
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElectrochemical rhodium catalysed alkyne annulation with pyrazoles through anodic oxidation – a metal oxidant/additive free methodology†
Subban
Kathiravan
 *a and
Prasad
Anaspure
b
*a and
Prasad
Anaspure
b
aAttana AB, Greta Arwidssons Väg 21, 11419 Stockholm, Sweden. E-mail: suppan.kathiravan@lnu.se
bBioorganic & Biophysical Chemistry Laboratory, Linnaeus University Centre for Biomaterials Chemistry, Department of Chemistry & Biomedical Sciences, Linnaeus University, Kalmar SE-39182, Sweden
First published on 9th February 2023
Abstract
Pyrazole and its derivatives are important azole heteroarenes prevalent in pharmaceutical compounds and have been used as ligands for protein binding, making them valuable targets for synthetic applications. Herein we disclose an electrochemical intermolecular C–H/N–H oxidative annulation of 2-phenylpyrazoles with alkynes using a rhodium(III) redox regime without any external metal oxidants in a water compatible solvent system. Both symmetrical and unsymmetrical alkynes were shown to be compatible with the optimized conditions.
Introduction
Transition metal catalysed regioselective transformations of inert C–H bonds to C–C/C–X bonds is of significant utility in the synthesis of pharmaceuticals and new materials due to their high atom economy, reduction in the number of synthetic steps and generally mild reaction conditions.1 A variety of metals have been explored for the elaboration of C–H bonds with saturated or unsaturated substituents.2 The development of efficient protocols using rhodium catalysts has been performed by the groups of Miura,3 Fagnou,4 Chang,5 Ellman,6 Rovis,7 Li8 and Glorius,9 among others.10 Rhodium catalysts exhibit high reactivities towards alkenes and alkynes, thus avoiding the need for prefunctionalized substrates in cross-coupling reactions. Alkyne annulations facilitated by RhIII/RhI catalytic regimes traditionally employ exogenous metal oxidants to recycle the active catalyst.11 From the pioneering work of Miura, it can be seen that stoichiometric amounts of metal oxidants are needed in C–H activation reactions, in particular in alkyne annulation for the synthesis of heterocycles.3 Among these metal oxidants, copper and silver are frequently used in the reoxidation step, despite their environmental and cost issues associated with by-product removal and waste disposal.The recent renascence arising from the marriage of transition metal catalysis with electrochemistry for electroorganic synthesis has provided environmentally benign redox processes that avoid the use of external toxic and expensive metal oxidants.12 This electrical control over the oxidation states of metals often provides alternative selectivities and can reduce the high-energy barrier associated with the reductive elimination step,13,14 as has been demonstrated by Ackermann,15 Xu,16 Lei,17 and Mei,18 among others,19 who have developed elegant metalla-electrocatalyzed C–H activation reactions. For example, Ackermann et al. have described the synthesis of polycyclic aromatic hydrocarbons by using double electrocatalysis-driven rhodaelectrocatalysis by annulative C–H activation (Scheme 1a).20 Furthermore, a scalable Rh(III) catalysed aryl C–H phosphorylation enabled by anodic oxidation-induced reductive elimination was reported by the Hu group.21 Very recently, Mei and co-workers have presented a divergent rhodium-catalysed electrochemical vinylic C–H annulation with alkynes.22 While significant development has been made using electricity in various Rh(III) catalysed C–H transformations, its use in annulation reactions using heterocycles prone to generate nitrogen-centred radicals though anodic oxidation is very scarce and alkyne annulation remains elusive.23 Herein we disclose a sustainable rhodaelectro-catalysed intermolecular C–H/N–H oxidative annulation of pyrazole with alkynes using a water-based solvent system (Scheme 1b).
 | ||
| Scheme 1 a) Previous work, (b) this work, (c) selected examples of bioactive compounds containing pyrazolo isoquinoline. | ||
Nitrogen containing azole heterocycles can be found in natural products and in biologically active compounds24 and are important building blocks in modern medicinal chemistry.25 Pyrazole-based six-membered azaheterocycles, while not naturally available, do contribute to a wide range of biological activities when used as motifs in drugs (Scheme 1c).26 Previous exploration of pyrazoles in C–H activation reactions has included alkyne annulations using conventional methods,27–29 though not with alternative sustainable electrochemical approaches.
Results & discussion
For the initial exploratory and control studies of the envisaged rhodium catalysed C–H activation, 2-phenylpyrazole (1a) and diphenylacetylene (2a) were used under reaction conditions previously developed by us for a copper catalysed C–H amination (Table 1).30 Upon comparison of rhodium, iridium, ruthenium and palladium catalysts, rhodium was found to be superior (Table 1, entries 1–4). A series of base additives were then examined, and KOAc was found to give better results than NaOPiv, KOPiv and LiOAc (Table 1, entries 5–8). In a screening of possible electrolytes, using KPF6 led to the product being formed in 34% yield (Table 1, entry 9); however, the use of tetrabutylammonium bromide, tetrabutylammonium acetate and tetrabutylammonium perchlorate as electrolytes provided little or no support for the reaction (Table 1, entries 10–12). Among the tested solvents, a protic solvent mixture containing tert-amyl alcohol mixed with water in a 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio (v/v) gave the best result. When trifluoroethanol was used, either alone or combined with tert-amyl alcohol and water, the product could not be isolated in a good yield (Table 1, entries 13–15). Alternative electrode materials were examined, with the best yield being obtained when using an RVC anode and nickel foam cathode rather than the more expensive platinum electrode containing configurations (Table 1, entries 16 and 17). To determine whether this reaction involves direct or indirect electrocatalysis, we performed the reaction in the presence of known electrochemical mediators.31 The use of ferrocene was found to be ineffective with no product formation (Table 1, entry 18). The quinone-based electrolytes 2,5-dimethylbenzoquinone, 2-chlorobenzoquinone, and benzoquinone provided the product with a very low yield (Table 1, entries 19–21). The use of 2,6-dichlorobenzoquinone resulted in no product formation (Table 1, entry 22), supporting direct electrocatalysis as the basis for the reaction. Further control experiments revealed that in the absence of electricity or the rhodium catalyst, no reaction was observed, and only traces of the product were obtained when the reaction was performed under air (Table 1, entries 23–25). We studied the effect of electricity on the annulation and determined that 4 mA is the optimal current by using various current values (Table 1, entries 26 and 27). Finally, reactions performed at 23, 60 and 80 °C resulted in the recovery of the starting material with no observed product formation (Table 1, entries 28–30).
:1 ratio (v/v) gave the best result. When trifluoroethanol was used, either alone or combined with tert-amyl alcohol and water, the product could not be isolated in a good yield (Table 1, entries 13–15). Alternative electrode materials were examined, with the best yield being obtained when using an RVC anode and nickel foam cathode rather than the more expensive platinum electrode containing configurations (Table 1, entries 16 and 17). To determine whether this reaction involves direct or indirect electrocatalysis, we performed the reaction in the presence of known electrochemical mediators.31 The use of ferrocene was found to be ineffective with no product formation (Table 1, entry 18). The quinone-based electrolytes 2,5-dimethylbenzoquinone, 2-chlorobenzoquinone, and benzoquinone provided the product with a very low yield (Table 1, entries 19–21). The use of 2,6-dichlorobenzoquinone resulted in no product formation (Table 1, entry 22), supporting direct electrocatalysis as the basis for the reaction. Further control experiments revealed that in the absence of electricity or the rhodium catalyst, no reaction was observed, and only traces of the product were obtained when the reaction was performed under air (Table 1, entries 23–25). We studied the effect of electricity on the annulation and determined that 4 mA is the optimal current by using various current values (Table 1, entries 26 and 27). Finally, reactions performed at 23, 60 and 80 °C resulted in the recovery of the starting material with no observed product formation (Table 1, entries 28–30).
|
|
||
|---|---|---|
| Yieldb (%) | ||
| a Reaction conditions: 1a (50 mg, 0.34 mmol), 2a (74 mg, 0.41 mmol), [Cp*RhCl2]2 (5 mol%), KOAc (2 equiv.), solvent (12 mL), 100 °C, 16 h, under N2. b Isolated yields. c KOAc was the base. NR = no reaction. | ||
| Effect of metal complexes | ||
| 1 | [Cp*RhCl2]2 | 73c |
| 2 | [Cp*IrCl2]2 | 35 |
| 3 | [(p-Cymene)RuCl2]2 | Trace |
| 4 | Pd(OAc)2 | NR |
| Effect of additives | ||
| 5 | NaOPiv | 64 |
| 6 | KOPiv | 65 |
| 7 | KOAc | 73 |
| 8 | LiOAc | 40 |
| Effect of electrolytes | ||
| 9 | KPF6 | 34 |
| 10 | n Bu4NBr | Trace |
| 11 | n Bu4NOAc | NR |
| 12 | n Bu4NClO4 | NR |
| Effect of solvents | ||
| 13 | TFE | Trace |
| 14 | TFE + t-AmylOH | 10 |
| 15 | TFE + t-AmylOH + H2O | 18 |
| Effect of electrodes | ||
| 16 | RVC/Pt | NR |
| 17 | Pt/Pt | NR |
| Effect of mediators | ||
| 18 | Ferrocene | NR |
| 19 | 2,5-Dimethylbenzoquinone | 33 |
| 20 | 2-Chlorobenzoquinone | 16 |
| 21 | Benzoquinone | 30 |
| 22 | 2,6-Dichlorobenzoquinone | NR |
| Control experiments | ||
| 23 | No electricity | 13 |
| 24 | No Rh catalyst | NR |
| 25 | Under air | Trace |
| Effect of current | ||
| 26 | 2 mA | 15 |
| 27 | 8 mA | 16 |
| Effect of temperature | ||
| 28 | 23 °C | NR |
| 29 | 60 °C | NR |
| 30 | 80 °C | NR |

With conditions for the electrochemical C–H/N–H alkyne annulation in hand, we proceeded to study the scope and versatility of the reaction using various pyrazole derivatives. The substrate scope screening revealed that both electron donating and electron withdrawing substituents at the para-position were well tolerated and delivered the products (3a–3f) in good yields (21–73%) (Scheme 2). It is noteworthy that meta-methyl substituted pyrazole gave one positional selective product (3g). The ortho, meta-dimethyl substituted pyrazole (1h) afforded product 3h in 72% yield, while the ortho-substituted pyrazole (1i) furnished product 3i in 21% yield. Interestingly, the 1-naphthyl pyrazole (1j) gave a single product, as the hypothetical 3j′ regioisomer would form an energetically less favored 7-membered ring, while the 2-naphthyl pyrazole (1k) gave two regioisomers in a 1:1 ratio (3j and 3k).
 | ||
| Scheme 2 Scope of pyrazoles with diphenylacetylene. Reaction conditions: 1a–k (0.34 mmol), 2a (0.41 mmol), [Cp*RhCl2]2 (5 mol%), KOAc (2 equiv.), solvent (12 mL), 100 °C, 16 h, under N2; aisolated yield. | ||
Subsequently, a variety of internal alkynes were tested for their suitability in the electro-redox C–H activation reaction (Scheme 3). Electronically biased substituted internal alkynes (4a–4d) delivered cyclized products (5a–5d) with moderate to good yields (33–51%). The tolerance of chloro, bromo, and fluoro at different positions on alkynes is highly appreciable as they can be useful for further cross-coupling reactions. A sterically bulky 2-naphthyl substituted internal alkyne (4e) also provided the product (5e) in 53% yield. A chlorinated alkyne at the meta-position gave the product (5f) in 30% yield. The use of 4-octyne (4h) and 5-decyne (4i) also successfully furnished the annulated products in good yields (5h and 5i). The unsymmetrical alkynes 4j–4n were then tested in this reaction and they successfully gave the expected products in moderate to excellent yields with high regioselectivity (Scheme 3). 1-Phenylpropyne (4j) gave one major regioisomer 5j in 29% yield. 1-Phenylbutyne (4k) also reacted with 2-phenylpyrazole (1) and provided the regioselective product (5k) in 33% yield. When we used 1-phenylpentyne (4l) as the coupling partner, we successfully isolated two possible regioisomers (5l + 5l′) in this reaction. 1-Phenylhexyne (4m) and 1-phenyl-2,2-dimethyl propyn-1-ol (4n) furnished regioselective products 5m (37%) and 5n (20%).
 | ||
| Scheme 3 Scope of alkynes with 2-phenylpyrazole. Reaction conditions: 1a (2 equiv.), 4a–n (50 mg, 0.24 mmol, 1 equiv.), [Cp*RhCl2]2 (5 mol%), KOAc (2 equiv.), solvent (12 mL), 100 °C, 16 h, under N2; aisolated yield. | ||
This electrochemical C–H activation could be readily scaled up to the 1 mmol scale in an undivided cell setup and provided the product (5i) in 60% yield (Scheme 4). To evaluate the efficacy of this green methodology for alkyne annulation with pyrazole, we calculated the E-factor associated with this reaction and found a total amount of 18.97, which is quite good in comparison with the conventional methods27 (see the ESI†).
 | ||
| Scheme 4 Reaction at the 1 mmol scale. | ||
Next, to elucidate the basis for the efficiency of this rhodo-electrocatalysis, we designed cyclic voltammetry studies under a nitrogen atmosphere with Ag/AgO-, nickel foam-, and RVC electrodes (Fig. 1). The slow conversion without electricity and the very good conversion after electrolysis support the hypothesis that this reaction follows an oxidation-induced reductive elimination pathway. Moreover, the peaks corresponding to the rhodium catalyst and 1a with a base and an electrolyte are indicative of the formation of a higher oxidation state for the rhodium catalyst and these two species are susceptible to electrochemical redox processes.32
 | ||
| Fig. 1 Cyclic voltammetry studies. | ||
Based on the observed experimental details and previously reported methods,15 we propose a plausible working mechanism for the electrochemical C–H activation reaction (Scheme 5). Initially, the [RhCp*Cl2]2 complex upon ligand exchange with KOAc gives RhCp*(OAc)2. The catalyst in its active form coordinates to the 2-phenylpyrazole and the subsequent C–H bond insertion gives cyclometallated rhodium complex A. The acetic acid formed in this step undergoes facile cathodic reduction and produces sustainable hydrogen as the only by-product. Complex A upon alkyne coordination results in the formation of B, which upon carbo-rhodation generates the cationic complex C. This cationic complex undergoes reductive elimination to deliver the annulated product along with reduced RhCp*L2(L = OAc). Anodic oxidation regenerates the active catalyst, which continues the cycle.
 | ||
| Scheme 5 Plausible rhoda-electrocatalytic C–H/N–H annulation. | ||
Conclusions
In summary, an unprecedented electrochemical intermolecular C–H/N–H oxidative annulation of alkynes with 2-phenylpyrazoles has been successfully achieved using electricity as the sustainable green oxidant with the implicit liberation of hydrogen as the environmentally friendly by-product, which indicates the significance of this protocol. The simple undivided cell setup in common reaction tubes, use of cheaper electrodes and a water-based solvent system, wide substrate scope and high regioselectivity showcase the novel features of this strategy. The overall waste generation with a low E-factor and a reaction at the 1 mmol scale were also explored. Further application of rhoda-electrocatalysis for the synthesis of heterocycles is ongoing in our lab and the results will be communicated in due course.Experimental details
General experimental procedure for the rhodium catalyzed electro-alkyne annulation
An undivided cell was used to carry out electrocatalysis, with an RVC anode (15 mm × 10 mm) and a nickel foam cathode (15 mm × 10 mm). 2-Phenylpyrazole (0.34 mmol), alkyne (0.41 mmol), KOAc (65 mg, 0.66 mmol), and [Cp*RhCl2]2 (10 mg, 5 mol%) were placed in a 50 mL cell and the cell was connected to a Schlenk line to add t-AmylOH + H2O (12 mL, 3:1). Electrocatalysis was performed at 100 °C with a constant current of 4 mA maintained for 16 h. Then the CC-power supply was stopped and the electrodes were washed with acetone and the combined liquids were evaporated under reduced pressure. The resulting residue was coated on preparative TLC plates (25 cm × 25 cm) and eluted with a petroleum ether and acetone mixture (100:5 mL/mL) to afford the corresponding products.
Experimental data
The general procedure was followed using 1a (50 mg, 0.34 mmol) and 2a (74 mg, 0.41 mmol) at 100 °C for 16 h. Purification using preparative TLC plates (petroleum ether/acetone = 100/5 mL/mL) afforded 3a (74 mg, 68%) as a brown solid. M. pt. 218 °C; 1H NMR (400 MHz, chloroform-d)δ 8.24 (d, J = 7.9 Hz, 1H), 8.02 (d, J = 2.2 Hz, 1H), 7.62 (ddd, J = 8.2, 6.4, 2.0 Hz, 1H), 7.51–7.43 (m, 2H), 7.37 (dtd, J = 5.7, 4.0, 3.4, 1.7 Hz, 2H), 7.31 (dddd, J = 11.3, 8.1, 4.4, 1.6 Hz, 6H), 7.24 (dd, J = 7.8, 1.9 Hz, 2H), 7.16 (d, J = 2.2 Hz, 1H). 13C NMR (101 MHz, CDCl3)δ 141.1, 138.6, 136.4, 136.2, 133.2, 131.6, 130.9, 130.0, 128.4, 128.0, 128.0, 127.8, 127.4, 127.2, 126.8, 124.1, 124.0, 123.6, 97.6. HRMS C23H16N2 [M + H]+: calculated 321.1392, found: 321.1389.

The general procedure was followed using 1b (55 mg, 0.34 mmol) and 2a (72 mg, 0.41 mmol) at 100 °C for 16 h. Purification using preparative TLC plates (petroleum ether/acetone = 100/5 mL/mL) afforded 3b (19 mg, 17%) as a brown solid. M. pt. 141 °C; 1H NMR (400 MHz, chloroform-d) δ 8.03 (s, 1H), 7.99 (d, J = 2.2 Hz, 1H), 7.39–7.35 (m, 2H), 7.33 (d, J = 4.5 Hz, 3H), 7.30 (dd, J = 7.2, 2.1 Hz, 5H), 7.27 (s, 1H), 7.25–7.17 (m, 3H), 7.13 (d, J = 2.2 Hz, 1H), 2.59 (s, 3H). 13C NMR (101 MHz, CDCl3)δ 140.9, 138.4, 137.4, 136.3, 135.5, 133.2, 131.6, 130.9, 129.3, 128.3, 127.9, 127.9, 127.7, 127.1, 126.6, 124.1, 123.9, 123.3, 97.4, 21.6. HRMS C24H18N2 [M + H]+: calculated 335.1548, found: 335.1543.

The general procedure was followed using 1c (68 mg, 0.34 mmol) and 2a (74 mg, 0.41 mmol) at 100 °C for 16 h. Purification using preparative TLC plates (petroleum ether/acetone = 100/5 mL/mL) afforded 3c (60 mg, 50%) as a brown solid. M. pt. 124 °C; 1H NMR (400 MHz, chloroform-d) δ 8.16 (d, J = 8.5 Hz, 1H), 8.01 (d, J = 2.2 Hz, 1H), 7.56 (dd, J = 8.5, 2.1 Hz, 1H), 7.41 (d, J = 2.1 Hz, 1H), 7.37–7.29 (m, 8H), 7.24–7.18 (m, 2H), 7.13 (d, J = 2.1 Hz, 1H). 13C NMR (101 MHz, CDCl3)δ 141.3, 138.0, 137.4, 135.4, 133.7, 132.8, 131.5, 131.3, 130.7 128.6, 128.2, 128.0, 127.8, 127.5, 126.0, 125.1, 123.1, 122.4, 97.8. HRMS C23H15ClN2 [M + H]+: calculated: 355.1002, found: 355.1001.

The general procedure was followed using 1d (50 mg, 0.45 mmol) and 2a (60 mg, 0.33 mmol) at 100 °C for 16 h. Purification using preparative TLC plates (petroleum ether/acetone = 87/13 mL/mL) afforded 3d (93 mg, 52%) as a brown solid. M. pt. 172 °C; 1H NMR (400 MHz, chloroform-d)δ 7.99 (d, J = 8.5 Hz, 1H), 7.91 (d, J = 2.2 Hz, 1H), 7.60 (dd, J = 8.5, 1.9 Hz, 1H), 7.47 (d, J = 2.0 Hz, 1H), 7.26–7.17 (m, 8H), 7.12–7.08 (m, 2H), 7.03 (d, J = 2.2 Hz, 1H). 13C NMR (101 MHz, CDCl3)δ 141.3, 138.0, 137.4, 135.3, 132.7, 131.6, 131.5, 130.7, 130.5, 129.1, 128.6, 128.2, 128.0, 127.5, 125.2, 123.0, 122.7, 121.9, 97.9. HRMS C23H15BrN2 [M + H]+: calculated: 401.0476, found: 401.0477.
The general procedure was followed using 1e (56 mg, 0.34 mmol) and 2a (72 mg, 0.41 mmol) at 100 °C for 16 h. Purification using preparative TLC plates (petroleum ether/acetone = 100/5 mL/mL) afforded 3e (25 mg, 21%) as a brown solid. M. pt. 121–123 °C; 1H NMR (400 MHz, chloroform-d)δ 8.11 (dd, J = 8.8, 5.5 Hz, 1H), 7.90 (d, J = 2.2 Hz, 1H), 7.25 (td, J = 4.9, 1.8 Hz, 4H), 7.22 (ddd, J = 6.5, 3.1, 1.4 Hz, 5H), 7.11 (dd, J = 7.6, 1.9 Hz, 2H), 6.99 (td, J = 5.5, 2.6 Hz, 2H). 13C NMR (101 MHz, CDCl3)δ13C NMR (101 MHz, CDCl3) δ 163.2, 160.8, 141.3, 138.2, 137.4, 135.6, 132.8, 132.0, 131.9, 131.4, 130.7, 128.5, 128.5, 128.2, 128.0, 127.4, 125.8, 125.8, 123.4, 123.3, 120.7, 120.7, 116.1, 115.9, 112.0, 111.8, 97.3. 19F NMR (376 MHz, CDCl3) δ −111.4, −111.4, −111.4, −111.4, −111.4, −111.4, −111.4, −111.4. HRMS C23H15FN2 [M + H]+: calculated: 339.1298, found: 339.1294.
The general procedure was followed using 1f (50 mg, 0.23 mmol) and 2a (63 mg, 0.35 mmol) at 100 °C for 16 h. Purification using preparative TLC plates (petroleum ether/acetone = 100/5 mL/mL) afforded 3f (50 mg, 56%) as a brown solid. (Rf = 0.53). M. pt. 86 °C; 1H NMR (400 MHz, chloroform-d)δ 8.34 (d, J = 8.4 Hz, 1H), 8.06 (d, J = 2.2 Hz, 1H), 7.82 (dd, J = 8.4, 1.8 Hz, 1H), 7.73 (d, J = 1.8 Hz, 1H), 7.34 (tt, J = 7.4, 2.1 Hz, 8H), 7.26–7.19 (m, 3H). 13C NMR (101 MHz, CDCl3)δ 141.5, 137.8, 137.7, 135.1, 132.6, 131.6, 131.5, 130.7, 129.8, 129.6 (q, J = 32.3 Hz), 128.7, 128.3, 128.1, 127.7, 124.4, 124.0 (q, J = 272.5 Hz), 124.0 (q, J = 4.2 Hz), 123.8, 123.5 (q, J = 3.5 Hz), 98.9. 19F NMR (376 MHz, CDCl3)δ −62.30. HRMS C24H15F3N2 [M + H]+: calculated: 389.1266, found: 389.1264.
The general procedure was followed using 1g (50 mg, 0.32 mmol) and 2a (74 mg, 0.41 mmol) at 100 °C for 16 h. Purification using preparative TLC plates (petroleum ether/acetone = 100/5 mL/mL) afforded 3g (80 mg, 70%) as a brown solid. (Rf = 0.8). M. pt. 143–145 °C; 1H NMR (400 MHz, chloroform-d)δ 8.14 (d, J = 8.1 Hz, 1H), 7.99 (d, J = 2.2 Hz, 1H), 7.46 (d, J = 1.7 Hz, 1H), 7.40–7.26 (m, 9H), 7.26–7.17 (m, 3H), 2.43 (s, 3H). 13C NMR (101 MHz, CDCl3)δ 141.0, 138.6, 137.8, 136.4, 136.3, 133.3, 131.6, 130.8, 130.0, 128.9, 128.3, 128.0, 127.9, 127.1, 126.3, 123.8, 123.5, 121.9, 97.0, 21.9. HRMS C24H18N2 [M + H]+: calculated: 335.1548, found: 335.1544.

The general procedure was followed using 1h (60 mg, 0.34 mmol) and 2a (72 mg, 0.41 mmol) at 100 °C for 16 h. Purification using preparative TLC plates (petroleum ether/acetone = 100/5 (mL/mL)) afforded 3h (85 mg, 72%) as a brown solid. M. pt. 158 °C; 1H NMR (400 MHz, chloroform-d)δ 7.91 (d, J = 2.3 Hz, 1H), 7.24–7.14 (m, 9H), 7.10 (dd, J = 7.8, 1.8 Hz, 2H), 7.07 (d, J = 2.3 Hz, 1H), 6.99 (s, 1H), 2.80 (s, 3H), 2.26 (s, 3H). 13C NMR (101 MHz, CDCl3)δ 140.7, 137.7, 136.9, 136.8, 136.4, 134.4, 133.7, 131.6, 131.3, 130.8, 128.2, 128.0, 127.9, 127.0, 124.6, 124.2, 121.6, 101.7, 23.8, 21.5. HRMS C25H20N2 [M + H]+: calculated: 349.1705, found: 349.1700.

The general procedure was followed using 1i (60 mg, 0.34 mmol) and 2a (74 mg, 0.41 mmol) at 100 °C for 16 h. Purification using preparative TLC plates (petroleum ether/acetone = 100/5 mL/mL) afforded 3i (25 mg, 21%) as a brown solid. M. pt. 137 °C; 1H NMR (400 MHz, chloroform-d)δ 7.93 (d, J = 2.1 Hz, 1H), 7.39 (d, J = 2.1 Hz, 1H), 7.32 (t, J = 8.1 Hz, 1H), 7.27–7.21 (m, 4H), 7.21–7.15 (m, 8H), 7.00 (d, J = 7.7 Hz, 1H), 6.93 (d, J = 8.2 Hz, 1H), 4.07 (s, 3H). 13C NMR (101 MHz, CDCl3)δ 155.8, 141.0, 136.9, 136.6, 135.4, 133.4, 131.9, 131.6, 130.7, 128.2, 127.9, 127.9, 127.7, 127.0, 123.6, 118.7, 115.0, 107.7, 102.8, 55.8. HRMS C24H18N2O [M + H]+: calculated: 351.1497, found: 351.1493.

The general procedure was followed using 1j (66 mg, 0.34 mmol) and 2a (72 mg, 0.41 mmol) at 100 °C for 16 h. Purification using preparative TLC plates (petroleum ether/acetone = 100/5 mL/mL) afforded 3j (30 mg, 24%) as a brown solid. M. pt. 191–192 °C; 1H NMR (400 MHz, chloroform-d)δ 9.14 (d, J = 8.6 Hz, 1H), 8.21 (d, J = 2.3 Hz, 1H), 8.02 (dd, J = 8.0, 1.4 Hz, 1H), 7.88–7.82 (m, 2H), 7.75–7.69 (m, 2H), 7.53 (d, J = 8.9 Hz, 1H), 7.42 (d, J = 2.0 Hz, 1H), 7.41–7.38 (m, 1H), 7.38 (s, 1H), 7.36 (d, J = 0.8 Hz, 1H), 7.35–7.34 (m, 2H), 7.33 (q, J = 1.6 Hz, 2H), 7.28 (d, J = 1.7 Hz, 2H). 13C NMR (101 MHz, CDCl3)δ 141.8, 137.3, 137.3, 136.6, 133.6, 132.6, 131.7, 130.7, 129.3, 129.0, 129.0, 128.5, 128.4, 128.1, 128.0, 127.5, 127.2, 126.5, 125.3, 124.7, 124.5, 120.6, 101.1. HRMS C27H18N2 [M + H]+: calculated: 371.1548, found: 371.1545.
The general procedure was followed using 1k (66 mg, 0.34 mmol) and 2a (93 mg, 0.52 mmol) at 100 °C for 16 h. Purification using preparative TLC plates (petroleum ether/acetone = 100/5 mL/mL) afforded 3k and 3k′ (18 mg, 15%) as a brown solid. M. pt. 164–166 °C; 1H NMR (400 MHz, chloroform-d)δ 8.72 (s, 1H), 8.26 (d, J = 8.7 Hz, 1H), 8.10–7.99 (m, 4H), 7.95–7.87 (m, 2H), 7.83 (d, J = 8.3 Hz, 1H), 7.57 (tt, J = 6.5, 1.3 Hz, 2H), 7.53–7.42 (m, 3H), 7.41 (d, J = 1.9 Hz, 1H), 7.39 (p, J = 1.8 Hz, 2H), 7.37 (d, J = 1.8 Hz, 1H), 7.35 (d, J = 2.8 Hz, 2H), 7.34–7.33 (m, 2H), 7.32 (d, J = 2.6 Hz, 4H), 7.30 (d, J = 2.1 Hz, 3H), 7.28 (d, J = 3.2 Hz, 3H), 7.27 (t, J = 1.9 Hz, 1H), 7.21 (td, J = 3.8, 1.9 Hz, 3H), 7.10 (ddd, J = 8.6, 6.9, 1.6 Hz, 1H). 13C NMR (101 MHz, CDCl3)δ 141.7, 140.8, 139.9, 139.0, 138.3, 137.9, 136.3, 136.1, 134.1, 133.6, 133.2, 132.6, 132.1, 131.7, 131.6, 131.4, 130.9, 130.8, 130.3, 129.6, 128.9, 128.5, 128.5, 128.5, 128.3, 128.2, 128.1, 128.1, 128.0, 127.9, 127.7, 127.5, 127.3, 127.1, 126.6, 126.2, 126.0, 125.6, 125.4, 124.0, 123.8, 123.3, 122.5, 122.2, 121.9, 99.2, 97.3. HRMS C27H18N2 [M + H]+: calculated: 371.1548, found: 371.1546.

The general procedure was followed using 1b (70 mg, 0.34 mmol) and 4a (50 mg, 0.41 mmol) at 100 °C for 16 h. Purification using preparative TLC plates (petroleum ether/acetone = 85/15 mL/mL) afforded 5a (31 mg, 38%) as a brown solid. M. pt. 180–182 °C; 1H NMR (400 MHz, chloroform-d)δ 8.33–8.15 (m, 1H), 8.01 (s, 1H), 7.60 (s, 1H), 7.46 (s, 2H), 7.39–7.23 (m, 3H), 7.15 (s, 6H), 2.37 (d, J = 11.5 Hz, 6H). 13C NMR (101 MHz, CDCl3)δ 140.9, 138.5, 138.0, 136.6, 136.5, 133.2, 131.4, 130.6, 130.3, 130.2, 128.7, 127.6, 127.2, 126.8, 124.0, 123.8, 123.5, 97.4, 21.4, 21.3. HRMS C25H20N2 [M + H]+: calculated: 349.1705, found: 349.1701.

The general procedure was followed using 1b (50 mg, 0.34 mmol) and 4b (50 mg, 0.23 mmol) at 100 °C for 16 h. Purification using preparative TLC plates (petroleum ether/acetone = 100/5 mL/mL) afforded 5b (26 mg, 33%) as a brown solid. M. pt. 160–162 °C; 1H NMR (400 MHz, chloroform-d)δ 7.91–7.86 (m, 1H), 7.50–7.45 (m, 4H), 7.35 (d, J = 8.5 Hz, 4H), 7.21 (d, J = 8.5 Hz, 1H), 7.10–7.03 (m, 2H), 7.01 (d, J = 8.5 Hz, 1H), 6.77 (d, J = 8.4 Hz, 1H). 13C NMR (101 MHz, CDCl3)δ 136.6, 134.5, 132.8, 132.4, 132.2, 131.1, 129.6, 128.9, 128.7, 128.3, 128.1, 127.7, 127.5, 125.4, 121.4, 89.1. HRMS C23H14N2Cl2 [M + H]+: calculated: 389.0612, found: 389.0610.

The general procedure was followed using 1b (67 mg, 0.34 mmol) and 4c (50 mg, 0.23 mmol) at 100 °C for 16 h. Purification using preparative TLC plates (petroleum ether/acetone = 100/5 mL/mL) afforded 5c (41 mg, 51%) as a brown solid. M. pt. 152–154 °C; 1H NMR (400 MHz, chloroform-d)δ 8.14 (dd, J = 7.7, 1.1 Hz, 1H), 7.90 (d, J = 2.2 Hz, 1H), 7.55–7.50 (m, 1H), 7.42–7.37 (m, 1H), 7.33–7.28 (m, 1H), 7.26–7.20 (m, 2H), 7.11–7.04 (m, 3H), 6.97–6.90 (m, 4H). 13C NMR (101 MHz, CDCl3)δ 162.6, 162.1, 160.2, 159.7, 140.1, 137.5, 134.6, 132.1, 132.0, 131.7, 131.6, 130.8, 130.8, 128.7, 127.9, 127.9, 126.9, 126.6, 125.4, 123.0, 122.6, 122.2, 114.4, 114.3, 114.2, 114.1, 96.8. 19F NMR (376 MHz, CDCl3)δ −112.2, −112.3, −112.3, −112.3, −112.3, −112.3, −112.3, −112.3, −114.2, −114.3, −114.3, −114.3, −114.3, −114.3. HRMS C23H14N2F2 [M + H]+: calculated: 357.1203, found: 357.1200.

The general procedure was followed using 1a (43 mg, 0.29 mmol) and 4d (50 mg, 0.15 mmol) at 100 °C for 16 h. Purification using preparative TLC plates (petroleum ether/acetone = 100/5 mL/mL) afforded 5d (28 mg, 40%) as a brown solid. M. pt. 227–230 °C; 1H NMR (400 MHz, chloroform-d)δ 8.26–8.20 (m, 1H), 8.00 (d, J = 2.2 Hz, 1H), 7.63 (ddd, J = 8.1, 7.1, 1.2 Hz, 1H), 7.49 (ddt, J = 7.2, 3.0, 1.7 Hz, 5H), 7.39 (dt, J = 8.3, 1.0 Hz, 1H), 7.26–7.21 (m, 2H), 7.15 (d, J = 2.2 Hz, 1H), 7.13–7.07 (m, 2H). 13C NMR (101 MHz, CDCl3)δ 141.2, 138.5, 135.2, 134.8, 133.1, 132.4, 131.7, 131.5, 131.5, 129.4, 128.0, 127.8, 126.4, 124.1, 123.7, 123.1, 122.9, 121.7, 97.9. HRMS C23H14N2Br2 [M + H]+: calculated: 478.9582, found: 478.9581.
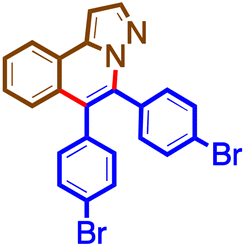
The general procedure was followed using 1b (48 mg, 0.33 mmol) and 4e (50 mg, 0.17 mmol) at 100 °C for 16 h. Purification using preparative TLC plates (petroleum ether/acetone = 85/15 mL/mL) afforded 5e (40 mg, 53%) as a brown solid. M. pt. 198–200 °C; 1H NMR (400 MHz, chloroform-d)δ 8.17 (d, J = 7.9 Hz, 1H), 7.91 (d, J = 2.1 Hz, 1H), 7.77 (s, 1H), 7.65 (dd, J = 13.6, 8.1 Hz, 6H), 7.57 (d, J = 8.0 Hz, 1H), 7.52 (ddd, J = 8.2, 6.4, 2.0 Hz, 1H), 7.46 (dd, J = 8.5, 1.7 Hz, 1H), 7.37–7.23 (m, 6H), 7.08 (d, J = 2.2 Hz, 1H). 13C NMR (101 MHz, CDCl3)δ 141.1, 138.6, 136.5, 133.6, 133.0, 132.9, 132.3, 130.6, 130.6, 130.5, 130.2, 129.3, 128.3, 128.0, 127.9, 127.8, 127.7, 127.6, 127.6, 127.4, 126.9, 126.4, 126.1, 126.1, 125.8, 124.2, 124.1, 123.6, 97.7. HRMS C31H22N2 [M + H]+: calculated: 421.1705, found: 421.1701.

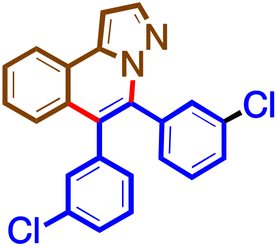
The general procedure was followed using 1b (50 mg, 0.34 mmol) and 4f (50 mg, 0.20 mmol) at 100 °C for 16 h. Purification using preparative TLC plates (petroleum ether/acetone = 100/5 mL/mL) afforded 5f (70 mg, 99%) as a brown solid. M. pt. 172–174 °C; 1H NMR (400 MHz, chloroform-d)δ 8.24 (dt, J = 8.0, 0.9 Hz, 1H), 8.01 (d, J = 2.1 Hz, 1H), 7.64 (ddd, J = 8.1, 7.1, 1.2 Hz, 1H), 7.51 (ddd, J = 8.4, 7.1, 1.3 Hz, 1H), 7.43–7.36 (m, 2H), 7.34–7.22 (m, 6H), 7.16 (d, J = 2.2 Hz, 1H), 7.12 (dt, J = 6.9, 1.7 Hz, 1H). 13C NMR (101 MHz, CDCl3)δ 141.4, 138.6, 137.6, 135.1, 134.3, 134.1, 134.0, 131.4, 130.9, 129.7, 129.5, 129.4, 129.3, 129.0, 128.9, 128.0, 127.9, 127.8, 126.5, 124.1, 123.7, 123.0, 97.9. HRMS C23H14N2Cl2 [M + H]+: calculated: 389.0612, found: 389.0606.
The general procedure was followed using 1b (129 mg, 0.89 mmol) and 4h (50 mg, 0.41 mmol) at 100 °C for 16 h. Purification using preparative TLC plates (petroleum ether/acetone = 85/15 mL/mL) afforded 5h (31 mg, 36%) as a yellow sticky solid. 1H NMR (400 MHz, chloroform-d)δ 7.57 (ddt, J = 9.1, 5.5, 1.7 Hz, 3H), 7.37–7.29 (m, 1H), 6.54 (d, J = 2.0 Hz, 1H), 6.08 (dd, J = 8.0, 6.9 Hz, 1H), 5.47 (t, J = 6.8 Hz, 1H), 2.42 (dq, J = 15.2, 7.6 Hz, 1H), 2.28 (dq, J = 14.9, 7.4 Hz, 1H), 1.81–1.70 (m, 1H), 1.66–1.55 (m, 1H), 1.37–1.23 (m, 3H), 1.15 (t, J = 7.5 Hz, 3H), 0.86 (t, J = 7.3 Hz, 3H). 13C NMR (101 MHz, CDCl3)δ 139.1, 132.8, 132.5, 131.8, 128.3, 127.9, 125.0, 124.9, 123.6, 100.2, 58.1, 38.7, 21.5, 18.7, 14.0, 13.8. HRMS C17H20N2 [M + NH3]+: calculated 269.1892, found: 269.1647.
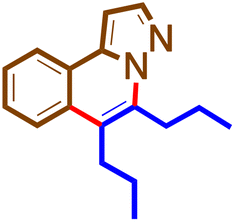
The general procedure was followed using 1b (50 mg, 0.34 mmol) and 4i (153 μL, 0.68 mmol) at 100 °C for 16 h. Purification using preparative TLC plates (petroleum ether/acetone = 100/5 mL/mL) afforded 5i (60 mg, 63%) as a yellow sticky solid. 1H NMR (400 MHz, chloroform-d)δ 8.13 (dd, J = 7.7, 1.6 Hz, 1H), 7.99 (d, J = 2.2 Hz, 1H), 7.94–7.85 (m, 1H), 7.56 (dddd, J = 21.3, 8.3, 7.1, 1.3 Hz, 3H), 7.02 (d, J = 2.2 Hz, 1H), 3.38–3.30 (m, 2H), 3.03–2.95 (m, 2H), 1.87–1.76 (m, 2H), 1.73–1.65 (m, 2H), 1.62–1.54 (m, 4H), 1.06 (d, J = 7.4 Hz, 3H), 1.02 (d, J = 7.5 Hz, 3H). 13C NMR (101 MHz, CDCl3)δ 139.8, 138.8, 137.7, 136.8, 129.0, 127.9, 127.6, 126.2, 126.2, 123.9, 123.8, 123.8, 123.6, 123.5, 119.0, 97.1, 32.7, 30.1, 27.9, 27.3, 23.1, 23., 14.0, 14.0, 13.9. HRMS C19H24N2 [M + NH4Cl]+: calculated: 333.1972, found: 333.2325.

The general procedure was followed using 1 (50 mg, 0.34 mmol) and 4j (86 μL, 0.68 mmol) at 100 °C for 16 h. Purification using preparative TLC plates (petroleum ether/acetone = 90/10 mL/mL) afforded 5j (25 mg, 29%) as a brown solid. M. pt. 80–82 °C; 1H NMR (400 MHz, chloroform-d)δ 8.12–8.08 (m, 1H), 7.86–7.82 (m, 1H), 7.81 (d, J = 2.1 Hz, 1H), 7.57–7.51 (m, 3H), 7.51–7.47 (m, 2H), 7.47–7.43 (m, 1H), 7.40 (dd, J = 8.1, 1.6 Hz, 2H), 6.96 (d, J = 2.2 Hz, 1H), 2.28 (s, 3H). 13C NMR (101 MHz, CDCl3)δ 140.4, 138.0, 135.9, 133.8, 130.4, 129.8, 128.9, 128.7, 127.8, 127.2, 124.4, 124.1, 123.8, 116.2, 97.3, 15.0. HRMS C18H14N2 [M + Na]+: calculated: 281.1055, found: 281.2008.

The general procedure was followed using 1 (50 mg, 0.34 mmol) and 4k (50 mg, 0.38 mmol) at 100 °C for 16 h. Purification using preparative TLC plates (petroleum ether/acetone = 100/5 mL/mL) afforded 5k (34 mg, 33%) as a brown solid. M. pt. 88–90 °C; 1H NMR (400 MHz, chloroform-d)δ 8.13–8.08 (m, 1H), 7.90–7.84 (m, 1H), 7.81 (d, J = 2.2 Hz, 1H), 7.56–7.42 (m, 5H), 7.42–7.36 (m, 2H), 6.96 (d, J = 2.2 Hz, 1H), 2.70 (q, J = 7.5 Hz, 2H), 1.14 (t, J = 7.5 Hz, 3H). 13C NMR (101 MHz, CDCl3)δ 139.5, 136.9, 134.7, 132.8, 128.8, 127.8, 127.8, 127.5, 126.7, 126.0, 123.6, 123.4, 123.1, 121.3, 96.1, 76.3, 75.9, 75.6, 20.5, 14.1. HRMS Calc'd for C19H16N2 [M + H]+: calculated 273.1386 found: 273.1388.

The general procedure was followed using 1 (50 mg, 0.34 mmol) and 4l (67 μL, 0.41 mmol) at 100 °C for 16 h. Purification using preparative TLC plates (petroleum ether/acetone = 100/5 mL/mL) afforded 5l (34 mg, 35%) and 5l′ (21 mg, 20%) as a brown solid. M. pt. 48–50 °C; 1H NMR (400 MHz, chloroform-d)δ 8.20–8.15 (m, 1H), 8.08 (d, J = 2.2 Hz, 1H), 7.58–7.49 (m, 4H), 7.45–7.38 (m, 1H), 7.38–7.35 (m, 2H), 7.21 (d, J = 8.1 Hz, 1H), 7.11 (d, J = 2.3 Hz, 1H), 3.04–2.97 (m, 2H), 1.83–1.77 (m, 2H), 0.92 (t, J = 7.4 Hz, 3H). 13C NMR (101 MHz, CDCl3)δ 140.2, 138.3, 137.2, 136.7, 130.9, 128.6, 127.7, 127.6, 126.6, 126.3, 123.4, 123.2, 97.5, 31.2, 21.2, 14.2. HRMS C20H18N2 [M + H]+: calculated: 287.1548, found: 287.1544.

Brown solid, M. pt. 104–106 °C; 1H NMR (400 MHz, chloroform-d)δ 8.24–8.17 (m, 1H), 7.99–7.87 (m, 2H), 7.68–7.52 (m, 5H), 7.52–7.45 (m, 2H), 7.06 (d, J = 2.2 Hz, 1H), 2.77–2.70 (m, 2H), 1.73–1.62 (m, 2H), 0.93 (t, J = 7.4 Hz, 3H). 13C NMR (101 MHz, CDCl3)δ 140.6, 138.0, 136.0, 133.9, 130.0, 128.9, 128.9, 128.7, 128.6, 127.7, 127.1, 124.6, 124.5, 124.1, 121.1, 97.2, 30.5, 24.0, 14.4. HRMS C20H18N2 [M + H]+: calculated: 287.1548, found: 287.1542.

The general procedure was followed using 1 (50 mg, 0.34 mmol) and 4m (122 μL, 0.68 mmol) at 100 °C for 16 h. Purification using preparative TLC plates (petroleum ether/acetone = 100/5 mL/mL) afforded 5m (38 mg, 37%) as a brown solid. M. pt. 48–50 °C; 1H NMR (400 MHz, chloroform-d)δ 8.13–8.07 (m, 1H), 7.85 (dd, J = 7.6, 2.0 Hz, 1H), 7.81 (d, J = 2.2 Hz, 1H), 7.55–7.42 (m, 5H), 7.41–7.36 (m, 2H), 6.95 (d, J = 2.1 Hz, 1H), 2.68–2.60 (m, 2H), 1.57–1.47 (m, 2H), 1.22 (dt, J = 14.8, 7.4 Hz, 2H), 0.75 (t, J = 7.3 Hz, 3H). 13C NMR (101 MHz, CDCl3)δ 140.6, 139.5, 137.9, 135.9, 133.8, 130.1, 130.0, 129.1, 128.3, 128.9, 128.8, 128.7, 128.6, 128.1, 127.7, 127.6, 127.1, 127.1, 124.6, 124.5, 124.4, 124.1, 123.7, 121.2, 97.2, 32.8, 28.0, 22.9, 13.7. HRMS C21H20N2 [M + H]+: calculated: 301.1705, found: 301.1707.

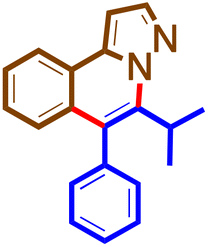
The general procedure was followed using 1 (67 mg, 0.46 mmol) and 4n (57 mg, 0.31 mmol) at 100 °C for 16 h. Purification using preparative TLC plates (petroleum ether/acetone = 100/5 mL/mL) afforded 5n (19 mg, 20%) as a brown solid. M. pt. 125–126 °C; 1H NMR (400 MHz, chloroform-d)δ 8.18–8.13 (m, 1H), 8.05 (d, J = 2.2 Hz, 1H), 7.58–7.48 (m, 4H), 7.41–7.32 (m, 3H), 7.14–7.07 (m, 2H), 1.66 (s, 1H), 1.53 (d, J = 7.1 Hz, 6H). 13C NMR (101 MHz, CDCl3)δ 141.1, 139.9, 138.9, 137.5, 130.6, 130.3, 128.6, 127.6, 127.4, 126.6, 126.5, 123.3, 123.1, 121.8, 96.8, 31.4, 30.2, 19.1. HRMS C20H18N2O [M + H]+: calculated: 303.1497, found: 303.1499.
Author contributions
SK conceived and designed the project and performed the bulk of the experimental studies. PA synthesized the 2-phenylpyrazoles and symmetrical alkynes. Both authors have contributed to the writing of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge funding from Linnaeus University, the Swedish Research Council (2014-4573), the Crafoord Foundation (2019-0925 and 2020-0775) and the Helge Ax:son Johnsons Foundation (2019-0318 and 2022-0317). We thank Dr Subramanian Suriyanarayanan (Linnaeus University) for help with cyclic voltammetry studies. We express our gratitude to Professor Ian A. Nicholls, the Dean of Chemistry and Biomedical Sciences at Linnaeus University, for his constant support.References
- D. A. Colby, R. G. Bergman and J. A. Ellman, Chem. Rev., 2010, 110, 624–655 CrossRef CAS PubMed; T. W. Lyon and M. S. Sanford, Chem. Rev., 2010, 110, 1147–1169 CrossRef PubMed; H. M. L. Davies and D. Morton, ACS Cent. Sci., 2017, 3, 936–943 CrossRef PubMed; R. Giri, B.-F. Shi, K. M. Engle, N. Maugel and J.-Q. Yu, Chem. Soc. Rev., 2009, 38, 3242–3272 RSC; T. Gensch, M. N. Hopkison, F. Glorius and J. Wencel-Delord, Chem. Soc. Rev., 2016, 45, 2900–2936 RSC; Z. Dong, Z. Ren, S. J. Thompson, Y. Xu and G. Dong, Chem. Rev., 2017, 117, 9333–9403 CrossRef PubMed; Y. Wei, P. Hu, M. Zhang and W. Su, Chem. Rev., 2017, 117, 8864–8907 CrossRef PubMed; Y. Yang, J. Lan and J. You, Chem. Rev., 2017, 117, 8787–8863 CrossRef PubMed.
- Z. Shi, C. Zhang, C. Tang and N. Jiao, Chem. Soc. Rev., 2012, 41, 3381–3430 RSC; O. Daugulis, J. Roane and L. D. Tran, Acc. Chem. Res., 2015, 48, 1053–1064 CrossRef CAS PubMed; J. F. Hartwig, J. Am. Chem. Soc., 2016, 138, 2–24 CrossRef PubMed; Y. Feng and G. Chen, Angew. Chem., Int. Ed., 2010, 49, 958–961 CrossRef PubMed; D. Dailler, G. Danoun and O. Baudoin, Angew. Chem., Int. Ed., 2015, 54, 4919–4922 CrossRef PubMed; L. M. Chapman, J. C. Beck, L. Wu and S. E. Reisman, J. Am. Chem. Soc., 2016, 138, 9803–9806 CrossRef PubMed; W. R. Gutekunsk and P. S. Baran, J. Am. Chem. Soc., 2011, 133, 19076–19079 CrossRef PubMed.
- T. Satoh and M. Miura, Chem. – Eur. J., 2010, 16, 11212–11222 CrossRef CAS PubMed.
- D. R. Stuart, P. Alsabeh, M. Kuhn and K. Fagnou, J. Am. Chem. Soc., 2010, 132, 18326–18339 CrossRef CAS PubMed.
- J. Kim, D. Kim and S. Chang, J. Am. Chem. Soc., 2020, 142, 19052–19057 CrossRef CAS PubMed.
- A. Wangweerawong, R. G. Bergman and J. A. Ellman, J. Am. Chem. Soc., 2014, 136, 8520–8523 CrossRef CAS PubMed.
- J. M. Neely and T. Rovis, J. Am. Chem. Soc., 2014, 136, 2735–2738 CrossRef CAS PubMed.
- G. Song, F. Wang and X. Li, Chem. Soc. Rev., 2012, 41, 3651–3678 RSC.
- F. W. Patureau, J. Wencel-Delord and F. Glorius, Aldrichimica Acta, 2012, 45, 31–41 CAS.
- S. Wu, R. Zeng, C. Fu, Y. Yu, X. Zhang and S. Ma, Chem. Sci., 2015, 6, 2275–2285 RSC; M. D. Wodrich, B. Ye, J. F. Gonthier, C. Corminboeuf and N. Cramer, Chem. – Eur. J., 2014, 20, 15409–15418 CrossRef CAS PubMed.
- J. Seo, Y. Park, I. Jeon, T. Ryu, S. Park and P. H. Lee, Org. Lett., 2013, 15, 3358–3361 CrossRef CAS PubMed; D. J. Burns, D. Best, M. D. Wieczysty and H. W. Lam, Angew. Chem., Int. Ed., 2015, 54, 9958–9962 CrossRef PubMed; S. Kathiravan and I. A. Nicholls, Chem. Commun., 2014, 50, 14964–14967 RSC.
- Z. J. Xu and X. Wang, Chem. – Eur. J., 2020, 26, 3897 CrossRef CAS PubMed; P. Gandeepan, L. H. Finger, T. H. Meyer and L. Ackermann, Chem. Soc. Rev., 2020, 49, 4254–4272 RSC; A. Jutand, Chem. Rev., 2008, 108, 2300–2347 CrossRef PubMed; M. Yan, Y. Kawamata and P. S. Baran, Chem. Rev., 2017, 117, 13230–133319 CrossRef PubMed; J.-I. Yoshida, K. Kataoka, R. Horcajada and A. Nagaki, Chem. Rev., 2008, 108, 2265–2299 CrossRef PubMed; S. R. Waldvogel, S. Lips, M. Selt, B. Riehl and C. J. Kampf, Chem. Rev., 2018, 118, 6706–6765 CrossRef PubMed; R. Feng, J. A. Smith and K. D. Moeller, Acc. Chem. Res., 2017, 50, 2346–2352 CrossRef PubMed; S. Tang, Y. Liu and A. Lei, Chem, 2018, 4, 27–45 Search PubMed; J. Parry, N. Fu and S. Lin, Synlett, 2018, 29, 257–265 CrossRef.
- Q.-L. Yang, Y.-K. Xing, X.-Y. Wang, H.-X. Ma, X.-J. Wang, X. Yang, H.-M. Guo and T.-S. Mei, J. Am. Chem. Soc., 2019, 141, 18970–18976 CrossRef CAS PubMed; J. E. Nutting, M. Rafiee and S. S. Stahl, Chem. Rev., 2018, 118, 4834–4885 CrossRef PubMed.
- X. Ye, C. Wang, S. Zhang, J. Wei, C. Shan, L. Wojtas, Y. Xie and X. Shi, ACS Catal., 2020, 10, 11693–11699 CrossRef CAS.
- Y. Qiu, C. Zhu, M. Stangier, J. Struwe and L. Ackermann, CCS Chem., 2020, 2, 1529–1552 Search PubMed.
- Z. W. Hou, Z. Y. Mao, H. B. Zhao, Y. Y. Melcamu, X. Lu, J. Song and H. C. Xu, Angew. Chem., Int. Ed., 2016, 55, 9168–9172 CrossRef CAS PubMed.
- Y. Yuan and A. Lei, Acc. Chem. Res., 2019, 52, 3309–3324 CrossRef CAS PubMed.
- K.-J. Jiao, Y.-K. Xing, Q.-L. Yang, H. Qiu and T.-S. Mei, Acc. Chem. Res., 2020, 53, 300–310 CrossRef CAS PubMed; C. Ma, P. Fang, D. Liu, K.-J. Jiao, P.-S. Gao, H. Qiu and T.-S. Mei, Chem. Sci., 2021, 12, 12866–12873 RSC.
- M. Elsherbini and T. Wirth, Acc. Chem. Res., 2019, 52, 3287–3296 CrossRef CAS PubMed; F. Marken and J. D. Wadhawan, Acc. Chem. Res., 2019, 52, 3325–3338 CrossRef PubMed.
- W.-J. Kond, Z. Shen, L. H. Finger and L. Ackermann, Angew. Chem., Int. Ed., 2020, 59, 5551–5556 CrossRef PubMed.
- Z.-J. Wu, F. Su, W. Lin, J. Song, T.-B. Wen and H.-C. Xu, Angew. Chem., Int. Ed., 2019, 58, 16770–16774 CrossRef CAS PubMed.
- Y.-K. Xing, X.-R. Chen, Q.-L. Yang, S.-Q. Zhang, H.-M. Guo, X. Hong and T.-S. Mei, Nat. Commun., 2021, 12, 930–939 CrossRef CAS PubMed.
- J. Wu, Y. Zhou, Y. Zhou, C.-W. Chiang and A. Lei, ACS Catal., 2017, 7, 8320–8323 CrossRef CAS.
- R. Hili and A. K. Yudin, Nat. Chem. Biol., 2006, 2, 284–287 CrossRef CAS PubMed; J. A. Halfen, Curr. Org. Chem., 2005, 9, 657–669 CrossRef; S. A. Lawrence, Amines: Synthesis, properties, and applications, Cambridge University Press, New York, 2004 Search PubMed.
- A. F. Pozharskii, A. T. Soldatenkov and A. R. Katritzky, Heterocycles in Life and Society, Wiley, Cichester, U. K., 1997 Search PubMed.
- J. Elguero, P. Goya, N. Jagerovic and A. M. S. Silva, Pyrazoles as Drugs: Facts and Fantasies, in Targets in Heterocyclic Systems, ed. O. A. Attanasi and D. Spinelli, Italian Society of Chemistry, Roma, 2002, vol. 6, pp. 52–98 CrossRef CAS PubMed; A. Dore, B. Asproni, A. Scampuddu, G. A. Pinna, C. T. Christoffersen, M. Langgård and J. Kehler, Eur. J. Med. Chem., 2014, 84, 181–193 CrossRef CAS PubMed; X.-H. Liu, P. Cui, B.-A. Song, P. S. Bhadury, H.-L. Zhu and S.-F. Wang, Bioorg. Med. Chem., 2008, 16, 4075–4082 CrossRef PubMed; S. Löber, H. Hubner and P. Gmeiner, Bioorg. Med. Chem. Lett., 1999, 9, 97–102 CrossRef PubMed; Z.-Y. Chen and J. Wu, Org. Lett., 2010, 12, 4856–4859 CrossRef PubMed; C. Custodi, R. Nuti, T. I. Opera and A. Macchiarulo, J. Mol. Graphics Modell., 2012, 38, 70–81 CrossRef PubMed.
- X. Li and M. Zhao, J. Org. Chem., 2011, 76, 8530–8536 CrossRef CAS PubMed.
- W. Ma, K. Graczyk and L. Ackermann, Org. Lett., 2012, 14, 6318–6321 CrossRef CAS PubMed.
- J. Huang, Y. Fu, Z. Deng, W. Chen, Z. Song and Y. Peng, Org. Biomol. Chem., 2020, 18, 9863–9872 RSC.
- S. Kathiravan and I. A. Nicholls, Org. Lett., 2019, 21, 1968–1972 CrossRef CAS PubMed.
- R. Francke and R. D. Little, Chem. Soc. Rev., 2014, 43, 2492–2521 RSC.
- S. Jin, J. Kim, D. Kim, J.-W. Park and S. Chang, ACS Catal., 2021, 11, 6590–6595 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental setup, spectra details, NMR & HRMS spectra. See DOI: https://doi.org/10.1039/d2ob02306g |
| This journal is © The Royal Society of Chemistry 2023 |