
Asymmetric synthesis of spirocyclic isobenzofuranones via a squaramide-catalysed sulfa-Michael desymmetrisation reaction†
Tamanna
,
Deepak
Sharma
and
Pankaj
Chauhan
 *
*
Department of Chemistry, Indian Institute of Technology Jammu, Nagrota, NH-44, (J&K), 181221, Jagti, India. E-mail: pankaj.chauhan@iitjammu.ac.in; Web: https://www.pankajchauhanresearch.com
First published on 27th February 2023
Abstract
Enantioselective synthesis of spirocyclohexenone isobenzofuranones has been achieved through an organocatalysed sulfa-Michael desymmetrisation reaction. A cinchona-derived squaramide effectively promotes the desymmetrisation of spirocyclic 2,5-cyclohexadienone isobenzofuranones via the controlled addition of various aryl thiols to generate two vicinal stereocenters with perfect diastereoselectivities and up to very good enantioselectivities.
Introduction
Synthesis of benzene-fused γ-lactones such as benzofuranones and isobenzofuranones has always attracted the attention of the organic synthesis community as they are the key structural units of several naturally occurring compounds and synthetic bioactive molecules.1 The isobenzofuranone unit, also known as phthalide, constitutes an important class of organic dyes, i.e. phthalein dyes.2 The isobenzofuranone framework is also an integral component of chiral natural products such as butylphthalide A, an essential aroma and taste constituent of celery oil and used for treating brain ischemia3 (Fig. 1). The tetra-substituted carbon-bearing phthalide dermacozine D4 (B) is a marine alkaloid, and isochlortetracycline5 (C) is a metabolite of chlortetracycline exhibiting antibiotic properties. The spirocyclic naturally occurring isobenzofuranones have also emerged as valuable motifs for biological evaluations. For example, (–)-arnottin II (D) possesses antibiotic activities,6 sinaspirolide (E) is an ingredient of traditional Chinese medicine and (–)-juglanaloid A (F) targets Alzheimer's disease.7 The naturally occurring spirocyclohexane isobenzofuranone ansaspirolide (G) is a serotonin receptor (5-HT7) binding compound, and the synthetic spirocyclohexane isobenzofuranone (H) is a Y-5 receptor antagonist.8 Due to these broad spectra of biological properties, the asymmetric synthesis of spirocyclic isobenzofuranone systems is highly desirable. | ||
| Fig. 1 Biologically active natural and synthetic products containing isobenzofuranone scaffolds. | ||
Asymmetric synthesis of benzofuranones has been widely explored compared to that of isobenzofuranones.9 The most common strategy for procuring enantioenriched isobenzofuranones involves the organo- or metal-catalysed nucleophilic attack from the γ-position of the 3-substituted phthalides (Scheme 1a).10 Another approach for the asymmetric synthesis of isobenzofuranones includes the halolactonization of styrene-type carboxylic acids (Scheme 1b).11 Besides, transition-metal catalysed asymmetric hydrogenation and organocatalysed nucleophilic addition/intramolecular substitution reactions have also evolved as alternative strategies to synthesize phthalide derivatives (Scheme 1c).12 In addition, one-pot aminolactonisation for the asymmetric synthesis of 3-substituted isobenzofuranone is also reported.13 Despite these elegant strategies, we realised that there is a need for the development of a generalised enantioselective route that would synthesize spiro-isobenzofuranones. To the best of our knowledge, the enantioselective synthesis of spiro-cyclohexenone isobenzofuranones remains unexplored.14
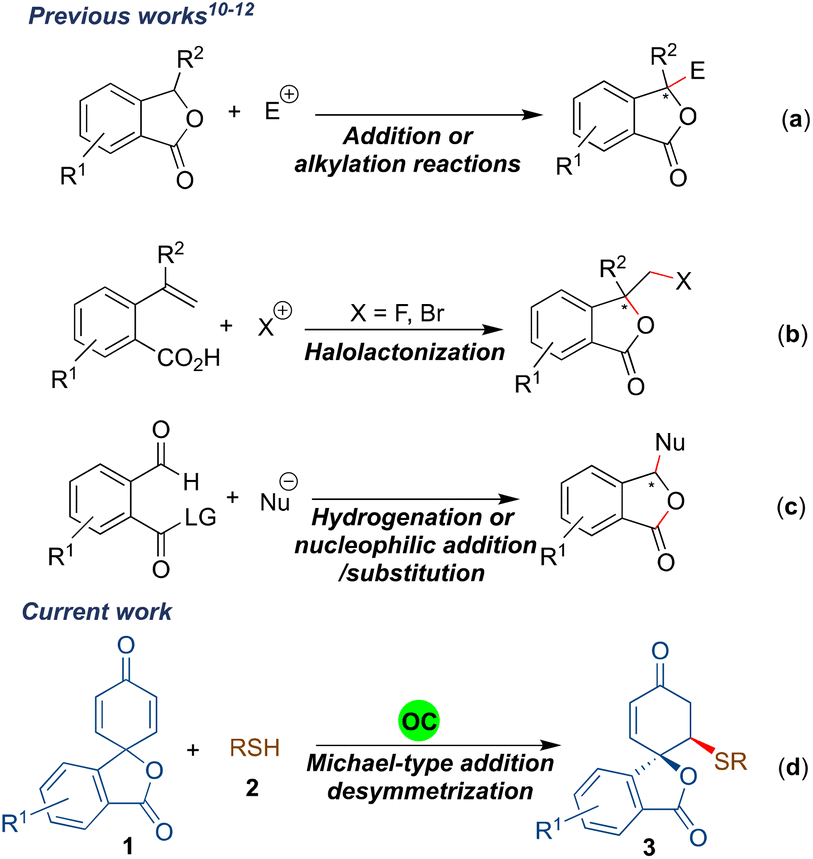 | ||
| Scheme 1 Enantioselective strategies for the synthesis of isobenzofuranones. | ||
Recently, transition metal- or organocatalysed desymmetrisations of 2,5-cyclohexadienones have emerged as excellent strategies for procuring valuable enantiopure entities.15 The majority of the organocatalytic 2,5-cyclohexadienone desymmetrisation reactions have been developed in an intramolecular manner; however, intermolecular desymmetrisations are less explored.16 In the search for an alternative protocol to synthesize stereopure spirocyclic isobenzofuranone derivatives and due to our interest in desymmetrisation reactions,17 we herein report the desymmetrisation of spirophthalide 2,5-cyclohexadienone via bifunctional hydrogen bonding organocatalyst promoted intermolecular sulfa-Michael addition to afford spirocyclohexenone isobenzofuranones bearing two stereogenic centres in a highly diastereo- and enantioselective fashion (Scheme 1d). It was envisaged that a bifunctional hydrogen-bonding organocatalyst would activate the dienone part of the substrate 1a through hydrogen bonding for the sulfa-Michael addition of the thiophenol derivative, which is in turn activated and directed by the tertiary amine moiety of the catalyst.
Results and discussion
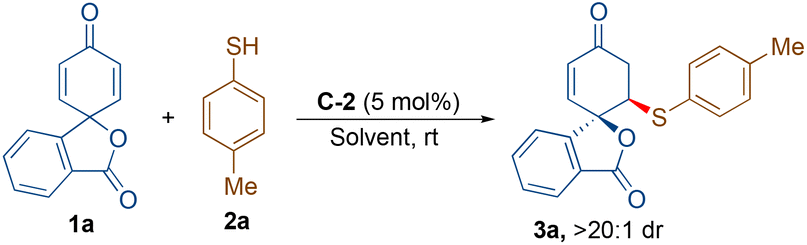
At the onset, we started our investigation by reacting 4-methyl thiophenol (2a) with spirocyclohexadienone 1a catalysed by 5 mol% of bifunctional hydrogen bonding organocatalysts in dichloromethane as the solvent (Scheme 2). Screening of cinchona-derived squaramides C-1 to C-4 revealed that the catalyst C-2 provided the best enantioselectivity (76% ee) of the desired product 3a. Screening of quinine (C-5) and benzyl cupreine C-6 resulted in lower yield and enantio-differentiation. The reason for the moderate to low yields of 3a is the formation of side-product 4avia the double sulfa-Michael addition reaction and unreactive starting materials 1a and 2a. For instance, the yield of 4a with the C-5 catalyst was only 15% and the reaction did not go to completion, as some of the unreactive starting materials 1a and 2a were observed. | ||
| Scheme 2 Catalyst screening. Reaction conditions: 1a (0.11 mmol), 2a (0.1 mmol) and C-1 to C-6 (5 mol%) in 1.0 mL of CH2Cl2 at rt. The minus (–) sign shows the formation of the enantiomer of 3a. Yield refers to the isolated yield of products 3a or ent-3a after column chromatography. Enantiomeric excess (ee) was determined by chiral HPLC. Diastereomeric ratio (dr) was determined by 1H NMR/HPLC. | ||
To improve the yield and ee of 3a, further optimisation of the reaction conditions was performed by solvent screening (Table 1, entries 1–7). In chlorinated solvents such as dichloromethane and chloroform (entries 1 and 2), 3a was isolated in moderate yields and enantioselectivities. The use of toluene as a solvent resulted in a slightly improved yield of 3a, although with a lower ee-value (entry 3). Even though the other ethereal solvents, such as tetrahydrofuran, MTBE and diethyl ether, resulted in lower yields and enantioselectivities of 3a (entries 4–6), 1,4-dioxane turned out to be the best solvent providing maximum enantioselectivity of 88% ee probably due to better H-bonding interactions between the catalyst and substrate molecules in this solvent (entry 7). However, no improvement in the yield of 3a was observed. When performing the reaction in 1,4-dioxane as the solvent, an attempt to improve the yield and enantioselectivity by lowering the reaction temperature to 15 °C did not work (entry 8). Applying molecular sieves (4 Å) as additives resulted in a decreased yield of 56% with an improvement of enantioselectivity to 92% ee (entry 9). It is believed that the molecular sieves help in eliminating the possibility of catalyst contamination with traces of water present in the solvent. Lowering the catalyst loading to 2.5 mol% resulted in a lower yield of 48% without affecting the enantioselectivity, 92% ee of 3a (entry 10). Increasing the equivalents of 1a (entries 11 and 12) and reducing the concentration to 0.05 M (by using 2.0 mL of solvent) resulted in the exclusive formation of 3a in a very good yield of 84% with 92% ee (entry 12).
|
|
||||
|---|---|---|---|---|
| Entry | Solvent | Time [h] | Yieldb [%] | eec [%] |
| a Reaction conditions: 1a (0.11 mmol), 2a (0.1 mmol) and catalyst C-2 (5 mol%) in 1.0 mL of solvent at rt. b Yield refers to the isolated yield of product 3a after flash column chromatography. c Enantiomeric excess (ee) was determined by chiral HPLC. d The reaction was carried out at 15 °C. e 4 Å molecular sieves (50 mg) were used as additives. f 2.5 mol% of C-2 was used. g The reaction was carried out with 2.0 eq. of 1a. h 2.0 mL of solvent was used. Diastereomeric ratio (dr) was determined by 1H NMR/HPLC. | ||||
| 1 | CH2Cl2 | 24 | 60 | 76 |
| 2 | CHCl3 | 24 | 71 | 62 |
| 3 | Toluene | 24 | 74 | 36 |
| 4 | THF | 24 | 68 | 60 |
| 5 | MTBE | 24 | 15 | 38 |
| 6 | Et2O | 24 | 12 | 0 |
| 7 | 1,4-Dioxane | 24 | 65 | 88 |
| 8d | 1,4-Dioxane | 24 | 48 | 84 |
| 9e | 1,4-Dioxane | 24 | 56 | 92 |
| 10e,f | 1,4-Dioxane | 24 | 48 | 92 |
| 11e,g | 1,4-Dioxane | 24 | 77 | 90 |
| 12 , , | 1,4-Dioxane | 48 | 84 | 92 |
With the optimised reaction conditions in hand (Table 1, entry 12), the substrate scope for this transformation was evaluated by screening various thiols 2 with spiro cyclohexadienone 1a (Scheme 3). The aryl thiols bearing electron-withdrawing and electron-releasing substituents at the C-4 position led to the formation of the desired products 3a–e in 62–89% yields with 72–92% ee; however, the 4-OMe substituted thiophenol gave the desired product 3f with a lower yield of 51% and 69% ee. Thiophenol provided the desired product 3g in 62% yield and 82% ee. The halogen substituents at the meta- and ortho-positions of thiophenols gave the desired products 3h and 3j in 86% ee and 84% ee, respectively, with somewhat lower yields of 62% and 59%. The meta-methoxy thiophenol gave the product 3i in a good yield of 85% with 84% ee. 2-Methyl thiophenol gave the product 3k in 39% yield and 65% ee. We further tested naphthalene thiol, which provided 3l with 64% yield and 84% ee. We have also tried aliphatic thiols, such as cyclohexane thiol, n-hexane thiol, ethanethiol, etc., as nucleophiles; however, the desired products were not observed. Furthermore, thiophene-2-thiol was also found to be an unfavourable nucleophile for this transformation.
 | ||
| Scheme 3 Substrate scope. Reaction conditions: 1 (0.2 mmol), 2 (0.1 mmol), 4 Å molecular sieves (50 mg) and C-2 (5 mol%) in 1,4-dioxane (2.0 mL) at 24 °C. Yield refers to the isolated yield of product 3a–o after column chromatography. Enantiomeric excess (ee) was determined by chiral HPLC. aee after re-crystallisation. Diastereomeric ratio (dr) was determined by 1H NMR/HPLC. | ||
The substrate scope was further evaluated by varying isobenzofuran 2,5-cyclohexadienone derivatives. The chloro-substituent at the C-5 position of the benzene ring gave the desired mono-substituted product 3m in a good yield of 65% and enantioselectivity of 89% ee. On the other hand, the methoxy group at the same position delivered the corresponding product 3n in a low yield of 44% and a moderate ee value of 64%. The strongly electronegative fluoro substituent at the C-4 position provided the desired product 3o in a low yield of 23% and a moderate enantioselectivity of 59% ee. In this case, the formation of side product 4b was observed in 42% yield with a poor ee of 20%. The fluoro-substituent at the C-6 position of the aryl moiety did not provide the corresponding mono-substituted product; however, it afforded the disubstituted product 4c in a lower yield of 33% and enantioselectivity of 44% ee. Under the optimised reaction conditions, the formations of spirocyclic isobenzofuran 2,5-cyclohexadienones 3p and 3q from their corresponding starting materials were not observed.
An attempt to obtain the di-substituted product 4a from 1a and 2a was also moderately successful by slightly modifying the reaction conditions, where thiol 2a was added in two installments (1.0 eq. at the beginning of the reaction and 1.2 eq. after 24 hours).18 However, the use of an excess of 2a (3.0 eq.) resulted in the deterioration of enantioselectivity to 42% ee with no improvement in the yield.
The enantioselectivity of products 3b and 4a could be enriched to >99% after re-crystallisation. The absolute configuration of the isobenzofuranone products 3a–o could be assigned by analogy to the single crystal X-ray structure of isobenzofuranone 3b (Fig. 2).
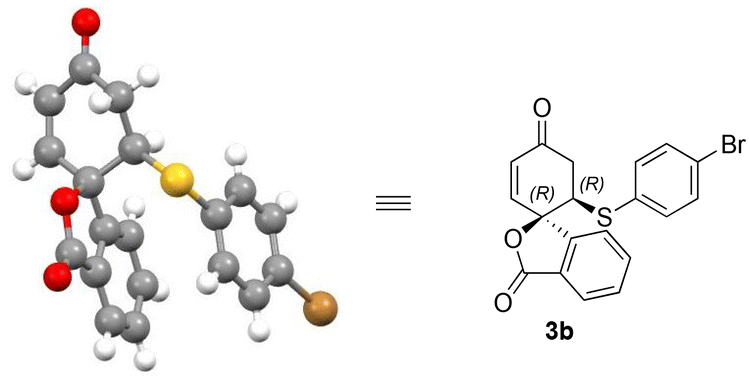
Based on the literature16b and the observed absolute configuration, the reaction was proposed to proceed via the activation of the dienone carbonyl through hydrogen bonding with the NH moiety of the squaramide. The simultaneous activation of the thiol nucleophile occurs by deprotonation through the tertiary amine group of the catalyst for the addition to the activated enone from the Re-face to afford (R,R)-3viaTS-1 (Scheme 4). The formation of (S,R)-3viaTS-2 is not favoured due to the steric interaction between the incoming nucleophile and the aryl moiety of the spiro 2,5-cyclohexadienone substrate 1a.
 | ||
| Scheme 4 Proposed mechanism. | ||
The synthetic utility of product 3a could be explored by the NaBH4-mediated reduction of the carbonyl group of the enone 3a to afford the desired allylic alcohol 5 bearing three stereocenters in excellent diastereoselectivity without much loss in the ee-value (Scheme 5). Additionally, bifunctional squaramide-catalyzed sulfa-Michael addition to 3a resulted in the formation of product 6 in good yield, excellent dr and good ee (84%). Some degree of racemization of 3a was observed as the ee value of 3a decreased from 92% to 87%. The ee-value >99% has been achieved after the re-crystallisation of 6. The one-pot synthesis of 6 from 1a and sequentially adding two thiols led to a poor yield of 6% with 86% ee.
 | ||
| Scheme 5 Synthetic transformations of 3a. aee after re-crystallisation. | ||
Conclusions
In conclusion, we have developed an organocatalytic asymmetric synthesis of spirocyclic isobenzofuranones via the squaramide-catalyzed desymmetrisation reaction. The reactions of spiro-phthalide 2,5-cyclohexadienones with aryl thiols are catalysed by a bifunctional squaramide catalyst, to provide isobenzofuranones bearing a cyclohexenone ring as single diastereomers in moderate to good enantioselectivities (44–92% ee). The virtually stereo-pure products could also be obtained after single-crystallisation. The synthetic applications of the products were also probed by a selective reduction of the carbonyl group to afford spirocyclic benzofuranone bearing allylic alcohol and additional sulfa-Michael addition to provide the disubstituted product bearing three stereogenic centers.Experimental section
General procedure for the synthesis of 3a–o
In a 10 mL reaction tube equipped with a magnetic stirring bar, isobenzofuranone 1 (2.0 eq., 0.2 mmol) and catalyst C-2 (5 mol%) were stirred in 1,4-dioxane (2.0 mL) in the presence of 4 Å molecular sieves (50 mg) at room temperature. After 5 minutes, thiol 2 (1.0 eq., 0.1 mmol) was added, and the stirring was continued at the same temperature. After the complete consumption of thiol 2 (indicated by TLC), the crude product was directly purified by flash column chromatography (n-hexane![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :EtOAc = 8:2) to afford products 3a–o.
:EtOAc = 8:2) to afford products 3a–o.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The generous financial support from the Council of Scientific and Industrial Research (CSIR) India (File No.: 02(0422)/21/EMR-II) and the Royal Society of Chemistry (RSC) UK (R21-7389181735) is highly acknowledged. Tamanna thank the Indian Institute of Technology Jammu and D. S. thank UGC-India for the fellowships.References
- For a review on the naturally occurring benzene-fused γ-lactones, see: (a) J. J. Beck and S. C. Chou, J. Nat. Prod., 2007, 20, 891 CrossRef PubMed. For selected examples, see: (b) S. Piacente, P. Montoro, W. Oleszek and C. Pizza, J. Nat. Prod., 2004, 67, 882 CrossRef CAS PubMed; (c) B. Sontag, M. Rüth, P. Spiteller, N. Arnold, W. Steglich, M. Reichert and G. Bringmann, Eur. J. Org. Chem., 2006, 1023 CrossRef CAS; (d) Y.-J. Kwon, M.-J. Sohn, C.-J. Zheng and W.-G. Kim, Org. Lett., 2007, 9, 2449 CrossRef CAS PubMed; (e) H. M. Ge, C. H. Zhu, D. H. Shi, L. D. Zhang, D. Q. Xie, J. Yang, S. W. Ng and R. X. Tan, Chem. – Eur. J., 2008, 14, 376 CrossRef CAS PubMed; (f) G. Ding, Z. Zheng, S. Liu, H. Zhang, L. Guo and Y. Che, J. Nat. Prod., 2009, 72, 942 CrossRef CAS PubMed; (g) R. Karmakar, P. Pahari and D. Mal, Chem. Rev., 2014, 114, 6213 CrossRef CAS PubMed; (h) R. Wang, L. Jing and D. Qin, Tetrahedron Lett., 2015, 22, 2867 CrossRef.
- R. W. Sabnis, Kirk-Othmer Encyclopedia of Chemical Technology, 2010, DOI:10.1002/0471238961.phthsabn.a01.
- D. H. R. Barton and J. X. de Vries, J. Chem. Soc., 1963, 1916 RSC.
- W. M. A. Mageed, B. F. Milne, M. Wagner, M. Schumacher, P. Sandor, W. Panthom-aree, M. Goodfellow, A. T. Bull, K. Horikoshi, R. Ebel, M. Diederich, H.-P. Fiedler and M. Jaspars, Org. Biomol. Chem., 2010, 8, 2352 RSC.
- D. G. Kennedy, R. J. McCracken, M. P. Carey, W. J. Blanchflower and S. A. Hewitt, J. Chromatogr. A, 1998, 812, 327 CrossRef CAS PubMed.
- M. J. Moschitto, D. R. Anthony and C. A. Lewis, J. Org. Chem., 2015, 80, 3339 CrossRef CAS PubMed.
- Z.-Y. Cheng, Y.-Q. Du, Q. Zhang, B. Lin, P.-Y. Gao, X.-X. Huang and S.-J. Song, Tetrahedron Lett., 2018, 59, 2050 CrossRef CAS.
- (a) S. Deng, S.-N. Chen, P. Yao, D. Nikolic, R. B. Breeman, J. L. Bolton, H. H. S. Fong, N. R. Farnsworth and G. F. Pauli, J. Nat. Prod., 2006, 69, 536 CrossRef CAS PubMed; (b) Y. Ogino, N. Ohtake, Y. Nagae, K. Matsuda, A. Ishikawa, H. Iwaasa, T. Ohe, A. Kanatani and T. Fukami, Bioorg. Med. Chem. Lett., 2008, 18, 4997 CrossRef CAS PubMed.
- Y. Li, X. Li and J. P. Cheng, Adv. Synth. Catal., 2014, 356, 1172 CrossRef CAS.
- For selected examples, see: (a) J. Luo, H. Wang, F. Zhong, J. Kwiatkowski, L.-W. Xu and Y. Lu, Chem. Commun., 2012, 48, 4707 RSC; (b) F. Zhong, J. Luo, G.-Y. Chen, X. Dou and Y. Lu, J. Am. Chem. Soc., 2013, 134, 10222 CrossRef PubMed; (c) Z.-P. Hu, Z. Zhuang and W.-W. Liao, J. Org. Chem., 2015, 80, 4627 CrossRef CAS PubMed; (d) J. Luo, H. Wang, F. Zhong, J. Kwiatkowski, L.-W. Xu and Y. Lu, Chem. Commun., 2013, 49, 5775 RSC; (e) J. Luo, C. Jiang, H. Wang, L.-W. Xu and Y. Lu, Tetrahedron Lett., 2013, 54, 5261 CrossRef CAS; (f) M. Sicignano, R. Schettini, G. Pierri, M. L. Marino, I. Izzo, F. D. Riccardis, L. Bernardi and G. D. Sala, J. Org. Chem., 2020, 85, 7476 CrossRef CAS PubMed.
- (a) H. Egami, J. Asada, K. Sato, D. Hashizume, Y. Kawato and Y. Hamashima, J. Am. Chem. Soc., 2015, 137, 10132 CrossRef CAS PubMed; (b) M. Okada, K. Kaneko, M. Yamanaka and S. Shirakawa, Org. Biomol. Chem., 2019, 17, 3747 RSC.
- For selected examples, see: (a) H.-T. Chang, M. Jeganmohan and C.-H. Cheng, Chem. – Eur. J., 2007, 13, 4356 CrossRef CAS PubMed; (b) B. Zhang, M.-H. Xu and G.-Q. Lin, Org. Lett., 2009, 11, 4712 CrossRef CAS PubMed; (c) H.-Y. Zhang, S.-L. Zhang, L. Liu, G.-S. Luo, W.-H. Duan and W. Wang, J. Org. Chem., 2010, 75, 368 CrossRef CAS PubMed.
- S. Hajra, S. M. S. Akhtar and S. M. Aziz, Chem. Commun., 2014, 50, 6913 RSC.
- For reviews on the asymmetric desymmetrization reactions of 2,5-cyclohexadienones, see: (a) G. Maertens, M.-A. Menard and S. Canesi, Synthesis, 2014, 1573 Search PubMed; (b) K. A. Kalstabakken and A. M. Harned, Tetrahedron, 2014, 70, 9571 CrossRef CAS PubMed; (c) B. Chen, C.-Y. He, W.-D. Chu and Q.-Z. Liu, Org. Chem. Front., 2021, 8, 825 RSC; (d) M. Escolano, D. Gaviña, J. Torres, S. Díaz-Oltra and C. del Pozo, Eur. J. Org. Chem., 2021, 2923 CrossRef CAS.
- K. Liu, H.-L. Teng, L. Yao, H.-Y. Tao and C.-J. Wang, Org. Lett., 2013, 15, 2250 CrossRef CAS PubMed.
- For selected examples, see: (a) L. Yao, K. Liu, H.-Y. Tao, G.-F. Qiu, X. Zhou and C.-J. Wang, Chem. Commun., 2013, 49, 6078 RSC; (b) P. Chauhan, S. Mahajan, U. Kaya, A. Valkonen, K. Rissanen and D. Enders, Adv. Synth. Catal., 2016, 358, 3173 CrossRef CAS.
- Tamanna, Y. Hussain, D. Sharma and P. Chauhan, J. Org. Chem., 2022, 87, 6397 CrossRef CAS PubMed.
- See the ESI.†.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2221710. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3ob00126a |
| This journal is © The Royal Society of Chemistry 2023 |