
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Furan-based inhibitors of pyruvate dehydrogenase: SAR study, biochemical evaluation and computational analysis†
Alex H. Y.
Chan‡
a,
Terence C. S.
Ho‡
a,
Daniel R.
Parle
ab and
Finian J.
Leeper
 *a
*a
aYusuf Hamied Department of Chemistry, University of Cambridge, Lensfield Road, Cambridge CB2 1EW, UK. E-mail: fjl1@cam.ac.uk
bCancer Research UK Cambridge Institute, University of Cambridge, Li Ka Shing Centre, Robinson Way, Cambridge CB2 0RE, UK
First published on 27th January 2023
Abstract
Suppression of pyruvate dehydrogenase complex (PDHc) is a mechanism for cancer cells to manifest the Warburg effect. However, recent evidence suggests that whether PDHc activity is suppressed or activated depends on the type of cancer. The PDHc E1 subunit (PDH E1) is a thiamine pyrophosphate (TPP)-dependent enzyme, catalysing the first and rate-limiting step of PDHc; thus, there is a need for selective PDH E1 inhibitors. There is, however, inadequate understanding of the structure–activity relationship (SAR) and a lack of inhibitors specific for mammalian PDH E1. Our group have reported TPP analogues as TPP-competitive inhibitors to study the family of TPP-dependent enzymes. Most of these TPP analogues cannot be used to study PDHc in cells because (a) they inhibit all members of the family and (b) they are membrane-impermeable. Here we report derivatives of thiamine/TPP analogues that identify elements distinctive to PDH E1 for selectivity. Based on our SAR findings, we developed a series of furan-based thiamine analogues as potent, selective and membrane-permeable inhibitors of mammalian PDH E1. We envision that our SAR findings and inhibitors will aid work on using chemical inhibition to understand the oncogenic role of PDHc.
Introduction
Pyruvate dehydrogenase complex (PDHc), catalysing the conversion of pyruvate to acetyl coenzyme A (acetyl CoA), is a gatekeeper in cellular energy metabolism.1–4 The E1 subunit (PDH E1) of PDHc catalyses the first and rate-limiting step of the complex, and uses thiamine pyrophosphate 1a (TPP, the bioactive form of thiamine 2) to reductively acetylate lipoamide (Fig. 1a). The attachment of pyruvate onto the nucleophilic centre (C2) of the thiazolium ylide 1b followed by decarboxylation yields a covalent intermediate. This intermediate then links covalently to lipoamide in a ring-opening reaction. The resultant acetylated dihydrolipoamide is recycled by other subunits of the complex (E2 and E3) giving acetyl CoA and NADH. Phosphorylation of PDH E1 by PDH kinases (PDKs) reduces the activity of PDHc. Suppression of PDHc activity through upregulation of PDKs is a common feature of cancer cells and inhibition of PDK has long been an anticancer strategy.3,5,6 However, since 2016 there has been growing evidence that PDHc overexpression and/or hyperactivity is associated with certain cancers,7,8 and that small-molecule PDH E1 inhibitors are a potential treatment for prostate cancer.9 The contrasting evidence on the role of PDHc in oncology suggests that the therapeutic effect of PDHc modulation is likely to be dependent on the tumour genetic background, tissue type, micro-environment, etc.7–9 One approach to unveiling its puzzling role is through chemical inhibition of PDH E1. Compounds that inhibit PDH E1 have been reported but are either specific towards PDH E1 from pathogenic micro-organisms10–12 or promiscuous, inhibiting all TPP-dependent enzymes.13 The lack of chemical probes specific for mammalian PDH E1 is attributed to the longstanding belief that cancer cells suppress PDHc5,6 so led to an inadequate understanding of the structure–activity relationship (SAR) of PDH E1 inhibitors.8 | ||
| Fig. 1 (a) Mechanism of pyruvate dehydrogenase complex E1 subunit (PDH E1) enzyme. The lipoyl group shown is attached to the E2 subunit. (b) Previously reported TPP analogues. (c) Reactions catalysed by the enzymes involved in this study. PDC, pyruvate decarboxylase; PO, pyruvate oxidase; OGDH E1, 2-oxoglutarate dehydrogenase complex E1 subunit. | ||
A common strategy in designing inhibitors of TPP-dependent enzymes lies in the use of TPP analogues.10–24 These mostly feature a central neutral ring in place of the thiazolium ring of TPP, e.g. deazathiamine pyrophosphate 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 18 (deazaTPP), triazole-TPP 419 and furan-TPP 520 (Fig. 1b). They form multiple polar interactions in the TPP-pockets: (1) the aminopyrimidine forms hydrogen-bonding interactions and π–π stacking interactions with some conserved residues; (2) the pyrophosphate has ionic interactions with the Mg2+ ion in the pyrophosphate pocket; and (3) the neutral central ring mimics the thiazolium ylide 1b (the catalytically active form of TPP 1a) and so captures the strong stabilising interactions between the enzyme and the high-energy TPP ylide. As a result, these TPP analogues are potent inhibitors with nanomolar or lower affinities towards most, if not all, TPP-dependent enzymes.13,14 However, they cannot be used in cellular studies due to their anionic pyrophosphate tail which makes them membrane-impermeable.14,25
18 (deazaTPP), triazole-TPP 419 and furan-TPP 520 (Fig. 1b). They form multiple polar interactions in the TPP-pockets: (1) the aminopyrimidine forms hydrogen-bonding interactions and π–π stacking interactions with some conserved residues; (2) the pyrophosphate has ionic interactions with the Mg2+ ion in the pyrophosphate pocket; and (3) the neutral central ring mimics the thiazolium ylide 1b (the catalytically active form of TPP 1a) and so captures the strong stabilising interactions between the enzyme and the high-energy TPP ylide. As a result, these TPP analogues are potent inhibitors with nanomolar or lower affinities towards most, if not all, TPP-dependent enzymes.13,14 However, they cannot be used in cellular studies due to their anionic pyrophosphate tail which makes them membrane-impermeable.14,25
While the two TPP-binding pockets (see Fig. 1a) are similar in all TPP-dependent enzymes, the C2-pocket where the substrate binds and biotransformation takes place must differ between different enzymes as they act on different substrates (Fig. 1c).1,4 In this work, we aimed to develop inhibitors that are both selective for mammalian PDH E1 and membrane permeable. We prepared a range of derivatives of thiamine analogues to systematically probe the three key pockets in order to identify electronic and steric elements that differentiate PDH E1 from other TPP-dependent enzymes. To attain membrane-permeability, we replaced the pyrophosphate moiety with non-charged moieties and throughout the iterative ligand design process, physiochemical properties of our inhibitors were calculated and monitored. This approach led to a series of potent, selective and membrane-permeable furan-based inhibitors of mammalian PDH E1. We anticipate that these inhibitors and our SAR findings will advance work on developing chemical probes for PDH E1 in cellular systems.
Results and discussion
Probing the pyrophosphate pocket
Our triazole-based thiamine analogues were very suitable for probing the pyrophosphate pocket because the parent alcohol 7 was easy to make in quantity (unlike the more synthetically demanding furan analogue, see below) and a large number of derivatives featuring different moieties on the hydroxyethyl tail could be readily prepared.19,22Scheme 1 outlines the construction of the central triazole scaffold by click chemistry between 3-butyn-1-ol and azide 6 (easily obtained from inexpensive thiamine hydrochloride). Triazole-thiamine 7 is however a weak binder to TPP-dependent enzymes. Pyrophosphorylation of the hydroxyethyl tail led to triazole-TPP 4, a potent inhibitor (with a KI value of 30 pM on a bacterial pyruvate decarboxylase19) but membrane-impermeable. In a recent paper,22 we reported seven monofunctionalised benzoyl esters 8a–g made by coupling the parent alcohol 7 with the corresponding carboxylic acids. Esters 8a–g were all stronger binders to PDH E1 than the parent alcohol 7 and among them, 8a was the best inhibitor (Table 1).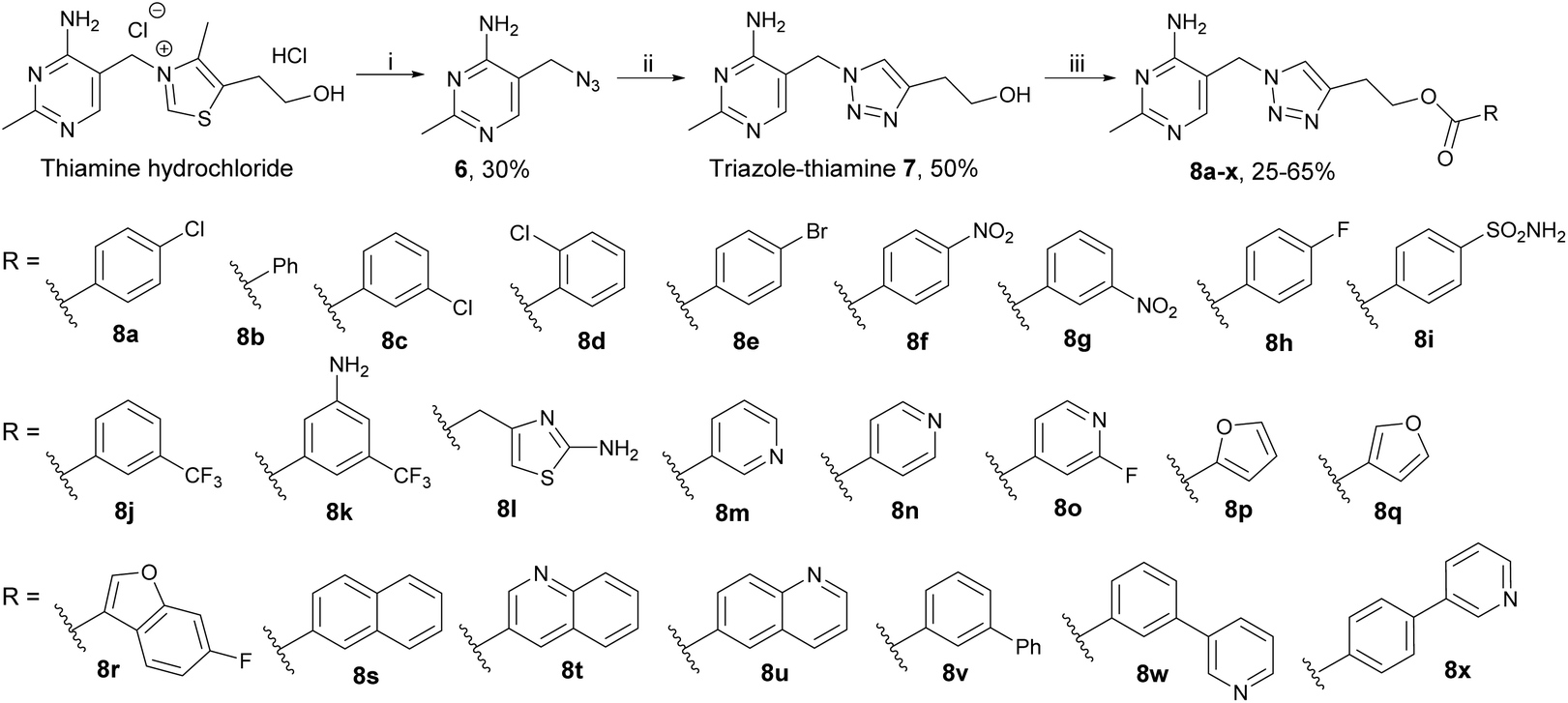 | ||
| Scheme 1 Synthesis of triazole-based esters 8a–x. Reagents and conditions: (i) NaN3, Na2SO3, H2O, 65 °C; (ii) 3-butyn-1-ol, CuSO4·5H2O, sodium ascorbate, t-BuOH, H2O, RT; (iii) carboxylic acid, DCC, DMAP, DMF, RT. | ||
P of triazole series 7 and 8a–x
| % Inhibition (2:1)a,b |
% Inhibition (5:1)a,c |
IC50a,d (μM ± SEM) |
cLogPe |
|
|---|---|---|---|---|
|
a Data are the means of measurements in three technical replicates.
b Percentage inhibition determined for compounds at 200 μM with [TPP] = 100 μM.
c Percentage inhibition determined for compounds at 200 μM with [TPP] = 40 μM.
d IC50 values determined at [TPP] = 10 μM.
e Calculated LogP determined using MarvinSketch 21.2.
f Compounds 7 and 8a–g from the previous study22 had their % inhibition re-determined in the current study for consistency.
|
||||
| 7 | 32 ± 5f | 58 ± 9 | 30 ± 5 | −0.45 |
| 8a | 58 ± 8f | 80 ± 10 | 10.6 ± 2.0 | 2.66 |
| 8b | 40 ± 6f | 73 ± 9 | 2.10 | |
| 8c | 48 ± 7f | 74 ± 10 | 2.66 | |
| 8d | 40 ± 5f | 62 ± 8 | 2.66 | |
| 8e | 44 ± 7f | 73 ± 9 | 2.87 | |
| 8f | 41 ± 6f | 55 ± 8 | 2.05 | |
| 8g | 38 ± 5f | 54 ± 8 | 2.05 | |
| 8h | 52 ± 7 | 72 ± 9 | 2.24 | |
| 8i | 70 ± 9 | >90 | 5.42 ± 0.63 | 0.77 |
| 8j | 52 ± 7 | 72 ± 9 | 2.98 | |
| 8k | 66 ± 7 | 88 ± 8 | 5.72 ± 0.80 | 2.19 |
| 8l | 30 ± 6 | 55 ± 6 | 0.65 | |
| 8m | 43 ± 4 | 67 ± 6 | 0.86 | |
| 8n | 36 ± 4 | 61 ± 6 | 0.86 | |
| 8o | 51 ± 5 | 70 ± 8 | 1.41 | |
| 8p | 71 ± 7 | >90 | 4.64 ± 0.47 | 1.15 |
| 8q | 70 ± 7 | >90 | 4.39 ± 0.51 | 1.28 |
| 8r | 76 ± 8 | >90 | 3.70 ± 0.42 | 2.52 |
| 8s | 45 ± 5 | 66 ± 6 | 3.10 | |
| 8t | 41 ± 5 | 72 ± 7 | 2.25 | |
| 8u | 52 ± 5 | 74 ± 7 | 2.25 | |
| 8v | 73 ± 6 | >90 | 3.96 ± 0.42 | 3.75 |
| 8w | 86 ± 7 | >90 | 2.95 ± 0.43 | 2.52 |
| 8x | 60 ± 5 | 77 ± 7 | 2.52 | |
To further probe the pyrophosphate pocket, 17 new esters 8h–x were synthesised and more substitution patterns were sampled, including mono- and di-substituted aryl and heteroaryl esters (8h–q), fused aryl esters (8r–u) and biaryl esters (8v–x). The seven previously reported esters 8a–g and two of the new ones, 8h and 8p, have been described in a patent and were tested for inhibition of bacterial and cyanobacterial growth.23 Compound potency on PDH E1 was evaluated on commercially available porcine PDH E1 which is a widely accepted22,26–29 alternative to human PDH E1 because the sequence of the two PDH E1 enzymes are >95% identical and the residues that differ are located away from the active site (Fig. S1†). Table 1 summarises the inhibitory activities and the calculated LogP values (cLogP) of esters 8a–x. In contrast to the previously reported series 8a–g, the more structurally diverse derivatives 8h–x in this work resulted in a wider range of activities (percentage inhibition at 2:1 ratio of [inhibitor] to [TPP] is 30–86% vs. 38–58%). Repeating the assays at lower [TPP] led to enhanced apparent inhibition, thus confirming their competitive relationship with TPP, as previously established for esters 8a–g.22 IC50 values were determined for compounds that inhibited more potently than 8a, among which esters 8i and 8w were of special interest: 8i being the most polar (with adequate aqueous solubility for all biological assays, see below) and 8w being the most potent. Due to the competitive nature of our analogues towards TPP, the IC50 value obtained will be proportional to the [TPP] used. Since assays for IC50 determination typically require high compound concentrations (>10 × IC50) for near-complete enzyme inhibition, a lower [TPP] was used to ensure our compounds remained soluble at the highest concentration needed.
Probing the aminopyrimidine pocket
The synthetic route to our furan scaffold20 enabled us to choose reagents that led to altered ring substituents on the aminopyrimidine ring, which were not accessible via the synthesis employed for the triazole scaffold. Scheme 2 outlines the synthesis of the furan-based derivatives with varying substituents in place of the methyl group on the pyrimidine ring. In brief, the TBDMS-protected 3-butyn-1-ol 9 and bis-TBDMS-protected dihydroxy-acetone 10 were coupled together to form tris-TBDMS-protected alkyne 11, which was then fully deprotected to yield alkyne triol 12. Hg(II)-catalysed cyclisation30 afforded furan diol 13 which was selectively oxidised by MnO2 to give aldehyde 14. After condensation with β-anilinopropionitrile, intermediate 15 was reacted with amidines or guanidine to yield furan-thiamine 16a as well as its derivatives 16b–d. Alcohols 16a–d were coupled with 4-sulfamoylbenzoic acid to afford esters 17a–d. Scheme 2 also outlines the synthesis of 4′-N-acetyl derivative 21. The hydroxylethyl tail of 16a was firstly protected with TBDMS and then amine 18 was acetylated to afford acetamide 19. After deprotection with TBAF, the hydroxyethyl tail of 20 was coupled with 4-chlorobenzoic acid to give ester 21. | ||
| Scheme 2 Synthesis of furan-based esters with a modified pyrimidine ring. Reagents and conditions: (i) TBDMS-Cl, imidazole, DCM, RT; (ii) n-BuLi, THF, RT; (iii) TBAF, THF, RT; (iv) HgCl2, MeCN, RT; (v) activated MnO2, CHCl3, RT; (vi) β-anilinopropionitrile, MeONa/MeOH, DMSO, 45 °C; (vii) amidine or guanidine, EtONa/EtOH, reflux; (viii) carboxylic acid, DCC, DMAP, DMF, RT; (ix) acetyl chloride, diisopropylethylamine, DCE, 45 °C. | ||
These furan-based derivatives together explored two positions on the pyrimidine ring. The analogues with modified pyrimidines 17b–d and 21 were compared with the corresponding compounds with unmodified pyrimidines, 17a and 22 (Scheme 3) respectively. As shown in Table 2, derivatives featuring larger (Et, 17b), electron-withdrawing (CF3, 17c) or electron-donating (NH2, 17d) substituents in place of the methyl group (17a) were all weaker inhibitors. This is consistent with reported docking studies where deazathiamine 3 derivatives bearing Et, iPr or CF3 at this position resulted in no reasonable docking pose.31 Acylation of the amino group (21) abolished inhibition. We concluded that the unmodified 2-methyl-4-aminopyrimidine ring best fits the aminopyrimidine pocket.
 | ||
| Scheme 3 Synthesis of C2-acylated furan-based esters. Reagents and conditions: (i) carboxylic acid, DCC, DMAP, DMF, RT; (ii) RCOCl, AlCl3, DCM, RT; (iii) acetic anhydride, pyridine, 40 °C; (iv) K2CO3, MeOH, RT. | ||
| Compound | % Inhibition (1:1)a,b |
|---|---|
|
a Data are the means of measurements in three technical replicates.
b Percentage inhibition determined for compounds at 100 μM with [TPP] = 100 μM. Relative to Table 1, assay conditions here had the ratio of [inhibitor] to [TPP] reduced from 2:1 to 1:1 as the furan is a better scaffold for binding to PDH E1.
|
|
| 17a | 85 ± 8 |
| 17b | 18 ± 3 |
| 17c | 16 ± 4 |
| 17d | 8 ± 2 |
| 21 | 6 ± 2 |
| 22 | 66 ± 7 |
C2-Acylation – probing the C2-pocket
The C2-pocket (where substrates bind and undergo transformation) is divergent in different enzymes and could well be key in determining the selectivity of inhibitors.1,4,27,28,32 We introduced substituents onto the furan-thiamine C2-position via Friedel–Crafts acylation reactions, Scheme 3. Furan-thiamine 16a was esterified with 4-chlorobenzoic acid and 3-(pyridin-3-yl)-benzoic acid to afford esters 22 and 23, respectively, followed by C2-acylation to yield 24a–i and 25. Since the acceptor substrate of PDH E1, lipoamide, is relatively hydrophobic (Fig. 1), all but one of the substituents attached to the C2-ketone were hydrocarbon-based, including alkyl chains of varying lengths and saturated rings of varying sizes. The sulfamoyl group was found to be susceptible to acylation under the Friedel–Crafts conditions, so the order of steps was changed for the synthesis of 26: alcohol 16a was firstly acetylated and then C2-acylated to give 27. The acetate ester was then hydrolysed and the alcohol finally esterified with 4-sulfamoylbenzoic acid to give 26.The C2-acylated compounds 24a–i were then assayed for inhibition of PDH E1 (Table 3). For the linear chains 24a–e the trend was that inhibition increased from acetyl (24a) to butanoyl (24c) but then decreased with further increase in length (24d and 24e). For the ring-bearing acyl groups (24f–h) the strongest inhibition was again with the four-carbon cyclopropanecarbonyl group (24f) and larger groups inhibited less well. 24i was an exception to this trend and Fig. S2† has a possible explanation from docking studies, in which the pyran oxygen hydrogen-bonds to the –OH of threonine-82. We conclude that the butanoyl derivative (24c) is the best in the series possibly because it maximises the hydrophobic contact without creating a steric clash with the C2-pocket.
| IC50a,b (μM ± SEM) |
Affinity relative to TPPc |
K
Id (nM ± SEM) |
|
|---|---|---|---|
| a Data are the means of measurements in three technical replicates. b IC50 values determined at [TPP] = 10 μM. c Compound affinity relative to TPP = [TPP]/IC50. d K I is based on the previously reported KM for TPP of 50 nM (ref. 14 and 33) using [TPP]/IC50 = KM(TPP)/KI. | |||
| 16a | 21.9 ± 3.4 | 0.45 | 111 ± 15 |
| 22 | 3.27 ± 0.40 | 3.1 | 16.1 ± 2.2 |
| 24a | 5.58 ± 0.58 | 1.8 | 27.8 ± 3.0 |
| 24b | 2.99 ± 0.40 | 3.4 | 15.2 ± 1.8 |
| 24c | 0.85 ± 0.22 | 11.8 | 4.24 ± 1.11 |
| 24d | 1.81 ± 0.20 | 5.5 | 9.09 ± 0.96 |
| 24e | 4.79 ± 0.53 | 2.1 | 23.8 ± 2.8 |
| 24f | 1.50 ± 0.20 | 6.7 | 7.46 ± 1.04 |
| 24g | 2.98 ± 0.28 | 3.4 | 14.7 ± 1.6 |
| 24h | 3.41 ± 0.49 | 2.9 | 17.2 ± 2.3 |
| 24i | 1.07 ± 0.13 | 9.3 | 5.38 ± 0.62 |
Also of note is that the IC50 of 22, which is not C2-acylated, is over three times lower than the IC50 of the corresponding triazole 8a. This shows that the furan is a better scaffold for binding to PDH E1, as well as having the advantage that it can be acylated at the 2-position, which the triazole cannot.
C2-Acylated furan-based thiamine analogues as potent and selective PDH E1 inhibitors
The next objective was to assemble the SAR findings from the three pockets (Tables 1–3). The preferred ester tails identified from Table 1 were added to our furan scaffold, with or without a C2-butanoyl group (Scheme 3). As summarised in Table 4, the potency of the benzoyl esters was p-chloro < p-sulfamoyl < m-pyridin-3-yl (22 < 17a < 23 and 24c < 26 < 25), as in the triazole series (8a < 8i < 8w). The matched pairs showed that the C2-butanoyl substituent led to a 3–4-fold increase in potency (22vs.24c, 17avs.26 and 23vs.25), and the furans bind 3–4-fold tighter than the triazoles (22vs.8a, 17avs.8i and 23vs.8w). We suggest that the more hydrophobic furan bears a closer resemblance to the high-energy TPP ylide 1b than the triazole. Comparing the most potent inhibitor 25 with its precursor furan-thiamine 16a, esterification and C2-functionalisation together have raised the affinity 170-fold (IC50 lowered from 22 to 0.13 μM). The TPP-competitive nature of the inhibitors was established by repeating the assays at higher [TPP], which decreased the inhibition. The affinity of 25 for PDH E1 was found to be 77 times higher than that of TPP and the calculated KI (based on KM of TPP33) is in the picomolar range (Table 4).| Mammalian PDH E1 | Yeast PDC | Bacterial PDC | Bacterial PO | Bacterial OGDC E1 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| % Inh. (5:1)a,b |
% Inh. (1:1)a,c |
IC50a,d,e (μM ± SEM) |
K
Ie (nM) |
L.E.f | % Inh. (1:1)a,g |
IC50a,h (μM ± SEM) |
% Inh. (1:1)a,c |
IC50a,i (μM ± SEM) |
% Inh. (1:1)a,c |
IC50a,i (μM ± SEM) |
% Inh. (1:1)a,c |
IC50a,i (μM ± SEM) |
|
| a Data are the means of measurements in three technical replicates. b Percentage inhibition determined for compounds at 50 μM with [TPP] = 10 μM. c Percentage inhibition determined for compounds at 50 μM with [TPP] = 50 μM. d IC50 values determined at [TPP] = 10 μM; compound affinity relative to TPP affinity = [TPP]/IC50. e K I is based on the previously reported KM for TPP of 50 nM (ref. 14 and 33) using [TPP]/IC50 = KM(TPP)/KI; numbers are rounded to two-significant figures. f Ligand efficiency (L.E.) = ΔG of binding (in kcal per mol)/number of non-hydrogen (heavy) atoms; preferred L.E. > 0.3. g Percentage inhibition determined for compounds at 200 μM with [TPP] = 200 μM. h IC50 values determined at [TPP] = 200 μM. i IC50 values determined at [TPP] = 40 μM. ND, not determined. Insol., compound not soluble under assay conditions. | |||||||||||||
| 16a | 66 ± 9 | 28 ± 5 | 22 ± 3.4 | 110 | — | 25 ± 6 | 638 ± 54 | ND | ND | ND | ND | 23 ± 6 | 134 ± 13 |
| 17a | 100 | 88 ± 9 | 1.5 ± 0.14 | 7.5 | 0.40 | 21 ± 3 | 827 ± 80 | 20 ± 3 | 250 ± 29 | 18 ± 8 | 258 ± 28 | 18 ± 4 | 231 ± 19 |
| 22 | >90 | 72 ± 9 | 3.3 ± 0.4 | 16 | 0.43 | 19 ± 4 | Insol. | ND | ND | ND | ND | 25 ± 6 | 185 ± 19 |
| 23 | 100 | 100 | 0.55 ± 0.08 | 2.7 | 0.39 | 14 ± 2 | Insol. | 7 ± 2 | Insol. | 8 ± 4 | Insol. | 6 ± 2 | Insol. |
| 24a | 85 ± 9 | 66 ± 8 | 5.6 ± 0.6 | 28 | 0.37 | 18 ± 5 | Insol. | ND | ND | ND | ND | 23 ± 5 | 189 ± 15 |
| 24c | 100 | >90 | 0.85 ± 0.2 | 4.2 | 0.38 | Insol. | Insol. | 2 ± 4 | ND | Insol. | Insol. | 27 ± 7 | Insol. |
| 25 | 100 | 100 | 0.13 ± 0.02 | 0.65 | 0.36 | Insol. | Insol. | 3 ± 6 | ND | 1 ± 4 | ND | 8 ± 3 | Insol. |
| 26 | 100 | 100 | 0.42 ± 0.07 | 2.0 | 0.36 | 1 ± 3 | ND | 1 ± 3 | ND | 2 ± 5 | ND | 17 ± 5 | 208 ± 20 |
In computational docking studies into the active site of human PDH E1 (Fig. 2a) 25 overlays well with TPP. Compared to the pyrophosphate of TPP, the bi-aryl ester extends deeper into the pyrophosphate pocket, which is considerably larger than needed to accommodate the pyrophosphate group. The benzene ring forms a pi-cation interaction with the Mg2+ and the nitrogen atom of the pyridine ring hydrogen bonds to the amide NH of an arginine residue, explaining why the m-pyridin-3-yl group of 8w binds better than the m-phenyl group of 8v. The C2-butanoyl group makes only hydrophobic contacts in the C2-pocket and the aminopyrimidine ring forms all the same hydrogen bonds and pi–pi interaction with surrounding residues that TPP does (Fig. 2b, see also Fig. S3†).
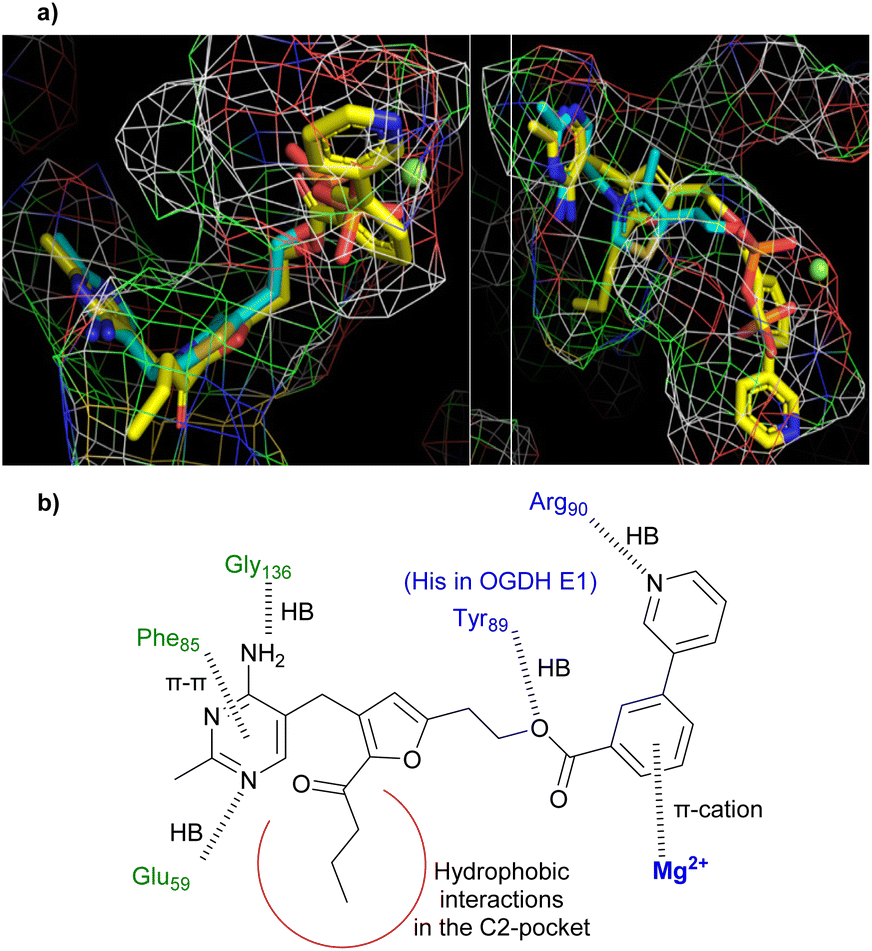 | ||
| Fig. 2 (a) Predicted binding mode of 25 (yellow carbons) in the active site of human PDH E1 (PDB: 6CFO), overlaid with the TPP binding pose (cyan). Left: View showing the characteristic V-shaped conformation of the pyrimidine-CH2-thiazolium motif of TPP. Right: View showing the coordination between the pyrophosphate of TPP and Mg2+ (green ball). (b) Predicted interactions (shown as dashed lines) between 25 and human PDH E1. | ||
To assess target specificity, compounds were tested on other TPP-dependent enzymes (Fig. 1c). Of the four enzymes, three use pyruvate as acyl-donor substrate but use different acceptor substrates, whereas the fourth OGDH E1 uses a different donor substrate but the same acceptor substrate as PDH E1. Thus, in using these four enzymes, we are exploring the binding sites for both donor and acceptor substrates. As shown in Table 4, introducing the C2-butanoyl group (17avs.26) completely abolished activities on PDC and PO but did not affect the inhibition of OGDH E1. PDC and PO use smaller acceptor substrates than PDH E1 and OGDH E1 (Fig. 1c), suggesting they may have smaller C2-pockets that cannot accept the larger butanoyl group of 26 and 25 for steric reasons (though polarity of the C2-pocket may also be a factor). These experiments show that introducing an appropriate C2-substituent can both improve affinity for the selected enzyme and introduce high levels of selectivity over other TPP-dependent enzymes.
Inhibition of OGDH E1 by 23 and 25 was much less than that of PDH E1, despite the fact that both enzymes use the same acceptor substrate and most active site residues are conserved (except Tyr89 replaced by His in OGDH, Fig. 2b). Docking studies suggested that the explanation is that the OGDH E1 pyrophosphate pocket is smaller than that of PDH E1 (which extends well beyond the Mg2+ and the pyrophosphate group of TPP) and so the selectivity has a steric origin (see Fig. S4† for more details). In support of this, the furan series (22 to 17a to 23) showed a trend of reducing affinity towards OGDH E1 as the esterifying group increases in size (Table 4).
Esters 23, 25 and 26 have been characterised as potent and selective TPP-competitive inhibitors of mammalian PDH E1, and marked the completion of our initial goal – to map the SAR of the active site. DeazaTPP 3 features a polyanionic tail (thus membrane-impermeable14) and potently but non-selectively inhibits all TPP-dependent enzymes.13 However, ester 25 is uncharged and selective to mammalian PDH E1, with potency four times greater than 3 (KI: 0.65 vs. 2.6 nM (ref. 14)) despite lacking the pyrophosphate moiety for binding the Mg2+ cation.
From esters to amides
Ester 25 has showcased the strategies to overcome the traditional barriers of applying TPP analogues in cell-based studies. However, we envisioned that the ester linkage of our thiamine analogues is likely to be a metabolic weakness given the high esterase activity within mammalian cells. Therefore, we prepared the amide equivalents of some selected esters (Scheme 4). The hydroxyethyl tail of furan 16a was firstly tosylated (28), then displaced by azide (29), and finally hydrogenated to the amine 30. Upon coupling with the corresponding benzoic acids, amides 31–33 were obtained. Starting from the triazole equivalent of 30,34 the same reactions gave amides 34–35. Furan-based 31 and 33 were C2-acylated with butyryl chloride to give amides 36 and 37, respectively. | ||
| Scheme 4 Synthesis of amides. Reagents and conditions: (i) TsCl, pyridine, RT or 0 °C; (ii) NaN3, DMF, RT; (iii) H2 (g), 10% Pd/C, MeOH, RT; (iv) carboxylic acid, DCC, DMAP, DMF, RT; (v) butyryl chloride, AlCl3, DCE, RT. | ||
Amides 31–37 were tested for their potency and selectivity (Table 5). In direct comparison (e.g.23vs.33), the amides were 2.7- to 6.4-fold less potent than their equivalent esters against PDH E1. However, amides 33 and 37 still bound to PDH E1 considerably more tightly than TPP (4- and 12-fold respectively) and remained highly selective, with no significant (<10%) inhibition of other TPP-dependent enzymes (Table 5). Docking studies suggested that the reduced affinity of the amides was due to the loss of the hydrogen bond between Tyr89 and the ester oxygen. The C2-functionalised 37 was slightly more potent than 33. The ligand efficiencies (L.E.s) of 33 and 37 were 0.36 and 0.33 (kcal mol−1 per heavy atom) respectively, implying that both are highly efficient binders,35 but the group efficiency of the C2-butanoyl group was only 0.14, implying that it is inefficiently contributing to the affinity for PDH E1,36 consistent with our prediction that it only contributes hydrophobic not polar interactions. The calculated physiochemical properties (Table 6) suggest that amides 33 and 37 are also drug-like36,37 but 37 has poor aqueous solubility.
| Mammalian PDH E1 | Yeast PDC | Bacterial PDC | Bacterial PO | Bacterial OGDH E1 | |||||
|---|---|---|---|---|---|---|---|---|---|
| % Inh. (5:1)a,b |
% Inh. (1:1)a,c |
IC50a,d (μM ± SEM) |
K
Ie (nM) |
L.E.f | % Inh. (1:1)a,g |
% Inh. (1:1)a,c |
% Inh. (1:1)a,c |
% Inh. (1:1)a,c |
|
| a Data are the means of measurements in three technical replicates. b Percentage inhibition determined for compounds at 50 μM with [TPP] = 10 μM. c Percentage inhibition determined for compounds at 50 μM with [TPP] = 50 μM. d IC50 values determined at [TPP] = 10 μM. e K I is based on the previously reported KM for TPP of 50 nM,14,33 and is rounded to two significant figures. f Ligand efficiency (L.E.) = ΔG of binding (kcal per mol)/number of non-hydrogen (heavy) atoms; preferred LE: >0.3. g Percentage inhibition determined for compounds at 200 μM with [TPP] = 200 μM. Insol., compound insoluble under assay conditions. ND, not determined. | |||||||||
| 31 | 84 ± 10 | 52 ± 7 | 9.17 ± 0.82 | 45 | 0.40 | 16 ± 3 | ND | ND | 22 ± 4 |
| 32 | >90 | 71 ± 9 | 4.14 ± 0.45 | 21 | 0.38 | 18 ± 3 | 16 ± 3 | 17 ± 4 | 16 ± 3 |
| 33 | 100 | 85 ± 9 | 2.52 ± 0.53 | 13 | 0.36 | 7 ± 3 | 6 ± 3 | 5 ± 1 | 8 ± 2 |
| 34 | 44 ± 7 | 15 ± 5 | 66.0 ± 6.5 | 333 | 0.35 | ND | ND | ND | ND |
| 35 | 63 ± 8 | 25 ± 5 | 26.7 ± 3.2 | 130 | 0.34 | ND | ND | ND | ND |
| 36 | >90 | 67 ± 9 | 4.52 ± 0.45 | 23 | 0.35 | Insol. | 3 ± 5 | 1 ± 3 | 25 ± 5 |
| 37 | 100 | >90 | 0.83 ± 0.14 | 4.2 | 0.33 | Insol. | 2 ± 4 | 6 ± 3 | 2 ± 3 |
a
| Calculated properties | Recommended values36 | 33 | 37 |
|---|---|---|---|
| a Using MarvinSketch 21.2. | |||
| Molecular weight (MW) | <500, prefer-ably <400 | 413 | 483 |
| Calculated LogP (cLogP) |
≤5, preferably ≤3–4 | 2.8 | 3.2 |
| Number of HB donors (HBDs) | ≤5 | 3 | 3 |
| Number of HB acceptors (HBAs) | ≤10 | 7 | 8 |
| Total polar surface area at pH = 7.4 (TPSA) | 75–140 Å2 | 107 | 124 |
| Number of rotatable bonds (RBs) | <10 | 7 | 10 |
| Aqueous solubility (mg mL−1) | — | Moderate (0.01–0.06) | Low (<0.01) |
Preliminary cell assays to assess membrane-permeability of 33
Although the main objectives of the current study were to study the SAR and develop compounds as potent and selective inhibitors of mammalian PDH E1, we wanted to confirm that our amides are indeed membrane-permeable. Of the seven amides (31–37), 33 and 37, with the m-pyridin-3-yl tail, were the most potent inhibitors. Although 37 is a slightly stronger inhibitor, its aqueous solubility is very low, so the more water-soluble 33 was selected and tested at 100 μM on four fast-growing human cell lines. Amide 33 showed no significant effect on cell-viability of any of the cell lines (Fig. S5†) but a marked cytostatic effect on two of the cell lines, growth-inhibition on a third and no effect on the fourth (Fig. 3). The cytostatic effect might be due to inhibition of PDHc, in which case greater suppression on cell growth in HEK and MDA-MB-231 cell lines (relative to the other two cell lines) may be because they rely more on PDHc-mediated oxygen-dependent metabolic pathways for providing energy to sustain their growth. This would be in accord with the previous contrasting findings on the roles of PDHc in different cancer cell lines.7–9 However, without showing dose-dependence nor confirming the intracellular biological target(s), it is premature to make any firm conclusion regarding the mode of action. Although preliminary, our cell-based data do suggest that amide 33 is membrane-permeable. | ||
| Fig. 3 Effects of amide 33 on cell growth for four fast-growing human cell lines, Colo-205 (colorectal cancer), HEK (embryonic kidney), MDA-MB-231 (breast cancer) and PANC-1 (pancreatic cancer). Cell count after a 48 h growth period is shown; the horizontal dashed lines show the starting cell density. Refer to Fig. S5† for full details. | ||
Conclusions
In response to the need for inhibitors to study the cellular roles of mammalian PDHc,7–9 structurally diverse derivatives of triazole- and furan-based thiamine analogues have been synthesised to elucidate their SAR towards PDH E1. Guided by the SAR data, we prepared a series of potent PDH E1 inhibitors; we showed their selectivity for PDH E1 over other TPP-dependent enzymes in biochemical assays and obtained insights into binding at the molecular level through computational modelling. We confirmed the competitive relationship between our inhibitors and the coenzyme TPP and determined their affinities to be least several times higher than that of TPP (77-fold stronger for ester 25). Given that the total plasma concentration of thiamine and its derivatives in humans is only ca. 0.1 μM,38–40 even if amide 33 (four-fold greater affinity than TPP) is dosed at just 1 μM, the binding to PDH E1 would be 40-fold greater than TPP. Although cell-based studies confirmed amide 33 is membrane-permeable, the exact mode of action has yet to be defined. While it is of interest to experimentally establish in the future the link between PDHc inhibition and the observed cytostatic effects, we believe that the SAR studies and PDH E1 inhibitors reported here can directly aid ligand design efforts to further develop inhibitors of mammalian PDH E1. Our rational inhibitor design through introducing hydrocarbon C2-groups to mimic the natural hydrophobic lipoamide substrate (as well as probing the other two pockets) has showcased the approach to overcoming the longstanding barriers (membrane-permeability and selectivity) of applying thiamine/TPP analogues to cell-based studies.Author contributions
AHYC synthesised all the compounds, TCSH performed most of the enzyme assays, DRP tested 33 on human cell lines, FJL supervised the project.Conflicts of interest
There are no conflicts to declare.Acknowledgements
AHYC and TCSH were funded by K. M. Medhealth and DRP by an EPSRC studentship.References
- R. A. W. Frank, F. J. Leeper and B. F. Luisi, Cell. Mol. Life Sci., 2007, 64, 892–905 CrossRef CAS PubMed.
- O. H. Wieland, Rev. Physiol., Biochem. Pharmacol., 1983, 94, 123–170 Search PubMed.
- S. Park, J.-H. Jeon, B.-K. Min, C.-M. Ha, T. Thoudam, B.-Y. Park and I.-K. Lee, Diabetes Metab. J., 2018, 42, 270 CrossRef PubMed.
- S. Prajapati, F. Rabe von Pappenheim and K. Tittmann, Curr. Opin. Struct. Biol., 2022, 76, 102441 CrossRef CAS PubMed.
- M. V. Liberti and J. W. Locasale, Trends Biochem. Sci., 2016, 41, 211–218 CrossRef CAS PubMed.
- G. Sutendra and E. D. Michelakis, Front. Oncol., 2013, 3, 38 Search PubMed.
- C. T. Hensley, B. Faubert, Q. Yuan, N. Lev-Cohain, E. Jin, J. Kim, L. Jiang, B. Ko, R. Skelton and L. Loudat, et al. , Cell, 2016, 164, 681–694 CrossRef CAS PubMed.
- S. M. Davidson, T. Papagiannakopoulos, B. A. Olenchock, J. E. Heyman, M. A. Keibler, A. Luengo, M. R. Bauer and A. K. Jha, et al. , Cell Metab., 2016, 23, 517–528 CrossRef CAS PubMed.
- J. Chen, I. Guccini, D. Di Mitri, D. Brina, A. Revandkar, M. Sarti, E. Pasquini, A. Alajati, S. Pinton, M. Losa and G. Civenni, et al. , Nat. Genet., 2018, 50, 219–228 CrossRef CAS PubMed.
- J. Feng, H. He, Y. Zhou, M. Cai, H. Peng, H. Liu, L. Liu, L. Feng and H. He, Bioorg. Med. Chem., 2019, 27, 115159 CrossRef CAS PubMed.
- X. X. Wang, H. Y. Qi, J. Chen, Y. Z. Yang, W. Qiu, W. Wang, P. Zou, B. Li, Y. L. Wang, H. W. He and G. C. Sun, J. Plant Pathol., 2019, 101, 59–69 CrossRef.
- Y. Zhou, M. Cai, H. Zhou, L. Hou, H. Peng and H. He, Pestic. Biochem. Physiol., 2021, 177, 104894 CrossRef CAS PubMed.
- S. Mann, C. Perez Melero, D. Hawksley and F. J. Leeper, Org. Biomol. Chem., 2004, 2, 1732 RSC.
- E. Grabowska, M. Czerniecka, U. Czyżewska, A. Zambrzycka, Z. Łotowski and A. Tylicki, J. Enzyme Inhib. Med. Chem., 2021, 36, 122–129 CrossRef CAS PubMed.
- T. Masini, B. Lacy, L. Monjas, D. Hawksley, A. R. de Voogd, B. Illarionov, A. Iqbal, F. J. Leeper, M. Fischer, M. Kontoyianni and A. K. H. Hirsch, Org. Biomol. Chem., 2015, 13, 11263–11277 RSC.
- L. J. Y. M. Swier, L. Monjas, A. Guskov, A. R. de Voogd, G. B. Erkens, D. J. Slotboom and A. K. H. Hirsch, ChemBioChem, 2015, 16, 819–826 CrossRef CAS PubMed.
- D. Zhu, S. Johannsen, T. Masini, C. Simonin, J. Haupenthal, B. Illarionov, A. Andreas, M. Awale, R. M. Gierse and T. van der Laan, et al. , Chem. Sci., 2022, 13, 10686–10698 RSC.
- D. Hawksley, D. A. Griffin and F. J. Leeper, J. Chem. Soc., Perkin Trans. 1, 2001, 144–148 RSC.
- K. M. Erixon, C. L. Dabalos and F. J. Leeper, Org. Biomol. Chem., 2008, 6, 3561 RSC.
- A. Iqbal, E.-H. Sahraoui and F. J. Leeper, Beilstein J. Org. Chem., 2014, 10, 2580–2585 CrossRef PubMed.
- A. H. Y. Chan, T. Ho, K. Agyei-Owusu and F. J. Leeper, Org. Biomol. Chem., 2022, 20, 8855–8858 RSC.
- A. H. Y. Chan, I. Fathoni, T. Ho, K. J. Saliba and F. J. Leeper, RSC Med. Chem., 2022, 13, 817–821 RSC.
- H. He, H. He, L. Feng, H. Peng and X. Tan, Chinese patentCN201510672520, 2017 Search PubMed.
- X. W. A. Chan, C. Wrenger, K. Stahl, B. Bergmann, M. Winterberg, I. B. Müller and K. J. Saliba, Nat. Commun., 2013, 4, 2060 CrossRef PubMed.
- E. S. Rudge, A. H. Y. Chan and F. J. Leeper, RSC Med. Chem., 2022, 13, 375–391 RSC.
- Y. Zhou, Y. Qin, H. Zhou, T. Zhang, J. Feng, D. Xie, L. Feng, H. Peng, H. He and M. Cai, Pestic. Biochem. Physiol., 2022, 105098 CrossRef CAS PubMed.
- S. Sanders, R. J. Vierling, D. Bartee, A. A. DeColli, M. J. Harrison, J. L. Aklinski, A. T. Koppisch and C. L. Freel Meyers, ACS Infect. Dis., 2017, 3, 467–478 CrossRef CAS PubMed.
- D. Bartee and C. L. Freel Meyers, Biochemistry, 2018, 57, 4349–4356 CrossRef CAS PubMed.
- D. Bartee, S. Sanders, P. D. Phillips, M. J. Harrison, A. T. Koppisch and C. L. Freel Meyers, ACS Infect. Dis., 2019, 5, 406–417 CrossRef CAS PubMed.
- K. Ravindar, M. Sridhar Reddy and P. Deslongchamps, Org. Lett., 2011, 13, 3178–3181 CrossRef CAS PubMed.
- L. Monjas, L. J. Y. M. Swier, A. R. de Voogd, R. C. Oudshoorn, A. K. H. Hirsch and D. J. Slotboom, MedChemComm, 2016, 7, 966–971 RSC.
- D. Bartee and C. L. Freel Meyers, Acc. Chem. Res., 2018, 51, 2546–2555 CrossRef CAS PubMed.
- D. A. Walsh, R. H. Cooper, R. M. Denton, B. J. Bridges and P. J. Randle, Biochem. J., 1976, 157, 41–67 CrossRef CAS PubMed.
- G. Mayer, C. E. Lünse, C. Suckling and F. Scott, WO2013117559, 2013.
- A. L. Hopkins, G. M. Keserü, P. D. Leeson, D. C. Rees and C. H. Reynolds, Nat. Rev. Drug Discovery, 2014, 13, 105–121 CrossRef CAS PubMed.
- N. A. Meanwell, Chem. Res. Toxicol., 2011, 24, 1420–1456 Search PubMed.
- C. A. Lipinski, Drug Discovery Today: Technol., 2004, 1, 337–341 CrossRef CAS PubMed.
- L. Bettendorff, Metab. Brain Dis., 1994, 9, 183–209 CrossRef CAS PubMed.
- M. Gangolf, J. Czerniecki, M. Radermecker, O. Detry, M. Nisolle, C. Jouan, D. Martin, F. Chantraine, B. Lakaye, P. Wins, T. Grisar and L. Bettendorff, PLoS One, 2010, 5, e13616 CrossRef PubMed.
- D. Coats, K. Shelton-Dodge, K. Ou, V. Khun, S. Seab, K. Sok, C. Prou, S. Tortorelli, T. P. Moyer, L. E. Cooper and T. P. Begley, et al. , J. Pediatr., 2012, 161, 843–847 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Methods and results for enzyme assays and computational docking; synthetic methods, compound characterisation and NMR spectra. See DOI: https://doi.org/10.1039/d2ob02272a |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2023 |