
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTotal synthesis of isoneoantimycin†
Yoshinosuke
Usuki
 *,
Yuka
Tanaka
,
Miyu
Morii
and
Tetsuya
Satoh
*,
Yuka
Tanaka
,
Miyu
Morii
and
Tetsuya
Satoh
Department of Chemistry, Graduate School of Science, Osaka Metropolitan University, 3-3-138 Sugimoto, Sumiyoshi, Osaka 558-8585, Japan. E-mail: usuki@omu.ac.jp
First published on 21st February 2023
Abstract
Antimycins are one of the well-known antifungal metabolites produced by Streptomyces bacteria. Neoantimycin and its analogues, the ring-expanded antimycins featuring a 15-membered tetraester ring, have been shown to be effective regulators of the oncogenic proteins GRP78/BiP and K-Ras. Isoneoantimycin was isolated from Streptomyces fradiae IFO12773 (ISP 5063) as a minor metabolite during the fermentation of neoantimycin and is the first reported antibiotic of the antimycin family without the macrolide core. In this study, we explored the total synthesis and stereochemical assignment of isoneoantimycin as an approach to perform structure–activity studies on neoantimycins. Taking the neoantimycin biosynthesis pathway into account, we presumed that the stereochemistry of isoneoantimycin is the same as that of neoantimycin. The synthesis of our target molecule with the (1S,2R,5S,6S,14R,15R,17S) configuration has been achieved by using chiral-pool building blocks. A comparison of the spectroscopic data between the synthetic and natural samples verified our presumption of the stereochemistry of natural isoneoantimycin.
Introduction
Isoneoantimycin (1) was isolated from Streptomyces fradiae IFO12773 (ISP 5063) as a minor metabolite produced during the fermentation of neoantimycin (2) by Takeda and co-workers in 1998.1 2D NMR analyses and EIMS fragmentation revealed that the planar structure of compound 1 comprises units identical to those of neoantimycin; isoneoantimycin is the first reported antibiotic of the antimycin family without the macrolide core and consists of five components, namely γ-butyrolactone, α-hydroxy-isovaleric acid, L-threonine, α-hydroxy-β-methyl-valeric acid, and 3-formylaminosalicylic acid, as shown in Fig. 1. However, the absolute configuration of 1 could not be elucidated at that time. | ||
| Fig. 1 Structures of isoneoantimycin (1), neoantimycin (2), and antimycin A (3). | ||
After the discovery of antimycin A in 1949,2 many antimycin-class depsipeptides have been reported.3 However, very few papers on compounds without a macrolactone structure have been published.1,4 The diverse biological activities of these antibiotics, including anti-cancer, antifungal, and immunosuppressant properties, have intrigued researchers and attracted significant attention from them.5 Neoantimycin and its analogues, the ring-expanded antimycins, have been shown to be effective regulators of the oncogenic proteins GRP78/BiP and K-Ras.6
During our exploration of antimycin-type antibiotics,7 we were engaged in studies directed toward the total synthesis and stereochemical assignment of 1 as an approach to perform structure–activity studies on neoantimycins. Herein, we report our success in these endeavours.
Results and discussion
The absolute configuration of 2 has been confirmed, as shown in Fig. 1, by total synthesis.7b Recently, the biosynthetic pathway for neoantimycin production was proposed, wherein 3-formylaminosalicylic acid is employed as the starting substrate in the hybrid NRPS (nonribosomal peptide synthetase)/PKS (polyketide synthase) machinery for the growth of the depsipepitidal chain.8 The subsequent macrolactonization and ketoreductase reduction provide neoantimycin. Taking the neoantimycin biosynthesis pathway into account, we presumed that the stereochemistry of 1 is the same as that of neoantimycin. Compound 4 was set as the target molecule for our synthesis (Scheme 1). Our synthetic strategy involves amide formation between amine 6 (four components linearly connected) and the protected 3-formylaminosalicylic acid 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 9 to afford the desired compound (1S,2R,5S,6S,14R,15R,17S)-4. Conceptually, 6 could be divided into left and right units; the left part 8 would be obtained by the condensation of N-Cbz L-threonine tert-butyl ester 910 and α-hydroxy carboxylic acid derivative 10.11 The right part 7 would be obtained by the condensation of α-hydroxy carboxylic acid 1112 with 3-hydoxy-γ-butyrolactone 12, which is derived from D-phenylalanine.
9 to afford the desired compound (1S,2R,5S,6S,14R,15R,17S)-4. Conceptually, 6 could be divided into left and right units; the left part 8 would be obtained by the condensation of N-Cbz L-threonine tert-butyl ester 910 and α-hydroxy carboxylic acid derivative 10.11 The right part 7 would be obtained by the condensation of α-hydroxy carboxylic acid 1112 with 3-hydoxy-γ-butyrolactone 12, which is derived from D-phenylalanine.
 | ||
| Scheme 1 Retrosynthesis of the target molecule (4). | ||
The Mukaiyama aldol reaction of aldehyde 14,13 derived from D-phenylalanine, with the commercially available silyl ketene acetal 13 in the presence of SnCl4 proceeded exclusively to afford the corresponding syn-15 in 83% yield.14 Hydrogenolysis of 15 with Pd(OH)2 in EtOH resulted in the removal of the benzyl ether protecting group and this was followed by a spontaneous intramolecular trans-esterification to provide 127b in 97% yield. The O-Bn protected acid 11,12 prepared from L-valine, was treated with oxalyl chloride and a catalytic amount of DMF to produce the corresponding acid chloride, and the subsequent condensation with 12 provided the corresponding 16 in 54% yield. Reductive deprotection of the benzyl group with Pd(OH)2 in AcOEt under a hydrogen atmosphere afforded the desired right fragment 7 in 90% yield (Scheme 2).
 | ||
| Scheme 2 Synthesis of the left fragment 7. | ||
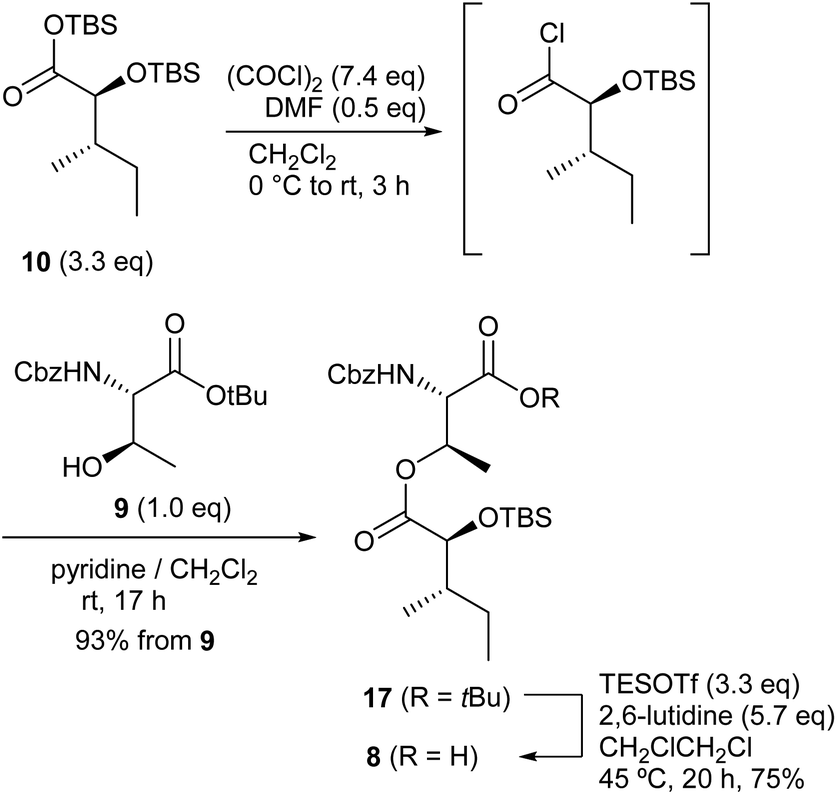
The commercially available N-Cbz L-threonine was converted into tert-Bu ester 910 in 97% yield. Bis-TBS protected 10, prepared from L-isoleucine, was treated with oxalyl chloride and a catalytic amount of DMF to produce the corresponding acid chloride intermediate and the subsequent condensation with 9 provided 17 in 93% yield. The tert-Bu group of 17 was selectively removed with TESOTf-2,6-lutidine in 1,2-dichloroethane to afford the left fragment 8 in 75% yield (Scheme 3).15
 | ||
| Scheme 3 Synthesis of the right fragment 6. | ||
Condensation of 7 and 8 with EDCI and DMAP provided the corresponding 18 in 67% yield (Scheme 4). In the endgame of the total synthesis of 4, the TBS group of 18 was removed with HF·Pyr in THF-Pyr to afford 19 in 95% yield. The subsequent hydrogenolysis with Pd(OH)2 in EtOAc provided amine 6. The subsequent condensation of 6 with benzyl ether 59 using EDCI, HOBt, and NMM in DCM provided the corresponding amide 20. Finally, the reductive removal of the benzyl ether protecting group was accomplished in the presence of a catalytic amount of Pd(OH)2 in EtOAc under a hydrogen atmosphere to provide synthetic isoneoantimycin (4) in 31% yield from 19.16
 | ||
| Scheme 4 Endgame in the synthesis of 4. | ||
The optical rotation of synthetic 4 ([α]D +40.9, c 0.51, EtOH) was consistent with that of the natural product ([α]D +53.8, c 0.26, EtOH).1 A comparison of the 1H NMR chemical shifts of synthetic 4 with those reported for the natural sample 1 showed that the chemical shifts of the two methine CH protons at C6 and C18 were misperceived in ref. 1 (Fig. 2(a) red bar). Swapping the ΔδH of C6 and the ΔδH of C18 resulted in a good agreement (Fig. 2(a) blue bar). The 13C NMR spectra of synthetic 4 was almost identical to those of the natural product (Fig. 2(b)). Two fragment ions were observed at m/z 320.1625 and 566.2268 in the HREIMS spectrum of 4, as before (Fig. S1†). However, chromatographic profiles have not been reported for the natural sample of isoneoantimycin as they not available at this moment. We could not ensure the absolute configuration of natural 1 any further.
 | ||
| Fig. 2 Differences between the NMR chemical shifts of natural isoneoantimycin (1) and synthetic 4. (a) ΔδH (in ppm) = δH of 4 – originally reported δH of 1 (red bar) or corrected δH of 1 (blue bar). (b) ΔδC (in ppm) = δC of 4 – originally reported δC of 1. | ||
Conclusions
In summary, the total synthesis of isoneoantimycin with the (1S,2R,5S,6S,14R,15R,17S) configuration has been achieved. A comparison of the spectroscopic data between synthetic 4 and natural 1 verified our presumption. Further studies on the biological activities of synthetic 4 are now in progress, and the results will be reported in due course.Experimental
General information and materials
1H NMR and 13C NMR spectra were recorded on a Bruker Biospin AVANCE III HD 400 (400 MHz) or JEOL JNM-ECZ400S (400 and 100 MHz) or Bruker Avance III 600 (600 and 150 MHz) instrument. Data are reported as follows: chemical shift, multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet and br = broad), coupling constant in Hz, integration. Coupling constants were determined directly from 1H NMR and 13C NMR spectra. The chemical shifts are reported in δ (ppm) values relative to CHCl3 (δ 7.26 ppm for 1H NMR and δ 77.0 ppm for 13C NMR) and to Me4Si (δ 0.00 ppm for 1H NMR). Mass spectra were obtained on a JEOL AccuTOF LC-plus JMS-T100LP (DART, ESI) spectrometer. HREIMS data were collected using a JEOL JMS-700 mass spectrometer. Infrared absorption spectra (IR) were measured using a JASCO FT/IR-4600 Fourier transform infrared spectrometer. UV spectra were recorded on a Shimadzu UV-1900 spectrophotometer. Optical rotations were measured on a JASCO P-2200 with a path length of 0.1 dm at ambient temperature; the concentrations are reported in g dL−1.All air and moisture-sensitive reactions were carried out in an argon-flushed 2-necked flask sealed with a rubber septum, and dried solvents and reagents were introduced using a syringe. Tetrahydrofuran (THF) was freshly distilled under an argon atmosphere from sodium benzophenone ketyl. Flash column chromatography was carried out using Kanto Chemical silica gel 60N (spherical, neutral, 40–50 μm), and pre-coated Merck silica gel plates (Art5715 Kieselgel 60F254, 0.25 mm) were used for thin-layer chromatography (TLC). TLC visualization was performed using UV (254 nm) or a charring solution (ethanoic p-anisaldehyde, ethanoic phosphomolybdic acid).
13 (1.47 g, 6.1 mmol) in CH2Cl2 (26 mL) was added SnCl4 (1.0 M CH2Cl2 solution, 7.4 mL, 7.4 mmol) dropwise at −78 °C. After being stirred for 40 min at −78 °C, commercially available ketene silyl acetal 13 (1.61 g, 9.2 mmol) was added and then the mixture was stirred for 4 h at −78 °C. The reaction mixture was allowed to warm up to room temperature and quenched with sat. aq. NaHCO3 (40 mL). The aqueous layer was extracted with Et2O. The combined organic layer was dried over Na2SO4, filtered and concentrated. The residue was purified by flash column chromatography (5% AcOEt in n-hexane) to give ester 15 (1.743 g, 5.1 mmol, 83%) as a colorless oil: 1H NMR (400 MHz, CDCl3) δ 7.43–7.10 (m, 10H), 4.55 (d, J = 10.7 Hz, 1H), 4.36 (d, J = 10.7 Hz, 1H), 3.70 (ddd, J = 9.0, 5.2, 1.3 Hz, 1H), 3.39 (d, J = 11.0 Hz, 1H), 3.32 (s, 3H), 3.26 (dd, J = 10.9, 1.3 Hz, 1H), 3.08 (dd, J = 13.1, 5.2 Hz, 1H), 3.00 (dd, J = 13.1, 9.0 Hz, 1H), 1.21 (s, 3H), 1.05 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 177.34, 138.10, 137.70, 129.51, 128.53, 128.27, 128.16, 127.75, 126.30, 79.21, 77.36, 71.75, 51.52, 44.79, 36.95, 23.79, 22.21; HRMS (ESI+) m/z calcd for C21H26NaO4 [M + Na]+ 365.17288, found 365.17324. IR (KBr): 3448, 3029, 2948, 2926, 1735, 1142, 1065, 743, 699 cm−1; [α]D = +14.0 (c 1.20, CHCl3).
12 (565 mg, 2.7 mmol) and DMF (2 drops) in CH2Cl2 (5 mL) was added oxalyl chloride (390 μL, 4.5 mmol) dropwise at 0 °C. The mixture was stirred at room temperature for 1.5 h. Then, the volatiles were removed in vacuo. To the crude carboxylic acid chloride thus obtained were added CH2Cl2 (1.0 mL), pyridine (370 μL, 4.5 mmol) and DMAP (30 mg, 0.25 mmol) at room temperature. After being stirred for 10 min, a solution of 12 (198 mg, 0.90 mmol) in CH2Cl2 (1.0 mL) was added and then stirred at room temperature for 3 h. The reaction mixture was quenched by adding brine (30 mL). The aqueous layer was extracted with AcOEt (60 mL × 3). The combined organic layer was washed with brine, dried over Na2SO4, filtered and concentrated. The residue was purified by flash column chromatography (30% AcOEt in n-hexane) to give 16 (0.200 g, 0.488 mmol, 54%) as a white solid.
1H NMR (400 MHz, CDCl3) δ 7.38–7.17 (m, 10H), 5.40 (d, J = 3.8 Hz, 1H), 4.80 (dt, J = 9.1, 4.0 Hz, 1H), 4.71 (d, J = 11.6 Hz, 1H), 4.40 (d, J = 11.6 Hz, 1H), 3.85 (d, J = 4.4 Hz, 1H), 2.99 (dd, J = 14.5, 9.1 Hz, 1H), 2.83 (dd, J = 14.5, 4.2 Hz, 1H), 2.23–2.11 (m, 1H), 1.34 (s, 3H), 1.21 (s, 3H), 1.04 (d, J = 6.9 Hz, 3H), 0.98 (d, J = 6.9 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ 179.24, 171.63, 137.38, 136.52, 129.22, 128.89, 128.59, 128.15, 128.10, 127.25, 82.70, 80.08, 78.26, 73.12, 44.84, 35.58, 31.62, 22.98, 19.57, 18.51, 17.01; HRMS (ESI+) m/z calcd for C25H30NaO5 [M + Na]+ 433.19909, found 433.19734; IR (KBr): 2965, 1750, 1497, 1457, 1387, 1347, 1190, 1133, 1109, 1073, 1003, 733, 697, 620 cm−1; [α]D = +32.9 (c 0.54, CH2Cl2).
IR (KBr): 3490, 2973, 2936, 2876, 1765, 1458, 1393, 1207, 1148, 2048, 1025, 1005, 979, 711, 624 cm−1; [α]D = +108.8 (c 1.78, CH2Cl2).
11 (1.10 g) and DMF (35 μL, 0.45 mmol) in CH2Cl2 (9.0 mL) was added oxalyl chloride (0.55 mL, 6.41 mmol) dropwise at 0 °C. The mixture was stirred at 0 °C for 2 h and then warmed to room temperature and stirred for an additional 1 h. Then, the volatiles were removed in vacuo. To the crude carboxylic acid chloride thus obtained were added a solution of alcohol 910 (269 mg, 0.869 mmol) in CH2Cl2 (0.5 mL) and pyridine (3.7 mL) dropwise at room temperature. After being stirred for 14 h, the reaction mixture was diluted with THF (120 mL) and filtered through a pad of Celite® and the filtrate was concentrated in vacuo. The crude residue was dissolved in AcOEt (50 mL) and then washed with H2O, sat. aq. NaHCO3 and brine, dried over Na2SO4, and concentrated in vacuo. Purification by flash column chromatography (10% AcOEt in n-hexane) afforded ester 17 as a colorless oil (436 mg, 0.81 mmol, 93%). 1H NMR (400 MHz, CDCl3) δ 7.42–7.28 (m, 5H), 5.43 (td, J = 6.4, 2.3 Hz, 1H), 5.39 (d, J = 9.8 Hz, 1H), 5.21–5.07 (m, 2H), 4.37 (dd, J = 9.7, 2.3 Hz, 1H), 4.18 (d, J = 3.7 Hz, 1H), 3.99 (d, J = 4.2 Hz, 1H), 1.83–1.69 (m, 1H), 1.57–1.46 (m, 1H), 1.44 (s, 9H), 1.29 (d, J = 6.4 Hz, 3H), 1.23–1.11 (m, 1H), 0.94 (s, 9H), 0.91 (s, 3H), 0.89 (s, 9H), 0.85 (t, J = 7.4 Hz, 3H), 0.13 (d, J = 7.3 Hz, 3H), 0.10 (s, 3H), 0.02 (s, 3H), 0.00 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 172.29, 168.82, 156.75, 136.36, 128.68, 128.35, 128.21, 82.91, 77.36, 76.41, 71.08, 67.36, 58.28, 39.65, 28.11, 25.85, 24.00, 18.33, 17.41, 15.78, 11.76, −4.67, −5.20; HRMS (ESI+) m/z calcd for C28H47NaO7Si [M + Na]+ 560.30197, found 560.29795. IR (KBr): 2960, 2933, 2858, 1734, 1517, 1461, 1255, 1143, 1065, 838, 778 cm−1; [α]D = −2.6 (c 1.06, CH2Cl2).
9(216 mg, 0.80 mmol), HOBt (109 mg, 0.81 mmol), EDCI·HCl (155 mg, 1.00 mmol), and NMM (87 μL, 0.79 mmol) successively. The reaction mixture was stirred at room temperature overnight, diluted with water, and extracted 3 times with AcOEt. The combined organic layers were washed with water and brine, dried over Na2SO4, and concentrated in vacuo. The resulting amide 20 was used for the next step without further purification.
To a solution of this material (401 mg) in AcOEt (15 mL) was added 20% Pd(OH)2 (90 mg). The inner atmosphere was purged with H2. After being stirred overnight at room temperature, the resulting mixture was filtered through a pad of Celite® and the filtrate was concentrated. The residue was purified by flash column chromatography (30% AcOEt in n-hexane) to give the white solid 4 (57.8 mg, 83 μmol, 31% for 3 steps) as a 6:1 mixture of rotamers; 1H NMR (600 MHz, CDCl3) Major isomer δ 12.54 (s, 1H), 8.54 (dd, J = 8.1, 1.3 Hz, 1H), 8.49 (d, J = 1.7 Hz, 1H), 7.91 (s, 1H), 7.34–7.24 (m, 3H), 7.22 (dd, J = 8.1, 1.3 Hz, 1H), 7.22 (dd, J = 8.1, 1.3 Hz, 1H), 7.01 (d, J = 8.8 Hz, 1H), 6.92 (t, J = 8.1 Hz, 1H), 5.68 (qd, J = 6.5, 3.0 Hz, 1H), 5.32 (d, J = 3.7 Hz, 1H), 5.14 (dd, J = 8.8, 3.1 Hz, 1H), 5.02 (d, J = 2.9 Hz, 1H), 4.77 (dt, J = 9.5, 3.5 Hz, 1H), 4.10 (d, J = 4.0 Hz, 1H), 3.02 (dd, J = 14.7, 9.5 Hz, 1H), 2.83 (dd, J = 14.6, 3.5 Hz, 1H), 2.37–2.31 (m, 1H), 1.84–1.77 (m, 1H), 1.42 (d, J = 6.4 Hz, 3H), 1.43–1.37 (m, 1H), 1.32 (s, 3H), 1.31–1.24 (m, 1H), 1.19 (s, 3H), 1.13 (d, J = 6.9 Hz, 3H), 1.00 (d, J = 6.9 Hz, 3H), 0.97 (d, J = 6.9 Hz, 3H), 0.91 (d, J = 7.4 Hz, 1H); minor isomer (diagnostic peaks only) δ 12.40 (s, 1H), 8.77 (d, J = 11.5 Hz, 1H), 7.78 (d, J = 11.4 Hz, 1H), 7.37 (d, J = 7.9 Hz, 1H), 1.51 (d, J = 6.4 Hz, 1H); 13C NMR (151 MHz, CDCl3) Major isomer δ 178.95, 173.96, 170.29, 169.03, 168.31, 159.10, 150.71, 136.67, 129.36, 128.86, 127.58, 127.20, 125.02, 120.20, 119.26, 112.90, 80.34, 79.45, 77.87, 74.59, 71.86, 55.44, 44.71, 39.13, 35.45, 30.01, 24.40, 22.92, 19.56, 18.43, 17.10, 16.36, 15.29, 11.97; minor isomer (diagnostic peaks only) δ 161.07, 151.43, 129.32, 127.30, 120.97, 120.54; HRMS (ESI+) m/z calcd for C36H46N2NaO12 [M + Na]+ 721.29484, found 721.29106; UV λEtOHmax nm (ε) 346 (3842), 182 (16384); IR (KBr): 3369, 2968, 2935, 2877, 1751, 1686, 1534, 1364, 1251, 1200, 1132, 1067 cm−1; [α]25D = +42.0 (c 0.19, EtOH).
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by a Grant-in-Aid for Scientific Research (C) (No. 22K06666). We are grateful to Dr Matsumi Doe, Analytical Division, Osaka Metropolitan University, for the 2D NMR measurements.References
- Isolation and structure elucidation of isoneoantimycin: Y. Takeda, T. Masuda, T. Matsumoto, Y. Takechi, T. Shingu and H. G. Floss, J. Nat. Prod., 1998, 61, 978 CrossRef CAS PubMed.
- (a) C. Leben and G. W. Keitt, Phytopathology, 1949, 39, 529 CAS; (b) B. R. Dunshee, C. Leben, G. W. Keitt and F. M. Strong, J. Am. Chem. Soc., 1949, 71, 2436 CrossRef CAS.
- For a recent review, see: J. Liu, X. Zhu, S. J. Kim and W. Zhang, Nat. Prod. Rep., 2016, 33, 1146 RSC.
- (a) Z. Han, Y. Xu, O. McConnell, L. Liu, Y. Li, S. Qi, X. Huang and P. Qian, Mar. Drugs, 2012, 10, 668 CrossRef CAS PubMed; (b) T. Nogawa, A. Okano, C. L. Lim, Y. Futamura, T. Shimizu, S. Takahashi, D. Ibrahim and H. Osada, J. Antibiot., 2017, 70, 222 CrossRef CAS PubMed.
- (a) C. J. Barrow, J. J. Oleynek, V. Marinelli, H. H. Sun, P. Kaplita, D. M. Sedlock, A. M. Gillum, C. C. Chadwick and R. Cooper, J. Antibiot., 1997, 50, 729 CrossRef CAS PubMed; (b) S. P. Tzung, K. M. Kim, G. Basanez, C. D. Giedt, J. Simon, J. Zimmerberg, K. Y. Zhang and D. M. Hockenbery, Nat. Cell Biol., 2001, 3, 183 CrossRef CAS PubMed; (c) L. S. Huang, D. Cobessi, E. Y. Tung and E. A. Berry, J. Mol. Biol., 2005, 351, 573 CrossRef CAS PubMed; (d) X. Lin, Y. Zhou, L. Liu, H. Zhu, Y. Chen, S. Wang, F. Sun, L. Chai, B. Liu, S. Xu and H.-W. Lin, Front. Chem., 2019, 7, 481 CrossRef CAS PubMed; (e) J. Seidel, Y. Miao, W. Porterfield, W. Cai, X. Zhu, S. J. Kim, F. Hu, S. Bhattarai-Kline, W. Min and W. Zhang, Chem. Commun., 2019, 55, 9379 RSC.
- (a) Y. Umeda, S. Chijiwa, K. Furihata, S. Sakuda, H. Nagasawa, H. Watanabe and K. Shin-ya, J. Antibiot., 2005, 58, 206 CrossRef CAS PubMed; (b) M. Izumikawa, J. Y. Ueda, S. Chijiwa, M. Takagi and K. Shin-ya, J. Antibiot., 2007, 60, 640 CrossRef CAS PubMed; (c) A. A. Salim, K. J. Cho, L. Tan, M. Quezada, E. Lacey, J. F. Hancock and R. J. Capon, Org. Lett., 2014, 16, 5036 CrossRef CAS PubMed.
- (a) Y. Usuki, H. Ogawa, K.-I. Yoshida, T. Inaoka and H. Iio, Asian J. Org. Chem., 2015, 4, 737 CrossRef CAS; (b) H. Ogawa, H. Iio and Y. Usuki, Chem. Lett., 2015, 44, 1214 CrossRef CAS; (c) Y. Usuki, C. Hamada and T. Satoh, Org. Biomol. Chem., 2017, 15, 7346 RSC; (d) C. Hamada, Y. Usuki, D. Takeuchi, H. Ogawa, R. Abe and T. Satoh, Org. Lett., 2019, 21, 965 CrossRef CAS PubMed.
- (a) X. Li, R. Zvanych, S. A. Vanner, W. Wang and N. A. Magarvey, Bioorg. Med. Chem. Lett., 2013, 23, 5123 CrossRef CAS PubMed; (b) W. Skyrud, J. Liu, D. Thankachan, M. Cabrera, R. F. Seipke and W. Zhang, ACS Chem. Biol., 2018, 13, 1398 CrossRef CAS PubMed; Y. Zhou, X. Lin, S. R. Williams, L. Liu, Y. Shen, S.-P. Wang, F. Sun, S. Xu, H. Deng, P. F. Leadlay and H.-W. Lin, ACS Chem. Biol., 2018, 13, 2153 Search PubMed.
- G. R. Pettit, T. H. Smith, S. Feng, J. C. Knight, R. Tan, R. K. Pettit and P. A. Hinrichs, J. Nat. Prod., 2007, 70, 1073 CrossRef CAS PubMed.
- I. Wilson and R. F. W. Jackson, J. Chem. Soc., Perkin Trans. 1, 2002, 2845 RSC.
- A. Murai, Y. Amino and T. Ando, J. Antibiot., 1985, 38, 1610 CrossRef CAS PubMed.
- G. J. Hanson, Q. Wei and M. Zhou, US Patent0163446, 2009 Search PubMed.
- (a) H. W. Yang and D. Romo, J. Org. Chem., 1998, 63, 1344 CrossRef CAS; H. Lubin, A. Tessier, G. Chaume, J. Pytkowicz and T. Brigaud, Org. Lett., 2010, 12, 1496 Search PubMed.
- For a review on the Mukaiyama aldol reaction, see: J.-I. Matsuo and M. Murakami, Angew. Chem., Int. Ed., 2013, 52, 9109 CrossRef CAS PubMed.
- A. B. Jones, A. Villalobos, R. G. Linde II and S. J. Danishefsky, J. Org. Chem., 1990, 55, 2786 CrossRef CAS.
- The 1H and 13C NMR spectra indicated that synthetic 4 existed as an ca. 6:1 mixture of two rotamers. This phenomenon was also observed in the total synthesis of 15-membered ring neoantimycin antibiotics.7a,b.
Footnote |
| † Electronic supplementary information (ESI) available: Comparisons of NMR data of natural and synthetic compounds (Table S1), HREIMS fragmentation (Fig. S1), experimental details for known compounds and NMR spectra for new compounds. See DOI: https://doi.org/10.1039/d3ob00099k |
| This journal is © The Royal Society of Chemistry 2023 |