
Visible/solar-light-driven thiyl-radical-triggered synthesis of multi-substituted pyridines†
Ashish Kumar
Sahoo
,
Amitava
Rakshit
,
Avishek
Pan
,
Hirendra Nath
Dhara
and
Bhisma K.
Patel
 *
*
Department of Chemistry, Indian Institute of Technology Guwahati, 781039, Assam, India. E-mail: patel@iitg.ac.in
First published on 24th January 2023
Abstract
A light-triggered synthesis of thio-functionalized pyridines is demonstrated using γ-ketodinitriles, thiols, and eosin Y as the photocatalyst. The reaction proceeds via the selective attack on one of the cyano groups by an in situ generated thiyl radical. The reaction also proceeds with nearly equal efficiency using direct sunlight. Large-scale synthesis and a few useful synthetic transformations of the substituted pyridines are also performed.
Photochemical transformations that take place by harnessing visible light have grabbed significant attention due to their sustainability, unique reactivity, and operational simplicity.1 Besides the use of LEDs as a source of visible light, efforts have been made to utilize renewable sources of energy such as sunlight in photochemical reactions.17a Usually, a photochemical reaction is triggered by visible light in combination with a suitable photocatalyst (organic dyes or transition-metal complexes), facilitating the SET process for the construction of various organic frameworks which otherwise require thermal conditions.2 Though transition-metal complexes are better for the excitation of organic molecules, the employment of organic dyes has also been useful in many photochemical transformations. In particular, eosin Y has become a competitor to metal catalysts in various reactions due to its cost effectiveness.3
Owing to their prevalence in various natural products and pharmaceuticals, pyridine frameworks have found applications in the fields of biology, medicinal sciences, advanced materials, etc.4 Moreover, pyridine-containing compounds are used in cancer therapy,5a asymmetric catalysis,5b and supramolecular chemistry.5c For instance, two well-known drugs Aciphex (I)6a and Nexium (esomeprazole) (II)6b are used for the treatment of duodenal ulcers and acid reflux, and both contain pyridine backbones (Fig. 1). There are other pyridine-bearing drugs such as amrinone D (Inocor) (III) and etoricoxib E (Arcoxia) (IV) used for the treatment of acute heart failure and arthritis.6c,d Avandia (rosiglitazone A) (V) and Actos (pioglitazone B) (VI) which are used as antidiabetic drugs also contain the pyridine backbone.6e,f
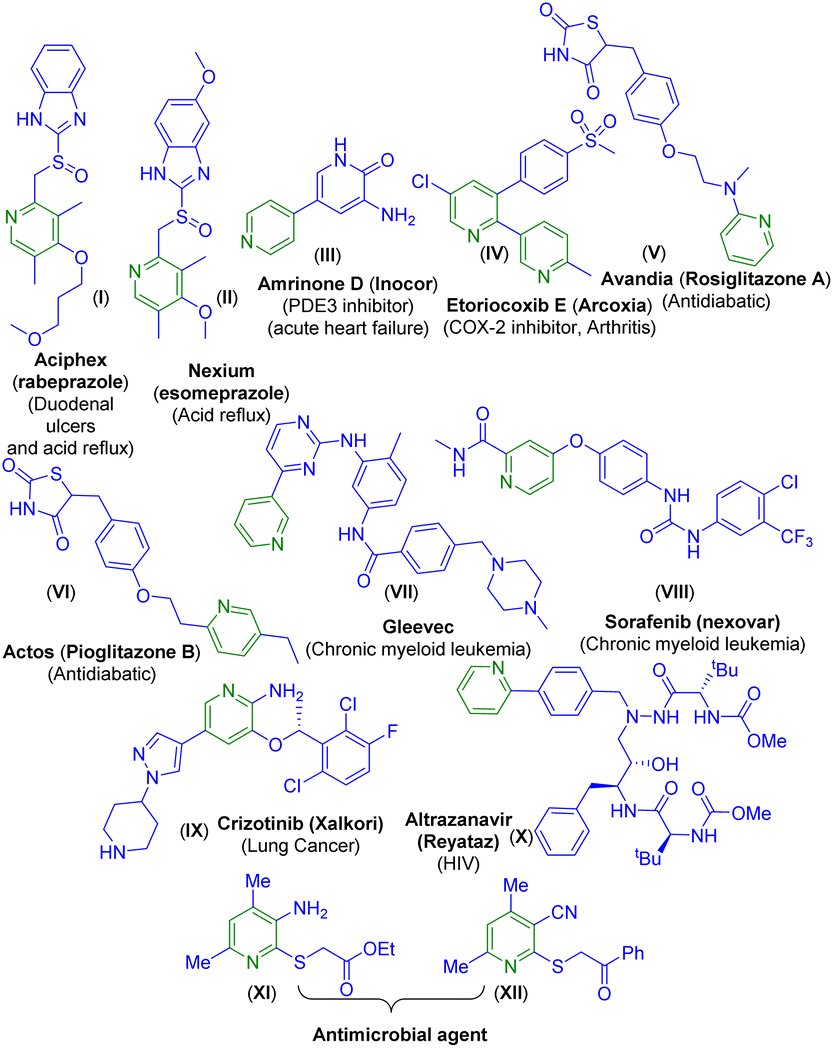 | ||
| Fig. 1 Drugs bearing the pyridine nucleus. | ||
Drugs such as Gleevec (VII), sorafenib (Nexavar) (VIII), crizotinib (Xalkori) (IX), and the antihuman immunodeficiency virus (anti-HIV) drug atrazanavir (Reyataz) (X) all contain a pyridine nucleus and are used as anticancer drugs. Furthermore, the thio-functionalized pyridines XI and XII have been reported to show antimicrobial activity (Fig. 1).7 Considering the multi-faceted applications of substituted pyridines, synthetic chemists are striving to develop alternative methods toward the pyridine core. There are well-established classical methods such as the Hantzsch method,8a Kröhnke8b pyridine synthesis and several others.9 The past years have reported a few transition-metal catalyzed [4 + 2] hetero Diels–Alder reactions of 1-azadienes with alkynes under thermal conditions.10a–c Recently, [2 + 2 + 2] cycloadditions of alkynes with nitriles under metal or metal-free catalysis have been reported.10d–g Besides these, there are methods that employ nitriles to synthesize nitrogenous heterocycles.11a–d Previously, the McQuade group reported a few methods for the synthesis of multisubstituted pyridines employing alkylidene malononitriles.11e–g
Furthermore, the Gao and Tang group reported a visible-light-induced phosphorylation with the 1,6-enyne moiety for the diverse and selective synthesis of phosphorylated polyheterocycles leading to phosphorylated aminophosphonates, iminophosphonates, and ketones.11h Recently, in 2022, the Chen and Yu group employed 1-acryloyl-2-cyanoindoles as the precursors to synthesize various sulfonated/thiocyanated pyrrolo[1,2-a]indolediones under visible-light irradiation.11i The Chen group in 2020 introduced a nickel(II)-catalyzed synthesis of pyrroles and pyridines via coupling of ketonitrile with aryl boronic acid (Scheme 1a).12a In 2019, Ji et al. reported a cascade cyclization of 3-isocyano-[1,1′-biphenyl]-2-carbonitriles with aryl boronic acids to synthesize pyrrolopyridines under Mn(III) catalysis (Scheme 1b).12b Previously, the Zhang and Yu group reported visible-light-mediated vinyl isocyanide insertions with Umemoto's reagent and electron-deficient bromides using Ir(III) (Scheme 1c).12c Though the above-mentioned methodologies are well established and utilize transition metals for the synthesis of pyridine frameworks, a metal-free approach is always desirable.13
 | ||
| Scheme 1 Synthesis of functionalized N-heterocycles. | ||
An intramolecular radical-triggered synthesis of heterocycles involves radical acceptors such as alkene, alkyne, nitrile, and isonitrile with appropriately placed radical precursors. In this context, nitrile has turned out to be an excellent radical acceptor to access various carbocycles and heterocycles.14 Basically a transition metal is required to activate the nitrile through coordination with the N-atom or the π-electrons of the C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N triple bond, making it a good acceptor for the nucleophilic/radical attack, followed by cyclization to access nitrogenous heterocycles.15 However, a metal-free photocatalytic approach employing nitrile as a radical acceptor would be interesting to generate poly-substituted pyridines. Previously, our group reported a nitrile-triggered synthesis of thio-substituted pyrroles with β-ketodinitriles as an acceptor and thiophenol as the radical donor (Scheme 1d).16a In continuation of our interest in nitrile-based precursors,15a,b,16a we hypothesized that a reaction is feasible with γ-ketodinitriles as the radical acceptors. To confirm our hypothesis, the CVs of γ-ketodinitrile (1) and thiophenol (a) were measured. The estimated E1/2 oxd of benzene thiol (+0.25 V vs. the SCE) is lower compared to the E1/2 oxd of γ-ketodinitriles (+2.13 V, +0.38 V vs. the SCE), making them a suitable radical donor and acceptor pair [Fig. 2(i)].
N triple bond, making it a good acceptor for the nucleophilic/radical attack, followed by cyclization to access nitrogenous heterocycles.15 However, a metal-free photocatalytic approach employing nitrile as a radical acceptor would be interesting to generate poly-substituted pyridines. Previously, our group reported a nitrile-triggered synthesis of thio-substituted pyrroles with β-ketodinitriles as an acceptor and thiophenol as the radical donor (Scheme 1d).16a In continuation of our interest in nitrile-based precursors,15a,b,16a we hypothesized that a reaction is feasible with γ-ketodinitriles as the radical acceptors. To confirm our hypothesis, the CVs of γ-ketodinitrile (1) and thiophenol (a) were measured. The estimated E1/2 oxd of benzene thiol (+0.25 V vs. the SCE) is lower compared to the E1/2 oxd of γ-ketodinitriles (+2.13 V, +0.38 V vs. the SCE), making them a suitable radical donor and acceptor pair [Fig. 2(i)].
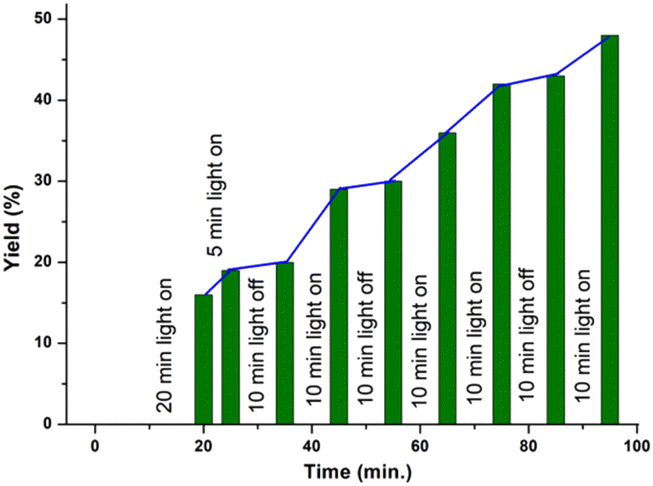 | ||
| Fig. 2 Stair type graph for on–off experiment. | ||
To verify our hypothesis, a preliminary reaction was carried out between 2-(3-oxo-1,3-diphenylpropyl)malononitrile (1, 1 equiv.) and thiophenol (a, 2 equiv.) in the presence of eosin Y (3 mol%) and K2CO3 (1 equiv.) in DMSO (1 mL) under the irradiation of 20 W (2 × 10 W) green LEDs (wavelength, 523 nm and flux, 39 mW cm−2) under a N2 atmosphere for 8 h. A new compound was obtained in 55% isolated yield along with the formation of diphenyl disulfide. From the spectroscopic (1H and 13C{1H} NMR) analyses, the structure of the product was determined and the product was identified as 4,6-diphenyl-2-(phenylthio)nicotinonitrile (1a). Furthermore, X-ray crystallography analysis of one of its analogs (1f) validated its structure (Fig. S2†).
Inspired by the present photocatalytic approach, further screening involving different catalytic systems, bases, light sources, and solvents was carried out. The employment of transition metal complexes such as [Ru(bpy)3]Cl2 and [Ru(bpy)3](PF6)2 provided 68% and 60% yields, respectively, which are inferior to those obtained employing eosin Y (Table 1, entries 2 and 3). The use of other organic dyes such as eosin B, rose bengal, and rhodamine B (Table 1, entries 4–6) did not improve the yield as compared to eosin Y. At 2 mol% eosin Y loading, 67% yield of the desired product (1a) was obtained (Table 1, entry 7), while increasing the catalyst loading to 5 mol% did not improve the yield significantly (Table 1, entry 8). Using other bases such as Na2CO3, Cs2CO3, KOH, and Et3N, instead of K2CO3 did not improve the yield (Table 1, entries 9–12). Furthermore, the use of one of the organic bases, DBU, failed to improve the yield (Table 1, entry 13). Among the solvents tested, DMSO was found to be the best as compared to other solvents such as DMF (58%), CH3CN (42%), EtOH (15%), and CH3OH (20%) (Table 1, entries 14–17). However, varying the equivalence of thiophenol from that used under the standard conditions failed to improve the yield (Table 1, entries 18 and 19). Thus, the use of 2 equivalents of thiol was found to be optimal. The standard reaction when carried out using 4 × 1 W blue (430 nm) and white LEDs did not improve the reaction yield beyond 70% (Table 1, entries 20 and 21). Thus, the use of green light is found to be the best option for the excitation of eosin Y, since both the absorption (543 nm) and emission (560 nm) spectra of eosin Y in DMSO coincide with the wavelength of green light (523 nm).16a Next, the use of 2 × 10 W green LEDs (bulb) did not prove to be much beneficial as compared to the use of 4 × 1 W green LEDs (523 nm) having a measured flux of 39 mW cm−2 (Table 1, entry 22). Furthermore, an increase in the reaction time did not result in any significant improvement in the reaction yield (45%) (Table 1, entry 23). A natural energy source (sunlight) was able to excite the catalyst and gave the desired product in a slightly less yield as compared to 4 × 1 W green LEDs (Table 1, entry 24). Control experiments revealed that eosin Y, a base, and a light source are indispensable for this protocol (Table 1, entries 25–27). However, the formation of 10% of the product in the absence of the catalyst eosin Y may be due to the possible competitive anionic pathway which is suppressed in the presence of eosin Y via the SET process. To see whether the desired product is formed under thermal conditions, the reaction of (1) with thiophenol (a) was carried out at 60 °C. The reaction failed to give the desired thio-functionalized pyridines and 1 decomposed to its corresponding chalcone and formed diphenyldisulfide (Table 1, entry 28).
|
|
||
|---|---|---|
| Entry | Variation from the optimal conditionsa | Yieldb (%) |
| a Reaction conditions: 1 (0.25 mmol), a (0.5 mmol), eosin Y (3 mol%), K2CO3 (1 equiv.), DMSO (1 mL), 3 h. b Isolated pure product. | ||
| 1 | None | 78 |
| 2 | [Ru(bpy)3]Cl2 instead of eosin Y | 68 |
| 3 | [Ru(bpy)3](PF6)2 instead of eosin Y | 60 |
| 4 | Eosin B instead of eosin Y | 62 |
| 5 | Rose bengal instead of eosin Y | 57 |
| 6 | Rhodamine B instead of eosin Y | 58 |
| 7 | 2 mol% eosin Y | 67 |
| 8 | 5 mol% eosin Y | 75 |
| 9 | Na2CO3 instead of K2CO3 | 65 |
| 10 | Cs2CO3 instead of K2CO3 | 59 |
| 11 | KOH instead of K2CO3 | 55 |
| 12 | Et3N instead of K2CO3 | 70 |
| 13 | DBU instead of K2CO3 | 52 |
| 14 | DMF instead of DMSO | 58 |
| 15 | CH3CN instead of DMSO | 42 |
| 16 | EtOH instead of DMSO | 15 |
| 17 | CH3OH instead of DMSO | 20 |
| 18 | 1 equiv. of thiol | 52 |
| 19 | 2.5 equiv. of thiol | 72 |
| 20 | 4 × 1 W blue LEDs | 65 |
| 21 | 4 × 1 W white LEDs | 70 |
| 22 | 2 × 10 W green LEDs | 68 |
| 23 | 8 h instead 2–3 h | 45 |
| 24 | Sunlight | 65 |
| 25 | Without eosin Y | 10 |
| 26 | Without a base | Trace |
| 27 | Reaction in the dark | 10 |
| 28 | Reaction at 60 °C without eosin Y | 00 |

After screening several parameters such as different catalytic systems, bases, light sources, and solvents, the optimal conditions for this transformation were found to be the combination of 2-(3-oxo-1,3-diphenylpropyl)malononitrile (1, 0.25 mmol), benzenethiol (a, 0.50 mmol, 2 equiv.), K2CO3 (0.25 mmol, 1 equiv.), and eosin Y (3 mol%) in DMSO (1 mL) under four 1 W green LED irradiation (Table 1, entry 1).
With the optimized conditions in hand, the present protocol was then used for the construction of multi-substituted pyridines using a variety of γ-ketodinitriles (Scheme 2). The γ-ketodinitrile carrying both unsubstituted phenyl rings (1) reacted with benzenethiol (a) giving the substituted pyridine (1a) in 78% yield. The γ-ketodinitriles having electron-donating substituents such as p-Me (2) and p-OMe (3) and electron-withdrawing substituents such as p-F (4) and p-Cl (5) on the aroyl ring provided the corresponding poly-functionalized pyridines (2a, 75%), (3a, 78%), (4a, 71%), and (5a, 74%) in good yields. Similarly, varying the substituents (EDGs or EWGs) on the α-phenyl ring of the malononitrile also resulted in smooth reactions with a, giving products (6a, 74%), (7a, 76%), (8a, 69%), (9a, 71%), and (10a, 71%) respectively. Besides this, heteroaromatic γ-ketodinitriles (11 and 12) reacted efficiently, giving poly-substituted pyridines (11a, 72% and 12a, 69%). In addition to this, γ-ketodinitriles containing a 1-naphthyl ring instead of an α-phenyl ring (13) and a 2-naphthyl ring and a 1-naphthyl ring on either side (14) also gave the desired products (13a, 79% and 14a, 81%). However, γ-ketodinitrile having the strong electron-withdrawing group o-NO2 (15) on the γ-aryl ring of the γ-ketodinitrile and the methyl group (16) instead of the γ-aryl ring of the γ-ketodinitrile failed to react. The failure of the electron-withdrawing substrate (15) may be due to both steric and electronic factors and for 16 due to the lower reactivity of the alkyl group containing substrate. The applicability of the protocol was further tested for gram-scale (5 mmol) synthesis, and product 1a was obtained in 64% yield. LEDs as an energy source for visible light have been frequently used in various photochemical reactions but the use of direct sunlight in photochemical reactions makes the protocol more sustainable. Sunlight is the combination of all possible wavelengths of light, where blue light accounts for 25%. Instead of LEDs as a source of light whether the reaction can be carried out under the sunlight.17a To check the compatibility of the reaction, γ-ketodinitriles 1, 2, 3, 4, 12 and 14 having EDGs and EWGs were reacted with thiophenol (a) under direct sunlight for 5 h. It was observed that all the substrates provided the corresponding pyridines (1a, 70%), (2a, 72%), (3a, 75%), (4a, 69%), (12a, 66%), and (14a, 79%) in good yields under irradiation with direct sunlight. The efficiency of the sunlight-mediated reaction was also demonstrated through a large-scale synthesis (5 mmol) yielding pyridine 1a in 65% yield.
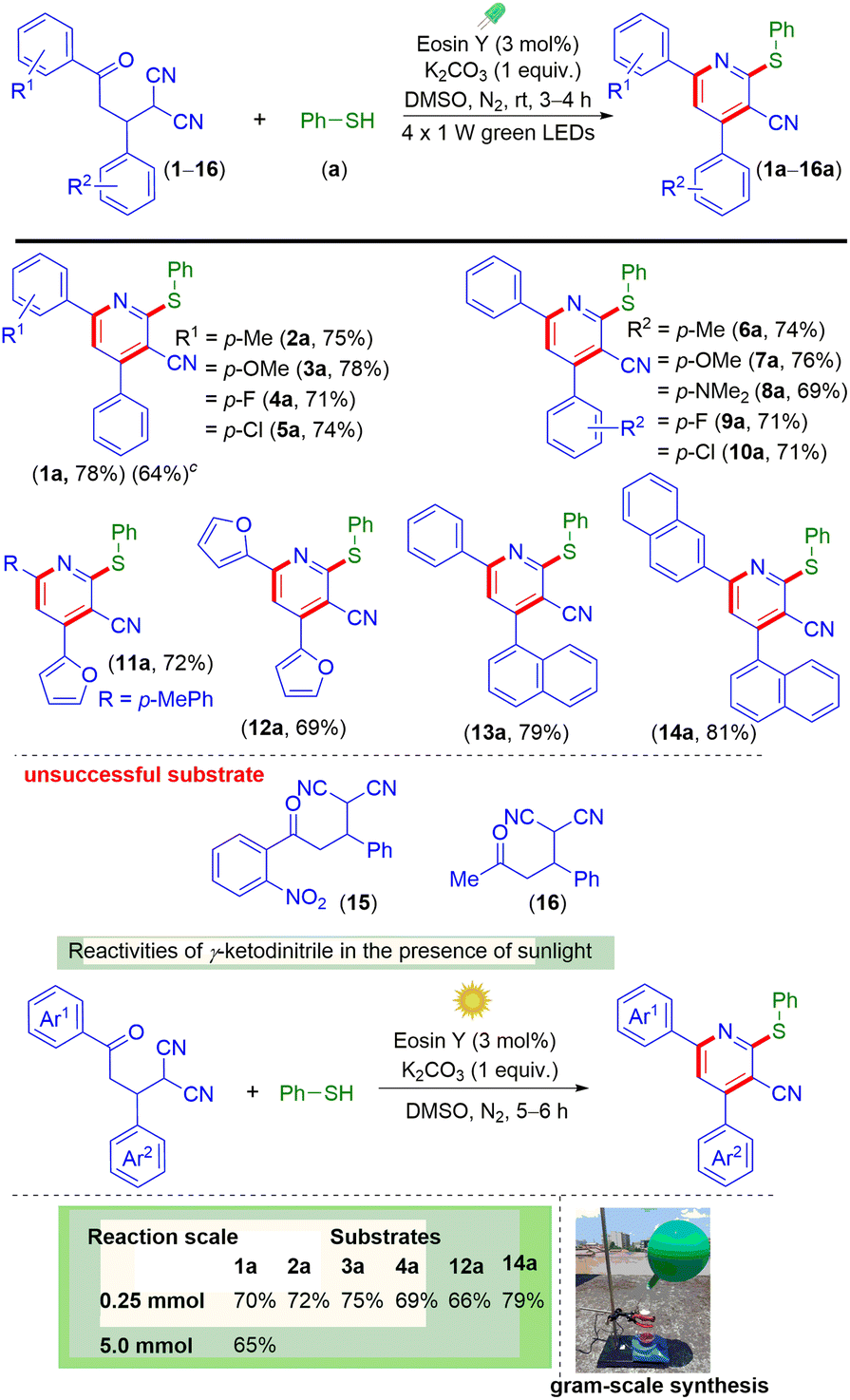 | ||
Scheme 2 Scope of multi-functionalized pyridines with different γ-ketodinitriles. a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Reaction conditions: 1–16 (0.25 mmol), a (0.5 mmol), K2CO3 (0.25 mmol), eosin Y (3 mol%), DMSO (1 mL), four 1 W green LEDs, under N2 for 3–4 h. bIsolated yield. 5 mmol scale. Reaction conditions: 1–16 (0.25 mmol), a (0.5 mmol), K2CO3 (0.25 mmol), eosin Y (3 mol%), DMSO (1 mL), four 1 W green LEDs, under N2 for 3–4 h. bIsolated yield. 5 mmol scale. | ||
Next, γ-ketodinitrile (1) was reacted with various thiophenols (b–k) having electron-donating groups (EDGs) and electron-withdrawing groups (EWGs), as shown in Scheme 3. Thiophenols having EDGs such as p-Me (b), o-Me (c), p-OMe (d), and m-OMe (e), and EWGs such as p-F (f), p-Cl (g), m-Cl (h), o-Br (i), and p-CF3 (j) were reacted efficiently to give the respective products (1b, 73%); (1c, 67%); (1d, 74%); (1e, 69%); (1f, 72%); (1g, 69%); (1h, 59%); (1i, 58%); and (1j, 59%) in good yields. Further, polyaromatic thiol such as 2-naphthyl thiol (k) underwent smooth reaction and gave the desired product (1k, 72%) in good yields. Besides aromatic thiols, heteroaromatic thiols such as thiophene 2-thiol (l) and aliphatic thiols such as cyclohexane thiol (m) reacted to give the desired pyridines 1l in 75% and 1m in 56% yields.
 | ||
| Scheme 3 Scope of multi-functionalized pyridines with different thiophenols. aReaction conditions: 1 (0.25 mmol), b–k (0.5 mmol), K2CO3 (0.25 mmol), eosin Y (3 mol%), DMSO (1 mL), four 1 W green LEDs, under N2 for 3–4 h. bIsolated yield. | ||
Furthermore, to see whether this methodology is compatible with acyclic alkyl thiols, γ-ketodinitrile (1) was reacted with ethyl thiol (n), propyl thiol (o), butyl thiol (p), and tert-butyl thiol (q) under the present photochemical conditions. All the alkyl thiols failed to give clean products, which is due to the inability of alkyl thiols to form the corresponding thiyl radicals. However, on prolonging the reaction time, the γ-ketodinitrile (1) started decomposing, giving a hydrogenated chalcone (1′) and the corresponding disulphides.16d After synthesizing a variety of poly-substituted pyridines, we also successfully performed the reactions under sunlight for a few thiophenols (b, d, g, i, k, and l). The reaction proceeded with thiophenols having different substituents. The γ-ketodinitrile 1 when reacted with thiophenols b, d, g, i, k, and l in the presence of sunlight for 5 h gave the corresponding products 1b, 1d, 1g, 1i, 1k, and 1l in 69%, 70%, 68%, 55%, 70%, and 72% yields.17b Hence it is concluded that sunlight can excite the catalyst eosin Y and shows almost equal efficacy compared to the use of green LEDs.
Next, some control experiments were carried out to decipher the mechanistic hypothesis. When the reaction was performed in the dark, the product (1a) was obtained in 10% yield [Scheme 4a (i)] and the yield remained unaltered upon prolonging the reaction to 24 h. This confirms the crucial role of light in initiating the reaction. Furthermore, the omission of the catalyst from the reaction provided only 10% of the product (1a), suggestive of its role in the formation of the thiyl radical [Scheme 4a (ii)]. The omission of the base resulted in a trace amount of the product (<10%) [Scheme 4a (iii)]. These experiments reveal that a photocatalyst, light, and a base are indispensable in this protocol. To see whether O2 in the form of a superoxide helps in the generation of the thiyl radical, the reaction was carried out in the absence of a base under an oxygen atmosphere.16b However, no desired product (1a) was obtained, confirming the non-involvement of oxygen [Scheme 4a (iv)]. Furthermore, the reaction in the absence of a photosensitizer under aerobic conditions resulted in a trace amount (<10%) of the product (1a) along with the formation of diphenyldisulfide and the decyanated product 4-oxo-2,4-diphenylbutanenitrile (16) [Scheme 4a (v)]. To ascertain the role of oxygen, if any, in the generation of the thiyl radical, the reaction was performed at 60 °C in the absence of a base under an air atmosphere. These conditions gave no desired product and most of the γ-ketodinitrile (1) remained unconsumed along with the formation of a substantial amount of diphenyldisulfide and a trace amount of the decyanated product (16) [Scheme 4a (vi)].16d,e The formation of the decyanated product (16) from the dicyano substrate (1) in a basic and aerobic medium is reported.16d
 | ||
| Scheme 4 Control experiments. | ||
Furthermore, it was observed that the use of 1,1-diphenyl ethylene as the radical scavenger resulted in a lower yield (<10%) of the product (1b) along with the trapped adduct (Q, 69%) and dimer (b′) of the corresponding thiol. This supports the intermediacy of the thiyl radical (Scheme 4b). The structure of adduct Q was confirmed by NMR (1H and 13C{1H}) and HRMS analyses (Fig. S4 and S5† respectively). Finally, an on–off experiment was performed to show that a continuous supply of light is necessary (Fig. 2) (for details see the ESI†).16,18
The Stern–Volmer (SV) fluorescence quenching experiment, which employed eosin Y as the probe and thiophenol (a) as the quencher, suggests a smooth electron transfer between thiophenol and eosin Y.16a For further confirmation, a fluorescence quenching experiment was performed taking eosin Y as the probe and the thiolate anion (thiol + base) as the quencher. It was observed that no uniform decrease in the intensity of fluorescence maxima was observed upon successive addition of the quencher thiolate anion even at higher concentrations. However, a uniform quenching pattern was observed with a lower concentration of thiophenol as the quencher. This suggests that there is a smooth electron transfer between the probe eosin Y and thiophenols compared to the thiolate anion [Fig. 3(i) and (ii)].
 | ||
| Fig. 3 (i) Fluorescence quenching of eosin Y with thiophenols as the quencher. (ii) Fluorescence quenching of eosin Y with the thiolate anion (thiophenols + base) as the quencher. | ||
To further confirm whether the generation of the thiyl radical is directly from thiophenols or from the thiolate anion, CV measurements were done. The CV measurements revealed that the oxidation potential of thiophenol (E1/2 ox = +0.25 V vs. SCE) is lower than the oxidation potential of eosin Y (E1/2 ox = +0.83 V vs. SCE for the excited state of eosin Y). This enables the oxidation of thiophenol by eosin Y and facilitates the formation of the thiyl radical (Fig. 4).
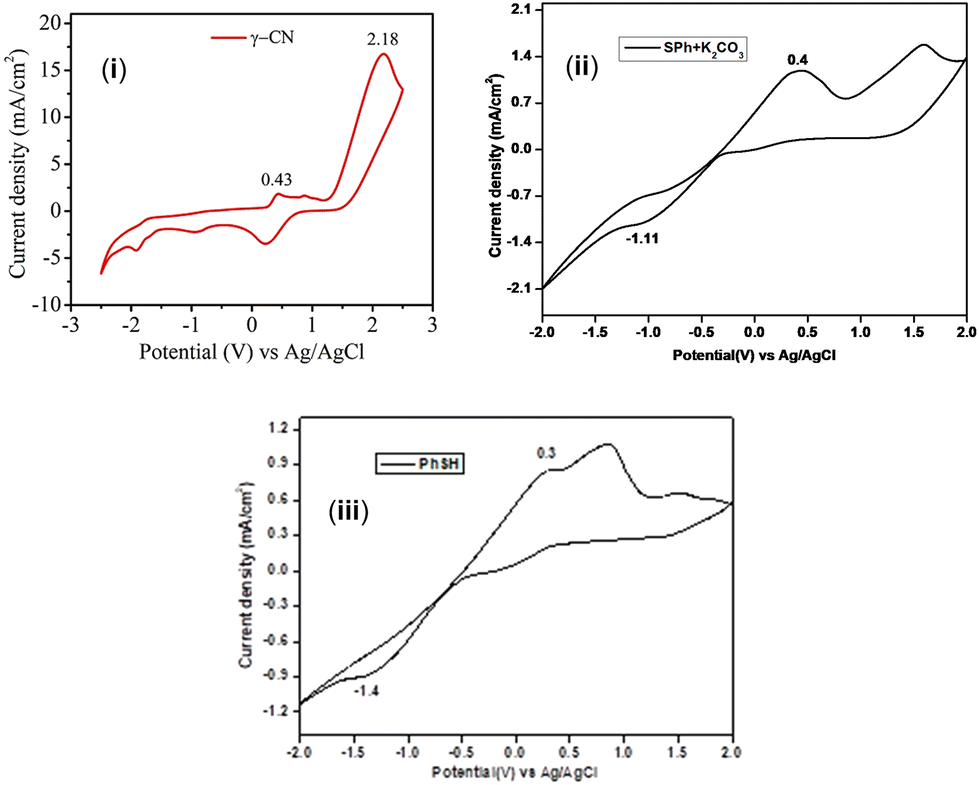 | ||
| Fig. 4 (i) CV graph of γ-ketodinitrile (1) and (ii) CV graph of benzene thiol (a) in the presence of a base. (iii) CV graph of benzene thiol. | ||
However, the possible formation of the thiyl radical from the thiolate anion cannot be completely ruled out. As is evident from the CV value, the thiolate anion (E1/2 ox = +0.35 V vs. SCE) has a higher tendency to gain an electron than PhSH (E1/2 ox = +0.25 V vs. SCE) or PhSH preferably loses an electron compared to the thiolate anion. When comparing these values with the oxidation potential of eosin Y (E1/2 ox = +0.83 V vs. SCE for the excited state of eosin Y), it is clear that EY* will better oxidize PhSH rather than the thiolate anion [Fig. 4(ii) and (iii)] (Fig. 4).16a
Based on control experiments and literature reports, a mechanistic path is suggested, as shown in Scheme 5.16,19 Initially, green LEDs excited eosin Y (EY) to EY*, which oxidizes the thiophenol (a) to the radical cation (a′). Next, the thiyl radical (a′′) is obtained via deprotonation from the radical cation (a′). The thiyl radical (a′′) undergoes addition with one of the nitrile groups of γ-ketodinitrile (1), generating intermediate A. Subsequently, A is converted to the anionic intermediate (B) via reductive quenching. Intermediate B upon protonation generates the imine intermediate (C) (detected by HRMS analysis of the reaction mixture, Fig. S19†).
 | ||
| Scheme 5 Plausible mechanism. | ||
The nucleophilic attack of imine (C) on the carbonyl group provided intermediate D. Finally, the loss of a water molecule from intermediate D gives intermediate E (detected by HRMS analysis of the reaction mixture, Fig. S19†), which upon tautomerization followed by oxidative aromatization produces pyridine 1a (Scheme 5).
To illustrate the utility of the present photochemical approach, pyridine (1a) was employed for some synthetic transformations (Scheme 6). The oxidation of 1a with mCPBA provided the sulfonylated pyridine (1aa) in 75% yield.20 Next, 1a was subjected to annulation using diphenyl acetylene which provided the C–H, N annulated product 1ab in 68% yield.21 The base hydrolysis of 1a gave 4,6-diphenyl-2-(phenylthio)nicotinamide (1ac) in 78% yield which is an analog of nicotinamide (a form of vitamin B3) used for the treatment of pellagra.22a,b
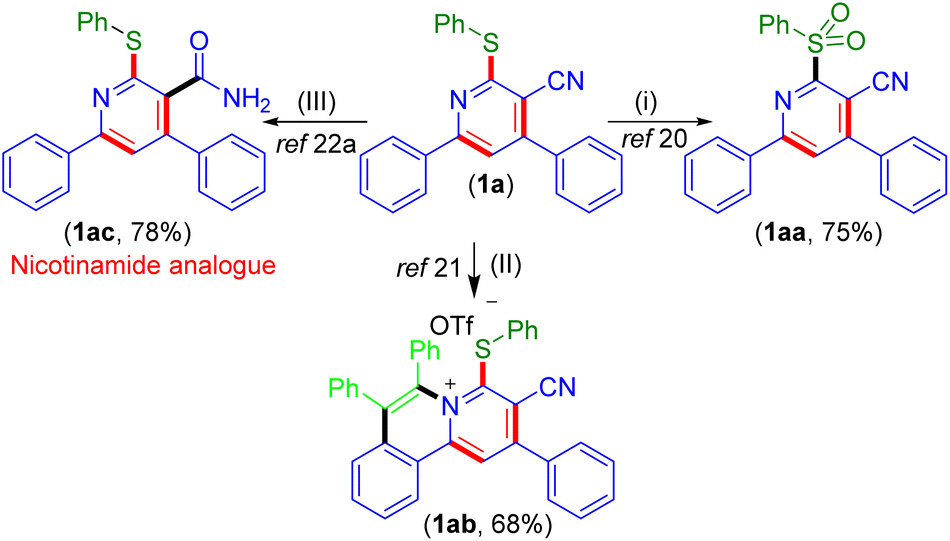 | ||
| Scheme 6 Post-synthetic transformation. aReaction conditions: (i) mCPBA (5 equiv.), DCM, rt, 5 h. (ii) [RuCl2(p-cymene)]2 (5 mol%), Cu(OAc)2·H2O (2.2 equiv.), diphenyl acetylene (1.2 equiv.), (TfOH (1.5 equiv.), DCE, 120 °C, 24 h, pressure tube. (iii) KOH (4 equiv.), EtOH, reflux, 5 h. bIsolated yield. | ||
In summary, a thiyl radical-triggered synthesis of multi-substituted pyridines is demonstrated using γ-ketodinitriles and thiols in the presence of eosin Y under metal-free conditions. This protocol simultaneously constructs C–N, C–S, and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds. This methodology can tolerate different substituents in both coupling partners. A few post-translational modifications and a scale-up reaction are illustrated to show the practical utility of the present protocol. The reaction also proceeds flawlessly when employing sunlight as a sustainable energy source.
C bonds. This methodology can tolerate different substituents in both coupling partners. A few post-translational modifications and a scale-up reaction are illustrated to show the practical utility of the present protocol. The reaction also proceeds flawlessly when employing sunlight as a sustainable energy source.
General information
All the reagents were of commercial grade and purified according to the established procedures. All the reactions were carried out in oven-dried glassware. The highest commercial quality reagents were purchased and were used without further purification unless otherwise stated. Reactions were monitored by thin-layer chromatography (TLC) on a 0.25 mm silica gel plates (60F254) and visualized under UV illumination at 365 nm. Organic extracts were dried over anhydrous sodium sulfate (Na2SO4). Solvents were removed using a rotary evaporator under reduced pressure. Column chromatography was performed to purify the crude products on a silica gel 60–120 mesh using a mixture of hexane and ethyl acetate as the eluent. All the isolated compounds were characterized by 1H and 13C{1H} NMR, HRMS-spectrometric and IR spectroscopic techniques. The NMR spectra for all the samples were recorded in deuterochloroform (CDCl3). 1H and 13C{1H} NMR spectra were recorded using a 600 (150) or 500 (125) or 400 (100) MHz spectrometer and were calibrated using tetramethylsilane or a residual undeuterated solvent for 1H NMR, and deuterochloroform for 13C NMR as an internal reference {Si(CH3)4: 0.00 ppm or CHCl3: 7.260 ppm for 1H NMR, 77.230 ppm for 13C NMR}. 19F NMR spectra were calibrated without any internal standard in CDCl3 in 400 MHz spectrometers. The chemical shifts are quoted in δ units, parts per million (ppm). 1H NMR data is represented as follows: chemical shift, multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, br = broad, and dd = doublet of doublet), integration and coupling constant(s) J in hertz (Hz). High-resolution mass spectra (HRMS) were recorded on a mass spectrometer using electrospray ionization-time of flight (ESI-TOF) reflection experiments. FT-IR spectra were recorded in KBr or neat and reported in the frequency of absorption (cm−1).General procedure for the synthesis of 2-(3-oxo-1,3-diphenylpropyl)malononitrile (1–14)
Compounds 1–14 were synthesized as per the method described in ref. 16d.Compounds 1–14 were synthesized by a slight modification of the literature procedure. To an oven-dried 50 mL round-bottom flask were added chalcone, 1,3-diphenyl-2-propen-1-one (1.04 g, 5.0 mmol.), malononitrile (0.66 g, 10.0 mmol), K2CO3 (1.38 g, 10.0 mmol), and DCE (5 mL). The reaction mixture was stirred at room temperature for 12 h. Then the reaction mixture was admixed with ethyl acetate (30 mL) and the organic layer was washed with water (10 mL). The organic layer was dried over anhydrous Na2SO4, and the solvent was evaporated under reduced pressure. The crude product thus obtained was purified over a column of silica gel (hexane/ethyl acetate, 9:1) to give pure 2-(3-oxo-1,3-diphenylpropyl)malononitrile (1) (1.30 g, 95%).
General procedure for the synthesis of 4,6-diphenyl-2-(phenylthio)nicotinonitrile (1a) from 2-(3-oxo-1,3-diphenylpropyl)malononitrile (1) and thiophenol (a)
To an oven-dried 10 mL borosilicate vial were added 2-(3-oxo-1,3-diphenylpropyl)malononitrile (1) (0.25 mmol, 68 mg), eosin Y (3 mol%, 5 mg), and K2CO3 (1 equiv., 34 mg). Then to the reaction mixture, benzene thiol (a) (0.5 mmol, 55 mg) in 1 mL of DMSO was added and stirred at room temperature under a N2 atmosphere for 3 h, tentatively at a distance of ∼1–2 cm from four 1 W green LEDs. After the completion of the reaction (monitored by TLC analysis), the mixture was diluted with water (10 mL) followed by extraction with ethyl acetate (20 mL). The organic layer was dried over anhydrous Na2SO4, and the solvent was evaporated under reduced pressure. The crude residue thus obtained was purified by column chromatography over silica gel (60–120 mesh) using hexane and ethyl acetate (98:2) as the eluent to afford 4,6-diphenyl-2-(phenylthio)nicotinonitrile (1a) in 78% yield. The identity and purity of the product were confirmed by spectroscopic analysis (Scheme S2†).
Spectral data
Conflicts of interest
There are no conflicts to declare.Acknowledgements
B. K. P. acknowledges the support from SERB (SCP/2022/000195), CSIR 02(0361/EMR-II). A. K. S., A. R., and H. N. D. thank the Institute for fellowships. We are grateful to MHRD for the 400 MHz NMR facility under the COE-FAST program (5-5/2014-TS VII), DST for providing the 500 MHz NMR under the DST-FIST program (SR/FST/CS-II/2017/23C), NECBH, IIT Guwahati, DBT, Govt. of India (BT/COE/34/SP28408/2018) and the Department of Chemistry, IIT Guwahati, for the XRD facility.References
- (a) K. Sun, Q.-Y. Lv, X.-L. Chen, L.-B. Qua and B. Yu, Green Chem., 2021, 23, 232–248 RSC; (b) Q.-Q. Zhou, Y.-Q. Zou, L.-Q. Lu and W.-J. Xiao, Angew. Chem., Int. Ed., 2019, 58, 1586–1604 CrossRef CAS PubMed; (c) G. Masson and B. König, Eur. J. Org. Chem., 2020, 1191–1192 CrossRef CAS; (d) G. E. M. Crisenza, D. Mazzarella and P. Melchiorre, J. Am. Chem. Soc., 2020, 142, 5461–5476 CrossRef CAS PubMed; (e) S. J. McCarver, J. X. Qiao, J. Carpenter, R. M. Borzilleri, M. A. Poss, M. D. Eastgate, M. M. Miller and D. W. C. MacMillan, Angew. Chem., Int. Ed., 2017, 56, 728–732 CrossRef CAS PubMed; (f) V. Srivastava, P. K. Singh, S. Tivaria and P. P. Singh, Org. Chem. Front., 2022, 9, 1485–1507 RSC; (g) A. Pokhriyal, B. S. Karki, R. Kant and N. Rastogi, J. Org. Chem., 2021, 86, 4661–4670 CrossRef CAS PubMed; (h) Q. Xiao, Q.-X. Tong and J.-J. Zhong, Molecules, 2022, 27, 619 CrossRef CAS PubMed; (i) A. A. Altaf, A. Shahzad, Z. Gul, N. Rasool, A. Badshah, B. Lal and E. Khan, J. Drug Des. Med. Chem., 2015, 1, 1–11 Search PubMed; (j) D. P. Hari and B. Konig, Chem. Commun., 2014, 50, 6688–6699 RSC; (k) V. Srivastavaa and P. P. Singh, RSC Adv., 2017, 7, 31377–31392 RSC.
- (a) N. Chalotra, M. A. Rizvi and B. A. Shah, Org. Lett., 2019, 21, 4793–4797 CrossRef CAS PubMed; (b) K. L. Skubi, T. R. Blum and T. P. Yoon, Chem. Rev., 2016, 116, 10035–10074 CrossRef CAS PubMed; (c) C. Zhu, H. Yue, L. Chu and M. Rueping, Chem. Sci., 2020, 11, 4051–4064 RSC; (d) X.-A. Liang, L. Niu, S. Wang, J. Liu and A. Lei, Org. Lett., 2019, 21, 2441–2444 CrossRef CAS PubMed; (e) A. Saha, S. Guin, W. Ali, T. Bhattacharya, S. Sasmal, N. Goswami, G. Prakash, S. K. Sinha, H. B. Chandrashekar, S. Panda, S. S. Anjana and D. Maiti, J. Am. Chem. Soc., 2022, 144, 1929–1940 CrossRef CAS PubMed; (f) J. Kumar, A. Ahmad, M. A. Rizvi, M. A. Ganie, C. Khajuria and B. A. Shah, Org. Lett., 2020, 22, 5661–5665 CrossRef CAS PubMed.
- (a) J. D. Bell and J. A. Murphy, Chem. Soc. Rev., 2021, 50, 9540–9685 RSC; (b) D.-M. Yan, J.-R. Chen and W. J. Xiao, Angew. Chem., Int. Ed., 2019, 58, 378–380 CrossRef CAS PubMed; (c) L. Xia, M. Jin, Y. Jiao and S. Yu, Org. Lett., 2022, 24, 364–368 CrossRef CAS PubMed; (d) A. Dahiya, B. Das, A. K. Sahoo and B. K. Patel, Adv. Synth. Catal., 2022, 364, 966–973 CrossRef CAS; (e) F. Wu, W. Dong, S. Fan, Y. Yuan, C. Liang, A. Chen, Z. Yin and Z. Zhang, J. Org. Chem., 2022, 87, 1302–1312 CrossRef CAS PubMed; (f) R. S. Rohokale, B. Koenig and D. D. Dhavale, J. Org. Chem., 2016, 81, 7121–7126 CrossRef CAS PubMed; (g) A. K. Sahoo, A. Dahiya, B. Das, A. Behera and B. K. Patel, J. Org. Chem., 2021, 86, 11968–11986 CrossRef CAS PubMed; (h) V. J. Roy, P. P. Sen and S. R. Roy, J. Org. Chem., 2021, 86, 16965–16976 CrossRef CAS PubMed.
- (a) T. Eicher, S. Hauptmann and A. Speicher, The Chemistry of Heterocycles, Wiley-VCH Verlag GmbH & Co., Weinheim, 2nd edn, 2003 CrossRef; (b) C. Allais, J.-M. Grassot, J. Rodriguez and T. Constantieux, Chem. Rev., 2014, 114, 10829–10868 CrossRef CAS PubMed; (c) M. D. Hill, Chem. – Eur. J., 2010, 16, 12052–12062 CrossRef CAS PubMed; (d) M. Stolar and T. Baumgartner, Chem. Commun., 2018, 54, 3311–3322 RSC.
- (a) H. Han and L. H. Hurley, Trends Pharmacol. Sci., 2000, 21, 136–142 CrossRef CAS PubMed; (b) G. Chelucci, Chem. Soc. Rev., 2006, 35, 1230–1243 RSC; (c) E. C. Constable, C. E. Housecroft, M. Neuburger, D. Phillips, P. R. Raithby, E. Schofield, E. Sparr, D. A. Tocher, M. Zehnder and Y. Zimmermann, J. Chem. Soc., Dalton Trans., 2000, 13, 2219–2228 RSC.
- (a) C. M. Baldwin and S. J. Keam, Drugs, 2009, 69, 1373–1401 CrossRef CAS PubMed; (b) A. F. Boushra, A. M. Elsayed, N. A. Ibrahim, M. K. Abdelwahed and E. I. A. Ahmed, Mol. Biol. Rep., 2019, 46, 4843–4860 CrossRef CAS PubMed; (c) J.-D. Charrier, A. Miller, D. P. Kay, G. Brenchley, H. C. Twin, P. N. Collier, S. Ramaya, S. B. Keily, S. J. Durrant, R. M. A. Knegtel, A. J. Tanner, K. Brown, A. P. Curnock and J.-M. Jimenez, J. Med. Chem., 2011, 54, 2341–2350 CrossRef CAS PubMed; (d) K. F. Croom and M. A. A. Siddiqui, Drugs, 2009, 69, 1513–1532 CrossRef CAS PubMed; (e) J. Liu, Z. Huang, W. Ma, S. Peng, Y. Li, K. M. Miranda, J. Tian and Y. Zhang, Eur. J. Med. Chem., 2019, 162, 650–665 CrossRef CAS PubMed; (f) S. A. Mosure, J. Shang, J. Eberhardt, R. Brust, J. Zheng, P. R. Griffin, S. Forli and D. J. Kojetin, J. Med. Chem., 2019, 62, 2008–2023 CrossRef CAS PubMed.
- (a) A. Takami, S. Ohtake, E. Morishita, Y. Terasaki, T. Fukushima, T. Kurokawa, N. Sugimori, S. Matono, K. Ohata, C. Saito, M. Yamaguchi, K. Hosokawa, H. Yamazaki, Y. Kondo and S. Nakao, Int. J. Hematol., 2012, 96, 357–363 CrossRef CAS PubMed; (b) F. Chen, Y. Fang, R. Zhao, J. Le, B. Zhang, R. Huang, Z. Chen and J. Shao, Eur. J. Med. Chem., 2019, 179, 916–936 CrossRef CAS PubMed; (c) J. E. Frampton, Drugs, 2013, 73, 2031–2051 CrossRef CAS PubMed; (d) K. F. Croom, S. Dhillon and S. J. Keam, Drugs, 2012, 69, 1107–1140 CrossRef PubMed.
- (a) A. Hantzsch, Ber. Dtsch. Chem. Ges., 1881, 14, 1637–1638 CrossRef; (b) F. Kröhnke, Synthesis, 1976, 1–24 CrossRef.
- (a) B. Nie, W. Wu, Q. Ren, Z. Wang, J. Zhang, Y. Zhang and H. Jiang, Org. Lett., 2020, 22, 7786–7790 CrossRef CAS PubMed; (b) F. Rammal, D. Gao, S. Boujnah, A.-C. Gaumont, A. A. Hussein and S. Lakhdar, Org. Lett., 2020, 22, 7671–7675 CrossRef CAS PubMed; (c) L. A. Hardegger, J. Habegger and T. J. Donohoe, Org. Lett., 2015, 17, 3222–3225 CrossRef CAS PubMed; (d) C. Sujatha, C. S. Bhatt, M. K. Ravva, A. K. Suresh and K. Namitharan, Org. Lett., 2018, 20, 3241–3244 CrossRef CAS PubMed; (e) D. Bai, X. Wang, G. Zheng and X. Li, Angew. Chem., Int. Ed., 2018, 57, 6633–6637 CrossRef CAS PubMed; (f) M. R. P. Heravi and A. Soufi, Chin. Chem. Lett., 2015, 26, 263–266 CrossRef.
- (a) J. M. Neely and T. Rovis, Org. Chem. Front., 2014, 1, 1010–1015 RSC; (b) D. L. Boger, Chem. Rev., 1986, 86, 781–793 CrossRef CAS; (c) J. Wu, W. Xu, Z.-X. Yu and J. Wang, J. Am. Chem. Soc., 2015, 137, 9489–9496 CrossRef CAS PubMed; (d) P. Kumar, S. Prescher and J. A. Louie, Angew. Chem., Int. Ed., 2011, 50, 10694–10698 CrossRef CAS PubMed; (e) G. Onodera, Y. Shimizu, J. Kimura, J. Kobayashi, Y. Ebihara, K. Kondo, K. Sakata and R. Takeuchi, J. Am. Chem. Soc., 2012, 134, 10515–10531 CrossRef CAS PubMed; (f) F. Ye and M. Haddad, Org. Lett., 2017, 19, 1104–1107 CrossRef CAS PubMed; (g) L.-G. Xie, S. Shaaban, X. Chen and N. Maulide, Angew. Chem., Int. Ed., 2016, 55, 12864–12867 CrossRef CAS PubMed.
- (a) A. Rakshit, H. N. Dhara, T. Alam, A. Dahiya and B. K. Patel, J. Org. Chem., 2021, 86, 17504–17510 CrossRef PubMed; (b) A. Rakshit, H. N. Dhara, A. K. Sahoo, T. Alam and B. K. Patel, Org. Lett., 2022, 24, 3741–3746 CrossRef CAS PubMed; (c) P. Natarajan, M. Muskan, N. K. Brar and J. J. Kaur, Org. Chem. Front., 2018, 5, 1527–1531 RSC; (d) Y. Zhang, K. Sun, Q. Lv, X. Chen, L. Qu and B. Yu, Chin. Chem. Lett., 2019, 30, 1361–1368 CrossRef CAS; (e) A. R. Longstreet, B. S. Campbell, B. F. Gupton and D. T. McQuade, Org. Lett., 2013, 20, 5298–5301 CrossRef PubMed; (f) A. R. Longstreet, D. Rivalti and D. T. McQuade, J. Org. Chem., 2015, 80, 8583–8596 CrossRef CAS PubMed; (g) J. M. de Souza, I. Abdiaj, J. Chen, K. Hanson, K. T. de Oliveira and D. T. McQuade, Org. Biomol. Chem., 2021, 19, 1991–1999 RSC; (h) S. Shi, Z. Zheng, Y. Zhang, Y. Yang, D. Ma, Y. Gao, Y. Liu, G. Tang and Y. Zhao, Org. Lett., 2021, 23, 9348–9352 CrossRef CAS PubMed; (i) A.-X. Huang, H.-L. Zhu, F.-L. Zeng, X.-L. Chen, X.-Q. Huang, L.-B. Qu and B. Yu, Org. Lett., 2022, 24, 3014–3018 CrossRef CAS PubMed.
- (a) Q. Zhen, R. Li, L. Qi, K. Hu, X. Yao, Y. Shao and J. Chen, Org. Chem. Front., 2020, 7, 286–291 RSC; (b) P. Xu, Y.-M. Zhu, F. Wang, S.-Y. Wang and S.-J. Ji, Org. Lett., 2019, 21, 683–686 CrossRef CAS PubMed; (c) K. Tong, T. Zheng, Y. Zhang and S. Yu, Adv. Synth. Catal., 2015, 357, 3681–3686 CrossRef CAS.
- (a) Z. Song, X. Huang, W. Yi and W. Zhang, Org. Lett., 2016, 18, 5640–5643 CrossRef CAS PubMed; (b) J. Dong, L. Bao, Z. Hu, S. Ma, X. Zhou, M. Hao, N. Li and X. Xu, Org. Lett., 2018, 20, 1244–1247 CrossRef CAS PubMed; (c) C. Allais, J.-M. Grassot, J. Rodriguez and T. Constantieux, Chem. Rev., 2014, 114, 10829–10868 CrossRef CAS PubMed; (d) J.-P. Wan, Y. Jing, C. Hu and S. Sheng, J. Org. Chem., 2016, 81, 6826–6831 CrossRef CAS PubMed.
- (a) K. Sun, Q.-Y. Lv, Y.-W. Lin, B. Yu and W.-M. He, Org. Chem. Front., 2021, 8, 445–465 RSC; (b) X. Li, X. Fang, S. Zhuang, P. Liu and P. Sun, Org. Lett., 2017, 19, 3580–3583 CrossRef CAS PubMed.
- (a) H. N. Dhara, A. Rakshit, T. Alam and B. K. Patel, Org. Biomol. Chem., 2022, 20, 4243–4277 RSC; (b) A. Rakshit, P. Sau, S. Ghosh and B. K. Patel, Adv. Synth. Catal., 2019, 361, 3824–3836 CrossRef CAS; (c) H.-D. Zuo, X.-S. Ji, C. Guo, S.-J. Hao, W.-J. Tu and B. Jiang, Org. Chem. Front., 2021, 8, 1496–1502 RSC.
- (a) A. K. Sahoo, A. Rakshit, A. Dahiya, A. Pan and B. K. Patel, Org. Lett., 2022, 24, 1918–1923 CrossRef CAS PubMed; (b) P. Natarajan, M. Manjeet, M. Muskan, N. K. Brar and J. J. Kaur, Org. Chem. Front., 2018, 5, 1527–1531 RSC; (c) Z. Xing, M. Yang, H. Sun, Z. Wang, P. Chen, L. Liu, X. Wang, X. Xie and X. She, Green Chem., 2018, 20, 5117–5122 RSC; (d) S. Lin, Y. Wei and F. Liang, Chem. Commun., 2012, 48, 9879–9881 RSC; (e) R. O. McCourt and E. M. Scanlan, Chem. – Eur. J., 2020, 26, 15804–15810 CrossRef CAS PubMed.
- (a) Y. Liu, B. Wang, X. Qiao, C.-H. Tung and Y. Wang, ACS Catal., 2017, 7, 4093–4099 CrossRef CAS; (b) Y. Su, L. Zhang and N. Jiao, Org. Lett., 2011, 13, 2168–2171 CrossRef CAS PubMed.
- (a) Y. Lv, J. Luo, M. Lin, H. Yue, B. Daia and L. A. He, Org. Chem. Front., 2021, 8, 5403–5409 RSC; (b) K. Gadde, P. Mampuys, A. Guidetti, H. Y. V. Ching, W. A. Herrebout, S. V. Doorslaer, K. A. Tehrani and B. U. W. Maes, ACS Catal., 2020, 10, 8765–8779 CrossRef CAS.
- (a) V. V. Pavlishchuka and A. W. Addison, Inorg. Chim. Acta, 2000, 298, 97–102 CrossRef; (b) A. G. Larsen, A. H. Holm, M. Roberson and K. Daasbjerg, J. Am. Chem. Soc., 2001, 123, 1723–1729 CrossRef CAS PubMed; (c) S. S. Zalesskiy, N. S. Shlapakov and V. P. Ananikov, Chem. Sci., 2016, 7, 6740–6745 RSC; (d) W. Wei, P. Bao, H. Yue, S. Liu, L. Wang, Y. Li and D. Yang, Org. Lett., 2018, 20, 5291–5295 CrossRef CAS PubMed; (e) R. Rahaman, S. Das and P. Barman, Green Chem., 2018, 20, 141–147 RSC; (f) G. Liang, J.-H. Wang, T. Lei, Y.-Y. Cheng, C. Zhou, Y.-J. Chen, C. Ye, B. Chen, C.-H. Tung and L.-Z. Wu, Org. Lett., 2021, 23, 8082–8087 CrossRef CAS PubMed; (g) A. M. Nair, S. Kumar and C. M. R. Volla, Adv. Synth. Catal., 2019, 361, 4983–4988 CrossRef CAS; (h) D. M. Lynch and E. M. Scanlan, Molecules, 2020, 25, 3094–3133 CrossRef CAS PubMed.
- A. A. Dar, N. Enjamuri, Md. Shadab, N. Ali and A. T. Khan, ACS Comb. Sci., 2015, 17, 671–681 CrossRef CAS PubMed.
- S. Ghosh, S. Pal, S. Rajamanickam, R. Shome, P. R. Mohanta, S. S. Ghosh and B. K. Patel, ACS Omega, 2019, 4, 5565–5577 CrossRef CAS.
- (a) J. H. Hall and M. A. Gisler, J. Org. Chem., 1976, 41, 3769–3770 CrossRef CAS; (b) A. C. Alport, M. D. Edin, P. G. M. D. Cairo and G. H. M. B. Cairo, Lancet, 1938, 24, 1460–1463 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, mechanistic investigation, and NMR spectra. CCDC 2201897. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3ob00009e |
| This journal is © The Royal Society of Chemistry 2023 |