
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis and applications of the sulfur containing analogues of cyclic carbonates
Carlos
Díez-Poza†
 ,
Lucía
Álvarez-Miguel†
,
Marta E. G.
Mosquera
* and
Christopher J.
Whiteoak
*
,
Lucía
Álvarez-Miguel†
,
Marta E. G.
Mosquera
* and
Christopher J.
Whiteoak
*
Universidad de Alcalá, Grupo SOSCATCOM, Departamento de Química Orgánica y Química Inorgánica, Facultad de Farmacia and Instituto de Investigación Química Andrés M. del Río (IQAR), Campus Universitario, Ctra. Madrid-Barcelona Km. 33600, 28871 Alcalá de Henares, Madrid, Spain. E-mail: christopher.whiteoak@uah.es; martaeg.mosquera@uah.es
First published on 31st March 2023
Abstract
Cyclic thiocarbonates are the sulfur containing analogues of the well-studied cyclic carbonates and are relatively poorly explored despite their potential applications and intriguing reactivities. To date, application of these organosulfur compounds has included their use as monomers for polythiocarbonate synthesis (their ring-opening is more readily achieved and more selective than the corresponding cyclic carbonates) and as reactive intermediates for the preparation of a range of higher-value sulfur containing compounds. Despite these uses, the synthesis of these compounds is far less explored and developed than their non-sulfur analogues. Here, we provide an overview of the state-of-the-art, both recent and historical, for the synthesis of a range of cyclic mono-, di- and tri-thiocarbonates (both five and six-membered rings), with selected examples of their reported applications also highlighted.
 Carlos Díez-Poza | Carlos Díez-Poza obtained his PhD from the University of Valladolid (2021), where he studied the use of organosilanes for the synthesis of heterocycles with potential biological properties, under the direction of Prof. Asunción Barbero. He had short stays in the group of Prof. Till Opatz (University of Mainz) and in the University of Ghent under the supervision of Prof. Matthias D'hooghe. He is now a postdoctoral researcher at the University of Alcalá, working in the SOSCATCOM research group, on the synthesis of non-isocyanate polyurethanes from renewable feedstocks. His research interests include sustainable chemistry, catalysis, heterocycle synthesis and organosilicon chemistry. |
 Lucía Álvarez-Miguel | Dr Lucía Álvarez Miguel obtained her PhD from University of Vallladolid (Spain) in 2018 (Supervisors; Prof. D. Miguel and Dr R. García). Thereafter, she joined the group of Dr F. Gloaguen as postdoctoral researcher at CNRS-UBO (Brest, France) developing new active electrocatalytic materials based on transition metal complexes. In October 2020, she moved to the SOSCATCOM group where her work focuses on the catalytic conversion of CO2 and bio-derived epoxides to form cyclic and polycarbonates. Her research interests also focus on the catalytic formation of cyclic thiocarbonates from the reaction of CS2/epoxides. |
 Marta E. G. Mosquera | Marta E. G. Mosquera is a full Professor in Inorganic Chemistry and co-leader of the SOSCATCOM research group (https://soscatcom.es/) at the Universidad de Alcalá. She obtained her PhD degree in Chemistry from the University of Oviedo. After her PhD, she moved to Cambridge University to join the group of Prof. D. S. Wright. In 2003 she moved to the Universidad de Alcalá where she has developed a research line focused on the synthesis and characterization of organometallic compounds based on group 13 and alkali metals, and the study of their activity in catalytic polymerization processes to achieve functionalized polymers and bioplastics. |
 Christopher J. Whiteoak | Christopher J. Whiteoak obtained his PhD from Imperial College London under the supervision of Prof. Vernon C. Gibson and George J. P. Britovsek. After postdoctoral positions in France (Dr Christophe Boisson; CPE-Lyon) and Spain (Prof. Arjan W. Kleij; ICIQ and Dr Xavi Ribas; Universitat de Girona), he was appointed as Lecturer and later, Senior Lecturer at Sheffield Hallam University (UK). In 2020 he joined the SOSCATCOM group at the Universidad de Alcala (Spain), where he currently holds a prestigious Atracción de Talento fellowship (Comunidad de Madrid). His research interests focus on the synthesis of heterocyclic compounds and bio-derived polymers. |
1. Introduction
The development of routes for the preparation of heterocyclic compounds is of significant interest to the synthetic chemistry community. This is predominately due to their prevalence in active pharmaceutical and agrochemical molecules, and also their potential for use as monomers in polymer synthesis, amongst many other applications. Organosulfur compounds are key molecules in both biology and many fields of chemistry. Whilst nature relies on a host of organosulfur compounds for essential conversions, they are also attracting increasing attention in chemistry for a range of applications. For example, many researchers are now focusing on their use as components in rechargeable lithium batteries due to their unique properties.1 As a result, it is clear that synthesis of novel, and improved access to known, organosulfur compounds could have significant impact on many fields of chemistry.Cyclic thiocarbonates, analogues of the well-known cyclic carbonates, are heterocycles which are typically five- or six-membered rings containing an incorporated carbonate functionality whereby one, or more, of the oxygen atoms has been replaced with a sulfur atom (Fig. 1, top and middle). Despite their widespread potential applications, they remain remarkably less well studied compared to the analogous cyclic carbonates. This difference in attention most likely results from the discovery that the traditional method for preparing cyclic carbonates from diols and toxic phosgene (Fig. 1, bottom, left) could be replaced by an atom-efficient reaction between an epoxide and carbon dioxide (CO2) (Fig. 1, bottom, right). This latter and more attractive process is usually carried out in the presence of a catalyst and to date a large number of compounds have been studied as catalysts for this conversion, both metal and organocatalysts.2 However, the field of cyclic thiocarbonate synthesis has been somewhat left behind, despite the interesting potential of these compounds.
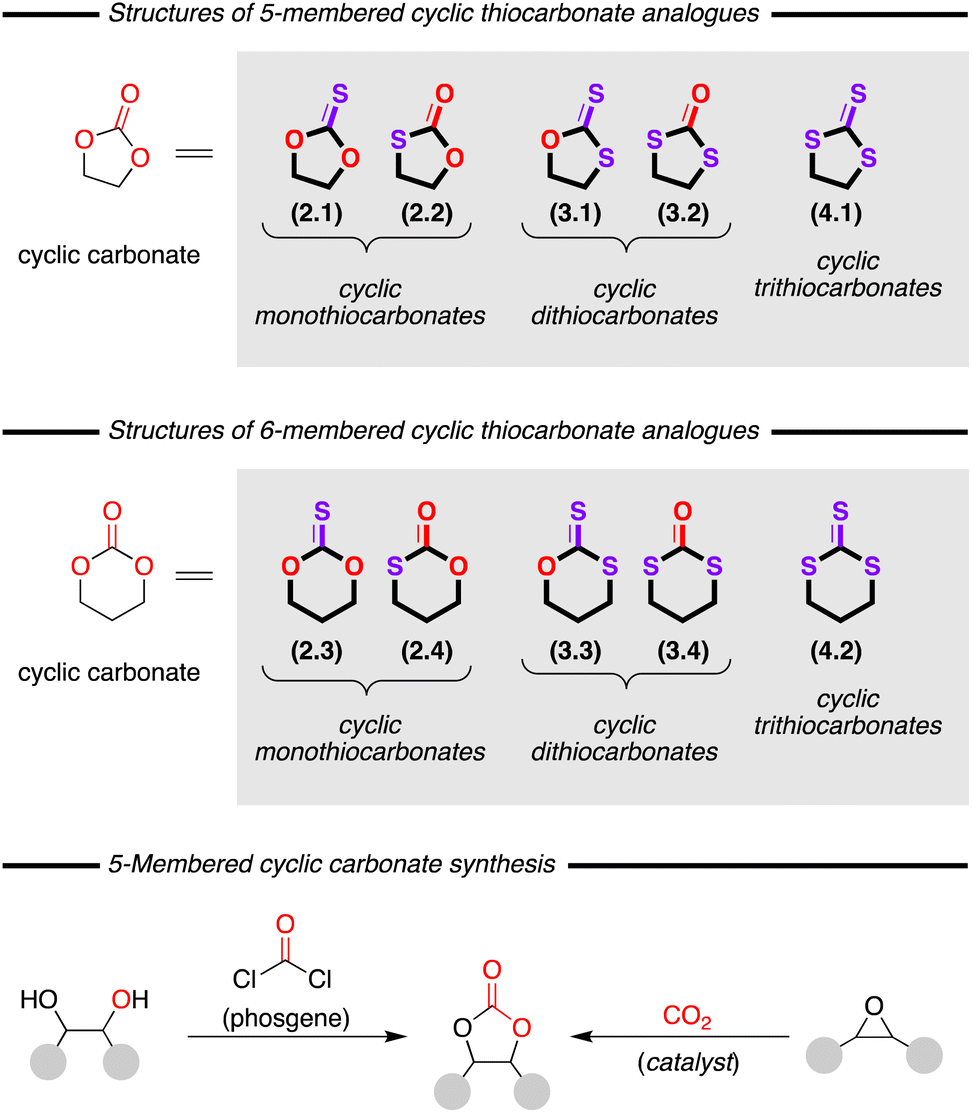 | ||
| Fig. 1 Top: structures of 5-membered cyclic thiocarbonates. Middle: structures of 6-membered cyclic thiocarbonates. Bottom: methods for synthesis of related cyclic carbonates. Note: numbers in parenthesis indicate the sections of this review where the compound is discussed. | ||
It should be noted that the synthesis of some cyclic thiocarbonates can be achieved using carbon disulfide (CS2). This is a toxic compound, mainly produced during the manufacture of viscose fibres and cellophane films.3 Having a relatively low boiling point of 46 °C, it is often inadvertently released into the atmosphere during these manufacturing processes, whereby it is converted into COS and SO2 which have well known negative environmental impacts. As a result, just like CO2, should these emitted CS2 gases have value/application, it would most likely result in an increased interest in their capture.
Several cyclic thiocarbonate analogues can be prepared; those including one sulfur atom (cyclic monothiocarbonates), two sulfur atoms (cyclic dithiocarbonates), and also those containing three sulfur atoms (cyclic trithiocarbonates). Obviously, this diversity has led to the requirement for the development of a wide range of protocols for their synthesis and herein within this review, we will attempt to provide an overview of the state-of-the-art concerning the synthesis and applications of the whole cyclic thiocarbonate family of compounds (Fig. 1, top and middle).
As has been mentioned above, cyclic thiocarbonates have been applied as monomers in Ring-Opening Polymerisation (ROP). Very recently, in 2023, Buchard and co-workers have systematically exchanged oxygen atoms for sulfur atoms in cyclic carbonates and studied its effect.4 The authors analysed a series of novel cyclic carbonate and thiocarbonate monomers derived from carbohydrate derivative D-glucal, in which the number and position of the sulfur atoms in the 6-membered ring was varied. All the monomers could be readily converted to the corresponding poly(thio)carbonate polymers, however, the different monomers presented distinct rates of polymerisation, arising from the differences in ring strain of the heterocycle. Introducing a sulfur atom inside the ring decreased the ring strain and leads to a decrease in the rate of polymerisation. It should be noted that as a result of the incorporation of sulfur into the polymer, the degradability of the linkages under UV light was improved, while physical properties, such as glass transition temperature and thermal degradation were decreased. These results clearly demonstrate both important reactivity and property differences between cyclic carbonates and cyclic thiocarbonates and their polymeric products.
In the following sections, methods for the synthesis of the cyclic thiocarbonate compounds will be presented, along with the mention of selected applications and interesting synthetic conversions that can be readily achieved when using them as chemical intermediates.
2. Cyclic monothiocarbonates
The first part of the review presents routes to the synthesis of cyclic thiocarbonates (both five- and six-membered; see Fig. 1) which contain a single S-atom and highlights some selected synthetic applications of these compounds.2.1. Synthesis and applications of 1,3-dioxolane-2-thione
Early reports of the synthesis of 1,3-dioxolane-2-thione came in the early 1960's from a publication by Corey/Winter.5 In this work, the authors presented two distinct approaches for the synthesis of the cyclic thiocarbonate (Fig. 2a). The first approach involved the reaction of diols with 1,1′-thiocarbonyldiimidazole (TCDI), which was inspired by the previous work of Staab/Walther, published the previous year, using other alcohols and phenols.6 The second approach proceeded through the reaction of a diol, first with CS2, and thereafter with nBuLi and MeI. Complete conversions were reported for the first route, but it was disclosed that the second route only provided a modest 50% yield. Despite being an early example of the synthesis of 1,3-dioxolane-2-thiones, the paper was actually focused on the conversion of these cyclic thiocarbonate to olefins through the reaction with trimethyl phosphite. In this context it is important to note that the original stereochemistry of the starting diol is presented in both the cyclic thiocarbonate and the final olefinic product. | ||
| Fig. 2 (a) Seminal synthesis from diols using two different approaches. (b) Reaction of tin dialkoxides with CS2. (c) Preparation through the reaction of germylated dioxolanes with thiophosgene. | ||
Beyond the use of simple diols there has been application of various non-conventional precursors. In this context, in 1970, Sakai and co-workers demonstrated that the reaction between linear tin dialkoxides and CS2 could also furnish the 1,3-dioxolane-2-thione products. In this work selectivity towards the cyclic thiocarbonate or a spirocyclic product was heavily dependent on the nature of the connecting R-group (Fig. 2b).7 Interestingly, in subsequent work the same research group showed that linear tin alkoxides could also afford the cyclic thiocarbonate. Meanwhile, cyclic dibutyltin dialkoxide starting materials resulted in the spiro-orthocarbonates, with no formation of the desired product.8 Later, Dousse and co-workers reported another synthesis involving the reaction of germane compounds and thionyl chloride (SOCl2; Fig. 2c).9,10 The reaction proceeds from germylated dioxolanes, which readily undergo oxygen/chlorine exchange under mild conditions (0 to 20 °C) in benzene or pentane.
Returning to the use of diols, in 1969 Jones reported a synthesis whereby a diol and thiophosgene were reacted together in the presence of K2CO3 as base (Fig. 3a(i)).11 The optimal method was reported to be addition of ethylene glycol to a boiling mixture of thiophosgene, dichloromethane and K2CO3. However, this procedure only furnished a relatively low yield of 33%. The compound was observed to be thermally unstable, decomposing appreciably at 100 °C, possibly explaining the low yield obtained. The cyclic thiocarbonate product was found to be reactive towards acids, bases and moisture. Interestingly, in terms of stability, it was found that halide ions isomerised the compound to form the more stable 1,3-oxathiolan-2-one. These highly reactive compounds were also found to undergo a range of synthetic conversions for upgrading to potentially useful compounds (Fig. 3b); reaction with alkyl halides allowed access to a range of unsymmetrical β-haloethyl thiolcarbonates. However, this conversion failed with unreactive halides like tert-butyl bromide (resulting in undesired polymerisation products). ROP was achieved under strongly acidic conditions (trifluoroacetic acid, TFA) at 25 °C, where notably the polymers obtained were found to be unstable above 200 °C. Finally, reaction with PPh3 at 60 °C produced CO2 and ethylene as decomposition products, a similar result to that observed earlier by Corey/Winter using trimethyl phosphite.5
 | ||
| Fig. 3 (a) Distinct synthetic approaches to the conversion of diols to form 1,3-dioxolane-2-thiones. (b) Examples of general-type conversions. (c) Cyclic thiocarbonate derivative as a key reaction intermediate for formation of glycofuranosyl azides and nucleosides. (d) Use of dicyclic thiocarbonates as monomers for non-isocyanate polythiourethane (NIPTU) synthesis. (e) Ring-Opening Polymerisation (ROP) of a functionalised 1,3-dioxolane-2-thione. | ||
Sometime later, in the mid 1980's, Kim and co-workers described the use of 1,1′-thiocarbonyldi-2(1H)-pyridone as reagent, which could be conveniently obtained directly from a rearrangement of di-2-pyridyl thionocarbonate after refluxing for 12 hours in toluene (Fig. 3a(ii)).12,13 With this novel reagent, cyclic thiocarbonates could be prepared, amongst other compounds like nitriles, carbodiimides and isothiocyanates. Starting from diols in refluxing toluene, the dipyridone acted as a thiocarbonyl transfer agent and afforded the desired products in excellent yields (89–93%). Some limitations were observed however, whereby the protocol gave good yields for monomethyl and monoaryl diols, however, this approach failed for tetramethylated diols. In some cases (R = Me, Ph), the addition of 0.1 equiv. of DMAP and changing solvent to dichloromethane at room temperature resulted in improved yields. This method provided significantly improved results and under much shorter reaction times compared to the use of thiophosgene reported by Jones (33% vs. 93%, respectively).
Since the 1980's the field has returned to relative dormancy. However, very recently, Qaroush and co-workers reported on the application of the Mukaiyama reagent (2-chloro-1-methylpyridinium iodide; CMPI) and triethylamine to produce 1,3-dioxolane-2-thiones from diols (Fig. 3a(iii)).14 This reaction was achieved at room temperature using CS2 as the S-source. The work was related to the results in the report by Massi/Dondoni and co-worker who previously reacted CS2 with monofunctionalised amines in the presence of Et3N and CMPI to prepare isothiocyanates.15
Several other notable reactions with 1,3-dioxolane-2-thiones have been studied. Robles and co-workers reported the ring-opening of these compounds for the synthesis of glycofuranosyl azides and nucleosides (Fig. 3c).16,17 Meanwhile, Endo and co-workers studied the synthesis of polyhydroxythiourethanes through polyaddition of a bis(cyclic thiocarbonate) species with a diamine (Fig. 3d).18 In this work the authors observed a higher polymerisation rate of the cyclic thiocarbonates compared to that of fully analogous cyclic carbonates. Finally, Endo and co-workers have studied the ROP of a 1,3-dioxolane-2-thione functionalised with an ester group (Fig. 3e). In this report, the corresponding polythiocarbonates were formed, with the final polymer structure influenced by an interesting effect of the neighbouring ester group, which will be discussed later in this review.19
2.2. Synthesis and applications of 1,3-oxathiolan-2-ones
As long ago as 1961, Reynolds and co-workers reported a straightforward synthesis of 1,3-oxathiolan-2-one. through reaction of a thiolcarbonate with p-toluenesulfonic acid (pTSA) in refluxing benzene (Fig. 4a).20 This procedure yielded the desired product in an attractive 70 to 77% yield. However, during this intramolecular transesterification reaction, small amounts of the rearranged thiol and polyethylene sulfide were formed. Reaction optimisation studies indicated that prolonged reaction times favoured the polymerisation of the thiol by-product, whilst maintaining the yield of the intended cyclic compound. | ||
| Fig. 4 (a) Intramolecular cyclisation of a thiolcarbonate to furnish 1,3-oxathiolan-2-one. (b) Ni-catalysed carbonylation of 2-mercaptoethanol. (c) Se-catalysed carbonylation of a diol thioether. | ||
Some years later, in 1974, Koch and co-worker described a rather distinct approach, applying a nickel-catalysed carbonylation of 2-mercaptoethanol (Fig. 4b).21 In this work, reaction of 2-mercaptoethanol with CO and O2 (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 at a pressure of 3 atm) in pyridine in the presence of a catalytic amount of Ni(CO)4, afforded the desired cyclic thiocarbonate product in 60% yield. A bis(mercaptoethanol)nickel(II) species was postulated as the key reaction intermediate after the authors successfully isolated this compound. In the same year, Koch also reported on a related selenium-catalysed carbonylation of a 2,2′-thiobis(ethan-1-ol) compound (Fig. 4c).22 Reaction of this diol thioether with a mixture of CO/O2 (10:1) with amorphous selenium as catalyst and Et3N as co-catalyst, over 20 hours afforded a 60% yield of the desired product. Since these reports little or no attention has been paid to the synthesis of these compounds, despite their interesting structures.
:2 at a pressure of 3 atm) in pyridine in the presence of a catalytic amount of Ni(CO)4, afforded the desired cyclic thiocarbonate product in 60% yield. A bis(mercaptoethanol)nickel(II) species was postulated as the key reaction intermediate after the authors successfully isolated this compound. In the same year, Koch also reported on a related selenium-catalysed carbonylation of a 2,2′-thiobis(ethan-1-ol) compound (Fig. 4c).22 Reaction of this diol thioether with a mixture of CO/O2 (10:1) with amorphous selenium as catalyst and Et3N as co-catalyst, over 20 hours afforded a 60% yield of the desired product. Since these reports little or no attention has been paid to the synthesis of these compounds, despite their interesting structures.
Even though there are relatively few examples of the preparation of this class of compound, they have found many applications. Various reports by Reynolds and co-workers have demonstrated how they can be further reacted (Fig. 5a). In one example, it is demonstrated how they can be readily decarboxylated to form ethylene sulfide when heated.23 Meanwhile, other synthetic transformations have also been studied; their reaction with amines results in mercaptoethylation,24 mercaptoethyl carboxylation,25 or polythioether formation25 depending on the reaction conditions applied.
 | ||
| Fig. 5 (a) Examples of conversions of 1,3-oxathiolan-2-one at elevated temperature or in the presence of amines. (b) Direct Ring-Opening Polymerisation (ROP) using a Ti-catalyst. (c) Polymerisation after the in situ formation of the cyclic thiocarbonate monomer. | ||
Similarly to most other cyclic thiocarbonates, 1,3-oxathiolan-2-ones have been applied as monomers in polymer chemistry. ROP of ethylene monothiocarbonate was originally reported by Soga and co-workers in 1975 (Fig. 5b).26 However, more recent methods for the preparation of these polymers prefer direct co-polymerisation of carbonyl sulfide (COS) with epoxides (Fig. 5c) as has been reported, for example, by Zhang/Darensbourg and co-worker,27 Ren and co-workers28 and most recently in 2022 by Zhang/Zhang and co-workers.29,30 In 2022, Coates and co-workers provided a good overview of this field of research in a review on multifunctional catalysts for ring-opening copolymerisations.31
In more detail, in the former two examples mentioned above, it is proposed that the catalysts produce the intermediate thiocarbonate in situ, which is then subsequently polymerised by ROP under the reaction conditions. This methodology has provided access to polythiocarbonates with an appealing perfectly alternating nature (regular structure) and with high molecular weights (Mw). Indeed, the work of Zhang/Zhang details the conversion of cyclic carbonates/COS to polythiocarbonates passing through the cyclic thiocarbonate intermediate compound.29 As an interesting extension, in the latter work by Zhang/Zhang,30 the authors reported terpolymerisation of propylene oxide, CO2 and COS. The terpolymers obtained exhibited a high refractive index of up to 1.55, and as a result the authors proposed applications in the field of optical materials. Upon varying the CO2 and COS feed ratios, the content of thiocarbonate units in the copolymer chain can be tuned from 27% to 81%.
The relatively safe nature of these compounds has led to the concept of thiocarbonate release in functional materials, for example, polymers that can be degraded under reducing conditions (Gillies and co-workers; 2010 and 2012).32,33 In these examples a disulfide bond is reduced, and the free thiol formed can thereafter cleave a neighbouring carbonate, thiocarbonate or urethane moiety, which breaks the parent polymer with release of the 1,3-oxathiolan-2-one (Fig. 6). The conditions required for this release are typical of those present in hypoxic tumour tissues. Indeed, this concept has already found wide application materials for drug delivery,34–39 hydrogel formation40 and in biological probing.41,42 In this latter field, Zhou/Zhang/Ye and co-workers reported the design and synthesis of trimodal redox-active imaging probes.41 Redox processes are linked with essential processes in biological systems, and changes in redox status are usually linked with many diseases including cancer, liver damage, and Alzheimer's disease. In this work, the use of a disulfide linker leads to thiocarbonate release and disassembly of the molecule. As a result, fluorescence, fluorine (19F) magnetic resonance spectroscopy or imaging (19F-MRS/19F-MRI) and proton nuclear magnetic resonance can be used for the detection of redox status in vitro or in vivo. Finally of note is that these thiocarbonates have also been long known to be suicide molecules for cyclohexanone oxygenase and so may present important medicinal applications.43
 | ||
| Fig. 6 Overview of drug/probe release under reducing conditions resulting in the formation of a cyclic thiocarbonate side product. | ||
2.3. Synthesis and applications of 1,3-dioxane-2-thiones
In comparison to other cyclic thiocarbonates, extremely little research has focused on the synthesis and applications of 1,3-dioxane-2-thiones. To the best of our knowledge there are only three reports on these compounds which date from 1998, 2000 and 2002, respectively. In the first work Kricheldorf and co-worker prepared these compounds through the reaction of 1,3-propanediol with thiophosgene and pyridine as base (Fig. 7a).44 Interestingly, the authors report that the compound spontaneously oligomerises at temperatures above 25 °C. The cationic polymerisation of this monomer was achieved by using methyl triflate (MeOTf) and boron trifluoride diethyl etherate (BF3·OEt2) (Fig. 7b). It was noted that it was extremely challenging to obtain high-molecular weight polymers from this monomer, with the best results obtained with a 100/1 ratio (monomer/catalyst) in 8 hours, and importantly, with freshly prepared monomer. These polymeric products possessed a poly(trimethylene thiocarbonate) backbone and proved to be rapidly crystallising polymers, in contrast to the analogous poly(trimethylene carbonate), derived from six-membered cyclic carbonates, which does not readily crystallise. Interestingly, during this study it was not possible to polymerise the 1,3-dioxane-2-thione monomers through an anionic mechanism. | ||
| Fig. 7 (a) Reaction of 1,3-propanediol with thiophosgene forming six-membered cyclic thiocarbonates. (b) Cationic Ring-Opening Polymerisation (ROP) of 1,3-dioxane-2-thiones. (c) A further example of the cationic ROP of 1,3-dioxane-2-thiones. (d) Anionic ROP of 1,3-dioxane-2-thiones. | ||
Endo and co-workers have also studied this reaction and reported results in 2000.45 In this work, it was found that the resulting polymer formed using cationic initiators provided polymers with narrow molecular weight distributions (Fig. 7c). Control of molecular weight could be achieved by modulating the ratio of monomer to initiator. In a more recent report, Endo and co-workers have also presented the anionic polymerisation of 1,3-dioxane-2-thiones bearing norbornene and norbornane groups, although no details on the synthesis of the monomers was provided (Fig. 7d).46 ROP of the monomer using DBU at 120 °C, resulted in the formation of a polymer with the same thiocarbonate backbone structure as previously reported by Kricheldorf and co-workers in Fig. 7b. The main difference between this work and the prior report is that the authors proposed an anionic ROP mechanism. The Mw/Mn's of the polymers were rather broad (1.40–1.48) with generally low yields, which was proposed to be due to favourable competing back-biting reactions, which limit polymer chain growth. Both of the obtained poly(trimethylene thiocarbonate)s in this work exhibited a higher thermal stability, but a lower Tg, compared to the corresponding poly(trimethylene carbonate).
2.4. Synthesis and applications of 1,3-oxathian-2-ones
Deliberate synthesis of 1,3-oxathian-2-ones has only once been reported. In 1999, Endo and co-workers reported on the anionic ROP of 1,3-oxathian-2-ones to form the corresponding poly(trimethylene thiocarbonate) product.47 It should be noted that this polymer is proposed to have the same backbone as the polymer derived from the 1,3-dioxane-2-thione monomers mentioned above. The reaction of 3-mercapto-1-propanol with triphosgene in the presence of 2.0 equiv. of 2,3-dimethyl-1-phenyl-3-pyrazolin-5-one in chloroform at room temperature for 18 hours furnished the desired product in a yield of 22% (Fig. 8a). This is indeed a low yield, however, to date no improved methodology appears to have been communicated. The anionic polymerisation of this monomer was performed using tBuOK as an initiator under a nitrogen atmosphere (Fig. 8b). The Mn of the resulting polymers ranged between 13–18000 g mol−1, with no clear correlation between Mn and temperature, likely as a result of the precipitation of the polymer part way through the reaction. However, it was clear that there was a decrease in the Mw/Mn ratio as the temperature was increased. It should be mentioned that Darensbourg and co-workers have observed 1,3-oxathian-2-ones as very minor by-products (up to 6%) in the co-polymerisation of oxetanes and COS using a Cr-salen based catalyst system, although no focus was made towards this product.48,49
 | ||
| Fig. 8 (a) Synthesis of 1,3-oxathian-2-ones through the reaction of triphosgene and 3-mercapto-1-propanol. (b) Anionic ROP of 1,3-oxathian-2-ones. | ||
3. Cyclic dithiocarbonates
This part of the review presents routes towards the synthesis of cyclic dithiocarbonates (both five- and six-membered; see Fig. 1). These are compounds which contain two S-atoms, either adjacent or separated by a carbonyl group. Herein, selected syntheses and applications of these compounds are highlighted.3.1. Synthesis and applications of 1,3-oxathiolane-2-thiones
Until the 1980s, only a few examples of the synthesis of 1,3-oxathiolane-2-thiones had been published. One of these early reports included the reaction of Et3N/CS2 with ethylene oxide to form the trithiocarbonate.50 Albeit the product in this work was a trithiocarbonate and not a dithiocarbonate, the 1,3-oxathiolane-2-thione was proposed to be a key intermediate in the mechanism, from which COS elimination provided the thiirane, which subsequently reacted again to form the reported trithiocarbonate product. An actual first example of the isolation of 1,3-oxathiolane-2-thiones was provided by Jones and co-worker in 1969.11 This procedure involved the reaction of thiophosgene with lead(II) mono(β-hydroxyethylmercaptide)monoacetate (formed from the reaction of lead(II) acetate with 2-mercaptoethanol) (Fig. 9a(i)), whereby the desired dithiocarbonate was obtained in a modest 46% yield. The product of this reaction was found to readily rearrange to form the more stable 1,3-dithiolan-2-one in the presence of KI at 60 °C (Fig. 9a(ii)).11 Later, Owen and co-workers described a significantly improved synthesis starting from a 1,2-dimethyl-substituted epoxide, proceeding via a mercaptoalcohol intermediate, and subsequent treatment of this with pyridine and TCDI (Fig. 9b).51 Interestingly, further reaction of this dithiocarbonate with mercury(II) acetate could provide access to the monothiocarbonate compound.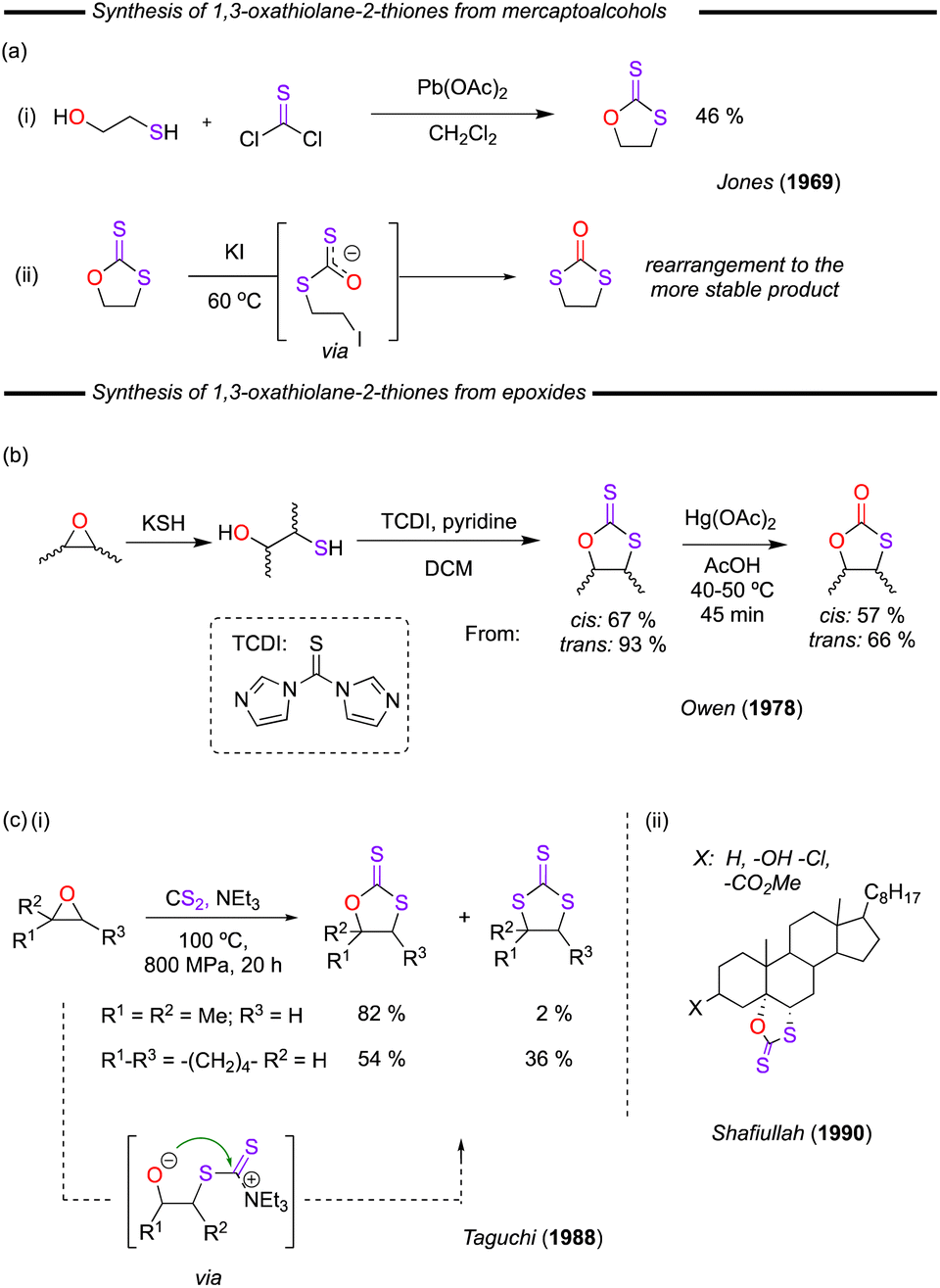 | ||
| Fig. 9 (a) (i) Pb mediated conversion of mercaptoalcohols and phosgene and (ii) rearrangement to the more stable 1,3-dithiolan-2-ones. (b) Two-step conversion of epoxides to cyclic dithiocarbonates. (c) Direct conversion of epoxides and CS2 to cyclic dithiocarbonates using NEt3 and an advanced example. | ||
It wasn't until 1988/9 when Taguchi and co-workers revisited the reaction of CS2 with epoxides that significant steps forward were made.52,53 This work is related to the aforementioned original report for the synthesis of trithiocarbonates using Et3N/CS2 (Fig. 9c(i)). The authors proposed a mechanism which includes the formation of a CS2–NEt3 adduct at high pressure and temperature (800 MPa, 100 °C). Selectivity towards the dithiocarbonate (1,3-oxathiolane-2-thione), rather than the trithiocarbonate was observed in two examples. One of these examples was the conversion of cyclohexene oxide, an internal epoxide, whilst the other example being isobutylene oxide, a 1,1′-substituted epoxide. Soon after, this methodology was employed for the preparation of steroidal dithiocarbonate derivatives by Ahmad and co-workers (Fig. 9c(ii)).54,55 These compounds were tested for and displayed genotoxic activity towards both Salmonella and E. coli. These works provide an early example of application of cyclic dithiocarbonates.
Soon after this report by Taguchi, Uenishi and co-workers established a different pathway to obtain cyclic dithiocarbonates; an intramolecular ring opening of an epoxide moiety from a 2,3-epoxyalcohol after reaction with stoichiometric amounts of NaH and CS2 (Fig. 10a and b).56,57 At low temperatures, chemo- and stereo-selective control over the final products was observed. Starting from primary or secondary epoxyalcohols the five-membered 1,3-oxathiolane-2-thiones could be selectively obtained, with the alcohol group being transposed from the terminal to internal position or vice versa. These reports also applied these cyclic dithiocarbonates as intermediates in the preparation of other valuable molecules. Meanwhile, their methanolysis to form episulfides with K2CO3 was also described by Uenishi and co-workers around the same time.58 Later, Vacher and co-worker provided a more complex example using a similar procedure (Fig. 10c).59
 | ||
| Fig. 10 (a) Synthesis from primary epoxyalcohols. (b) Synthesis from secondary epoxyalcohols. (c) Preparation of cyclohexane restrained products. (d) Use of levoglucosenone as starting material. (e) Conversion of α,β-unsaturated ester. | ||
Most recently in 2018, 2,3-epoxy alcohols derived from levoglucosenone and methyl α-D-glucopyranoside were used as substrates by Mangione/Spanevello and co-workers (Fig. 10d).60,61 In these reports the cis-epoxy alcohol formed the expected xanthate leaving the epoxide ring unreacted, whilst the trans-epoxy alcohol yielded the desired 1,3-oxathiolane-2-thione product with migration of the xanthate group. The authors proposed a rational mechanism to explain these observations, based on the inherent stereochemistry of the starting material.
A similar, yet innovative approach was employed to form a key intermediate towards the total synthesis of namenamicin, an antitumor antibiotic of marine origin (Fig. 10e).62 In this work, Nicolaou and co-worker took advantage of dithiocarbonate chemistry to selectively craft the highly unusual quaternary C-4 centre of the heterocyclic-ring of namenamicin. More specifically, a xanthate anion formed from the reaction of the C-4 hydroxyl and CS2 in the presence of NaH/imidazole reacted with the neighbouring alkene, forming the cyclic dithiocarbonate product in an impressive 83% yield.
At this point, most of these routes have relied on stoichiometric approaches. In 1995 Endo and co-workers developed the first catalytic system based on alkali metal halides to selectively form 1,3-oxathiolane-2-thiones from epoxides and CS2 (Fig. 11a).63 In this work, 5.0 mol% of LiBr in THF furnished high conversions from monosubstituted epoxides but relatively low yields were obtained in the case of 1,1′- and 1,2-disustituted substrates. The protocol is selective and proceeds by ring-opening at the least hindered position of the epoxide. This procedure is also notable as it operates at room temperature. Further to this, the catalytic system was able to successfully provide the cyclic dithiocarbonate derived from cyclohexene oxide in an 83% yield. Notably, only styrene oxide provided significant amounts of both regioisomers. In 2005, the same research group also described an approach to form a cyclic dithiocarbonate with a hydroxy group functionality (Fig. 11b), although the alcohol had to be protected before the reaction with LiBr/CS2 and deprotected afterwards, as the direct reaction with the substrate bearing the free-alcohol group failed.64
 | ||
| Fig. 11 (a) Use of metal halides as catalysts for the synthesis of cyclic dithiocarbonates. (b) Indirect route necessary for the conversion of glycidol. | ||
After this important contribution, surprisingly, few further advances were made into the design and development of new catalysts for the synthesis of 1,3-oxathiolane-2-thiones. This most likely results from the concurrent discovery that cyclic carbonates could be obtained from the coupling of CO2 and epoxides in a similar reaction. This preference for research focused on CO2 utilisation appears to have left the corresponding CS2 reaction in relative dormancy, despite the potential applications of these compounds.
However, a limited number of catalyst systems have been developed over the years and important advances have been made. Examples of these are summarised in Fig. 12a and will be discussed herein. In 2001, Endo and co-workers described the activation of oxetanes (4-membered oxacycles) using a Ti-based catalyst in order to prepare six-membered trithiocarbonates.65 Despite the evident focus of this report, the authors also reported one example of a quantitative conversion of isobutylene oxide into the corresponding dithiocarbonate after 48 hours, using 2.0 mol% of the catalyst at elevated temperature (120 °C). Under the same conditions, the corresponding 1-substituted epoxide, propylene oxide provided the trithiocarbonate, indicating the importance of the inclusion of the extra methyl group in the reactivity profile. Indeed, the authors again proposed that the dithiocarbonate species is the intermediate on the way to the final trithiocarbonate product, thus suggesting that this extra methyl inhibits/slows further reaction of this intermediate, most likely through steric prevention of the ring-opening of the dithiocarbonate compound.
 | ||
| Fig. 12 (a) Examples of catalysts reported for the synthesis of cyclic dithiocarbonates from epoxides and CS2. (b) Proposed mechanism for Al(salen) catalysts. (c) Proposed mechanism using LiOtBu as catalyst. | ||
Later, in 2004, Shi and co-workers reported an organocatalytic conversion that provided a moderate 45% yield of the dithiocarbonate from propylene oxide and CS2. In this example p-methoxyphenol and DMAP were employed as the catalyst system, however, again at a high temperature of 120 °C.66
Aside from homogeneous catalysis, an example of a heterogeneous catalytic process was described by Maggi and co-workers.67 In this report, commercially available hydrotalcite MG30 (a material containing magnesium oxide/alumina) was used as catalyst and was able to afford exclusively the 1,3-oxathiolane-2-thione regioisomer, with yields of 75–99% starting from a range of different epoxide at 50 °C. Notable results are that 1,2-epoxydodecane gave a low 25% yield, but still resulted in perfect regioselectivity. Meanwhile, styrene oxide provided a lower selectivity, furnishing a 62:38 mixture of the two possible regioisomers. This challenging regioselectivity is a theme which continues through many examples in this section with this substrate.
Stoichiometric strong bases had previously also been applied by Uenishi, but it was not until a report by Yavari and co-workers that a catalytic approach was developed, employing 10 mol% of NaH and MeOH.68 The in situ generated methoxide promoted a clean and facile reaction with various epoxides at room temperature, with high yields obtained from common monosubstituted epoxides and even from a challenging internal 1,2-diphenylepoxide. A similar approach was reported by Saidi and co-workers who also carried out the reaction with a range of monosubstituted epoxides using 10 mol% of DMAP in water, at room temperature for 20 hours. Again, as with many other catalyst examples, regioselectivities obtained from styrene oxide were somewhat disappointing.69
In many cases, conversion of CS2 into the corresponding cyclic di- and tri-thiocarbonates became an extension of the reactivity studies for several of the catalysts developed for cyclic carbonate formation. This is particularly the case of the North group who used their bimetallic [Al(salen)O]2 complex to convert epoxides/CS2.70,71 The same catalyst that had been previously reported for as able to efficiently provide cyclic carbonates from epoxides/CO2 was transferred to the corresponding conversion using CS2. In this case, at 50 °C, using 5.0 mol% of the Al(salen) catalyst and 5.0 mol% of the co-catalyst (Bu4NBr), cyclic dithiocarbonates were produced in good yields. High selectivities for dithiocarbonates over trithiocarbonate products were observed at this temperature, however, when performing the same reaction at 90 °C, the selectivity is inversed and trithiocarbonates are preferentially obtained. North proposed for the first time, a mechanism in which the rate-determining step was the Al(salen)-epoxide intermediate formation (Fig. 12b). The studies carried out with monometallic Al(salen) analogues provided evidence that the bimetallic nature of the complex was not essential for the catalytic activity itself. Furthermore, replacing the Al by Ti, another active catalyst was developed, which was active at 0.5 mol% catalyst loading at 90 °C.72 The Ti-based complex afforded good conversions and moderate selectivities towards the dithiocarbonate compounds, in contrast to those observed for the Al congeners at elevated temperature. Most recently in 2022, using the same ligand, Darensbourg presented a Cr-based salen catalyst system that was able to realise the 1,3-oxathiolane-2-thione derived from a bio-based eugenol substrate, using 0.4 mol% of the Cr(salen) complex along with an equimolar loading of bis(triphenylphosphine)iminium chloride (PPNCl) co-catalyst, at only 40 °C.73
Shi/Cao and co-workers have described a catalyst system based on an N-heterocyclic carbene (NHC) which was prepared in situ through the reaction of 4,5-dihydro-1,3-bis(2,6-diisopropylphenyl)-1H-imidazolinium chloride with K2CO3.74 With a 5.0 mol% loading of the NHC it was possible to obtain moderate to high selectivities from several monosubstituted epoxides towards the dithiocarbonate over the trithiocarbonate product, at 80 °C. Again, in this case, as in many examples, styrene oxide proved to be a challenging substrate to convert selectively to the dithiocarbonate, where in this case selective formation of the undesired cyclic trithiocarbonate product was observed.
A few years after this report, Cao/Shi described a catalyst system comprising of 5.0 mol% of LiCl, 10 mol% of [Bmim]Br (1-butyl-3-methyl-1H-imidazol-3-ium bromide) and 5.0 mol% of NaHCO3, which could be applied under neat conditions.75 This catalyst system was able to convert a range of terminal epoxides, selectively providing the 1,3-oxathiolane-2-thione in excellent yields at 40 °C. The work also described how the reaction conditions could be altered to selectively form the trithiocarbonate analogues.
In a slightly different approach, a procedure which permits both regio- and stereo-selective formation of dithiocarbonates has been recently described by Werner and co-workers using the strong base LiOtBu.76,77 In these reports a wide range of 1,3-oxathiolane-2-thiones were readily prepared. In the case of monosubstituted substrates, they were efficiently synthesised with a 5.0 mol% loading of LiOtBu within 5 hours at room temperature, with yields of up to 90%. This catalyst system operates under relatively mild reaction conditions and without solvents (neat) and given its wide substrate scope and operational simplicity, it is probably one of the most significant contributions to the field to date. Notably, vinyl and styrene oxides led to the formation of mixtures of the two regioisomers due to the stabilisation of the carbocation resulting from the epoxide ring-opening step. The authors also studied more demanding internal epoxy-substrates, obtaining moderate to high conversions and isolated yields after increased reaction times and/or temperatures. Notably, cis-2,3-epoxybutane furnished exclusively the trans-dithiocarbonate product and the related trans-epoxide afforded the cis-dithiocarbonate product. However, it should be noted that in both these cases, detectable amounts of a thiirane by-product was formed. Based on all these observations, a plausible mechanism for stereoselective conversion of internal epoxides, which proceeds via inversion of the configuration. Formation of thiiranes was proposed to proceed by rearrangement of the intermediates (Fig. 12c).
In the same year Ghazanfarpour-Darjani and co-worker, reported on a catalyst system based on NEt3 and nitromethane.78 Various 1,3-oxathiolane-2-thiones were prepared, including those starting from internal epoxides such as cyclohexene oxide. Meanwhile, in 2020, Endo and co-worker discovered that six-membered cyclic amidines efficiently catalyse (1.0–3.0 mol%) the cycloaddition of monosubstituted epoxides with CS2 at ambient temperature.79 An extensive substrate scope was reported, which demonstrated the wider applicability of this catalyst system.
The most recent report of a catalyst system for the formation of 1,3-oxathiolane-2-thiones was disclosed by Whiteoak/Mosquera and co-worker in 2022.80 This catalyst system is based on the use of a Ga-aminotrisphenolate catalyst in combination with a nBu4NX (X = halide) co-catalyst. This catalyst system was previously found to be highly active for the synthesis of cyclic carbonates from epoxides/CO2.81 In this more recent work, both terminal and internal epoxides derived from the bio-based substrate erucic acid, which comes from rapeseed oils, were converted into the corresponding dithiocarbonates. The Ga-catalyst provided full conversion and excellent yields when a 0.5 mol% of catalyst and 2 mol% of TBAI reacted with the terminal epoxide at 50 °C. Only the cyclic dithiocarbonate product and a single regioisomer with the O-atom located nearest to the R group of the epoxide substrate were obtained (Fig. 13). However, the internal epoxide cycloaddition with CS2 proved to be more challenging, requiring 5.0 mol% of the Ga-catalyst and 5.0 mol% of TBACl, 90 °C and 3 days to achieve full conversion and high selectivity towards the dithiocarbonate. It was not possible to identify the two distinct cyclic dithiocarbonates arising from the nucleophilic attack to the two almost equivalent carbon atoms of the internal epoxide. Significantly, after optimisation, the catalyst system avoids the formation of the undesired ketone by-products resulting from the Meinwald rearrangement that have been reported in many internal epoxide/Lewis acid reactions.
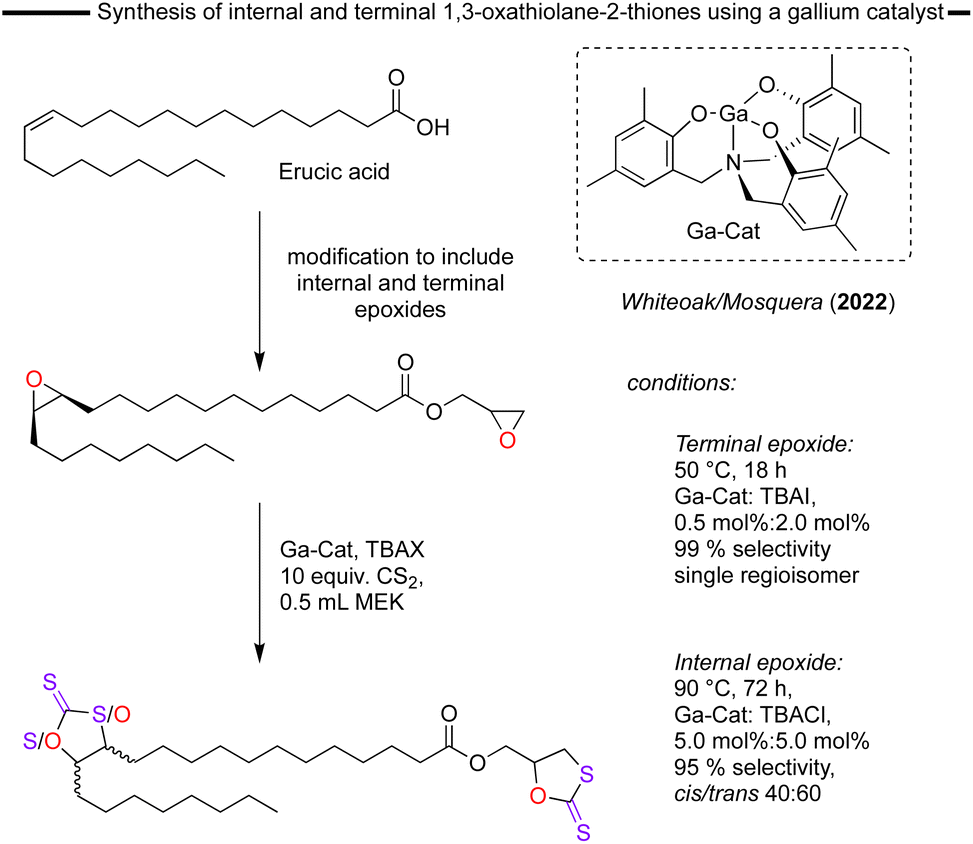 | ||
| Fig. 13 Overview of the synthesis of cyclic dithiocarbonates using a substrate derived from erucic acid. | ||
In a remarkably different approach, in 2016, Yadav and co-worker described the first visible light-based protocol for the regioselective synthesis of 1,3-oxathiolane-2-thiones.82 The one-pot multicomponent cyclisation strategy was performed with styrenes (as has been mentioned, problematic substrates for the direct epoxide/CS2 reaction) and CS2, in the presence of MeOH and Cs2CO3 (a reaction which forms an activated CS2 species; Fig. 14a). In this example eosin Y acts as the organophotoredox catalyst, with the procedure affording very good yields with only a 2.0 mol% loading for monosubstituted, 1,2-di- and even tri-substituted olefins (Fig. 14b). The authors demonstrated that the presence of O2 (from air) was also essential: only traces of the product were obtained if the reaction was performed under an inert atmosphere. Thereafter, in 2020, Hosseini-Sarvari and co-worker also accomplished the selective conversion of substituted styrenes using 10 mol% of CdS nanoparticles under mild conditions (Fig. 14c).83 In this work, the CS2 is again activated by Cs2CO3 forming a xanthate anion that thereafter reacts with the double bond under blue light this time. Interestingly, as a consequence of olefin photo-oxidative cleavage, small amounts of aryl carbonyls also appeared as by-products. In the absence of CS2, under similar conditions (CdS nanoparticles, blue LEDs, but solvent-free), aldehydes were obtained as major products. Finally, it should be mentioned that as part of a study of the reaction of potassium xanthates with five-membered cyclic carbonates, Rumyantsev and co-workers observed the formation of cyclic thiocarbonate products.84
 | ||
| Fig. 14 (a) Activation of CS2 using Cs2CO3. (b) Synthesis of cyclic dithiocarbonates from styrene's, using eosin Y and activated CS2. (c) Synthesis of cyclic dithiocarbonates from styrene's, using CdS and activated CS2. | ||
In terms of applications of these dithiocarbonate compounds, over the past few years, much attention has been paid to their use as monomers for direct polymerisation and this subject has been recently well-reviewed and so will not be discussed in detail here.85,86 A representative example comes from 1998, when Endo and co-workers reported on the selective ring-opening reaction of terminal 1,3-oxathiolane-2-thiones in the presence of a range of Lewis acids (ZnCl2, SnCl4, TfOMe, TfOEt), and protic acids (TfOH, CH3SO3H).87 Depending on the cationic carbenium or oxonium intermediate formed (Fig. 15a), the system evolved directly to the S–O–S polymer through an S–O isomerisation, or provided the corresponding 1,3-dithiolan-2-one compound. TfOMe or TfOEt promoted the ROP at 60 °C, whilst using ZnCl2 and TfOH, the isomerisation reaction takes place instead, and 1,3-dithiolan-2-one is formed, in the bulk or with solvent at 60–80 °C. In addition, the poly(dithiocarbonate) produced was detected to be degrading to a 1,3-dithiolan-2-one after the reaction with TfOEt. Later, the same group confirmed the control over cationic ROP with monomers through the neighbouring group interaction (Fig. 15b). The esters,88 urethanes89 or tertiary amines90 polymerised via a conjugated π-system, whereby the neighbouring group stabilised the intermediate cations. Other 1,3-oxathiolane-2-thione monomers, containing spiro thia-heterocycles have also been studied by Do and co-workers,91 which provided an interesting ring contraction during the polymerisation.
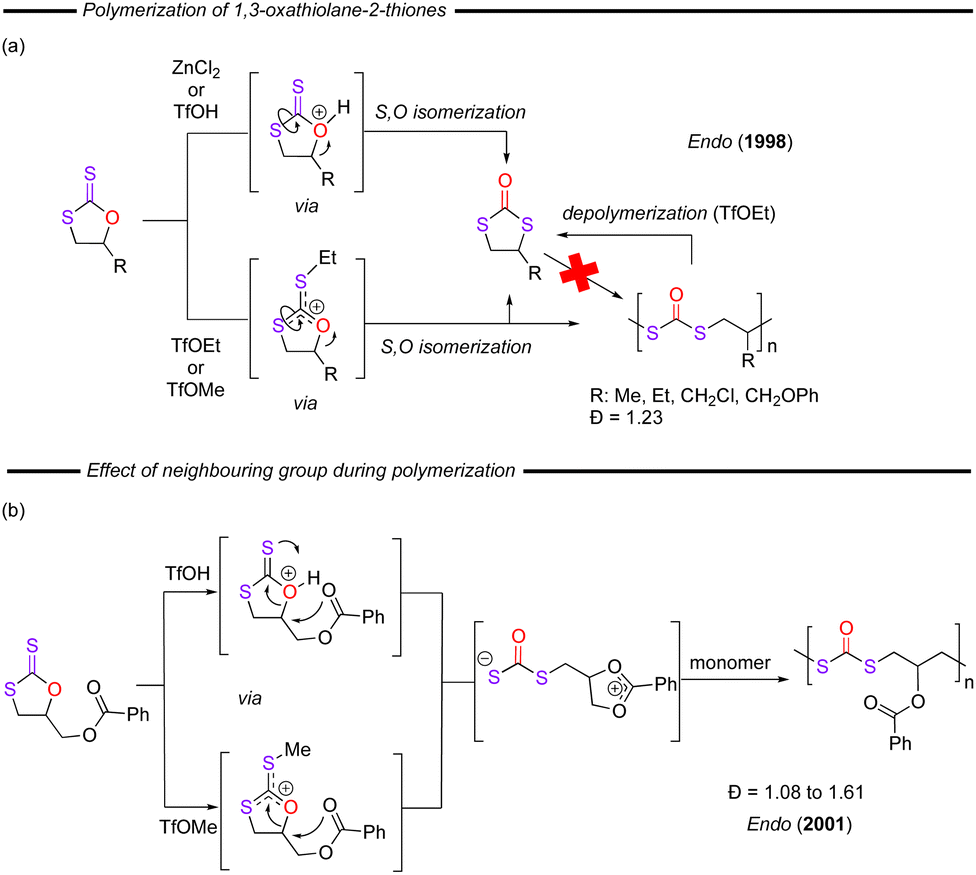 | ||
| Fig. 15 (a) Polymerisation of 1,3-oxathiolane-2-thiones and the potential isomerisation steps highlighted. (b) Example of neighbouring group interaction in the polymerisation process. | ||
Bis-cyclic carbonates are attracting increasing interest as they can be used as monomers for the synthesis of non-isocyanate polyurethanes (NIPU's), polymers which can replace traditional polyurethanes. These NIPU's are prepared through the reaction of a bis-cyclic carbonate and a diamine.92 One of the limitations with this reaction is the non-selective ring-opening of the cyclic carbonate by the diamine which can result in the formation of both primary and secondary alcohol functionalities along the chain. In this context, as long ago as 1995, Endo and co-worker reported the reaction of cyclic dithiocarbonates with amines.93 It was found to be both more efficient than the analogous reaction with cyclic carbonates, whilst also displaying a higher level of cyclic dithiocarbonate ring-opening selectivity. The reaction selectively forms mercaptothiourethanes, without formation of hydroxydithiourethanes, a result which is proposed to arise from the increased stability of thiolate anion intermediates over the analogous alkoxide (Fig. 16). This was applied to bis(dithiocarbonates) and diamines for the synthesis of poly(mercaptothiourethanes).94 This selectivity is rather interesting and may be a route towards the formation of NIPU's with more controlled microstructures.
 | ||
| Fig. 16 Explanation of the selectivity observed in the reaction of amines with cyclic dithiocarbonates. | ||
3.2. Synthesis and applications of 1,3-dithiolan-2-ones
1,3-Dithiolan-2-ones have been prepared through a variety of approaches. In addition, the crystal structure of this compound has also been reported.95 The structure confirms the pseudo-twofold symmetry and consists of a twisted five-membered ring of three C and two S atoms, with a ketone O atom in an equatorial position.As mentioned above, as long ago as 1969, Jones and Andreades presented a method for the synthesis of 1,3-dithiolan-2-one.11 The authors studied in detail the potential isomerisation of the proposed product, observing that it actually formed the 1,3-dithiolan-2-one instead (Fig. 17a). A similar unexpected product outcome has also previously been observed by others for related reactions; in 2021, Zhang and co-workers reacted ethylene oxide with COS in the presence of DBU, whereby at reaction temperatures of up to 80 °C the 1,3-dithiolane-2-one product was predominantly the species formed (Fig. 17b).96 The formation of the 1,3-dithiolan-2-one is proposed to arise from the cycloaddition of COS to ethylene oxide via a zwitterionic-based mechanism (Fig. 17f).
 | ||
| Fig. 17 (a) Synthesis from mercaptoalcohol. (b) Reaction of epoxides and COS to form cyclic mono and dithiocarbonates. (c) Use of dithiols and phosgene. (d) Use of CMPI as a key reagent. (e) Use of in situ formed COS and 1,2-dibromoethane. (f) Proposed mechanism for the conversion in reaction (b). | ||
In 1973, Satsumabayashi first reported the phosgene-based synthesis of 1,3-dithiolane-2-one through the reaction of 1,2-dithioethane in the presence of an organic base, pyridine (Fig. 17c).97 The reaction proceeded in high yield at 0 °C, however, significant amounts of polymeric products were observed when the temperature was increased above 30 °C.
Very recently, in 2022, Qaroush/Eftaiha and co-workers have described the synthesis of 1,3-dithiolan-2-one, amongst other related compounds, through the reaction of 1,2-dithioethane with triethylamine, sodium hydride and CO2 to form an intermediate carbonothiolate adduct (Fig. 17d).14 This intermediate then readily reacts with the Mukaiyama reagent (CMPI) to furnish the final cyclic product. This is an attractive methodology as all the reagents are readily available and the procedure also uses CO2 as reagent. In 1990, Mizuno and co-workers reported on a route to provide S,S-dialkyl dithiocarbonates from a two-step reaction from elemental sulfur, triethylamine, carbon monoxide (CO) (Fig. 17e, step 1) and thereafter reacting the product with alkyl halides in water/THF using DBU as base (Fig. 17e, step 2).98 In this procedure the required equivalents of COS are obtained from the reaction of the CO with elemental sulfur. In the context of this discussion, the authors applied 1,2-dibromoethane as precursor, enabling the synthesis of the corresponding cyclic 1,3-dithiolane-2-one product, albeit in a very moderate yield of around 35%.
In a different approach, many groups have sought to convert other cyclic carbonates and trithiocarbonates to the corresponding 1,3-dithiolane-2-one product. In 1982, Fujinami and co-workers reported that the reaction of ethylene carbonate with CS2 using a catalyst system composed of hexabutyldistannathiane (5.0 mol%)/18-crown-6 (10 mol%)/K2CO3 (5.0 mol%) in MeCN furnished a 17% yield of 1,3-dithiolane-2-one (Fig. 18a).99 It should be noted that without the addition of 18-crown-6 only trace product was obtained. Further optimisation or understanding of the reaction was not pursued and so this serves as an early example that cyclic carbonates can be ring-opened with carbon disulfide, with the reaction furnishing the 1,3-dithiolane-2-one product. A few years later, Barbero described how 1,3-dithiolan-2-ones could also be obtained by reacting of 1,3-dithiolane-2-thiones and epoxides in the presence of HBF4·Et2O at low temperatures (Fig. 18b).100 The reaction was found to proceed via an intriguing 1-oxa-4,6,9-trithiaspiro[4.4]nonane intermediate. As part of the work, the authors were able to amplify the substrate scope and produce a range of substituted 1,3-dithiolan-2-ones for the first time.
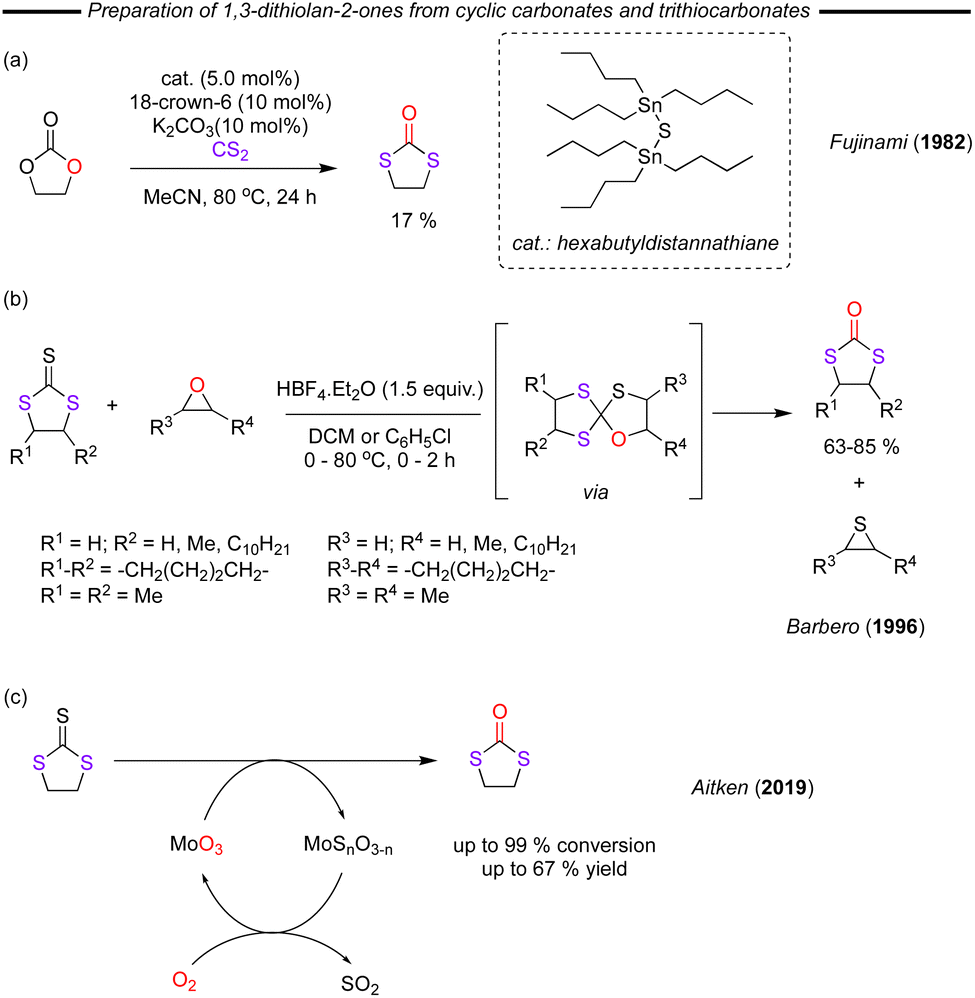 | ||
| Fig. 18 (a) Stannane conversion of cyclic carbonates to 1,3-dithiolan-2-ones. (b) Conversion of cyclic trithiocarbonates to 1,3-dithiolan-2-ones. (c) Molybdenum oxide catalysed conversion of cyclic trithiocarbonates to 1,3-dithiolan-2-ones. | ||
In a relatively recent contribution, Aitken and co-workers reported on the gas-phase reaction of 1,3-dithiolane-2-thione over molybdenum trioxide (MoO3) supported on pumice stone to form 1,3-dithiolan-2-one (Fig. 18c).101 The MoO3 was readily regenerated on exposure to air and thus acts as a catalyst for the overall conversion of the thione and O-atom from the air. Quantitative conversions with excellent selectivity were observed. Nevertheless, isolated yields were lower, due to partial decomposition of the product.
In 2020 Dove/Sardon and co-workers described the depolymerisation of bisphenol A-based polycarbonate (BPA-PC), using various reagents in combination with a triazabicyclodecene (TBD) and methanesulfonic acid (MSA) based catalyst system (Fig. 19).102 One of reagents studied was 1,2-dithioethane, which readily reacted with the polymer forming 1,3-dithiolane-2-one in 92% yield after 30 minutes of reaction at 90 °C. A similar result was previously obtained using NaOH as catalyst by Oku and co-workers in 2003.103 Substrate scoping with this cyclic dithiocarbonates is rather weakly explored with mainly only the parent compound reported to date.
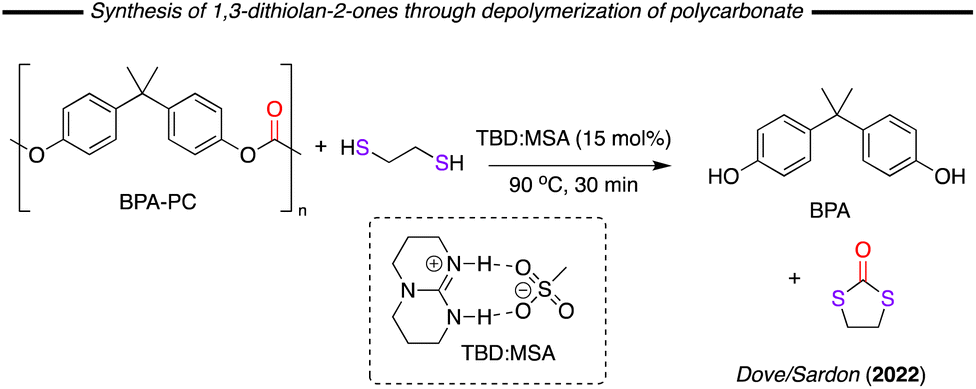 | ||
| Fig. 19 Formation of 1,3-dithiolan-2-ones through the depolymerisation of polycarbonates with dithiols. | ||
In an attempt to develop novel compounds from the parent 1,3-dithiolane-2-one, Fuchigami and co-worker presented the anodic α-fluorination of these compounds (Fig. 20).104 The authors measured the oxidation potential of a range of thiocarbonate compounds and found them to be lower in comparison to the analogous cyclic carbonates. This fact was explained by the presence of easily oxidisable sulfur atoms and indeed, the oxidation potential was observed to decrease as the number of sulfur atoms in the compound was increased. Meanwhile, when the number of sulfur atoms is the same, thiocarbonates with a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S bond showed lower oxidation potentials. The reason for this latter observation is likely that the electron transfer from the CS pi-bond is easier than those from the lone paired electrons of sulfur atoms in the heterocycle ring. The anodic fluorination of the 1,3-dithiolane-2-one proceeded in up to 48% yield using MeCN as solvent with Et3N·5HF as both supporting electrolyte and F-source.
S bond showed lower oxidation potentials. The reason for this latter observation is likely that the electron transfer from the CS pi-bond is easier than those from the lone paired electrons of sulfur atoms in the heterocycle ring. The anodic fluorination of the 1,3-dithiolane-2-one proceeded in up to 48% yield using MeCN as solvent with Et3N·5HF as both supporting electrolyte and F-source.
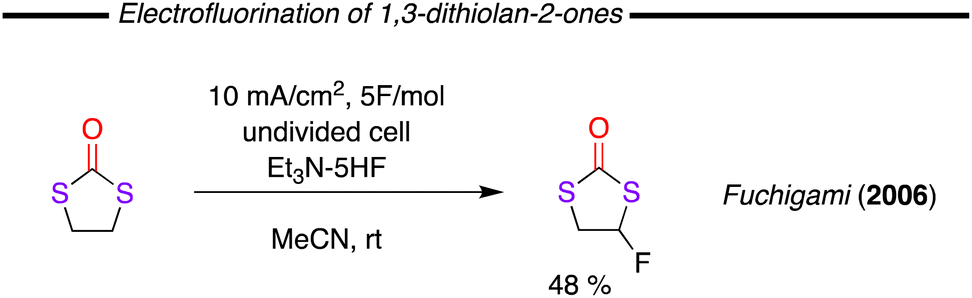 | ||
| Fig. 20 Direct fluorination of 1,3-dithiolan-2-ones using electrochemical methodology. | ||
3.3. Synthesis and applications of 1,3-oxathiane-2-thiones
Very little attention has been paid towards this type of cyclic dithiocarbonate. In 1994, Uenishi and co-workers reported an approach for the synthesis of a six-membered cyclic dithiocarbonate (Fig. 21a).57 The formation of the alkoxide-epoxide intermediate after the addition of NaH followed by the reaction with CS2 resulted in a moderate final product yield of 44%. As was previously mentioned for the related 5-membered dithiocarbonate analogue, the mechanism proceeded through an acid–base reaction and a subsequent xanthate migration step. Relatively few other examples exist, other than in an interesting contribution by Buchard and co-workers in 2018.105 In this report cyclothiocarbonation of 2-deoxy-D-ribose- and D-xylose-biobased diols with CS2 was performed (Fig. 21b). Both the trans- and cis-fused products (in relation to the furanose) were isolated in relatively low yields, whereby these products were later polymerised using organocatalytic ROP methods (TBD catalyst with 4-MeBnOH as initiator). The 2-deoxy-D-ribose-derived substrate afforded poly(xanthate) species as a consequence of a regio-regular opening process whereas in contrast the D-xylose-derived substrate provided a polymeric structure with both trithiocarbonate and thionocarbonate linkages resulting from the alternating opening of the monomer. Most recently, in 2022, Buchard and co-workers prepared a 1,3-oxathiane-2-thione derived from tri-O-acetyl-D-glucal (Fig. 21c).106 The authors then copolymerised this bio-derived cyclic thiocarbonate with L-lactide (a bio-derived monomer), providing bio-derived polymers which showed enhanced UV-degradability compared to the corresponding polylactide (PLA). It was proposed that this novel approach has the potential to increase the environmental degradability of PLA without significantly affecting its properties as only 3% of sulfur-containing linkages were required for a mass loss of 40% within 6 hours. | ||
| Fig. 21 (a) Synthesis from epoxyalcohols using a NaH/CS2 approach. (b) Synthesis from diols and polymerisation to form polydithiocarbonates. (c) Ring-opening co-polymerisation of cyclic dithiocarbonates with L-lactide. | ||
3.4. Synthesis and applications of 1,3-dithian-2-ones
As in the case of five-membered 1,3-dithian-2-ones synthesis, the most logical route for the six-membered cycles is through the direct reaction of 1,3-propanedithiol with phosgene. This reaction was first reported by Satsumabayashi and co-workers as long ago as 1979 (Fig. 22a).107 Through this approach, the authors were able to achieve a 72% isolated yield when working on a 0.1 mole scale in only 3 hours of reaction making it suitable for laboratory preparation, but as is typical with phosgene-based reactions, unfavourable for industrial scale application. In 1972, Adam and co-worker reported the reaction of substituted dithioethylenes with singlet oxygen which was generated photochemically from triplet molecular oxygen by dinaphthylthiophen sensitisation at −78 °C (Fig. 22b).108 The authors proved the necessity of the photochemistry and singlet oxygen as the dithioethylene substrates were stable towards the photolysis conditions in the absence of oxygen and towards molecular oxygen in the dark. During these studies some photo-oxidation of the sulfur was observed, but in all cases the 1,3-dithian-2-ones were the major products. A decade later, Foote and co-workers identified the key species in this reaction by characterising a key dioxetane compound that was proposed to be the reaction intermediate. This intermediate was demonstrated to evolve to the final 1,3-dithian-2-one product.109 During studies attempting to generate bis-alkyldithiocarbenes from (3,4-diaza-2,2-dimethyl-1-oxa-6,10-dithiaspiro[4.5]dec-3-ene), Schreiner and co-workers observed that when the compound was irradiated at 313 nm, the desired thiocarbene product was not obtained (Fig. 22c).110 Instead, the 1,3-dithian-2-ones and 2-diazopropane was observed, providing an interesting reactivity, which could make the starting compounds useful sources of in situ generated 1,3-dithian-2-ones or reactive diazo compounds.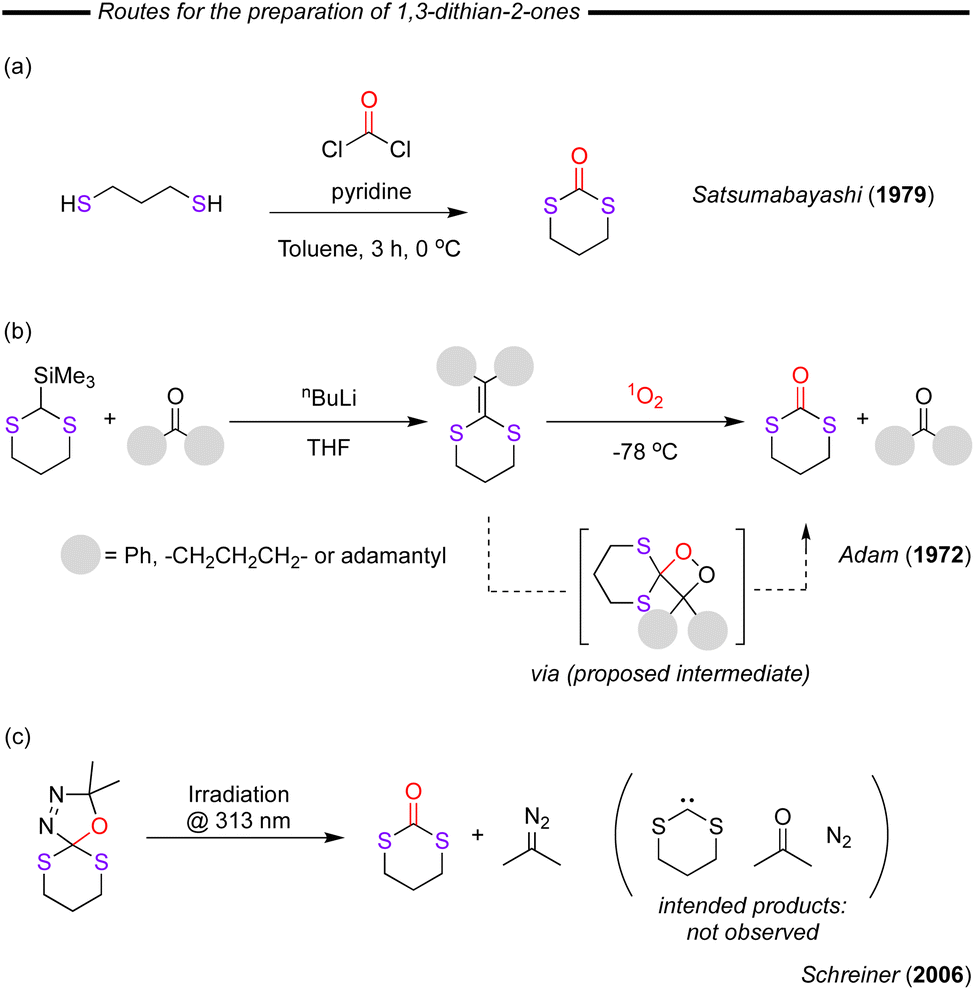 | ||
| Fig. 22 (a) Synthesis from dithiols and phosgene. (b) Reaction of dithioethylene functionalities with singlet oxygen to form 1,3-dithian-2-ones. (c) Unexpected photochemical reaction of spiro compounds forming the 1,3-dithian-2-one product. | ||
Depolymerisation of polycarbonates using nucleophiles has also been successful in generating synthetically useful yields of 1,3-dioxane-2-thiones. In 2003, Oku and co-workers attempted the depolymerisation of pure polycarbonate pellets with a range of nucleophile reagents, one of them being 1,3-propanedithiol. This reagent was reacted with the polymer forming 1,3-dithian-2-ones which could be readily isolated.103 Indeed, yields of up to 75% of the desired 1,3-dithian-2-ones under reaction conditions of 100 °C for only 3 h with an aqueous NaOH catalyst (1.5 mol%) were achieved.
In terms of their applications, 1,3-dithian-2-ones have been employed in the field of polymerisation chemistry in quite an innovative and distinct way. In 1997, Kricheldorf and co-workers studied the reaction of 2,2-dibutyl-2-stanna-1,3-dioxacycloalkanes, which were obtained directly from the reaction of dibutyltin oxide and diols, with lactones and thiolactones, which resulted in ring expansion of the heterocycles (Fig. 23a).111 In the case of the thiolactones the greater stability of the resulting Sn–S bond allows for stepwise expansion, meanwhile the insertion of lactones provides a new Sn–O bond which has a similar reactivity to the parent compound and thus results in polymerisation forming macrocyclic polymers. In contrast, reaction with 1,3-dioxane-2-thione allowed for unexpected selective removal of the dibutyltin group. The authors reasoned this as a result of ring contraction which arises from the intramolecular coordination of the S–CO sulfur with a free orbital of the Sn atom, followed by an intramolecular transesterification. Taking advantage of this reactivity the authors polymerised lactones and just before the reaction was completed added the 1,3-dithian-2-ones to eliminate the potentially toxic Sn compound (Fig. 23b). This allowed for the realisation of perfectly non-toxic (non-tin containing) biodegradable macrocyclic esters when ε-caprolactone was utilised as monomer. This presents a particularly interesting application of the 1,3-dithian-2-ones.
 | ||
| Fig. 23 (a) Reaction of cyclic organostannane forming cyclic carbonate and transfer product. (b) Removal of stannanes from cyclic polycarbonates using 1,3-dithian-2-ones. | ||
In 1993, Fournet and Goré described the ring expansion of 1,3-dithian-2-ones with lithium acetylides to furnish 4-substituted-7,8-dihydro-2H,6H-1,5-dithiocin-2-ones (Fig. 24a).112 In this study the authors were actually attempting to prepare the corresponding α-acetylenic thiocarboxylic-S-esters, but after repeated efforts, only ring-expansion products were obtained. The authors proposed that the observed product arises from the Michael addition of the thiolate intermediate to the activated triple bond. Interestingly, the analogous reaction with 1,3-dithiolan-2-ones does not proceed. 1,3-Dithian-2-ones can also be readily alkenylated to from ketene dithioacetals. Takai and co-workers presented an alkylidenation of 1,3-dithian-2-one with various 1,1-dibromalkanes mediated by a mixture of Zn/TiCl4/TMEDA in THF at room temperature providing several examples of potentially useful ketene dithioacetals (Fig. 24b).113 In 1996, Takai revisited the reaction and PbCl2 was found to act as catalyst.114 After analysis of the original procedure, it is likely that trace impurities of lead compounds were present in the zinc and this actually catalysed the reaction. When high-purity zinc was employed in this latter study the reaction did not proceed, however in the presence of added PbCl2 the reaction worked.
 | ||
| Fig. 24 (a) Ring-expansion with lithiated alkynes. (b) Reaction with 1,1′-substituted dibromides (note: presence of Pb found to be important). | ||
4. Cyclic trithiocarbonates
This class of compound mimics the cyclic carbonates, but with all the O-atoms changed to S-atoms (see Fig. 1). It should be noted that the synthesis of this class of compounds was reviewed in the recent past by Fallah-Mehrjardi.115 Herein, synthesis of both 5- and 6-membered cyclic trithiocarbonates will be described, along with selected applications to highlight their potential utility.4.1. Synthesis and applications of 1,3-dithiolane-2-thiones
In 2007, Lee/Chan and co-worker described a protocol for the reaction of 1,2-dibromoethane and CS2 to form the cyclic trithiocarbonate.116 The catalyst system comprised of 3.0 mol% of tetrabutylammonium hydrogen sulfate (nBu4NHSO4) as an effective phase-transfer catalyst in combination with 33% aqueous NaOH, providing a 77% yield after 24 h (Fig. 25a(i)). In a similar approach, Soleiman-Beigi and co-worker proposed the use of imidazole as an inexpensive, non-toxic, and readily available catalyst (Fig. 25a(ii)).117 The reaction of 1,2-diiodoethane with CS2 in aqueous DMSO was promoted by imidazole under mild reaction conditions to form the desired product, with the proposed reaction mechanism summarised in Fig. 25b. Another starting material that has been used is 1,2-ethanedithiol. In this context Qaroush/Eftaiha and co-workers have reported that the Mukaiyama reagent (CMPI) can promote the reaction of this substrate with CS2 at ambient temperature and pressure (Fig. 25a(iii)).14 In this example, treatment with triethylamine and sodium hydride and subsequent addition of CMPI in acetonitrile afforded the cyclic trithiocarbonate in only a few hours. | ||
| Fig. 25 (a) Synthesis methods employing CS2 as reagent. (b) Mechanism proposed for the imidazole promoted reaction between alkyl halides and CS2. | ||
Aside from these examples starting from alkyl-dihalides and dithiols, most methods focus instead on the addition of CS2 to epoxides. Depending on the reaction conditions cyclic dithiocarbonates or trithiocarbonates can be selectively formed from this type of reaction, as was mentioned earlier in this review. In this section, the cyclic trithiocarbonate synthesis is consider rather than the dithiocarbonate preparation.
In 2001, Endo and co-workers described an approach for synthesis of cyclic trithiocarbonates from epoxides and CS2, catalysed by a titanium complex.65 Here, the reaction of epoxides and CS2 at 120 °C catalysed by a (2-propanolato)titanatrane complex selectively afforded the desired five-membered cyclic product (Fig. 26a). Additionally, the reaction with oxetanes (four-membered oxacycles) resulted in six-membered trithiocarbonate products. This latter outcome was the focus of the work and will be described in greater detail later. Meanwhile, a single example starting from propylene oxide resulted in the corresponding cyclic trithiocarbonate. Interestingly, as mentioned in the appropriate section above, the reaction from isobutylene oxide stopped at the cyclic dithiocarbonate, resulting in a quantitative yield of this compound. The authors proposed a mechanism involving the activation of the oxirane and reaction with CS2 furnishing the corresponding dithiocarbonate. Further reaction with CS2 then provided the desired trithiocarbonate (Fig. 26e). A similar approach was also reported by North and co-worker, using a titanium(salen) catalyst (Fig. 26b).72 In this example an ample substrate scope was provided, whereby reactions between a range of epoxides and CS2 were successfully catalysed by a bimetallic titanium(salen) complex, [Ti(salen)]2O and tetrabutylammonium bromide (TBAB) as co-catalyst. However, the results showed that the selectivity towards either the dithio- or trithio-carbonate was relatively poor, except in the case of styrene oxide. The reaction mechanism was similar to that proposed by Endo. The selectivity towards the trithiocarbonate in this work is poorer than the previously reported Al(salen)-based catalyst system that also struggled to provide selectivity for either the dithio- or trithiocarbonate, except when styrene oxide was employed.71
 | ||
| Fig. 26 (a) Use of a titanium catalyst for the synthesis of cyclic trithiocarbonates. (b) Application of a Ti(salen)-based catalyst. (c) Use of an N-heterocyclic carbene to activate CS2. (d) Use of a combined LiBr and N-heterocyclic carbene catalyst system. (e) Proposed mechanism for the titanium-catalysed reaction of epoxides and CS2. | ||
N-heterocyclic carbenes (NHCs) have also found their application in the synthesis of trithiocarbonates (Fig. 26c).74 An example reported by Shi/Cao and co-workers, in 2015, involves the in situ formation of a carbene–CS2 adduct. Thereafter, addition of this adduct to a solution of epoxides in DMSO furnished the desired dithio- and trithiocarbonates. However, the only case that gave full selectivity towards the cyclic trithiocarbonate was styrene oxide. In other cases, either cyclic dithiocarbonates or a mixture of both products was obtained.
A more recent report from, Shi/Cao and co-workers described the combined use of an NHC/LiBr catalyst system (Fig. 26d).75 In this procedure, various terminal epoxides were converted to the corresponding 1,3-dithiolane-2-thiones in excellent yields, with high selectivity, a significant improvement on their previous report. The recyclability of the catalytic system LiBr/[Bmim]Br/KOH was also examined, where the results indicated that the catalyst could be used for three times without loss of the catalytic activity; however, the product yield decreased gradually in subsequent runs after the third reaction.
In a rather different approach, Samoshin and co-workers reported on the synthesis of 1,3-dithiolane-2-thiones by reaction between epoxides and the commercially available potassium ethyl xanthogenate, KSC(S)OEt.118 A range of trans-trithiocarbonates derived from the corresponding cyclohexene oxide substrates were obtained in good to moderate yields by reaction in methanol at 35–45 °C under an Ar atmosphere (Fig. 27a). Treatment of the trithiocarbonate product with Br2, NBS, or DDQ led to formation of the corresponding cyclic dithiocarbonates instead and can be considered as an alternative route to the synthesis of these compounds (Fig. 27b).
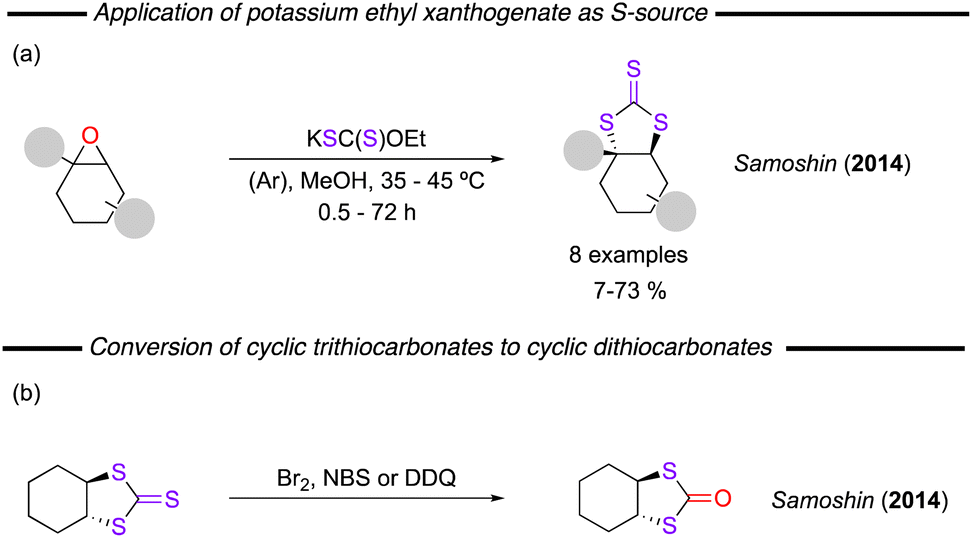 | ||
| Fig. 27 (a) Reaction of epoxides and potassium ethyl xanthogenate to form 1,3-ditholane-2-thiones. (b) Reaction of 1,3-dithiolane-2-ones to form 1,3-dithiolan-2-ones. | ||
Synthesis of cyclic trithiocarbonates has also been described starting from highly reactive thiiranes. Thiiranes are interesting substrates as they have significantly lower ring-opening barriers than the corresponding epoxides. This makes them susceptible to polythioether formation rather than cyclic thiocarbonate formation. In this context, Endo and co-worker reported on the use of cyclic and linear amidines as catalysts for the reaction of thiiranes and CS2 (Fig. 28a).119 The high basicity of the amidine proved to be beneficial, whereby 1,2-dimethyl-1,4,5,6-tetrahydropyrimidine was found to be the optimal catalyst. The reaction worked either under solventless conditions or in toluene and several examples were provided.
 | ||
| Fig. 28 (a) Use of a cyclic amidines as catalysts for the cycloaddition of thiiranes and CS2. (b) Application of LiOtBu as catalyst. (c) Selective formation of 1,3-dithiolane-2-thiones or polytrithiocarbonates depending on the reaction conditions applied. | ||
Meanwhile, Werner and co-worker reported the synthesis of a limited number of examples of trithiocarbonates, in their contribution focused on the synthesis of dithiocarbonates using LiOtBu as catalyst (Fig. 28b).77 In the case of trithiocarbonates, the reaction uses thiiranes and CS2. Under solvent-free conditions, 5.0 mol% of LiOtBu was found to be optimal for the formation of the desired products, although only low to moderate isolated yields were obtained. Interestingly, the reaction starting from the thiirane analogue of epichlorohydrin failed, whereas the synthesis of the dithiocarbonate using the same protocol starting from epichlorohydrin provided almost quantitative yield.
Most recently, in 2022, Gnanou/Feng and co-worker disclosed an approach which described how the selection of catalyst system could allow for selective formation of either poly(trithiocarbonates) or cyclic trithiocarbonates (Fig. 28c).120 In this metal-free process, the cyclic trithiocarbonates can be selectively obtained by reaction of the thiirane and carbon disulfide using tetrabutylammonium fluoride (TBAF) at 80 °C. It was indicated that above 60 °C when using fluoride as nucleophile, back-biting reactions largely predominate over propagation, resulting in the formation of the cyclic product. Meanwhile, using PPNCl or phosphazene benzoxide resulted in the polymeric product. This selectivity was reported to be independent of the nature of the substituents of the starting thiirane.
An interesting report into the application of five-membered cyclic trithiocarbonates was presented as long ago as 1982 by Hatanaka/Tanimoto.121 This work details how a range of alkyl vinyl trithiocarbonates can be readily obtained by reaction of the 1,3-dithiolane-2-thiones with lithium diisopropylamide, followed by treatment with an alkyl halide (Fig. 29a). Cyclic trithiocarbonates can also be key intermediates for the synthesis of tetrathiafulvalene derivatives, like tetrathiafulvallene tetracarboxylate (Fig. 29b).122 These compounds have shown many applications over the last few decades; as field-effect transistors,123,124 conductive porous materials,125 among others. Other specific examples include a report from Inigo/Skabara and co-workers who incorporated this moiety into terthiophene–diketopyrrolopyrrole co-polymers for air stable solution processable organic field effect transistors,126 meanwhile, Devic and co-workers prepared 3D-coordination polymers of tetrathiafulvalenetetracarboxylate that exhibited a semiconducting behaviour.127 In 2014, D'Alessandro and co-workers installed tetrathiafulvallene carboxylate as donor in a metal–organic framework.128 This application allowed charge separation to be controlled using N,N′-di-(4-pyridyl)-1,4,5,8-naphthalenetetracarboxydiimide as an acceptor, whereby the authors achieved a partial charge transfer, useful for conductive and photoactive materials. Most recently, Gu and co-workers synthesised a Covalent Organic Framework (COF) based on tetraformyl-tetrathiafulvalenes with enhanced electrochemical properties, that could be an efficient material for supercapacitor applications.129
 | ||
| Fig. 29 (a) Preparation of alkyl vinyl trithiocarbonates. (b) Use as reagents for the synthesis of tetrathiafulvalene derivatives. (c) Products obtained after reaction with anilines. | ||
In the context of polymerisation applications, it is interesting to note that cyclic five- and six-membered trithiocarbonates do not appear to undergo ROP, whilst seven-membered variants are able to. In this context, in 2022, Ren and co-workers reported the ROP of 7-membered trithiocarbonates which could be readily recycled under pyrolysis.130 Furthermore, the obtained polythiocarbonate was superior to the corresponding analogous oxygen-containing polycarbonate in terms of its thermal, mechanical, and optical properties.
Nevertheless, the corresponding polymers can be obtained by other means, as has been demonstrated by Gnanou/Feng and co-workers.120 In this work, the obtained polymers also underwent complete unzipping when treated with radicals or under UV light irradiation to form the cyclic trithiocarbonates. This unexpected activity was exploited to prepare degradable polymers using oligotrithiocarbonates as self-immolating linkers between dithiol alkyl precursors.
Finally, Gu/Guo and co-workers have described the reaction of 1,3-dithiolane-2-thione with aniline which produced [1,3]dithiolan-2-ylidene-phenylamine instead of the expected 3-phenylthiazolidine-2-thione (Fig. 29c).131 These unexpected dithiocarbonimidates are interesting, as compounds containing this scaffold have previously shown effective anti-inflammatory properties.132
4.2. Synthesis and applications of 1,3-dithiane-2-thiones
These compounds are the six-membered analogues of the 1,3-dithiolane-2-thiones and are the final compound-type to be discussed in this review. In comparison to other cyclic thiocarbonates their synthesis has been poorly studied, with only a few reports available, which are highlighted herein.As was mentioned above, in 2001 when Endo and co-workers performed the reaction of oxetanes and CS2 catalysed by titanium complex which afforded the desired six-membered cyclic trithiocarbonate products from 4 differently substituted oxetane substrates (Fig. 30a).65 Meanwhile, recently, Devdutt and co-workers presented a simple procedure using Cs2CO3 and CS2 to convert alkyldibromides to the cyclic trithiocarbonate products in DMSO (Fig. 30b).133 This protocol is operative at room temperature and requires short reaction times (3 to 6 hours), furnishing the desired products in attractively high yields. Imidazole has also been applied as catalyst for the conversion of 1,3-dibromopropane and CS2 by Soleiman-Beigi and co-worker (Fig. 30c).117 It should be noted here that this work also discloses preparation of a larger ring-sized cyclic trithiocarbonate (obtained from the reaction of 1,3-dibromobutane) in a slightly lower yield of 70%, albeit an interesting example of further application of the developed protocol.
 | ||
| Fig. 30 (a) Reaction of oxetanes and CS2 to form 1,3-dithiane-2-thiones. (b) Use of dibromoethanes as starting materials using CS2CO3. (c) Imidazole promoted synthesis of 6- and 7-membered cyclic trithiocarbonates. (d) Synthesis of cyclic trithiocarbonates based on carbohydrate scaffolds. (e) Preparation of cyclic trithiocarbonates fused to thieto-imidazoles. | ||
In 2003, Voelter and co-workers reported a synthesis of cyclic trithiocarbonates forming part of a carbohydrate scaffold (Fig. 30d).134 Reaction of sodium thiocarbonate and epoxytriflate pentoses resulted in the desired product, although in relatively low yields. The procedure is regio- and stereoselective, presenting an interesting advanced application of the developed procedures for preparation of more complex 1,3-dithiane-2-thiones. Another example of inclusion of these compounds into more complex molecules was reported by Yadav and co-worker (Fig. 30e).135 In this work, a fused thieto-imidazole was synthesised and reacted with CS2 using a catalytic amount of LiI to obtain fused trithiocarbonate-imidazoles. This work presents a substrate scope whereby the aromatic functionalities have been systematically varied. In similarity to their synthesis, application of six-membered trithiocarbonates has also received relatively little attention. Some synthetic transformations have been reported. The sulfur version of the Reformatsky reaction was reported by Ila/Junjappa and co-workers (Fig. 31a),136 whilst their reaction with m-CPBA to access trithiocarbonate oxides and their reaction with organolithium reagents and subsequent rearrangement to disulfide thioformates has also been demonstrated (Fig. 31b).137 In addition, they have also been found to react with lithium reagents, as was reported by Sugawara and co-workers (Fig. 31c), where attack at the carbon atom at room temperature and at the sulfur at −78 °C was observed. The corresponding intermediates could then be alkylated with alkyl halides, providing a range of potentially useful synthons.138
 | ||
| Fig. 31 (a) Reaction of 1,3-dithiane-2-thiones with organometallic zinc compounds. (b) Oxidation with m-CPBA/reaction with MeLi. (c) Temperature dependent outcome during the reaction with nBuLi and subsequent reaction with alkyl halides. | ||
5. Conclusions
In conclusion, this review has presented an overview of a wide array of synthetic routes towards the synthesis of both five- and six-membered mono-, di- and trithiocarbonates. Whilst some of the methodologies shown are not synthetically scalable due to the use of unusual starting materials, it is important to highlight how these cyclic products can be formed through non-conventional approaches. Meanwhile, in many cases, it is clear that there are still opportunities to develop more efficient (including catalytic, rather than stoichiometric) methods of synthesis in order to improve their availability. This is particularly true for 1,3-oxathian-2-ones, which have only been prepared in modest yield to date, despite their potential applications.In addition to highlighting synthetic methods for the preparation of cyclic thiocarbonates, examples of applications of these compounds have also been presented. These include both their uses as monomers and as synthetic intermediates in the preparation of more complex and valuable molecules. Specific applications, such as, use as precursors for the synthesis of tetrathiafulvalene derivatives, compounds which present potential as field-effect transistors, conductive porous materials, have also been detailed. Another particularly interesting application that has been described, exemplifies how cyclic dithiocarbonates can be used to remove potentially toxic stannanes from macrocyclic polymers, due to the increased affinity that stannanes have for these cyclic thiocarbonates over their cyclic carbonate O-containing analogues. In summary, whilst it is clear that cyclic thiocarbonates already present many potential applications, it is likely that increased interest will result in improved synthetic access and the development of further and more varied applications.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
CW would like to thank the Comunidad de Madrid (Spain) for funding (Programa de Atracción de Talento 2019: Modalidad 1; award number 2019-T1/AMB-13037, and CM/JIN/2021-018). All authors would like to acknowledge funding from the Spanish Government (PID2021-122708OB-C31/AEI/10.13039/501100011033 and PID2020-113046RA-I00/AEI/10.13039/501100011033).References
- For an overview of organosulfur compounds in rechargeable batteries, see: (a) Z. Shadike, S. Tan, Q.-C. Wang, R. Lin, E. Hu, D. Qu and X.-Q. Yang, Mater. Horiz., 2021, 8, 471–500 RSC; (b) P. Sang, Q. Chen, D.-Y. Wang, W. Guo and Y. Fu, Chem. Rev., 2023, 123, 1262–1326 CrossRef CAS PubMed.
- For an overview of catalysts for the synthesis of cyclic carbonates from epoxides and CO2, see: (a) G. A. Bhat and D. J. Darensbourg, Green Chem., 2022, 24, 5007–5034 RSC; (b) P. P. Pescarmona, Curr. Opin. Green Sustainable Chem., 2021, 29, 100457 CrossRef CAS; (c) H. Büttner, L. Longwitz, J. Steinbauer, C. Wulf and T. Werner, Top. Curr. Chem., 2017, 375, 50 CrossRef PubMed; (d) C. Martín, G. Fiorani and A. W. Kleij, ACS Catal., 2015, 5, 1353–1370 CrossRef.
- D. Majumadar, A. Bhanarkar, C. Rao and D. Gouda, Atmos. Environ.: X, 2022, 13, 100157 Search PubMed.
- C. Hardy, G. Kociok-Köhn and A. Buchard, Polym. Chem., 2023, 14, 623–632 RSC.
- E. J. Corey and R. A. E. Winter, J. Am. Chem. Soc., 1963, 85, 2677–2678 CrossRef CAS.
- H. A. Staab and G. Walther, Justus Liebigs Ann. Chem., 1962, 657, 98–103 CrossRef CAS.
- S. Sakai, Y. Kiyohara, K. Itoh and Y. Ishii, J. Org. Chem., 1970, 35, 2347–2350 CrossRef CAS.
- S. Sakai, Y. Kobayashi and Y. Ishii, J. Chem. Soc. D, 1970, 235–236 RSC.
- G. Dousse, H. Lavayssière and J. Satgé, J. Organomet. Chem., 1975, 88, C35–C37 CrossRef CAS.
- G. Dousse, H. Lavayssière and J. Satgé, Synth. React. Inorg. Met.-Org. Chem., 1989, 19, 49–74 CrossRef CAS.
- F. N. Jones and S. Andreades, J. Org. Chem., 1969, 34, 3011–3014 CrossRef CAS.
- S. Kim and K. Y. Yi, J. Org. Chem., 1986, 51, 2613–2615 CrossRef CAS.
- S.-G. Kim and K.-Y. Yi, Bull. Korean Chem. Soc., 1987, 8, 466–470 CAS.
- A. K. Qaroush, A. F. Eftaiha, A. H. Smadi, K. I. Assaf, F. M. Al-Qaisi and F. Alsoubani, ACS Omega, 2022, 7, 22511–22521 CrossRef CAS PubMed.
- R. F. Barghash, A. Massi and A. Dondoni, Org. Biomol. Chem., 2009, 7, 3319–3330 RSC.
- L. Álvarez de Cienfuegos, C. Rodríguez, A. J. Mota and R. Robles, Org. Lett., 2003, 5, 2743–2745 CrossRef PubMed.
- R. Robles, C. Rodríguez, L. Álvarez de Cienfuegos and A. J. Mota, Tetrahedron: Asymmetry, 2004, 15, 831–838 CrossRef CAS.
- H. Tomita, F. Sanda and T. Endo, Macromolecules, 2001, 34, 727–733 CrossRef CAS.
- N. Nemoto, X. Xu, F. Sanda and T. Endo, Macromolecules, 2001, 34, 7642–7647 CrossRef CAS.
- D. D. Reynolds, D. L. Fields and D. L. Johnson, J. Org. Chem., 1961, 26, 5122–5124 CrossRef CAS.
- P. Koch and E. Perrotti, J. Organomet. Chem., 1974, 81, 111–114 CrossRef CAS.
- P. Koch and E. Perrotti, Tetrahedron Lett., 1974, 34, 2899–2900 CrossRef.
- D. D. Reynolds, J. Am. Chem. Soc., 1957, 79, 4951–4952 CrossRef CAS.
- D. D. Reynolds, M. K. Massad, D. L. Fields and D. L. Johnson, J. Org. Chem., 1961, 26, 5109–5110 CrossRef CAS.
- D. D. Reynolds, D. L. Fields and D. L. Johnson, J. Org. Chem., 1961, 26, 5111–5115 CrossRef CAS.
- K. Soga, H. Imamura and S. Ikeda, Makromol. Chem., 1975, 176, 807–811 CrossRef CAS.
- M. Luo, X.-H. Zhang and D. J. Darensbourg, Acc. Chem. Res., 2016, 49, 2209–2219 CrossRef CAS PubMed.
- W.-M. Ren, T.-J. Yue, M.-R. Li, Z.-Q. Wan and X.-B. Lu, Macromolecules, 2017, 50, 63–68 CrossRef CAS.
- Y. Wang, Y. Xia, Z. Hua, C. Zhang and X. Zhang, Polym. Chem., 2022, 13, 5397–5403 RSC.
- J. Kiriratnikom, J. Guo, X. Cao, M. U. Khan, C. Zhang and X. Zhang, J. Polym. Sci., 2022, 60, 3414–3419 CrossRef CAS.
- C. A. L. Lidston, S. M. Severson, B. A. Abel and G. W. Coates, ACS Catal., 2022, 12, 11037–11070 CrossRef CAS.
- M. A. Dewit, A. Beaton and E. R. Gillies, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 3977–3985 CrossRef CAS.
- E. K. Y. Chen, R. A. McBride and E. R. Gillies, Macromolecules, 2012, 45, 7364–7374 CrossRef CAS.
- A. K. Jain, M. G. Gund, D. C. Desai, N. Borhade, S. P. Senthilkumar, M. Dhiman, N. K. Mangu, S. V. Mali, N. P. Dubash, S. Halder and A. Satyam, Bioorg. Chem., 2013, 49, 40–48 CrossRef CAS PubMed.
- D. He, W. Zhang, H. Deng, S. Huo, Y.-F. Wang, N. Gong, L. Deng, X.-J. Liang and A. Dong, Chem. Commun., 2016, 52, 14145–14148 RSC.
- P. Botella, C. Muniesa, V. Vicente and A. Cabrera-García, Mater. Sci. Eng., C, 2016, 58, 692–699 CrossRef CAS PubMed.
- K. Li, W. Dong, L. Qiu, Q. Liu, G. Lv, Y. Peng, M. Xie and J. Lin, Eur. J. Med. Chem., 2019, 181, 111582 CrossRef CAS PubMed.
- Q. Yang, Z. Deng, D. Wang, J. He, D. Zhang, Y. Tan, T. Peng, X.-Q. Wang and W. Tan, J. Am. Chem. Soc., 2020, 142, 2532–2540 CrossRef CAS PubMed.
- W. Zhang, J. Song, B. Zhang, L. Liu, K. Wang and R. Wang, Bioconjugate Chem., 2011, 22, 1410–1415 CrossRef CAS PubMed.
- Q. Liu, C. Ou, C. Ren, L. Wang, Z. Yang and M. Chen, New J. Chem., 2012, 36, 1556–1559 RSC.
- M. A. Miller, R. A. Day, D. A. Estabrook and E. M. Sletten, Synlett, 2022, 450–454 Search PubMed.
- M. Zheng, Y. Wang, H. Shi, Y. Hu, L. Feng, Z. Luo, M. Zhou, J. He, Z. Zhou, Y. Zhang and D. Ye, ACS Nano, 2016, 10, 10075–10085 CrossRef CAS PubMed.
- J. A. Latham and C. Walsh, J. Am. Chem. Soc., 1987, 109, 3421–3427 CrossRef CAS.
- H. R. Kricheldorf and D.-O. Damrau, Macromol. Chem. Phys., 1998, 199, 2589–2596 CrossRef CAS.
- N. Nemoto, F. Sanda and T. Endo, Macromolecules, 2000, 33, 7229–7231 CrossRef CAS.
- K. Kakimoto, N. Nemoto, F. Sanda and T. Endo, Chem. Lett., 2002, 31, 156–157 CrossRef.
- F. Sanda, J. Kamatani and T. Endo, Macromolecules, 1999, 32, 5715–5717 CrossRef CAS.
- M. Luo, X.-H. Zhang and D. J. Darensbourg, Macromolecules, 2015, 48, 5526–5532 CrossRef CAS.
- H.-L. Wu, J.-L. Yang, M. Luo, R.-Y. Wang, J.-T. Xu, B.-Y. Du, X.-H. Zhang and D. J. Darensbourg, Macromolecules, 2016, 49, 8863–8868 CrossRef CAS.
- J. A. Durden, H. A. Stansbury and W. H. Catlette, J. Am. Chem. Soc., 1960, 82, 3082–3084 CrossRef CAS.
- R. C. Forster and L. N. Owen, J. Chem. Soc., Perkin Trans. 1, 1978, 822–829 RSC.
- Y. Taguchi, K. Yanagiya, I. Shibuya and Y. Suhara, Bull. Chem. Soc. Jpn., 1988, 61, 921–925 CrossRef CAS.
- Y. Taguchi, M. Yasumoto, I. Shibuya and Y. Suhara, Bull. Chem. Soc. Jpn., 1989, 62, 474–478 CrossRef CAS.
- S. A. Qadri, S. Islam and M. Ahmad, Mutat. Res., Genet. Toxicol., 1992, 298, 53–60 CrossRef CAS PubMed.
- S. A. Qadri and M. Ahmad, Mutat. Res., Fundam. Mol. Mech. Mutagen., 1994, 311, 199–208 CrossRef CAS PubMed.
- J. Uenishi, M. Motoyama, Y. Nishiyama and S. Wakabayahi, J. Chem. Soc., Chem. Commun., 1991, 1421–1422 RSC.
- J. Uenishi, M. Motoyama, Y. Nishiyama, Y. Hirota, Y. Kubo and H. Akashi, Heteroat. Chem., 1994, 5, 51–60 CrossRef CAS.
- J. Uenishi, M. Motoyama and Y. Kubo, Heteroat. Chem., 1994, 5, 529–532 CrossRef CAS.
- T. W. Hart and B. Vacher, Tetrahedron Lett., 1992, 33, 3009–3012 CrossRef CAS.
- M. B. Comba, A. G. Suárez, A. M. Sarotti, M. I. Mangione, R. A. Spanevello and E. D. Giordano, Org. Lett., 2016, 18, 1748–1751 CrossRef CAS PubMed.
- M. B. Comba, M. I. Mangione, A. G. Suárez, A. M. Sarotti and R. A. Spanevello, Eur. J. Org. Chem., 2018, 6848–6856 CrossRef CAS.
- D. S. Weinstein and K. C. Nicolaou, J. Chem. Soc., Perkin Trans. 1, 1999, 545–558 RSC.
- N. Kihara, Y. Nakawaki and T. Endo, J. Org. Chem., 1995, 60, 473–475 CrossRef CAS.
- S. Motokucho, Y. Itagaki, A. Sudo and T. Endo, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 3711–3717 CrossRef CAS.
- S. Motokucho, D. Takeuchi, F. Sanda and T. Endo, Tetrahedron, 2001, 57, 7149–7152 CrossRef CAS.
- Y.-M. Shen, W.-L. Duan and M. Shi, Eur. J. Org. Chem., 2004, 3080–3089 CrossRef CAS.
- R. Maggi, C. Malmassari, C. Oro, R. Pela, G. Sartori and L. Soldi, Synthesis, 2008, 53–56 CrossRef CAS.
- I. Yavari, M. Ghazanfarpour-Darjani, Z. Hossaini, M. Sabbaghan and N. Hosseini, Synlett, 2008, 889–891 CrossRef CAS.
- A. Z. Halimehjani, F. Ebrahimi, N. Azizi and M. R. Saidi, J. Heterocycl. Chem., 2009, 46, 347–350 CrossRef CAS.
- M. North and P. Villuendas, Synlett, 2010, 623–627 CrossRef CAS.
- W. Clegg, R. W. Harrington, M. North and P. Villuendas, J. Org. Chem., 2010, 75, 6201–6207 CrossRef CAS PubMed.
- C. Beattie and M. North, ChemCatChem, 2014, 6, 1252–1259 CAS.
- M. Sengoden, G. A. Bhat and D. J. Darensbourg, Green Chem., 2022, 24, 2535–2541 RSC.
- J. Cao, M. Yu, H. Li, L. Wang, X. Zhu, G. Wang, Y. Shi and C. Cao, Res. Chem. Intermed., 2014, 41, 5323–5330 CrossRef.
- C. Mei, X. Li, L. Liu, C. Cao, G. Pang and Y. Shi, Tetrahedron, 2017, 73, 5706–5714 CrossRef CAS.
- J. Diebler, A. Spannenberg and T. Werner, ChemCatChem, 2016, 8, 2027–2030 CrossRef CAS.
- J. Diebler, A. Spannenberg and T. Werner, Org. Biomol. Chem., 2016, 14, 7480–7489 RSC.
- M. Ghazanfarpour-Darjani and A. Khodakarami, Monatsh. Chem., 2015, 147, 829–835 CrossRef.
- N. Aoyagi and T. Endo, Synlett, 2020, 92–96 CAS.
- L. Álvarez-Miguel, M. E. G. Mosquera and C. J. Whiteoak, Org. Biomol. Chem., 2022, 20, 9629–9638 RSC.
- L. Álvarez-Miguel, J. Damián Burgoa, M. E. G. Mosquera, A. Hamilton and C. J. Whiteoak, ChemCatChem, 2021, 13, 4099–4110 CrossRef.
- A. K. Yadav and L. D. S. Yadav, Green Chem., 2016, 18, 4240–4244 RSC.
- S. Firoozi and M. Hosseini-Sarvari, Eur. J. Org. Chem., 2020, 3834–3843 CrossRef CAS.
- M. Rumyantsev, I. A. Korablev and S. Rumyantsev, RSC Adv., 2020, 10, 36303–36316 RSC.
- N. M. Bingham, Z. Abousalman-Rezvani, K. Collins and P. J. Roth, Polym. Chem., 2022, 13, 2880–2901 RSC.
- V. B. Purohit, M. Pieta, J. Pietrasik and C. M. Plummer, Polym. Chem., 2022, 13, 4858–4878 RSC.
- W. Choi, F. Sanda and T. Endo, Macromolecules, 1998, 31, 2454–2460 CrossRef CAS.
- A. Steblyanko, W. Choi, F. Sanda and T. Endo, J. Polym. Sci., Part A: Polym. Chem., 2001, 39, 3967–3980 CrossRef CAS.
- S. Motokucho, A. Sudo and T. Endo, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 4459–4464 CrossRef CAS.
- S. Krishnamurthy, Y. Yoshida and T. Endo, Polym. Chem., 2022, 13, 267–274 RSC.
- J. Y. Do, S. B. Shin, S. M. Jeong and M.-Y. Jung, Eur. Polym. J., 2020, 131, 109689 CrossRef CAS.
- For an overview, see: (a) L. Maisonnueve, O. Lamarzelle, E. Rix, E. Grau and H. Cramail, Chem. Rev., 2015, 115, 12407–12439 CrossRef PubMed; (b) G. Rokicki, P. G. Parzuchowski and M. Mazurek, Polym. Adv. Technol., 2015, 26, 707–761 CrossRef CAS.
- N. Kihara, H. Tochigi and T. Endo, J. Polym. Sci., Part A: Polym. Chem., 1995, 33, 1005–1010 CrossRef CAS.
- T. Moriguchi and T. Endo, Macromolecules, 1995, 28, 5386–5387 CrossRef CAS.
- E. Reinheimer, J. Bacsa and K. R. Dunbar, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2004, 60, o1206–o1207 CrossRef CAS.
- J. Kiriratnikom, X. Zhang, X. Cao, B. Chu, C. Zhang and X. Zhang, J. Polym. Sci., 2022, 60, 2262–2268 CrossRef CAS.
- S. Satsumabayashi, H. Takahashi, T. Tanaka and S. Motoki, J. Org. Chem., 1973, 38, 3953–3954 CrossRef CAS.
- T. Mizuno, T. Yamaguchi, I. Nishiguchi, T. Okushi and T. Hirashima, Chem. Lett., 1990, 19, 811–812 CrossRef.
- T. Fujinami, S. Sato, N. Uchida and S. Sakai, Bull. Chem. Soc. Jpn., 1982, 55, 1174–1177 CrossRef CAS.
- M. Barbero, I. Degani, S. Dughera, R. Fochi and L. Piscopo, J. Chem. Soc., Perkin Trans. 1, 1996, 289–294 RSC.
- R. A. Aitken, T. E. Curzon and M. J. Andrews, Front. Chem., 2019, 7, 204 CrossRef CAS PubMed.
- C. Jehanno, J. Demarteau, D. Mantione, M. C. Arno, F. Ruipérez, J. L. Hedrick, A. P. Dove and H. Sardon, ACS Macro Lett., 2020, 9, 443–447 CrossRef CAS PubMed.
- S. Hata, H. Goto, S. Tanaka and A. Oku, J. Appl. Polym. Sci., 2003, 90, 2959–2968 CrossRef CAS.
- Y. Cao and T. Fuchigami, Electrochim. Acta, 2006, 51, 2477–2482 CrossRef CAS.
- E. M. López-Vidal, G. L. Gregory, G. Kociok-Köhn and A. Buchard, Polym. Chem., 2018, 9, 1577–1582 RSC.
- C. Hardy, G. Kociok-Köhn and A. Buchard, Chem. Commun., 2022, 58, 5463–5466 RSC.
- S. Satsumabayashi, S. Motoki and H. Takahashi, Synthesis, 1979, 184–185 CrossRef CAS.
- W. Adam and J.-C. Liu, J. Chem. Soc., Chem. Commun., 1972, 73–74 RSC.
- G. G. Geller, C. S. Foote and D. B. Pechman, Tetrahedron Lett., 1983, 24, 673–676 CrossRef CAS.
- P. R. Schreiner, H. P. Reisenauer, J. Romanski and G. Mloston, Angew. Chem., Int. Ed., 2006, 45, 3989–3992 CrossRef CAS PubMed.
- H. R. Kricheldorf, S.-R. Lee and N. Schittenhelm, Macromol. Chem. Phys., 1998, 199, 273–282 CAS.
- G. Fournet and J. Goré, Tetrahedron Lett., 1993, 34, 7057–7058 CrossRef CAS.
- K. Takai, O. Fujimura, Y. Kataoka and K. Utimoto, Tetrahedron Lett., 1989, 30, 211–214 CrossRef CAS.
- K. Takai, Y. Kataoka, J. Miyai, T. Okazoe, K. Oshima and K. Utimoto, Org. Synth., 1996, 73, 73 CrossRef CAS.
- M. Fallah-Mehrjardi, Monatsh. Chem., 2018, 149, 1931–1944 CrossRef CAS.
- A. W. M. Lee, W. H. Chan and H. C. Wong, Synth. Commun., 1988, 18, 1531–1536 CrossRef CAS.
- M. Soleiman-Beigi and Z. Taherinia, J. Sulphur Chem., 2014, 35, 470–476 CrossRef CAS.
- I. A. Dotsenko, Q. Zhao, A. H. Franz, P. Batoon and N. M. Samoshin, ARKIVOC, 2014, 5, 16–41 Search PubMed.
- N. Aoyagi and T. Endo, Tetrahedron Lett., 2018, 59, 1702–1704 CrossRef CAS.
- C. Chen, Y. Gnanou and X. Feng, Polym. Chem., 2022, 13, 3471–3478 RSC.
- K. Hatanaka and S. Tanimoto, Tetrahedron Lett., 1982, 23, 425–426 CrossRef CAS.
- C. U. Pittman, Jr., M. Narita and Y. F. Liang, Macromolecules, 1976, 9, 360–361 CrossRef.
- N. M. Tucker, A. L. Briseno, O. Acton, H.-L. Yip, H. Ma, S. A. Jenekhe, Y. Xia and A. K.-Y. Jen, ACS Appl. Mater. Interfaces, 2013, 5, 2320–2324 CrossRef CAS PubMed.
- M. Scheele, D. Hanifi, D. Zherebetskyy, S. T. Chourou, S. Axnanda, B. J. Rancatore, K. Thorkelsson, T. Xu, Z. Liu, L.-W. Wang, Y. Liu and A. P. Alivisatos, ACS Nano, 2014, 8, 2532–2540 CrossRef CAS PubMed.
- M. Vicent-Morales, M. Esteve-Rochina, J. Calbo, E. Ortí, I. J. Vitórica-Yrezábal and G. Mínguez Espallargas, J. Am. Chem. Soc., 2022, 144, 9074–9082 CrossRef CAS PubMed.
- D. Cortizo-Lacalle, S. Arumugam, S. E. T. Elmasly, A. L. Kanibolotsky, N. J. Findlay, A. R. Inigo and P. J. Skabara, J. Mater. Chem., 2012, 22, 11310–11315 RSC.
- T. L. A. Nguyen, R. Demir-Cakan, T. Devic, M. Morcrette, T. Ahnfeldt, P. Auban-Senzier, N. Stock, A.-M. Goncalves, Y. Filinchuk, J.-M. Tarascon and G. Férey, Inorg. Chem., 2010, 49, 7135–7143 CrossRef CAS PubMed.
- C. F. Leong, B. Chan, T. B. Faust and D. M. D'Alessandro, Chem. Sci., 2014, 5, 4724–4728 RSC.
- T. Li, X. Yan, W.-D. Zhang, W.-K. Han, Y. Liu, Y. Li, H. Zhu, Z. Li and Z.-G. Gu, Chem. Commun., 2020, 56, 14187–14190 RSC.
- J.-Z. Zhao, T.-J. Yue, B.-H. Ren, Y. Liu, W.-M. Ren and X.-B. Lu, Macromolecules, 2022, 55, 8651–8658 CrossRef CAS.
- B. Wang, S. Yang, L. Min, Y. Gu, Y. Zhang, X. Wu, L. Zhang, E. H. M. Elageed, S. Wu and G. Gao, Adv. Synth. Catal., 2014, 356, 3125–3134 CrossRef CAS.
- N. S. Doherty, T. H. Beaver, G. L. Westrich, F. P. Miller and L. E. Roebel, Drug Dev. Res., 1989, 16, 31–44 CrossRef CAS.
- S. Nitin, K. Ram and C. Devdutt, Res. J. Chem. Environ., 2021, 25, 142–148 CrossRef CAS.
- M. Saeeda, M. Abbas, R. J. Abdel-Jalil, M. Zahid and W. Voelter, Tetrahedron Lett., 2003, 44, 315–317 CrossRef.
- L. D. S. Yadav and S. Singh, Synthesis, 2003, 340–342 CrossRef.
- M. Chandrasekharam, L. Bhat, H. Ila and H. Junjappa, Tetrahedron Lett., 1993, 34, 6439–6442 CrossRef CAS.
- C. Leriverend, P. Metzner and A. Capperucci, Tetrahedron, 1997, 53, 1323–1342 CrossRef CAS.
- A. Sugawara, R. Sugawara, H. Itoh, H. Tanaka, T. Segawa and R. Sato, Chem. Lett., 1991, 20, 1315–1318 CrossRef.
Footnote |
| † These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2023 |