
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDevelopment of a new synthetic method for oligodeoxynucleotides using 3′-H-phosphonamidate derivatives†
Taiki
Tsurusaki
 ,
Kazuki
Sato
and
Takeshi
Wada
*
,
Kazuki
Sato
and
Takeshi
Wada
*
Department of Medicinal and Life Sciences, Faculty of Pharmaceutical Sciences, Tokyo University of Science, 2641 Yamazaki, Noda, Chiba 278-8510, Japan. E-mail: twada@rs.tus.ac.jp
First published on 13th January 2023
Abstract
In this study, we developed a new approach for the solution-phase synthesis of oligodeoxynucleotides (ODNs) using nucleoside 3′-H-phosphonamidate derivatives as monomers. The H-phosphonamidate monomers having a heterocyclic amino group as the leaving group reacted with an alcohol to form an internucleotidic H-phosphonate diester under mild basic conditions without using additives. The resultant internucleotidic linkage was converted into a more stable linkage, such as an S-cyanoethyl phosphorothioate diester. Moreover, under the conditions for detritylation, the unreacted H-phosphonamidate monomer was converted into a water-soluble compound, which was easily removed by extraction. Thus, only simple extractions were required to purify intermediates, and the solution-phase synthesis of trithymidine diphosphorothioate from the monomer was achieved with only one silica gel column chromatography purification. This method was applied to deoxyadenosine, deoxycytidine, and deoxyguanosine derivatives. This strategy enables us to reduce the number of reagents and simplify the purification process.
Introduction
A wide variety of P-modified oligodeoxynucleotides (ODNs) have been developed and used as antisense oligonucleotides,1 and there is an expanding demand for efficient synthetic methods for diverse kinds of P-modified ODNs. The phosphoramidite method is the most frequently used for synthesizing ODNs.2 In the phosphoramidite method, phosphoramidite derivatives bearing a 4,4-dimethoxytrityl (DMTr) group on the 5′-hydroxy group are used as monomers. To form an internucleotidic linkage, the phosphoramidite monomer is condensed with a 5′-hydroxy group of an oligonucleotide with an acidic activator, such as tetrazole. The phosphoramidite method affords a high condensation efficiency and is the most versatile method for obtaining ODNs.The H-phosphonate method is well-known to be used for the synthesis of ODNs.3–5 In the H-phosphonate method, 3′-H-phosphonate monoester derivatives bearing a DMTr group on the 5′-hydroxy group are used as monomers. The H-phosphonate monomer is condensed with a 5′-hydroxy group of another nucleoside or nucleotide using a condensing reagent to form an H-phosphonate diester linkage. The internucleotidic H-phosphonate diester linkages of oligomers are converted into a variety of P-modified linkages, such as phosphate,3 phosphorothioate,6 phosphoramidate,7 and alkylphosphonate.8 Therefore, the H-phosphonate method is useful for synthesizing a variety of P-modified ODNs.
From the utility of an H-phosphonate diester intermediate, we attempted to develop an efficient synthetic method for phosphorodiamidate morpholino oligonucleotides using an H-phosphonamidate derivative (1) as a monomer. However, compound 1 was unstable and hydrolyzed into a nucleoside (2) and phosphonic acid. From a different viewpoint, a morpholino nucleoside would be a good leaving group. Thus, we used H-phosphonamidate derivatives having a heterocyclic amino group as monomers to synthesize ODNs (Scheme 1). In 1986, H-phosphonamidate derivatives were synthesized by Van Boom et al. for the first time.9 Thereafter, a synthetic method for H-phosphonamidate derivatives was well established by Stawinski et al.;10–13 however, reports on the use of an H-phosphonamidate derivative as a monomer are scarce. In 1990, we reported a synthetic method using a nucleoside 3′-N,N-diisopropylphosphonamidate as a monomer; however, the amino group was not a leaving group in the construction of an internucleotidic H-phosphonate diester linkage.14,15 In this study, 3′-N,N-diisopropylphosphonamidate nucleosides are reacted with a chlorinating reagent to generate the corresponding aminophosphorochloridites without cleaving the P–N bond. The aminophosphorochloridites are reacted with an alcohol to obtain phosphoramidites. Afterward, the P–N bond of the phosphoramidites is hydrolyzed to afford an H-phosphonate diester linkage. Starting from the H-phosphonamidate derivatives, three steps are required for the construction of an H-phosphonate diester linkage.
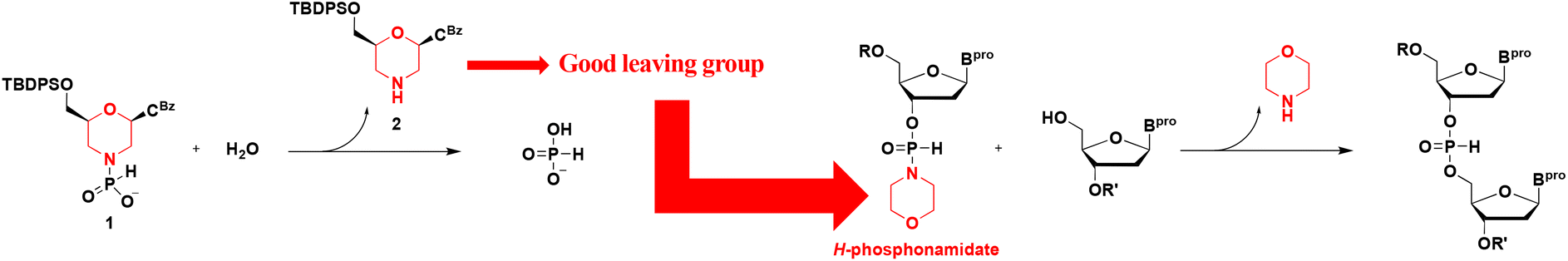 | ||
| Scheme 1 Nature of a morpholino nucleoside and a new concept for the synthesis of ODNs. | ||
In this study, we developed a new synthetic method for ODNs using 3′-H-phosphonamidate monomers bearing a heterocyclic amine as the leaving group. In this method, the H-phosphonate diester linkage was obtained from the H-phosphonamidate derivatives in only one step.
Results and discussion
Synthesis of nucleoside 3′-H-phosphonamidate monomers
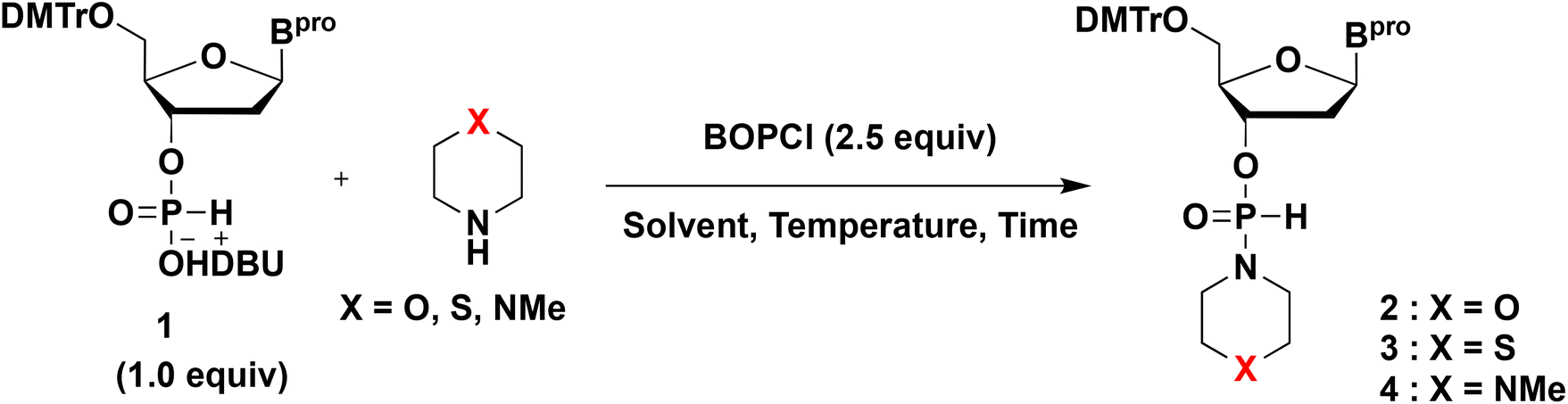
First, we investigated the synthesis of 3′-H-phosphonamidate derivatives having a heterocyclic amino group. The synthetic method for the H-phosphonamidate derivatives was reported by Stawinski et al. Two synthetic routes were developed to synthesize H-phosphonamidate derivatives from an H-phosphonate monoester and an amine, depending on the steric hindrance of amines (Scheme 2). To introduce less-hindered amines, such as butylamine, isopropylamine, and piperidine, H-phosphonamidate derivatives were synthesized using aryl-H-phosphonate intermediates, which were obtained by the condensation of an H-phosphonate monoester and a phenol derivative using a condensing reagent (route A). Conversely, a highly sterically hindered amine, such as diisopropylamine, was directly condensed with an H-phosphonate monoester using a condensing reagent (route B). Heterocyclic amines, such as morpholine, thiomorpholine, and N-methylpiperadine, were classified as less-hindered amines. Therefore, we attempted to synthesize an H-phosphonamidate having a heterocyclic amine via route A. | ||
| Scheme 2 Two routes for the synthesis of H-phosphonamidate derivatives. | ||
H-Phosphonamidate (2t) was synthesized using an aryl-H-phosphonate intermediate, which was obtained by condensing an H-phosphonate monoester (1t) and 2,4,6-trichlorophenol using pivaloyl chloride as a condensing reagent. Although 2t was obtained as the main product, 2t and 2,4,6-trichlorophenol were not separable by silica gel column chromatography.
Therefore, we attempted to synthesize 2tvia route B (Table 1). The H-phosphonate monoester (1t) was condensed with 2 equiv. of morpholine using bis(2-oxo-3-oxazolidinyl)phosphinic chloride (BOPCl) as a condensing reagent (Table 1, entry 1). Fortunately, the desired 2t was obtained as the main product (Fig. S1†). The H-phosphonamidate (2t) was purified by silica gel column chromatography and isolated with a yield of 43%. H-Phosphonamidate derivatives 3t and 4t, which have thiomorpholino and N-methylpiperadino groups, respectively, were synthesized via the same method (entries 2 and 3, Fig. S2 and S3†), and the former was isolated in 60% yield. The higher yield of 3t than that of 2t might reflect the higher stability of 3t than that of 2t. Conversely, because H-phosphonamidate 4t was unstable in silica gel, it was used without silica gel column chromatography purification. Next, we optimized the reaction conditions for synthesizing H-phosphonamidate monomers having the thiomorpholino group and bearing other nucleobases. For the thymidine and deoxyguanosine derivatives, side reactions with the nucleobases occurred under the conditions in entry 2. The side reactions were suppressed by reducing the amount of thiomorpholine (1.0 equiv.) and lowering the reaction temperature to 0 °C. All the H-phosphonamidate monomers were purified by silica gel column chromatography and isolated with yields of 53%–68%.
|
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | Bpro | X | Amine (equiv.) | Solvent | Temp. (°C) | Time (min) | Product | Isolated yield (%) |
| a 2t, 3t, and 4t: Bpro = thymine, 3a : Bpro = N6-benzoyladenine, 3c : Bpro = N4-isobutyrylcytosine, and 3g: Bpro = N2-isobutyrylguanine. | ||||||||
| 1 | T | O | 1 to 2 | CH2Cl2–pyridine (99![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v) :1, v/v) |
rt | 50 | 2t | 43 |
| 2 | T | S | 1 to 2 | CH2Cl2–pyridine (99:1, v/v) |
rt | 50 | 3t | 60 |
| 3 | T | NMe | 1 to 2 | CH2Cl2–pyridine (99:1, v/v) |
rt | 50 | 4t | Not isolated |
| 4 | Abz | S | 1 to 2 | CH2Cl2–pyridine (99:1, v/v) |
rt | 50 | 3a | 53 |
| 5 | Ci–bu | S | 1 to 2 | CH2Cl2–pyridine (99:1, v/v) |
rt | 50 | 3c | 58 |
| 6 | Gi–bu | S | 1 | MeCN–pyridine (1:1, v/v) |
0 | 20 | 3g | 53 |
| 7 | T | S | 1 | MeCN–pyridine (1:1, v/v) |
0 | 20 | 3t | 68 |
Through a literature survey, we found an example that used a dimethylamino H-phosphonamidate derivative to construct an H-phosphonate diester linkage in the field of carbohydrate chemistry.16 Thus, a dimethylamino H-phosphonamidate derivative was synthesized; however, the compound was unstable and decomposed during the work-up. This result indicated that a dimethylamino H-phosphonamidate derivative was not suitable as a monomer for synthesizing ODNs.
Synthesis of dithymidine H-phosphonate diester derivative (TPHT)
Next, a dithymidine H-phosphonate diester derivative was synthesized using the obtained H-phosphonamidate 2t and the thymidine derivative 5t bearing a 5′-OH group (Table 2). We first activated the morpholino group using 1H-tetrazole, which was used as an acid activator in the phosphoramidite method; however, the reaction did not proceed. We reported that an H-phosphonamidate derivative having a diisopropylamino group was stable under acidic conditions, such as 0.5% trifluoroacetic acid (TFA) in CHCl3.15 Thus, the result of this study was in good agreement with that in our previous report. Next, we conducted a condensation reaction under basic conditions. Surprisingly, H-phosphonamidate 2t reacted with 5t to form an H-phosphonate diester during azeotropic manipulation with pyridine at approximately 40 °C (Fig. S4†). Moreover, H-phosphonamidates 3t and 4t reacted with 5t under the same conditions (Fig. S5 and S6†). Although the order of reactivity was 2 > 3 > 4, from the perspective of both stability and reactivity, H-phosphonamidate 3 was chosen as the monomer for further investigation. H-Phosponamidate 3t and a small excess amount of 5t were reacted under the conditions of constant temperature, concentration, and pressure. The conversion rate was estimated by the 31P nuclear magnetic resonance (NMR) analysis of the reaction mixture, and the results are shown in Table 2.
When the condensation reaction was conducted at rt using 0.05 M 3t in pyridine, the reaction did not proceed (entry 1). Increasing either the concentration or temperature resulted in no reaction or sluggish reaction, respectively (entries 2 and 3). Conversely, the reaction at 40 °C with a 0.20 M concentration of 3t for 1 h afforded a 72% NMR yield of the product (entry 4). The extension of the reaction time to 2 h improved the NMR yield to 85% (entry 5). These results and the fact that the condensation reaction proceeded during azeotropic manipulation with pyridine at higher than rt showed that both the temperature and concentration were important factors for the condensation. Notably, the condensation reaction proceeded without using additives such as an acidic activator and a condensing reagent. Although the condensation afforded the H-phosphonate diester 6tt as the main product, byproduct 7 was obtained (10%, determined by 31P NMR). Compound 7 was characterized by 31P NMR (δ 9.6 ppm) and mass spectrometry analysis (high-resolution mass spectroscopy (electrospray ionization–time-of-flight) (HRMS (ESI-TOF)) m/z calcd for C52H63N4NaO11PSi2+ [M + Na]+, 1029.3662; found 1029.3669). A plausible mechanism for the formation of byproduct 7 is shown in Scheme 3. After the H-phosphonate diester 6tt was formed, the H-phosphonate diester linkage of 6tt was attacked by the 5′-OH group of 5t, which produced the 5′, 5′-symmetrical compound 7 and a thymidine derivative bearing a 3′-OH group (Scheme 3).
 | ||
| Scheme 3 A plausible mechanism of the side reaction that affords 5′,5′-symmetrical compound 7. | ||
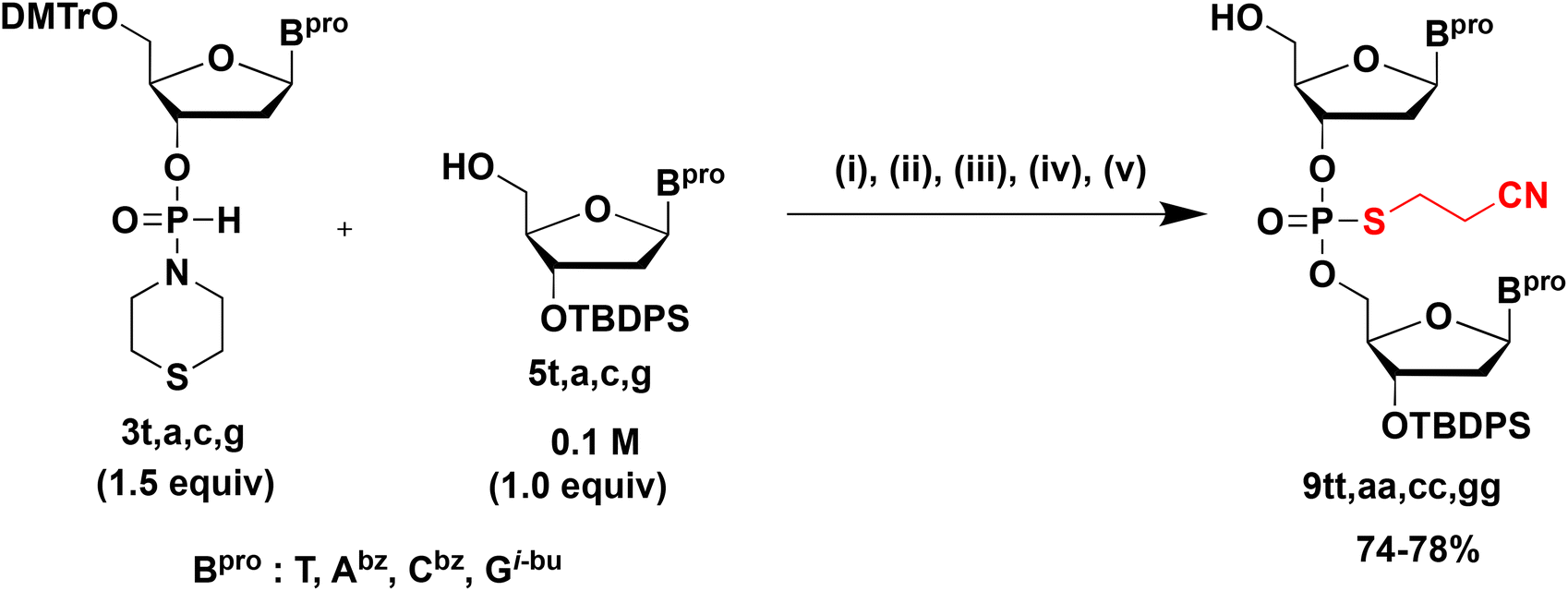
Synthesis of S-cyanoethyl phosphorothioate diester (NPSCEN) 9
Next, we converted the H-phosphonate diester linkage into an S-cyanoethyl phosphorothioate diester linkage because the H-phosphonate diester linkage was unstable under the basic conditions as mentioned above (Scheme 4). | ||
| Scheme 4 Synthesis of S-cyanoethyl phosphorothioate diester (TPSCET) 9tt. | ||
Thus, 1.5 equiv. of H-phosphonamidate 3t was condensed with 5t bearing the 5′-OH group. Thereafter, the H-phosphonate diester linkage was sulfurized using the sulfurizing reagent 8.17 After the sulfurization reaction, dimethyl phosphonate was added to the reaction mixture to scavenge the excess amounts of sulfurizing reagent 8. These three steps were conducted as a one-pot reaction. After the mixture was concentrated, a detritylation using 1% TFA in CHCl3 and a simple extraction afforded 9tt. The excess amount of 3t was completely removed by a simple extraction because the H-phosphonamidite monomer 3t was converted into a water-soluble derivative by the detritylation reaction. Although the sulfurization reaction would be accelerated by adding N,O-bis(trimethylsilyl)acetamide (BSA) as a silylating reagent,18,19 BSA helped to convert the H-phosphonamidate monomer 3t to the phosphorothioamidate monoester derivative, which was not removed by extraction after detritylation (Fig. S13†). It has been shown that an H-phosphonate diester linkage is more reactive to an electrophile than an H-phosphonamidate linkage.15 Owing to the difference in the reactivities of sulfurizing reagents, only the H-phosphonate diester linkage was sulfurized without a silylating reagent.
Using the above-mentioned reaction conditions, dimers bearing all nucleobases were synthesized using H-phosphonamidate derivatives 3t, 3a, 3c, or 3g bearing the thiomorpholino group and 5′-OH derivatives 5t, 5a, 5c, or 5g (Table 3). All dimers were obtained in 74%–78% isolated yields, indicating that this strategy could be applied to all nucleobases.
|
|
||||
|---|---|---|---|---|
| Entry | H-Phosphonamidate monomer | 5′-OH nucleoside | Product | Isolated yield (%) |
| Reagents and conditions: (i) pyridine, MS4 Å, 40 °C, 2 h; (ii) sulfurizing reagent 8 (1.2 equiv.), pyridine, rt, 1 h; (iii) 1% TFA in CHCl3, rt, 10 min; (iv) extraction with a saturated aqueous solution of NaHCO3; (v) silica gel column chromatography. | ||||
| 1 | 3t | 5t | 9tt | 74 |
| 2 | 3a | 5a | 9aa | 75 |
| 3 | 3c | 5c | 9cc | 75 |
| 4 | 3g | 5g | 9gg | 78 |
Synthesis of TPSTPST trimer 10
Finally, we attempted to synthesize the TPSTPST trimer (10) using a new synthetic method (Scheme 5). The H-phosphonamidate monomer 3t was condensed with 5t using pyridine as a solvent. Thereafter, the internucleotidic linkage was sulfurized by treatment with the sulfurizing reagent 8. An excess amount of sulfurizing reagent 8 was scavenged using dimethyl phosphonate. These steps were conducted as a one-pot reaction. After evaporating the mixture to remove pyridine, detritylation was conducted by treatment with 1% TFA in CHCl3. Afterward, compounds derived from the detritylation of H-phosphonamidate monomer 3t were removed by extraction. Chain elongation was conducted by repeating this cycle. After obtaining the trimer, the cyanoethyl groups of the internucleotide linkages were removed by treatment with 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) using BSA, followed by the removal of the 3′-TBDPS group using TBAF. Thereafter, the resultant crude reaction mixture was analyzed by reversed-phase high-performance liquid chromatography (RP-HPLC), and the condensation yields were estimated by the area ratios of the trimer to the unreacted dimer and thymidine. The profile of RP-HPLC is shown in Fig. 1. | ||
| Scheme 5 Synthesis of trimer (TPSTPST) 10. | ||
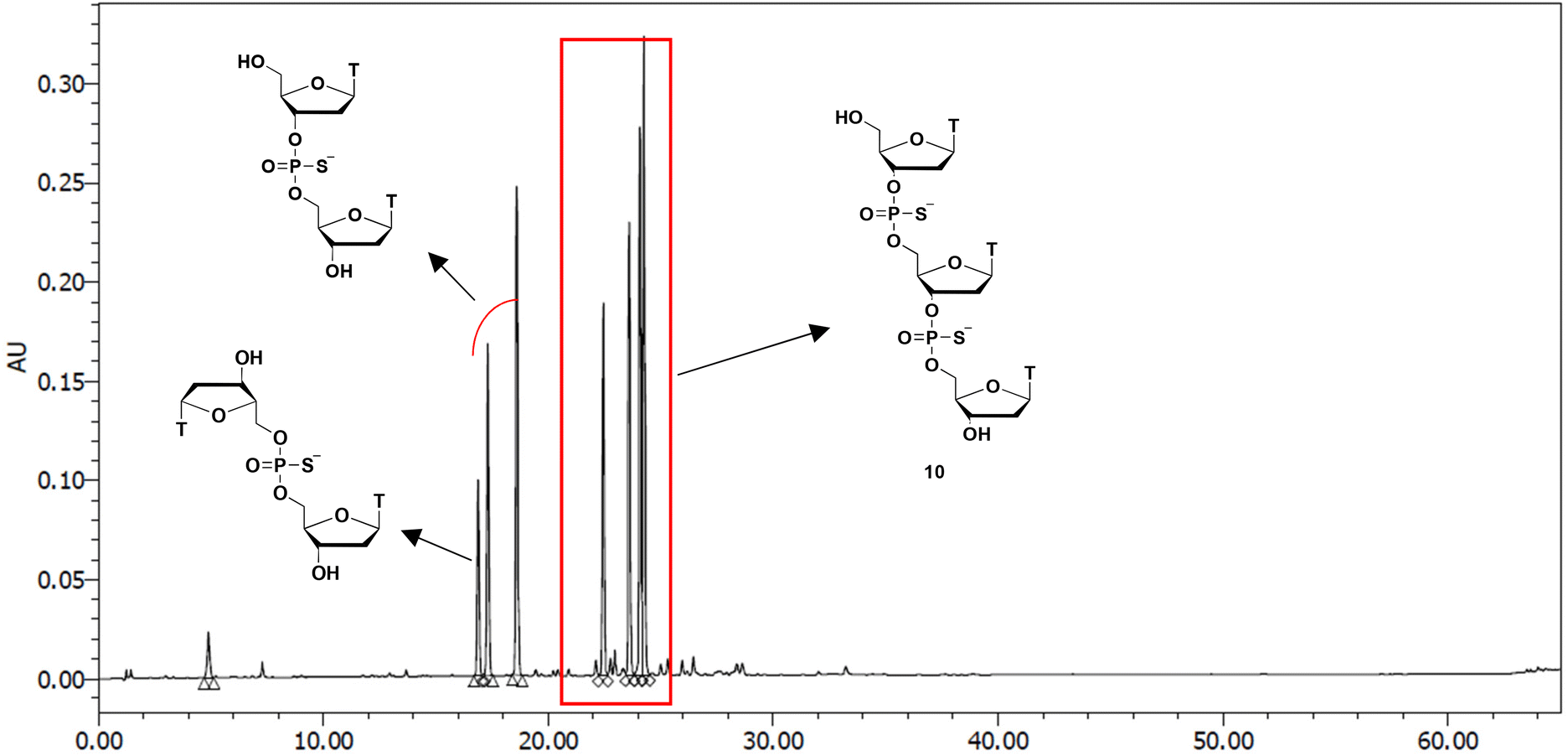 | ||
| Fig. 1 RP-HPLC profile of crude TPSTPST; RP-HPLC was performed with a linear gradient of 0%–30% CH3CN in 0.1 M triethylammonium acetate (TEAA) buffer (pH 7.0) over 60 min at 50 °C at a rate of 0.5 mL min−1. | ||
The trimer TPSTPST (10) was obtained in 70% HPLC yield and isolated in 32% yield by silica gel column chromatography. The HPLC result showed that the excess amount of the H-phosphonamidate monomer was completely removed by simple extraction. This result indicated that only simple extraction was required to purify the intermediates. Conversely, a non-negligible number of peaks corresponding to two kinds of dimers was detected. The formation of the 5′, 5′-symmetrical dimer was attributed to the side reaction during the first condensation reaction (Scheme 3). The other dimer was unreacted in the second condensation reaction.
Conclusion
We have developed a new synthetic method for the solution-phase synthesis of ODNs using 3′-H-phosphonamidate derivatives having a heterocyclic amino group as monomers. H-Phosphonamidate derivatives 2t, 3t, and 4t, which have morpholino, thiomorpholino, and N-methylpiperadino groups, respectively, were synthesized using the H-phosphonate monoester 1 and amines. Among them, the H-phophonamidate monomer having a thiomorpholino group was chosen as a monomer because of its stability and reactivity. The condensation reaction proceeded under mild basic conditions without using additives. Four kinds of S-cyanoethyl phosphorothioate diesters, 9tt, 9aa, 9cc, and 9gg, were synthesized in three steps of condensation, sulfurization, and detritylation in good yields. Notably, because the H-phosphonamidate monomer (3) was converted into a water-soluble derivative in the detritylation step, the excess amount of 3 was completely removed by simple extraction. TPSTPST was successfully synthesized from the monomer by only one silica gel column chromatography step. This strategy can avoid the excess use of reagents and tedious purification manipulations. Additionally, because this strategy affords the H-phosphonate diester as an intermediate, a wide variety of P-modified ODNs would be obtained by an appropriate transformation of the intermediate. The synthesis of relatively long ODNs using this strategy is in progress.Author contributions
T. T. established the research plans. T. T. conducted the experiments. All authors analyzed the data. T. T. wrote the manuscript. K. S. and T. W. revised the manuscript. All authors have approved the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Dr Yayoi Yoshimura (Tokyo University of Science) for the mass spectral measurements. We would like to thank Enago (https://www.enago.jp) for the English language review. This work was supported by JST, the establishment of university fellowships towards the creation of science technology innovation, Grant Number JPMJFS2144 and AMED under Grant Number JP21ae0121025.References
- S. T. Crooke, J. L. Witztum, C. F. Bennett and B. F. Baker, Cell Metab., 2018, 27, 714–739 CrossRef CAS PubMed.
- S. L. Beaucage and M. H. Caruthers, Tetrahedron Lett., 1981, 22, 1859–1862 CrossRef CAS.
- B. C. Froehler and M. D. Matteucci, Tetrahedron Lett., 1986, 27, 469–472 CrossRef CAS.
- P. J. Garegg, I. Lindh, T. Regberg, J. Stawinski, R. Strömberg and C. Henrichson, Tetrahedron Lett., 1986, 27, 4055–4058 CrossRef CAS.
- J. Stawinski and A. Kraszewski, Acc. Chem. Res., 2002, 35, 952–960 CrossRef CAS PubMed.
- J. Stawinski and M. Thelin, J. Org. Chem., 1991, 56, 5169–5175 CrossRef CAS.
- B. Froehler, P. Ng and M. Matteucci, Nucleic Acids Res., 1988, 16, 4831–4839 CrossRef CAS PubMed.
- A. Kers and J. Stawiński, Tetrahedron Lett., 1999, 40, 4263–4266 CrossRef CAS.
- J. E. Marugg, A. Burik, M. Tromp, G. A. Van der Marel and J. H. Van Boom, Tetrahedron Lett., 1986, 27, 2271–2274 CrossRef CAS.
- A. Sobkowska, M. Sobkowski, J. Cieślak, A. Kraszewski, I. Kers and J. Stawiński, J. Org. Chem., 1997, 62, 4791–4794 CrossRef CAS.
- A. Sobkowska, M. Sobkowski, J. Stawiski and A. Kraszewski, Nucleosides, Nucleotides Nucleic Acids, 1995, 14, 703–706 CrossRef CAS.
- I. Kers, J. Stawiński and A. Kraszewski, Tetrahedron, 1999, 55, 11579–11588 CrossRef CAS.
- A. Kraszewski, M. Sobkowskia and J. Stawiński, J. Chem. Soc., Perkin Trans. 1, 1993, 1699–1704 RSC.
- T. Wada, K. Ishikawa and T. Hata, Tetrahedron Lett., 1990, 31, 6363–6366 CrossRef CAS.
- T. Wada, K. Ishikawa and T. Hata, Tetrahedron, 1993, 49, 2043–2054 CrossRef CAS.
- D. V. Khodarev, T. S. Kukhareva, L. K. Vasyanina and E. E. Nifant'ev, Russ. J. Gen. Chem., 2006, 76, 529–534 CrossRef CAS.
- N. Iwamoto, D. C. D. Butler, N. Svrzikapa, S. Mohapatra, I. Zlatev, D. W. Y. Sah, Meena, S. M. Standley, G. Lu, L. H. Apponi, M. Frank-Kamenetsky, J. J. Zhang, C. Vargeese and G. L. Verdine, Nat. Biotechnol., 2017, 35, 845–851 CrossRef CAS PubMed.
- T. Wada, Y. Sato, F. Honda, S.-I. Kawahara and M. Sekine, J. Am. Chem. Soc., 1997, 119, 12710–12721 CrossRef CAS.
- E. De Vroom, C. Dreef, H. Van den Elst, G. Van der Marel and J. Van Boom, Recl. Trav. Chim. Pays-Bas, 1988, 107, 592–595 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ob02292c |
| This journal is © The Royal Society of Chemistry 2023 |