rf = 0.57 (CHCl3/MeOH, 8:2, v/v); 1H NMR (700 MHz, D2O) δ (ppm): 3.89 (dd, 1H, J = 2.7 Hz, J = 13.0 Hz, H5′′), 4.05 (dd, 1H, J = 2.5 Hz, J = 13.0 Hz, H5′), 4.18–4.22 (m, 1H, H4′), 4.29 (dd, 1H, J = 5.0 Hz, J = 7.2 Hz, H3′), 4.38 (dd, 1H, J = 2.5 Hz, J = 5.0 Hz, H2′), 5.93 (d, 1H, J = 2.5 Hz, H1′), 8.77 (s, 1H, H6); 13C NMR (176 MHz, D2O) δ (ppm): 59.35 (C5′), 68.26 (C3′), 74.34 (C2′), 83.76 (C4′), 90.39 (C1′), 104.36 (q, J = 33.26 Hz, C5), 122.15 (q, J = 268.75 Hz, CF3), 142.90 (q, J = 5.28 Hz, C6), 150.68 (C2), 161.54 (C4); 19F NMR (376 MHz, D2O) δ: −63.26; HRMS (ESI) calcd for C10H10N2O6F3 [M − H]− 311.0490, found 311.0496 (ESI, Fig. S1–S3†).
Synthesis of 5-trifluoromethylcytidine (2b)
Cytidine 1b (1 g, 4.11 mmol) was dissolved in H2O (12 mL); then CF3SO2Na (1.86 g, 12.33 mmol) was added and the solution was stirred at 0 °C. After 10 min, t-BuOOH (70% in H2O, 2.82 mL, 20.55 mmol) was added dropwise, and the reaction mixture was warmed to rt and stirred for 4 h. Then the reaction mixture was evaporated under reduced pressure and co-evaporated with anhydrous toluene (3 × 10 mL). The crude product was purified by column chromatography (silica gel, 0–18% MeOH in CHCl3) to give compound 2b as a yellow solid (0.92 g, 72%). TLC: Rf = 0.34 (CHCl3/MeOH, 8:2, v/v); 1H NMR (700 MHz, D2O) δ (ppm): 3.89 (dd, 1H, J = 2.8 Hz, J = 13.3 Hz, H5′′), 4.07 (dd, 1H, J = 2.8 Hz, J = 13.3 Hz, H5′), 4.19 (dt, 1H, J = 2.8 Hz, J = 8.0 Hz, H4′), 4.25 (dd, 1H, J = 4.9 Hz, J = 8.0 Hz, H3′), 4.32 (dd, 1H, J = 2.1 Hz, J = 4.9 Hz, H2′), 5.90 (d, 1H, J = 2.1 Hz, H1′), 8.76 (s, 1H, H6); 13C NMR (176 MHz, D2O) δ (ppm): 59.18 (C5′), 67.90 (C3′), 74.54 (C2′), 83.27 (C4′), 91.08 (C1′), 97.69 (q, J = 35.02 Hz, C5), 122.84 (q, J = 269.45 Hz, CF3), 143.43 (q, J = 6.16 Hz, C6), 156.27 (C2), 161.30 (C4); 19F NMR (376 MHz, D2O) δ: −62.70; HRMS (ESI) calcd for C10H11N3O5F3 [M − H]− 310.0651, found 310.0660 (ESI, Fig. S4–S6†).
Synthesis of 5-carboxyuridine (3a) and 5-carboxycytidine (3b)
Nucleoside 2a/2b (1 g, 3.2 mmol) was treated with 0.5 M aq. NaOH solution (100 mL) and incubated at 60 °C for 4 h (compound 2a) or 1 h (compound 2b). The reaction mixture was diluted with water and passed through cation exchange resin (Dowex 50WX2-100, H+ form). The fraction containing the nucleoside was concentrated under reduced pressure and lyophilized to give compound 3a in 90% yield. TLC: Rf = 0.16 (BuOH/H2O, 85:15, v/v); 1H NMR (700 MHz, D2O) δ (ppm): 3.89 (dd, 1H, J = 3.5 Hz, J = 12.6 Hz, H5′′), 4.05 (dd, 1H, J = 2.8 Hz, J = 13.3 Hz, H5′), 4.21–4.23 (m, 1H, H4′), 4.29 (dd, 1H, J = 5.1 Hz, J = 7.0 Hz, H3′), 4.41 (dd, 1H, J = 2.8 Hz, J = 5.1 Hz, H2′), 5.96 (d, 1H, J = 2.8 Hz, H1′), 9.08 (s, 1H, H6); 13C NMR (176 MHz, D2O) δ (ppm): 59.66 (C5′), 68.47 (C3′), 74.36 (C2′), 83.95 (C4′), 90.65 (C1′), 103.22 (C5), 149.20 (C6), 150.43 (C2), 164.33 (C4), 166.08 (COOH); HRMS (ESI) calcd for C10H11N2O8 [M − H]− 287.0515, found 287.0522; IR (ATR) cm−1: 3354 (O–H), 1712 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) (ESI, Fig. S7–S9†).
O) (ESI, Fig. S7–S9†).
Compound 3b was obtained in 85% yield. TLC: Rf = 0.38 (1) CHCl3/MeOH, 9:1, v/v, (2) EtOAc/Me2CO/AcOH/H2O, 5:3:1:1, v/v/v/v; 1H NMR (700 MHz, D2O) δ (ppm): 3.89 (dd, 1H, J = 3.5 Hz, J = 13.3 Hz, H5′′), 4.07 (dd, 1H, J = 2.8 Hz, J = 13.3 Hz, H5′), 4.22–4.24 (m, 1H, H4′), 4.28 (dd, 1H, J = 4.9 Hz, J = 7.7 Hz, H3′), 4.40 (dd, 1H, J = 2.8 Hz, J = 4.9 Hz, H2′), 5.95 (d, 1H, J = 2.8 Hz, H1′), 9.24 (s, 1H, H6); 13C NMR (176 MHz, D2O) δ (ppm): 62.33 (C5′), 71.11 (C3′), 76.92 (C2′), 86.63 (C4′), 93.44 (C1′), 102.72 (C5), 151.45 (C6), 151.65 (C2), 161.65 (C4), 169.82 (COOH); HRMS (ESI) calcd for C10H12N3O7 [M − H]− 286.0675, found 286.0684; IR (ATR) cm−1: 3226 (O–H), 1726 (CO) (ESI, Fig. S23–S25†).
Synthesis of 5-methoxycarbonyluridine (4a)
Nucleoside 2a (1 g, 3.2 mmol) was treated with 50 mM K2CO3/MeOH (140 mL) and the mixture was stirred for 72 h at 60 °C. The amount sufficient for spectral analysis was evaporated under reduced pressure and purified by column chromatography (silica gel, 0–15% MeOH in CHCl3) affording 5-(trimethoxymethyl)uridine. TLC: Rf = 0.33 (BuOH/AcOH/H2O, 5:1:4, v/v/v); 1H NMR (700 MHz, D2O) δ (ppm): 3.21 (s, 9H, 3 × O–CH3), 3.86 (m, 1H, J = 3.5 Hz, J = 12.6 Hz, H5′′), 4.01 (dd, 1H, J = 2.8 Hz, J = 12.6 Hz, H5′), 4.16–4.19 (m, 1H, H4′), 4.26 (dd, 1H, J = 4.9 Hz, J = 7.0 Hz, H3′), 4.37 (dd, 1H, J = 2.8 Hz, J = 4.9 Hz, H2′), 5.94 (d, 1H, J = 2.8 Hz, H1′), 8.50 (s, 1H, H6) (ESI, Fig. S10†). The residual part of the reaction was diluted with anhydrous methanol and acidified with cation exchange resin (Dowex 50WX2-100, H+ form). After resin filtration, the mixture was evaporated under reduced pressure and purified by column chromatography (silica gel, 0–15% MeOH in CHCl3) affording compound 4a in 90% yield. TLC: Rf = 0.41 (CHCl3/MeOH, 8:2, v/v), Rf = 0.2 (BuOH/AcOH/H2O, 5:1:4, v/v/v); 1H NMR (700 MHz, D2O) δ (ppm): 3.88–3.91 (m, 4H, O–CH3, H5′′), 4.07 (dd, 1H, J = 2.8 Hz, J = 13.0 Hz, H5′), 4.20–4.22 (m, 1H, H4′), 4.29–4.31 (m, 1H, H3′), 4.39–4.40 (m, 1H, H2′), 5.96 (d, 1H, J = 2.5 Hz, H1′), 9.10 (s, 1H, H6); 13C NMR (176 MHz, D2O) δ (ppm): 52.43 (CH3–O), 59.48 (C5′), 68.35 (C3′), 74.39 (C2′), 83.78 (C4′), 90.58 (C1′), 104.17 (C5), 149.13 (C6), 150.61 (C2), 162.15 (C4), 164.73 (O–CO); HRMS (ESI) calcd for C11H13N2O8 [M − H]− 301.0672, found 301.0680; IR (ATR) cm−1: 2946 (C–H), 1681 (CO), 1087 (C–O–C) (see ESI, Fig. S11–S13†).
Synthesis of 5-methoxycarbonylcytidine (4b)
Nucleoside 2b (1 g, 3.2 mmol) was treated with 50 mM K2CO3/MeOH (140 mL) and the mixture was stirred for 72 h at 60 °C. The amount sufficient for spectral analysis was evaporated under reduced pressure and purified by column chromatography (silica gel, 0–20% MeOH in CHCl3), affording 5-(trimethoxymethyl)cytidine. TLC: Rf = 0.43 (CHCl3/MeOH, 8:2, v/v); 1H NMR (700 MHz, D2O) δ (ppm): 3.23 (s, 9H, 3 × O–CH3), 3.84–3.88 (m, 1H, H5′′), 4.04 (dd, 1H, J = 2.8 Hz, J = 12.6 Hz, H5′), 4.16–4.19 (m, 1H, H4′), 4.24 (dd, 1H, J = 4.9 Hz, J = 7.7 Hz, H3′), 4.31 (dd, 1H, J = 2.8 Hz, J = 4.9 Hz, H2′), 5.92 (d, 1H, J = 2.8 Hz, H1′), 8.51 (s, 1H, H6) (see ESI, Fig. S26†). To the residual part of the reaction, 0.5 M aq. HCl was added dropwise. The mixture was stirred at rt for 12 h, concentrated under reduced pressure and purified by column chromatography (silica gel, 0–20% MeOH in CHCl3) to give compound 4b in 72% yield. TLC: Rf = 0.38 (CHCl3/MeOH, 8:2, v/v); 1H NMR (700 MHz, D2O) δ (ppm): 3.88–3.91 (m, 4H, O–CH3, H5′′), 4.08–4.10 (m, 1H, H5′), 4.18–4.20 (m, 1H, H4′), 4.24–4.27 (m, 1H, H3′), 4.31–4.33 (m, 1H, H2′), 5.91 (m, 1H, H1′), 9.21 (s, 1H, H6); 13C NMR (176 MHz, D2O) δ (ppm): 52.52 (CH3–O), 59.03 (C5′), 67.79 (C3′), 74.53 (C2′), 83.24 (C4′), 91.25 (C1′), 96.90 (C5), 149.32 (C6), 154.33 (C2), 162.53 (C4), 165.77 (O–CO); HRMS (ESI) calcd for C11H14N3O7 [M − H]− 300.0832, found 300.0842; IR (ATR) cm−1: 2940 (C–H), 1710 (CO), 1098 (C–O–C) (see ESI, Fig. S27–S29†).
Synthesis of 5-cyanouridine (5a) and 5-cyanocytidine (5b)
Nucleoside 2a/2b (1 g, 3.2 mmol) was treated with 30% aq. NH3–EtOH (140 mL, 3:1, v/v). The reaction mixture was stirred in a sealed vial tube at 60 °C for 4 h (compound 2a) or 3 h (compound 2b). Then the mixture was cooled to rt and evaporated under reduced pressure. Compound 5a was purified by column chromatography (silica gel, 0–10% MeOH in CHCl3) to obtain a white foam in 70% yield. TLC: Rf = 0.78 (H2O/EtOH/Me2CO/AcOEt, 0.5:1:1:6, v/v/v/v); 1H NMR (700 MHz, D2O) δ (ppm): 3.88 (dd, 1H, J = 3.5 Hz, J = 12.6, H5′′), 4.03 (dd, 1H, J = 2.1 Hz, J = 12.6 Hz, H5′), 4.19–4.21 (m, 1H, H4′), 4.24 (dd, 1H, J = 5.6 Hz, J = 7.0 Hz, H3′), 4.37 (dd, 1H, J = 2.8 Hz, J = 4.9 Hz, H2′), 5.89 (d, 1H, J = 2.8 Hz, H1′), 8.83 (s, 1H, H6); 13C NMR (176 MHz, D2O) δ (ppm): 59.90 (C5′), 68.48 (C3′), 74.27 (C2′), 84.08 (C4′), 89.01 (C5), 90.77 (C1′), 113.91 (CN), 150.09 (C2), 150.37 (C6), 162.40 (C4); HRMS (ESI) calcd for C10H10N3O6 [M − H]− 268.0570, found 268.0579; IR (ATR) cm−1: 2241 (C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N) (see ESI, Fig. S14–S16†).
N) (see ESI, Fig. S14–S16†).
Compound 5b was purified by column chromatography (silica gel, 0–12% MeOH in CHCl3) to obtain a white foam in 70% yield. TLC: Rf = 0.47 (H2O/EtOH/Me2CO/AcOEt, 0.5:1:1:6, v/v/v/v); 1H NMR (700 MHz, D2O) δ (ppm): 3.89 (dd, 1H, J = 3.5 Hz, J = 13.3 Hz, H5′′), 4.06 (dd, 1H, J = 2.1 Hz, J = 13.3 Hz, H5′), 4.20–4.21 (m, 2H, H3′, H4′), 4.31–4.32 (m, 1H, H2′), 5.86 (d, 1H, J = 2.1 Hz, H1′), 8.78 (s, 1H, H6); 13C NMR (176 MHz, D2O) δ (ppm): 59.84 (C5′), 68.26 (C3′), 74.47 (C2′), 81.17 (C5), 83.61 (C4′), 91.45 (C1′), 114.26 (CN), 150.66 (C6), 155.24 (C2), 163.31 (C4); HRMS (ESI) calcd for C10H11N4O5 [M − H]− 267.0729, found 267.0724; IR (ATR) cm−1: 2225 (CN) (see ESI, Fig. S30–S32†).
Synthesis of 5-N-methylcarbamoyluridine (6a) and 5-N-methylcarbamoylcytidine (6b)
Nucleoside 2a/2b (1 g, 3.2 mmol) was treated with 4% aq. MeNH2 solution (140 mL) and incubated in a sealed vial tube for 20 h at 60 °C. The reaction mixture was cooled to rt and evaporated under reduced pressure. The obtained residue was purified by column chromatography (silica gel, 0–30% MeOH in CHCl3) to give product 6a in 65% yield. TLC: Rf = 0.56 (BuOH/AcOH/H2O, 5:1:4, v/v/v); 1H NMR (700 MHz, D2O) δ: 2.93 (s, 3H, NH–C![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) 3), 3.87 (dd, 1H, J = 4.2 Hz, J = 12.6 Hz, H5′′), 4.01 (dd, 1H, J = 2.8 Hz, J = 12.6 Hz, H5′), 4.19–4.21 (m, 1H, H4′), 4.27–4.29 (m, 1H, H3′), 4.39 (dd, 1H, J = 3.5 Hz, J = 5.2 Hz, H2′), 5.97 (d, 1H, J = 3.5 Hz, H1′), 8.81 (s, 1H, H6); 13C NMR (176 MHz, D2O) δ (ppm): 25.86 (NH–CH3), 60.29 (C5′), 69.05 (C3′), 74.25 (C2′), 84.19 (C4′), 90.17 (C1′), 106.11 (C5), 146.24 (C6), 150.88 (C2), 163.89 (C4), 164.53 (CO–NH); HRMS (ESI) calcd for C11H14N3O7 [M − H]− 300.0832, found 300.0839; IR (ATR) cm−1: 3313 (N–H), 2936 (C–H), 1680 (CO) (see ESI, Fig. S17–S19†).
3), 3.87 (dd, 1H, J = 4.2 Hz, J = 12.6 Hz, H5′′), 4.01 (dd, 1H, J = 2.8 Hz, J = 12.6 Hz, H5′), 4.19–4.21 (m, 1H, H4′), 4.27–4.29 (m, 1H, H3′), 4.39 (dd, 1H, J = 3.5 Hz, J = 5.2 Hz, H2′), 5.97 (d, 1H, J = 3.5 Hz, H1′), 8.81 (s, 1H, H6); 13C NMR (176 MHz, D2O) δ (ppm): 25.86 (NH–CH3), 60.29 (C5′), 69.05 (C3′), 74.25 (C2′), 84.19 (C4′), 90.17 (C1′), 106.11 (C5), 146.24 (C6), 150.88 (C2), 163.89 (C4), 164.53 (CO–NH); HRMS (ESI) calcd for C11H14N3O7 [M − H]− 300.0832, found 300.0839; IR (ATR) cm−1: 3313 (N–H), 2936 (C–H), 1680 (CO) (see ESI, Fig. S17–S19†).
Compound 6b was purified by column chromatography (silica gel, 0–30% MeOH in CHCl3) in 60% yield. TLC: Rf = 0.36 (BuOH/AcOH/H2O, 5:1:4, v/v/v); 1H NMR (700 MHz, D2O) δ (ppm): 2.87 (s, 3H, C3–NH), 3.89 (dd, 1H, J = 3.5 Hz, J = 12.6 Hz, H5′′), 4.06 (dd, 1H, J = 2.8 Hz, J = 12.6 Hz, H5′), 4.18–4.20 (m, 1H, H4′), 4.27 (dd, 1H, J = 4.9 Hz, J = 7.7 Hz, H3′), 4.33 (dd, 1H, J = 2.8 Hz, J = 5.6 Hz, H2′), 5.88 (d, 1H, J = 2.1 Hz, H1′), 8.60 (s, 1H, H6); 13C NMR (176 MHz, D2O) δ (ppm): 26.04 (CH3–NH), 59.76 (C5′), 68.19 (C3′), 74.37 (C2′), 83.48 (C4′), 90.95 (C1′), 101.44 (C5), 143.70 (C6), 156.10 (C2), 163.55 (C4), 167.14 (CONH); HRMS (ESI) calcd for C11H15N4O6 [M − H]− 299.0992, found 299.0998; IR (ATR) cm−1: 3302 (N–H), 2925 (C–H), 1646 (CO) (see ESI, Fig. S33–S35†).
Synthesis of 5-N-methylcarbamoylcytidine (6b)
Nucleoside 2b (0.15 g, 0.48 mmol) was dissolved in 40% aq. MeNH2 solution (10 mL) and incubated in a sealed vial tube for 2 h at 60 °C. The reaction mixture was concentrated under reduced pressure. The crude residue was purified by column chromatography (silica gel, 0–30% MeOH in CHCl3) to give product 6b in 65% yield. Spectral data were identical to those described above for 6b.Synthesis of 5-(N,N′-dimethylamidinyl)uridine (7a)
Nucleoside 2a (0.15 g, 0.48 mmol) was treated with 40% aq. MeNH2 solution (21 mL) and incubated in a sealed vial tube for 3 h at 60 °C. The reaction mixture was cooled to rt and evaporated under reduced pressure. The obtained residue was purified by column chromatography (silica gel, CHCl3/MeOH/H2O, 65:25:4 v/v/v) to give product 7a in 82% yield. TLC: Rf = 0.13 (BuOH/AcOH/H2O, 5:1:4, v/v/v); 1H NMR (700 MHz, D2O) δ: 2.99 (s, 3H, NH–C3), 3.02 (s, 3H, N–CH3), 3.85 (dd, 1H, J = 2.8 Hz, J = 13.0 Hz, H5′′), 4.00 (dd, 1H, J = 2.8 Hz, J = 13.3 Hz, H5′), 4.16–4.20 (m, 1H, H4′), 4.25–4.27 (m, 1H, H3′), 4.36 (dd, 1H, J = 2.8 Hz, J = 4.9 Hz, H2′), 5.92 (d, 1H, J = 2.8 Hz, H1′), 8.46 (s, 1H, H6); 13C NMR (176 MHz, D2O) δ: 28.63 (NH–CH3), 30.91 (N–CH3), 59.91 (C5′), 68.56 (C3′), 74.29 (C2′), 83.80 (C4′), 90.49 (C1′), 103.74 (C5), 144.22 (C6), 148.29 (C2), 155.60 (C4), 160.18 (NH–CN); HRMS (ESI) calcd for C12H17N4O6 [M − H]− 313.1148, found 313.1153; IR (ATR) cm−1: 3201 (N–H), 2925 (C–H), 1644 (CN) (see ESI, Fig. S20–S22†).
Author contributions
G. L. designed and supervised the study; K. P. performed the experiments and characterization of novel compounds; A. K. and S. D. assisted with chemical work; K. P. prepared the initial draft including data presentation; and G. L. wrote the manuscript with input from all the authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by a grant UMO-2021/43/B/ST4/01570 from the National Science Centre to G. L. This paper was completed while the first author (K. P.) was a doctoral candidate in the Interdisciplinary Doctoral School at the Lodz University of Technology, Poland.References
- P. Boccaletto, F. Stefaniak, A. Ray, A. Cappannini, S. Mukherjee, E. Purta, M. Kurkowska, N. Shirvanizadeh, E. Destefanis, P. Groza, G. Avsar, A. Romitelli, P. Pir, E. Dassi, S. G. Conticello, F. Aguilo and J. M. Bujnicki, Nucleic Acids Res., 2022, 50, 3523 CrossRef PubMed.
- M. A. Machnicka, A. Olchowik, H. Grosjean and J. M. Bujnicki, RNA Biol., 2014, 11, 1619 CrossRef PubMed.
- T. Suzuki, Nat. Rev. Mol. Cell Biol., 2021, 22, 375 CrossRef CAS PubMed.
- M. Duechler, G. Leszczynska, E. Sochacka and B. Nawrot, Cell. Mol. Life Sci., 2016, 73, 3075 CrossRef CAS PubMed.
- L. Shen, C. X. Song, C. He and Y. Zhang, Annu. Rev. Biochem., 2014, 83, 585 CrossRef CAS.
- B. Delatte, F. Wang, L. V. Ngoc, E. Collignon, E. Bonvin, R. Deplus, E. Calonne, B. Hassabi, P. Putmans, S. Awe, C. Wetzel, J. Kreher, R. Soin, C. Creppe, P. A. Limbach, C. Gueydan, V. Kruys, A. Brehm, S. Minakhina, M. Defrance, R. Steward and F. Fuks, Science, 2016, 351, 282 CrossRef CAS.
- I. A. Roundtree, M. E. Evans, T. Pan and C. He, Cell, 2017, 169, 1187 CrossRef CAS.
- L. Fu, C. R. Guerrero, N. Zhong, N. J. Amato, Y. Liu, S. Liu, Q. Cai, D. Ji, S. G. Jin, L. J. Niedernhofer, G. P. Pfeifer, G. L. Xu and Y. Wang, J. Am. Chem. Soc., 2014, 136, 11582 CrossRef CAS PubMed.
- J. Mathlin, L. Le Pera and T. Colombo, Int. J. Mol. Sci., 2020, 21, 4684 CrossRef CAS PubMed.
- W. Huang, M. D. Lan, C. B. Qi, S. J. Zheng, S. Z. Wei, B. F. Yuan and Y. Q. Feng, Chem. Sci., 2016, 7, 5495 RSC.
- (a) G. Leszczynska, M. Cypryk, B. Gistynski, K. Sadowska, P. Herman, G. Bujacz, E. Lodyga-Chruscinska, E. Sochacka and B. Nawrot, Int. J. Mol. Sci., 2020, 21, 2882 CrossRef CAS PubMed; (b) E. Sochacka, E. Lodyga-Chruscinska, J. Pawlak, M. Cypryk, P. Bartos, K. Ebenryter-Olbinska, G. Leszczynska and B. Nawrot, Nucleic Acids Res., 2017, 45, 4825 CAS; (c) G. Leszczynska, K. Sadowska, P. Bartos, B. Nawrot and E. Sochacka, Eur. J. Org. Chem., 2016, 3482 CrossRef CAS.
- (a) M. Tardu, J. D. Jones, R. T. Kennedy, Q. Lin and K. S. Koutmou, ACS Chem. Biol., 2019, 14, 1403 CrossRef CAS PubMed; (b) W. Kozak, S. Demkowicz, M. Dasko, J. Rachon and J. Rak, Russ. Chem. Rev., 2020, 89, 281 CrossRef CAS; (c) D. A. Harki, J. D. Graci, J. E. Galarraga, W. J. Chain, C. E. Cameron and B. R. Peterson, J. Med. Chem., 2006, 49, 6166 CrossRef CAS.
- Z. Wen, S. H. Suzol, J. Peng, Y. Liang, R. Snoeck, G. Andrei, S. Liekens and S. F. Wnuk, Arch. Pharm., 2017, 350, e1700023 CrossRef PubMed.
- (a) M. Johar, T. Manning, D. Y. Kunimoto and R. Kumar, Bioorg. Med. Chem., 2005, 13, 6663 CrossRef CAS PubMed; (b) Y. F. Shealy, C. A. O'Dell, W. M. Shannon and G. Arnett, J. Med. Chem., 1983, 26, 156 CrossRef CAS PubMed; (c) J. A. Mekras, D. A. Boothman and S. B. Greer, Cancer Res., 1985, 11, 5270 Search PubMed.
- (a) K. H. Scheit, Chem. Ber., 1966, 99, 3884 CrossRef CAS; (b) K. Ikeda, S. Tanaka and Y. Mizuno, Chem. Pharm. Bull., 1975, 23, 2958 CrossRef CAS PubMed; (c) H. Sajiki, A. Yamada, K. Yasunaga, T. Tsunoda, M. F. A. Amer and K. Hirota, Nucleic Acids Symp. Ser., 2002, 2, 13 CrossRef CAS PubMed; (d) Q. Dai, C.-X. Song, T. Pan and C. He, J. Org. Chem., 2011, 76, 4182 CrossRef CAS PubMed.
- (a) S. S. Jones, C. B. Reese and A. Ubasawa, Synthesis, 1982, 259 CrossRef CAS; (b) C. B. Reese and Y. S. Sanghvi, J. Chem. Soc., Chem. Commun., 1984, 62 RSC; (c) R. J. Capon and J. K. MacLoed, J. Org. Chem., 1987, 52, 560 CrossRef.
- (a) G. J. Crouch and B. E. Eaton, Nucleosides Nucleotides, 1994, 13, 939 CrossRef CAS; (b) N. Karino, Y. Ueno and A. Matsuda, Nucleic Acids Res., 2001, 29, 2456 CrossRef CAS PubMed; (c) Y. Liang, J. Gloudeman and S. F. Wnuk, J. Org. Chem., 2014, 79, 4094 CrossRef CAS PubMed; (d) K. Podskoczyj, K. Kulik, J. Wasko, B. Nawrot, T. Suzuki and G. Leszczynska, Chem. Commun., 2021, 57, 12540 RSC.
- (a) H. Tanaka, H. Hayakawa, K. Obi and T. Miyasaka, Tetrahedron, 1986, 42, 4187 CrossRef CAS; (b) R. W. Armstrong, S. Gupta and F. Whelihan, Tetrahedron Lett., 1989, 30, 2057 CrossRef CAS; (c) B. Nawrot and A. Malkiewicz, Nucleosides Nucleotides, 1989, 8, 1499 CrossRef CAS.
- Y. Fu, Q. Dai, W. Zhang, J. Ren, T. Pan and C. He, Angew. Chem., Int. Ed., 2010, 49, 8885 CrossRef CAS PubMed.
- (a) N. Isenberg and C. Heidelberger, J. Med. Chem., 1967, 10, 970 CrossRef CAS PubMed; (b) R. Borowski, A. Dziergowska, E. Sochacka and G. Leszczynska, RSC Adv., 2019, 9, 40507 RSC.
- Y. H. Kim, D. H. Lee and S. G. Yang, Tetrahedron Lett., 1995, 36, 5027 CrossRef CAS.
- Y. Ji, T. Brueckl, R. D. Baxter, Y. Fujiwara, I. B. Seiple, S. Su, D. G. Blackmond and P. S. Baran, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 14411 CrossRef CAS PubMed.
- (a) D. Musumeci, C. Irace, R. Santamaria and D. Montesarchio, MedChemComm, 2013, 4, 1405 RSC; (b) J. Barletta and S. B. Greer, Antiviral Res., 1992, 1, 1 CrossRef PubMed.
- M. Chrominski, J. Kowalska and J. Jemielity, Curr. Protoc. Nucleic Acid Chem., 2020, 83, e118 CAS.
- (a) Y. Ito, M. Matsuo, T. Osawa and Y. Hari, Chem. Pharm. Bull., 2017, 65, 982 CrossRef CAS PubMed; (b) L. Beigelman, R. Pandey, V. K. Rajwanshi, D. B. Smith, L. M. Blatt, J. Hong and M. E. Fitzgerald, WIPO, WO2021119325A1, 2021 Search PubMed.
- (a) T. Yamakawa, K. Yamamoto, D. Uraguchi and K. Tokuhisa, WIPO, WO2007055170A1, 2007 Search PubMed; (b) D. Uraguchi, K. Yamamoto, Y. Ohtsuka, K. Tokuhisa and T. Yamakawa, Appl. Catal., A, 2008, 342, 137–143 CrossRef CAS; (c) T. Yamakawa, K. Yamamoto, D. Uraguchi and K. Tokuhisa, US Pat, US7884202B2, 2011 Search PubMed; (d) T. Yamakawa, K. Yamamoto, D. Uraguchi and K. Tokuhisa, European Pat, EP1947092B1, 2015 Search PubMed.
- (a) L. Li, X. Mu, W. Liu, Y. Wang, Z. Mi and C.-J. Li, J. Am. Chem. Soc., 2016, 138, 5809 CrossRef CAS PubMed; (b) P. Liu, W. Liu and C.-J. Li, J. Am. Chem. Soc., 2017, 139, 14315–14321 CrossRef CAS PubMed; (c) I. Ghosh, J. Khamrai, A. Savateev, N. Shlapakov, M. Antonietti and B. König, Science, 2019, 365, 360 CrossRef CAS PubMed.
- (a) V. Guerniou, D. Gasparutto, S. Sauvaigo, A. Favier and J. Cadet, Nucleosides, Nucleotides Nucleic Acids, 2003, 22, 1073 CrossRef CAS PubMed; (b) M. J. Jones, J. Pharm. Pharmacol., 1981, 33, 274 CrossRef CAS PubMed; (c) B. S. Ross, R. H. Springer, G. Vasquez, R. S. Andrews, P. D. Cook and O. L. Acevedo, J. Heterocycl. Chem., 1994, 31, 765 CrossRef CAS.
- Y. Ito, M. Matsuo, K. Yamamoto, W. Yamashita, T. Osawa and Y. Hari, Tetrahedron, 2018, 74, 6854 CrossRef CAS.
- (a) R. G. Jones, J. Am. Chem. Soc., 1947, 69, 2346 CrossRef CAS PubMed; (b) H. J. Nestler and E. R. Garrett, J. Pharm. Sci., 1968, 57, 1117 CrossRef CAS PubMed; (c) Y. Kobayashi and I. Kumadaki, Acc. Chem. Res., 1978, 11, 197 CrossRef CAS.
- K. J. Ryan, E. M. Acton and L. Goodman, J. Org. Chem., 1966, 31, 1181 CrossRef CAS PubMed.
- (a) R. Mechoulam, S. Cohen and A. Kaluszyner, J. Org. Chem., 1956, 21, 801 CrossRef CAS; (b) K. Ramig, M. Englander, F. Kallashi, L. Livchits and J. Zhou, Tetrahedron Lett., 2002, 43, 7731 CrossRef CAS.
- J. C. Markley, P. Chirakul, D. Sologub and S. Th. Sigurdsson, Bioorg. Med. Chem. Lett., 2001, 11, 2453 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Spectral characterization of nucleosides 2a–7a and 2b–6b. See DOI: https://doi.org/10.1039/d3ob00161j |
| This journal is © The Royal Society of Chemistry 2023 |