
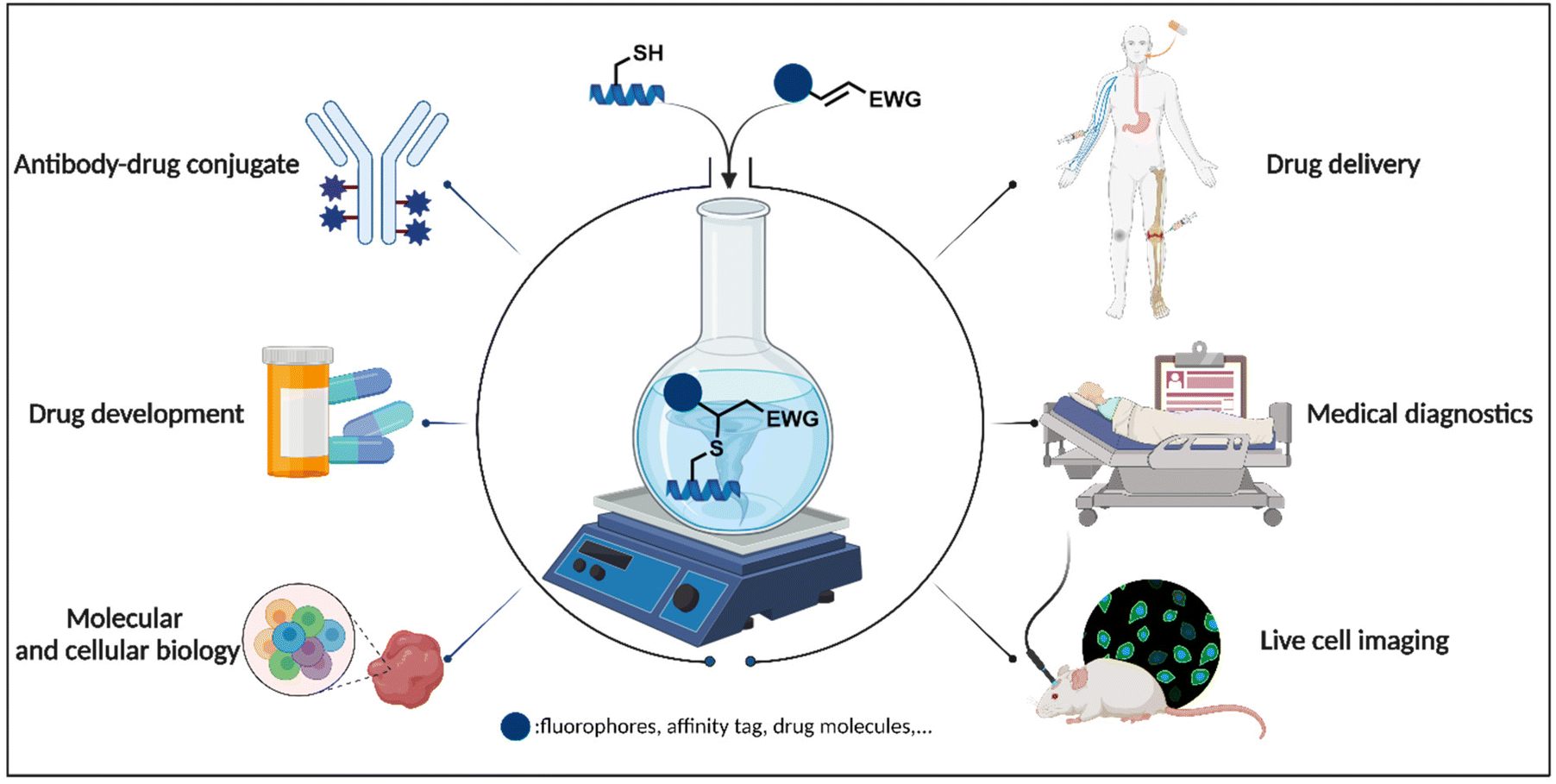
Thia-Michael addition: the route to promising opportunities for fast and cysteine-specific modification
Marzieh
Ahangarpour
 ab,
Iman
Kavianinia
bc and
Margaret A.
Brimble
*abc
ab,
Iman
Kavianinia
bc and
Margaret A.
Brimble
*abc
aSchool of Chemical Sciences, The University of Auckland, 23 Symonds Street, Auckland 1010, New Zealand. E-mail: m.brimble@auckland.ac.nz
bMaurice Wilkins Centre for Molecular Biodiscovery, The University of Auckland, 3 Symonds Street, Auckland, New Zealand
cSchool of Biological Sciences, The University of Auckland, 3A Symonds Street, Auckland 1010, New Zealand
First published on 23rd March 2023
Abstract
Over the last few decades, design and discovery of chemical reactions that enable modification of proteins at pre-determined sites have been the focus of synthetic organic chemists. As an invaluable tool, the site-and chemoselective functionalization of peptides and proteins offers an exciting opportunity for creating high-value multicomponent conjugates with diverse applications in life sciences and pharmacology. In recent years, multiple strategies have emerged that target natural amino acids directly or convert them into other reactive species for further ligations. However, reactivity and selectivity are still key issues in the current state of chemical modification methodologies. Cysteine is one of the least abundant amino acids and exhibits unique chemistry of the thiol or thiolate group which makes it susceptible to a series of post-translational modifications. The thia-Michael “click” addition reactions, which can proceed under facile conditions provide a promising way for thiol-selective modification of cysteine-containing proteins. In this review, we summarize various reactions for cysteine-selective peptide and protein modification, focus on thia-Michael “click” addition reactions, elaborate on their historical perspective and mechanism, and highlight their applications in modifying biomolecules in a site-specific way.
 Marzieh Ahangarpour | Marzieh Ahangarpour studied organic chemistry at the Sharif University of Technology. Fascinated by the art of “Organic Synthesis”, she worked on the application of transition metal spinels in carbon–carbon bond formation with a focus on natural products and organic synthesis. Following this, Marzieh became interested in the amalgamation of organic chemistry and biology. As a result, she pursued a Ph.D. degree at the University of Auckland under the supervision of Dist. Prof. Dame. Margaret Brimble to undertake research on the chemical, site-selective modification of peptides and proteins and their applications in drug discovery. She is currently a Research Fellow at the University of Auckland focusing on the site-specific functionalization of antibodies for the development of antibody–drug conjugates. |
 Iman Kavianinia | Iman Kavianinia is currently a Senior Research Fellow at the University of Auckland. He received his PhD in Medicinal chemistry. His main research interests and expertise are in the area of antibody–drug conjugates, including the development of potent drug payloads and novel linker systems. He also has an interest in total synthesis and structure–activity relationship studies of naturally-occurring peptides with anticancer and antimicrobial activity. |
 Margaret A. Brimble | Margaret Brimble is Director of Medicinal Chemistry and a Distinguished Professor at the University of Auckland where her research focuses on the synthesis of bioactive natural products and peptide chemistry. She was elected a Fellow of the Royal Society London in 2018 and awarded the RSC Pedler Award for organic chemistry (2022). In 2012 she was awarded the 2012 Royal Society of New Zealand Rutherford, MacDiarmid and Hector Medals and conferred the Queen's Honour DNZM in 2019. She is Past-President of IUPAC Division III and an Associate Editor for Organic Letters. She discovered the first drug “trofinetide” that was given FDA approval on march 10, 2023 for treatment of Rett syndrome for people 2 and over under the tradename DaybueTM https://www.neurenpharma.com. She also co-Founded the company SapVax to develop self-adjuvanting cancer vaccines (https://sapvaxllc.com). |
1. Introduction
“Biology is moving in the direction of chemistry. Much of the understanding of biology that exists now is based on the structure of molecules and the properties of molecules in relation to their structure. If you have that basis, then biology isn't just a collection of disconnected facts”.1 This quote is from Linus Pauling, double Nobel Prize laureate in 1954 and 1962, who showed that no discipline is an isolated island and helped to define modern biology.1Modern biology overlaps with chemistry and takes advantage of what chemists can do best. This nexus is not only necessary but essential for providing new tools, new insights, and solutions to the unresolved questions of biology as well as deriving a deeper understanding of biological systems.2
Bioconjugation takes place at the crossroads of biology and chemistry. Covalent derivatization of biomolecules to form stable conjugates while preserving their function and structural integrity is broadly labelled as “bioconjugation” chemistry. Furthermore, chemo- and regioselective conjugation of the reactive group to the desired positions on a polypeptide or protein is known as site-selective bioconjugation which confers additional functional and structural properties, leads to specific functional outcomes, and enables the interrogation of biological systems (Fig. 1).3
 | ||
| Fig. 1 Tailoring peptides/proteins with selective reactions in the presence of all the functionality found within living systems finds diverse applications, created with BioRender.com (accessed 1 November 2022). | ||
The early application of chemical modification methodologies for modifying proteins was mostly based on empirical evidence, with minimal chemistry knowledge. For example, historically, formaldehyde has been used as a tanning agent for the hardening of leather products due to its ability to crosslink proteins; though the process was not entirely clear at the time.4 In addition, formaldehyde is also used in detoxification processes, converting toxins to non-toxic products for vaccine applications.4 During the 1920s the vaccine toxin was incubated with formaldehyde yielding a non-toxic but still antigenic moiety for developing immunization studies which is a method that is still utilized for the production of non-toxic protein products.5
Early studies of medical application of bio-chemical modification can be traced back to research by Avery and Goebel during the late 1920s and early 1930s.6,7 They reported an immune response of bacterial capsular polysaccharide in rabbits upon cross coupling with a foreign protein carrier, though this carbohydrate alone is non-immunogenic in rabbits.7 Later, the first conjugate vaccine used in humans became available in 1987 when the deficiencies of the polysaccharide vaccine led to the production of the conjugate Hib (Haemophilus influenzae type b) polysaccharide-protein vaccine.8 In this period, PEGylated drugs were also brought to the market.9 Monomethoxy-polyethylene glycol (PEG) is a synthetic water soluble, non-immunogenic, biocompatible polymer available as linear or branched derivatives in a variety of molecular weights and widely accepted in therapeutics.10 The chemical modification of covalently attaching non-toxic and non-immunogenic PEG to a biomolecule known as PEGylation was first reported in 1977 by Abuchowski et al.11
PEGylation of therapeutic proteins often imparts several significant pharmacological advantages over the unmodified form. It is a potent method for increasing the half-life of the molecule in vivo, protecting proteins from proteolytic degradation, and improving water solubility and stability of the biomolecule.12
In parallel, the concept of antibody–drug conjugates (ADC) for site-specific drug delivery revolutionalized the field of cancer targeted therapy (Fig. 2).13 ADCs use antibodies to deliver a potent cytotoxic compound selectively to tumour cells, thus improving the therapeutic index of chemotherapeutic agents.14 For example, in Gemtuzumab ozogamicin (Mylotarg®, Wyeth-Pfizer) which was introduced into the market in 2000 for the treatment of acute myeloid leukemia (AML), solvent accessible lysine residues on the monoclonal antibody (mAb) were used for linker-payload conjugation.15 On the other hand, in brentuximab vedotin (Adcetris®, Seagen), approved in 2011 for Hodgkin lymphoma (HL), cysteine based conjugation was used for payload attachment to mAb.16
 | ||
| Fig. 2 Antibody–drug conjugates (ADC) show therapeutic benefit to patients with cancer, created with BioRender.com (accessed 1 November 2022). | ||
Research over the past two decades has embarked on characterizing the chemistry of chemical modification of peptides and proteins, thereby providing the framework for the chemical methods that are commonly used today.17–26 Such methods for tethering of drugs, carbohydrates, peptides, and other synthetic polymers into a biomolecule have resulted in a major leap in the pharmacological and medical applications and have continued to expand and develop.
In recent years, multiple strategies have emerged that target natural amino acids directly or convert them into other reactive species for further ligations, among which, site-selectivity has been troublesome when there are multiple competing nucleophilic functional groups with different reactivity and relative abundance, leading to highly heterogeneous products with varying properties and functions (Fig. 3).3
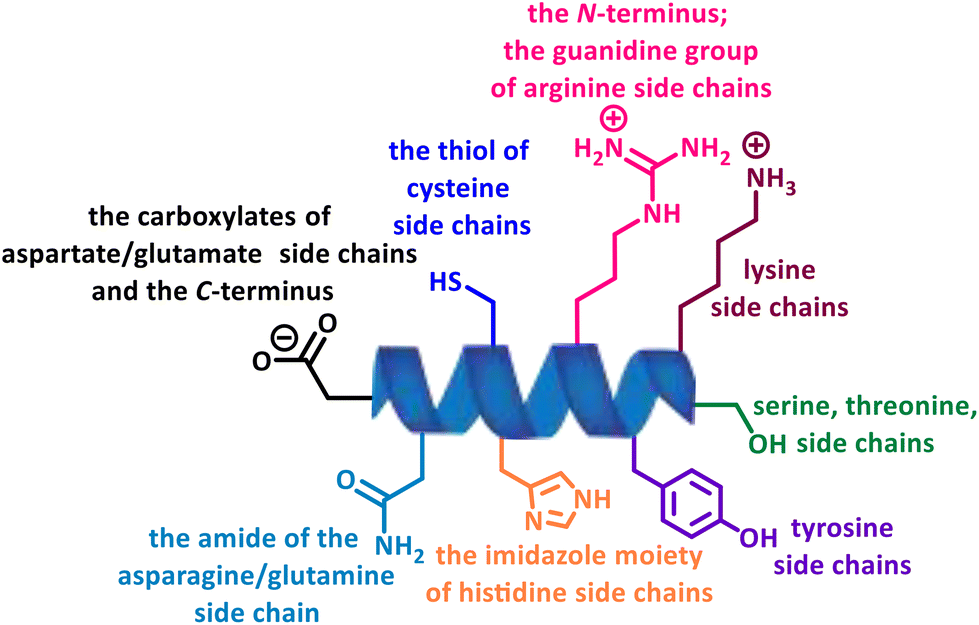 | ||
| Fig. 3 Various competing nucleophilic residues present on the surface of proteins. | ||
This challenge gives rise to discussion as to how introduction of a chemical modification at a single, distinct site on the unprotected polypeptide is crucial and requires prioritization for the formation of a homogeneous and well-defined bioconjugate.
Given the stringent demands, in the last few decades, many studies have been published on the development of chemical tools for the modification of amino acids with a high degree of chemoselectivity and regioselectivity.18,20–23
Among the 20 canonical amino acids, cysteine (Cys) is perhaps the most popular target for site-specific modification due to its low abundance and selective reactivity of its thiol group, which is characterized by a strong nucleophilicity (Fig. 4).20
 | ||
| Fig. 4 Chemical protein modification through cysteine, created with BioRender.com, accessed 1 November 2022. | ||
For the purposes of this review, focus will remain on the enduring utility of cysteine in selective peptide/protein modification with emphasis on the thia-Michael addition reaction, a notable tool for controlled functionalization of biomolecules in the context of “bioorthogonal chemistry” to show how this combination has had a huge impact on the toolbox of bioconjugation chemistry. This review helps clarify future directions for the development of peptide/protein modification through cysteine modification in the context of thia-Michael addition reactions.
2. Targeting cysteine residues
Among the various remarkable conjugation strategies, functionalization of the side chains of the nucleophilic amino acid cysteine, is a popular strategy.20 The cysteine residue offers a unique, reactive handle within proteins and is among the most widely studied natural amino acids for manipulating and modifying with chemical reagents in a selective manner. Cysteine has a low natural abundance on protein surfaces (<1.7%) which is helpful for maintaining homogeneity. The intrinsic nucleophilicity of the cysteine thiolate enables rapid reactivity with electrophiles to form thioethers under physiological conditions.19,27One of the most commonly used methods for cysteine modification is alkylation with a wide range of electrophiles. The first reagents to be reported were α-halocarbonyl compounds such as iodoacetamide reagents to form S-carboxyamidomethyl-cysteine (Scheme 1a).28 This methodology is frequently employed as an identity test for proteins in mass spectrometry as the protein molecular mass will increase by 57 Da for each cysteine derivative.29
 | ||
| Scheme 1 Site-selective methods for the modification of cysteine by (a) α-halocarbonyls, (b) alkylation through a SN2 reaction with haloalkyl reagents, (c) sulfhydryl-disulfide (SH/SS) exchange, (d) disulfide formation at cysteine, (e) dehydroalanine formation and its modification with functionalized thiols, (f) dibromomaleimides as useful scaffolds for peptide modification. | ||
Aminoethylation of cysteine to form lysine or methylated lysine analogues was first described seventy years ago.30 Since then, aminoethylation has been developed into a chemical method for generating residues (‘thia-lysines’) to study the function of active site lysines (Scheme 1b).31
Disulfide bonds, generated from the oxidation of thiol groups, frequently play an important role in providing stability to a protein by reducing the entropy of unfolded forms and preserving protein tertiary and quaternary structure.32,33 Sulfhydryl-disulfide (SH/SS) exchange is another protocol for peptide and protein modification, a transformation that can be easily reversed by adding excess of a thiol such as 2-mercaptoethanol and dithiothreitol (DTT).34 The generated disulfides are unstable in the cytoplasm of cells due to the high concentration of the reducing agent glutathione in cells. As a result, this strategy has applications in drug delivery systems (Scheme 1c).
Sulfenyl halides are suitable reagents for mixed disulfide bond formation,35 as well as selenenyl sulfides as reactive precursors.36 Methanethiosulfonate and phenylthiosulfonates (SO2R, R = Me, Ph) also are increasingly common for thiol modification (Scheme 1d).37
The oxidative elimination of cysteine to dehydroalanine (Dha), a reactive α,β-unsaturated electrophilic amino acid, has enabled access to a distinct approach for post-translational cysteine modification.38,39 Dha formation is favoured by exposure to heat and alkali. Dha can be used as a Michael acceptor and the site of modification is especially useful where innumerable possibilities for electrophilic alkylation of cysteine exist or when a suitable electrophile cannot be generated. The addition of functionalized thiols to Dha is rapid under mild reaction conditions leading to selective modification. This reaction has been used to install a range of post-translational modifications such as lipidation, glycosylation, phosphorylation and lysine methylation/acetylation (Scheme 1e).40 Caddick et al. introduced the use of maleimides with good leaving groups to react sequentially with two nucleophilic thiol groups such as two cysteines (Scheme 1f).41 Use of bromomaleimides incorporating a bromo leaving group on the maleimide has also been used as a possible way to effect reversible conjugation for modifying cysteine in a selective and reversible way without any racemisation at the cysteine residue.42,43
Since Caddick's studies, an enormous amount of research on site-selective modification of cysteine has been undertaken. Examples of many such methods that have been developed over the last 20 years include metal-catalyzed cysteine modification where allyl sulfides undergo efficient cross-metathesis using a ruthenium catalyst reported by Davis et al.,44 a gold-mediated oxidative allene–thiol coupling reaction reported by Che et al.,45 a new permanent chemical ligation method developed by Hutton et al. in which thiol groups react with allyl selenosulfate salts,46 a chemoselective Pd-catalyzed prenylation, functionalization, and stapling of cysteine by Breinbauer et al.,47 and a precise lysine modification in native proteins using a cysteine-based chemoselective linchpin by Rai et al.48
Thia-Michael addition reactions in particular have garnered significant attention in the context of powerful tools for the modification of peptides and proteins. As a result, the discussion henceforth will concentrate on the journey to the development of new and more effective applications of this prized tool for cysteine modification.
2.1. Thia-Michael addition “click” reaction, historical perspective, and application in thiol-selective bioconjugation reactions
Nowadays, there are numerous methods for the chemical modification of peptides and proteins. The simplest and potentially most used methods take advantage of the chemical reactivity of nucleophilic amino acid side chains with electrophiles resulting in specific and unique functional outcomes.49The sulfhydryl group of the cysteine residue in peptides and proteins has remained an attractive target for site-selective modification. At physiological pH, cysteine reacts faster than lysine as the thiolate state is prevalent hence the thiolate addition to electrophiles proceeds unimpeded by competition from other nucleophiles.3
A reliable way to selectively modify the nucleophilic cysteine side chain is via the conjugate addition of cysteine thiols to Michael-type acceptors (Fig. 5). This occurs at neutral pH and is a simple, robust and efficient reaction to form a C–S bond under facile reaction conditions. A large number of α,β-unsaturated Michael acceptors have been exploited and applied to cysteine-selective peptide and protein modification.
 | ||
| Fig. 5 The thia-Michael addition reaction creates a hinge to connect chemistry, biology, and medicine, created with BioRender.com, accessed 1 November 2022. | ||
The highly modular thia-Michael addition reaction is now 136 years old and adopted the “click” nomenclature due to the highly reactive nature of thiols, its high selectivity, the versatility of functional group and catalysts/initiators used, and its ability to progress in a wide range of solvents as well as under solvent-free conditions. This special type of 1,4-addition reaction using a thiol species as a nucleophile reacting with an electron deficient carbon–carbon double bond was discovered in 1887 by Arthur Michael50 and proceeds with high conversion, under clean and mild reaction conditions, with a rapid reaction rate and minimal by-product formation even under dilute conditions and results in the formation of an anti-Markovnikov addition product.51–53 In a thia-Michael addition reaction, vinyl groups attached to electron withdrawing groups are more reactive and base and nucleophile catalysis are often required. The nature, pKa, and steric accessibility of the thiol, basicity, nucleophilicity, and concentration of the catalyst, polarity of the solvent, electron deficiency and steric accessibility of Michael acceptors and alkene components should be selected with care for optimal conditions since they affect the reaction rate, product specificity, conversion, and side reactions.54,55
The thia-Michael addition reaction has become a valuable tool for substrate modification in a range of burgeoning fields in chemical and biological systems (Fig. 5).
Efforts have been devoted to the design of an efficient catalyst system for achieving a high yield of hydrothiolation (SH addition) to an activated C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond (Michael acceptor) for decades. Well-studied examples have highlighted that basic and nucleophilic catalysts are extremely rapid among the variety of catalytic systems studied resulting in minimal by-product formation affording high yields of products under ambient reaction conditions.51
C bond (Michael acceptor) for decades. Well-studied examples have highlighted that basic and nucleophilic catalysts are extremely rapid among the variety of catalytic systems studied resulting in minimal by-product formation affording high yields of products under ambient reaction conditions.51
The reaction between a thiol and an electron-deficient CC bond can be facilitated using a catalytic amount of base. The base catalyzed thia-Michael addition reaction starts with the spontaneous formation of a thiolate and conjugate acid from the deprotection of a thiol (Brønsted acid) proton by the catalyst (Brønsted base) (Scheme 2a).56,57 The pKa of the thiol and strength of the base plays a crucial role in this reaction. The thiolate anion which is generally unstable and highly reactive then attacks the more favourable electron-deficient carbon–carbon beta-position of the alkene to form a carbanion intermediate. The final product, a thioether, is generated when the carbanion intermediate abstracts a proton from the conjugated acid, regenerating the thiolate (Scheme 2c).
 | ||
| Scheme 2 The mechanism of base- and nucleophile-catalyzed thiol Michael addition. | ||
The nucleophilic attack of a thiolate anion on the electron-deficient alkene is commonly determined as the rate-controlling step of the thia-Michael addition reaction pathway (Fig. 6, blue curve). However, a different perspective demonstrates that the protonation of the enolate is the rate-limiting step (Fig. 6, pink curve).58
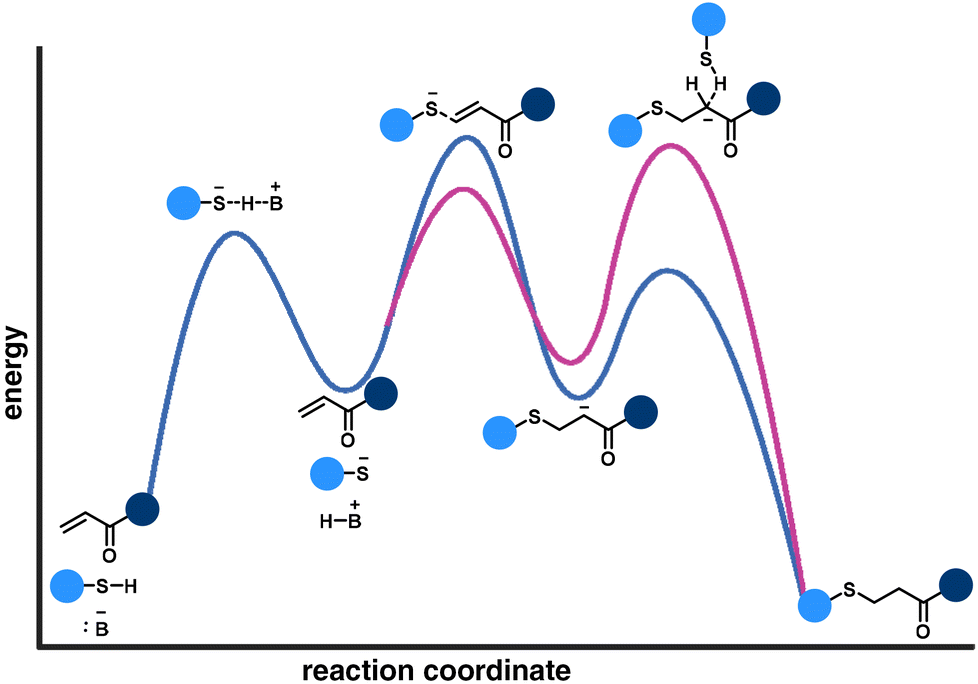 | ||
| Fig. 6 Two different energy perspectives for base-catalyzed-thia Michael additions. | ||
The reaction dependency on the pKa of the thiol may be negated by using a strong nucleophile as a catalyst instead of a base (Scheme 2b). The process of nucleophile-catalyzed thia-Michael reaction starts with the attack of a nucleophilic catalyst to the CC double bond affording an intermediate zwitterion. The enolate base then abstracts the thiol proton, generating the thiolate anion which adds to the activated alkene group affording the thioether adduct (Scheme 2c).59
The relative rates of Michael addition to α,β-unsaturated esters, amides, and ketones are readily understood by consideration of the electrophilicity of the enoyl α-carbon, which tracks the energy of the transition state for formation of the initial enolate intermediate upon addition of the nucleophile to the Michael acceptor. The relative energy of the enolate species is highly dependent on the inductive and/or resonance effects of the substituents bonded to the carbonyl carbon. Considerable experiments and computational studies have been dedicated to elucidating the factors that influence the addition of thiolates to Michael acceptors.55,60,61
Haddleton et al. described an in-depth evaluation of the catalytic behaviour of primary amines, tertiary amines, and phosphines in the Michael reaction using different thiols 1–9 (Scheme 3).56 They found that triethylamine 12 which is less nucleophilic than primary and secondary amines leads to the formation of expected thia-Michael adduct without forming any side product even when used in excess, although a prolonged reaction time was needed to increase the conversion. On the other hand, they observed rapid formation of the thia-Michael adduct alongside phospha- or aza-Michael side products when phosphine based catalysts such as DMPP 13 or TCEP 14 and primary amines such as pentylamine 10 and hexylamine 11, were used. Haddleton et al. also demonstrated the role of solvents on reaction conversion and rate and found that DMSO with higher polarity compared to acetonitrile and acetone results in a higher reaction rate as it allows a better stabilization of thiolate anion thus favouring its formation.56
 | ||
| Scheme 3 Summary of the different thiols and catalysts investigated by Haddleton et al. | ||
In another study, Bowman et al. investigated the kinetic behaviour of organocatalysts, amines and tertiary phosphines (Scheme 4) as nucleophile-initiators of the thia-Michael addition reaction between hexanethiol and hexyl acrylate.59
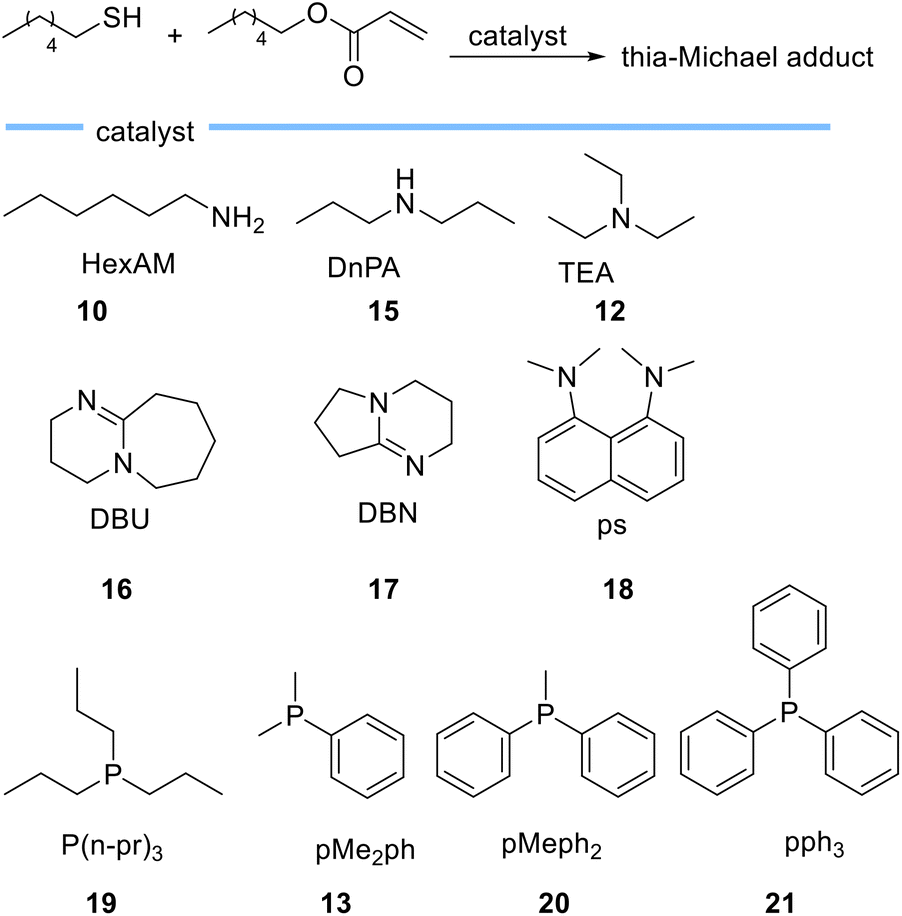 | ||
| Scheme 4 Chemical structures of bases employed as catalysts for thia-Michael reactions by Bowman et al. | ||
They disclosed that the activity of the catalyst is more related to the nucleophilic character of the catalyst rather than basicity and rejected the hypothesis of purely base-mediated process. This interesting conclusion is drawn from the following observations. Based on the comparison between 1°, 2°, and 3° amines, hexylamine 10 (pKa = 10.6)-mediated thia-Michael reaction, leads to a higher conversion when compared to di-n-propylamine (DnPA) 15 (pKa = 11) and triethylamine (TEA) 12 (pKa = 10.75) which both have higher pKa compared to hexylamine 10. On the other hand, when using the more basic bicyclic amidines 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) 16 (pKa = 11.6) and 1,5-diazabicyclo[4.3.0]non-5-ene (DBN) 17 (pKa = 13.5) the reaction rate was much faster than when using 1°, 2°, and 3° amines. This behaviour was attributed to the nucleophilic character of DBN 17 and DBU 16. The use of N,N,N′,N′-tetramethyl-1,8-naphthalenediamine (PS) 18 with a pKa of 12.1, which is between the pKa of DBU 16 and DBN 17 showed very low activity as a catalyst, even less than triethylamine. Tertiary phosphines 19 (pKa = 8.64), 13 (pKa = 6.49), 20 (pKa = 4.59), and 21 (pKa = 2.73) which are weak bases allowed the reaction to occur extremely fast even at low catalyst loadings thereby establishing the crucial role of catalyst nucleophilicity.59
Thia-Michael additions pose highly challenging reactions for computational methods. There are a few papers reporting computational or kinetic investigations on the thia-Michael reactions.
Roush et al. assessed the relative rates of TEA 12-catalyzed Michael additions of 2-phenylethanethiol 22 to different vinyl sulfonyl acceptors 23–29 to evaluate the effect of the R-substituent on the rate of the Michael additions (Scheme 5).62 The results showed that rates of addition varied by up to 3 orders of magnitude from highly reactive phenyl vinyl sulfonate ester 23 with σ-electron-withdrawing inductive effects to N-benzyl vinyl sulfonamide 29. The data for compound pairs 26/27 and 28/29 show that N-methylated sulfonamides affect the reaction rate by a factor of 3. The N-alkoxy sulfonamides 26 and 27 have higher Michael reactivities than 28 and 29, possibly due to the lower electronegativity of nitrogen relative to oxygen.62
 | ||
| Scheme 5 Relative pseudo-first-order rate constants of Michael addition of 2-phenylethanethiol 22 to various Michael acceptors 23–29. | ||
Houk et al. performed a theoretical calculation showing how the substituents on a Michael acceptor affect the kinetics and thermodynamics of thiol additions, contributing to the degree of reversibility of the thiol adduct. Density functional theory (DFT) calculations showed that Michael acceptors containing an electron-withdrawing group (EWG) at the α-position conjugated with a suitable C-β substituent such as aryl or branched-chain alkyl make the kinetics of addition and elimination both fast (Scheme 6).63 Not only does the α-EWG group stabilize the anionic intermediate of the Michael addition, but also destabilizes the thia-Michael adduct which makes the reverse reaction both fast and thermodynamically favourable.63–65
 | ||
| Scheme 6 Reversible covalent modifiers of protein cysteines. | ||
Taunton et al. also studied the kinetic stability of a range of acrylonitrile-based Michael acceptors, activated by aryl or heteroaryl electron-withdrawing groups at rates that span more than 3 orders of magnitude (Scheme 7).66
 | ||
| Scheme 7 Design of reversible cysteine-targeted Michael acceptors. | ||
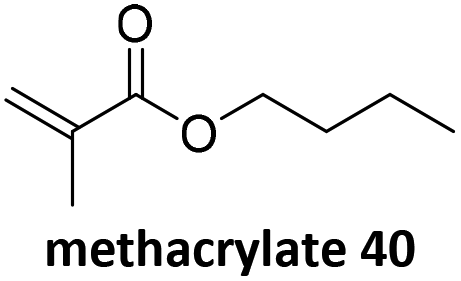
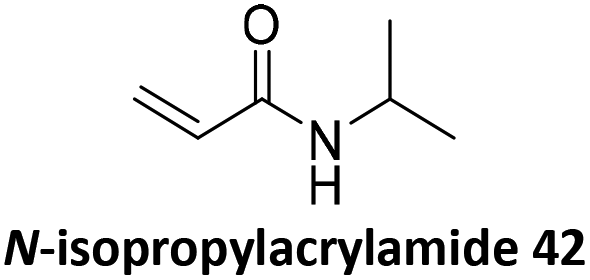
Prez et al. also presented kinetic comparison of the addition of 6-methylheptyl 3-mercaptopropanoate, a model thiol, to nine thiol-Michael acceptors using online FT-IR and real-time monitoring of the peak that corresponds to the S–H stretch. Effects of solvents and different catalysts (amine or phosphine) were also studied.67 A summary of the conversions for equimolar reactions of the thiol and the nine substrates in DMF at various conditions is shown in Table 1. In terms of reactivity of the thiol in DMF, the overall comparison is as follows. Maleimide 35 (no catalyst) > vinyl sulfone 36 (phosphine catalyzed) ≥ acrylate 37 (phosphine catalyzed) > isocyanate 38 (amine catalyzed) > isothiocyanate 39 (amine catalyzed) > vinyl sulfone 36 (amine catalyzed) > acrylate 37 (amine catalyzed) > methacrylate 40 (phosphine catalyzed) > N,N-dimethylacrylamide 41 (amine catalyzed) > N-isopropylacrylamide 42 (amine catalyzed) > methacrylate 40 (amine catalyzed).
| Thia-Michael acceptor | Catalyst/initiator | Time (min) | Conv. (%) |
|---|---|---|---|

|
No catalyst | <1 | 100 |
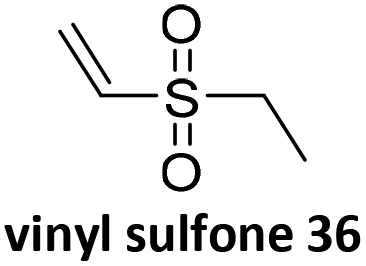
|
Phosphine | 1 | 100 |

|
Phosphine | 1 | 100 |

|
NEt3 | 2 | 100 |
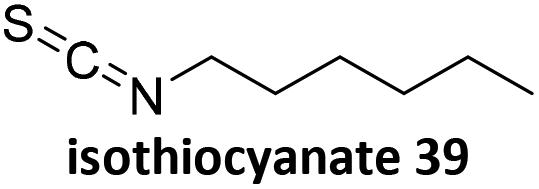
|
NEt3 | 25 | 100 |
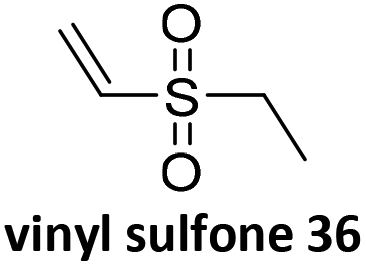
|
NEt3 | 40 | 100 |

|
NEt3 | 140 | 100 |
|
Phosphine | 840 | 60 |
| DBU | 24 | 97 | |
| NEt3 | 840 | 10 | |

|
NEt3 | 825 | 33 |
|
NEt3 | 820 | 19 |
Over the last decade, the Michael reaction has claimed undisputable capability in the design and synthesis of complex small organic molecules as well as opened new avenues for diverse challenging chemistry projects. The Michael reaction takes place under mild conditions and gives high yields with little or no by-products and benefits from high functional group tolerance. Various factors such as the nucleophile/base type, solvent and substrates should be taken into account when optimizing the Michael addition protocol.
Loh et al. developed a rapid, selective, and irreversible strategy to modify cysteine residues in fully unprotected peptides, and proteins using a novel orthogonal handle, C-substituted terminal allenamides 43, via a Michael addition reaction (Scheme 8). Allenamide reagents were confirmed to be stable at room temperature in aqueous conditions and inert towards other encountered nucleophilic sidechains demonstrating the applicability of the allenamide moieties for the site-specific labelling of cysteine-containing proteins.69
 | ||
| Scheme 8 Allenamides as orthogonal handles for selective modification of cysteine thiols. | ||
Following this Cameron and Brimble developed a robust platform to introduce allenamides on resin while assembling the peptide followed by preparing modified cyclic peptides by intramolecular thia-Michael addition (Scheme 9).70
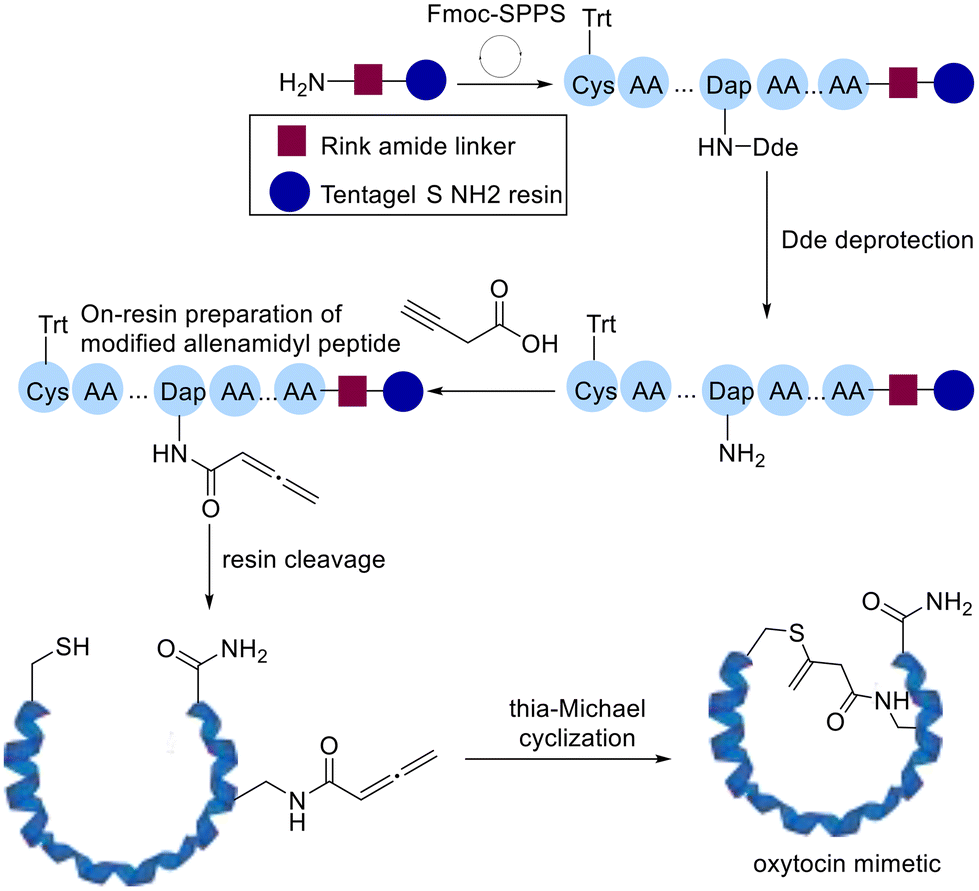 | ||
| Scheme 9 A robust platform for cyclization of allenamidyl peptide developed by Cameron and Brimble. | ||
Vilarrasa et al. developed a simple thiol-Michael-type addition for the biorthogonal conjugation of cysteine derivatives using activated triple bond linkers, propynamides 51, under aqueous biologically-friendly conditions (Scheme 10). This strategy afforded carboxamido groups with high chemical stability.71
 | ||
| Scheme 10 Addition of the thiol group of cysteines to propynamides 51. | ||
Bernardes et al. described a new class of Michael acceptors, carbonylacrylic reagents, for chemoselective cysteine bioconjugation under biocompatible conditions (Scheme 11). They designed three derivatives 52a–c of which benzoyl acrylamide 52b was experimentally identified as the best candidate. They also demonstrated the utility of their cysteine-bioconjugation method in protein conjugation and precisely linked a fluorescent tag and PEG to furnish functionalized benzoyl acrylamides at pre-determined sites on proteins. These conjugates afforded fully stable products in the presence of excess external thiol and in human plasma when compared with the thiosuccinimidyl derivative of maleimide.72 Following the development of this new strategy, Bernardes et al. used a minimal excess of carbonylacrylic reagents functionalized with a synthetic molecule of interest such as cytotoxic drugs for the irreversible and selective modification of antibodies containing native or engineered cysteine residues to yield cysteine-tagged antibodies with high quality, homogeneity, and plasma stability.73
 | ||
| Scheme 11 Carbonylacrylic derivatives for reaction with cysteine. | ||
Bernardes and co-workers also developed simple warhead-like structures, quaternized 2-vinylpyridines 58, that are exceedingly reactive and selective toward cysteine (Scheme 12). Nitrogen quaternization converted the poorly reactive 2-vinylpyridine to an ultrafast reactive reagent toward thiols that were present in four different protein scaffolds 54–46 and 59. Introduction of the +1 charge of the Michael acceptor per cysteine residue to the overall net charge of the protein also modulates its properties by enhancing their hydrophilicity, half-life in circulation, or internalization rates.74
 | ||
| Scheme 12 Cysteine modification using quaternized vinylpyridines 58. | ||
Chiba et al. introduced a new approach for efficient and selective dual modification of cysteine residues of peptides and bovine serum albumin (BSA) 63 using 2-azidoacrylate derivatives 60a–e armed with a synthetic molecule of interest such as PEG and fluorescent dyes (Scheme 13). From simple thia-Michael addition to the 2-azidoacrylate moiety, conjugates anchoring an azido group were afforded. Subsequent azide–alkyne reaction with functionalized alkynes afforded the second modification at the same position of the target proteins.75
 | ||
| Scheme 13 Site-specific dual functionalization of cysteine residue with 2-azidoacrylates. | ||
Fox et al. used cyclopropenyl ketone 64, a new class of Michael acceptor, to participate in a strain-driven reaction with the redox active protein, thioredoxin (Trx), that is both fast and irreversible (Scheme 14). Further stability studies demonstrated full resistance towards retro-Michael addition in the presence of excess external thiol in phosphate-buffered saline (PBS) and human plasma.76
 | ||
| Scheme 14 Strain-driven reactions of cyclopropenylketones with cysteine residues. | ||
Wong et al. reported that isoxazoliniums 66 chemoselectively modifies cysteine residues in peptides and proteins to form a stable thioether linkage (Scheme 15). If an alkyne handle 66h or 66i is introduced, the modified peptide can rapidly reverse the reaction and form a thioaldehyde under UV irradiation (λmax = 365 nm) via a Norrish type II photolysis reaction. Reduction of the thioaldehyde regenerates the original cysteine residue.77
 | ||
| Scheme 15 Cysteine modification using isoxazoliniums. | ||
Bernardes et al. site-selectively introduced robust α,β-unsaturated diazoketones 72 onto cysteine-containing proteins and antibodies under biological conditions at near-neutral pH (Scheme 16). Subsequent 1,3-cycloaddition reactions with strained alkynes gave homogeneous conjugates in high yields that provide opportunities to use diazoketones as probes to label cysteines with an in-cell click reaction using strained and non-strained alkynes.78
 | ||
| Scheme 16 Diazocarbonylacrylic 72 for cysteine modification. | ||
Cysteine-specific protein modification using 5-hydroxy-pyrrolones 75 was reported by Madder et al. to site selectivity introduce two different functional groups on peptides and proteins to yield thiol conjugates with superior stability and resistance to thiol-exchange (Scheme 17).79
 | ||
| Scheme 17 Cysteine multi-functionalization using 5-hydroxy-pyrrolone based building blocks 75. | ||
Yin et al. used highly reactive 4-substituted cyclopentenone 76 for cysteine modification proceeding via Michael addition/elimination under physiological conditions to yield a stable product with minimal effects on the structure and function of the protein (Scheme 18). The original protein can be released with a Michael addition donor demonstrating 4-substituted cyclopentenones as traceless, removable and valuable bioconjugation tools for the controlled release of a conjugate.80
 | ||
| Scheme 18 Cleavable cysteine-specific modification via 4-substituted cyclopentenone 76. | ||
5-Methylene pyrrolones 80 are highly cysteine-specific thia-Michael acceptors that afford stable conjugates at neutral pH although they are cleavable in alkaline buffer (pH 9.5) or via thiol exchange at pH 7.5. Zhou et al. introduced cysteine specific and tracelessly removable bioconjugation via Michael addition to 5-methylene pyrrolones. An application of 5-methylene pyrrolones for cysteine modification was illustrated using E. coli. acetohydroxyacid synthase isozyme I (Scheme 19).81
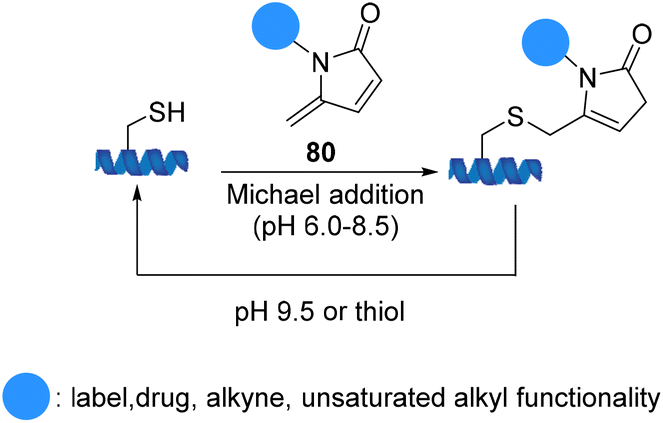 | ||
| Scheme 19 Michael addition to 5-methylene pyrrolones. | ||
Hackenberger et al. demonstrated phosphonamidate electrophiles 81 as a new type of thiol-specific bioconjugation tool in aqueous systems to facilitate the attachment of functional reagents such as biotin, fluorophores, and peptides to proteins and antibodies with superior stability towards thiol-exchange (Scheme 20).91
 | ||
| Scheme 20 Cysteine-selective phosphonamidate electrophiles. | ||
Vinyl sulfones 82 (α,β-unsaturated sulfones) readily react with a variety of nucleophiles via a Michael-type 1,4-addition due to the electron-withdrawing capability of sulfones (Scheme 21).82 The ability to perform reactions under physiological conditions that maintain the biological characteristics of the proteins, the lack of catalysts and by-products, and formation of stable functionalized adducts led the introduction of vinyl sulfones to the field of protein modification. In addition, they have exceptional biomedical significance due to their ability to serve as irreversible inhibitors by conjugating to the thiol group of a cysteine residue. Comparative studies on the relative nucleophilic reactivities of amino and thiol groups have been performed. The studies showed that the rates of reaction depend on the pKa of the nucleophile and steric environment. Vinyl sulfones react with thiols selectively and significantly faster than reaction with amines or other nucleophiles.83 The application of these findings provides a framework for using vinyl sulfones to effect chemoselective chemistry of cysteine-containing peptides and proteins.
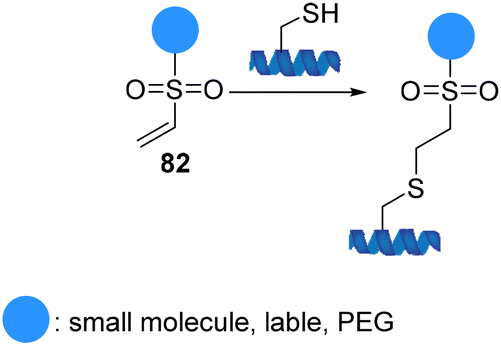 | ||
| Scheme 21 Cysteine modification using vinyl sulfones. | ||
Maleimide motifs 83 are highly reactive conjugate acceptors, commonly used for modification of cysteine residues in proteins (Scheme 22). The selective addition of thiol to the double bond of the Michael acceptor maleimide yields a succinimide thioether that has been known for more than a century. The importance of maleimide chemistry was demonstrated in 1949 when the reaction between substituted maleimides and thiols proceeded rapidly, efficiently, in high-yield, and at room temperature.84
 | ||
| Scheme 22 Cysteine conjugate addition to a maleimide Michael acceptor. | ||
The first conjugation reaction between a maleimide and a cysteine residue within a protein was published by Moore and Ward in 1956 in which N,N′-(1,3-phenylene)-bis-maleimide 84 was used as a crosslinking agent (Scheme 23).85
 | ||
| Scheme 23 The first deployment of bis-maleimides as a crosslinking agent published by Moore and Ward. | ||
Over the next few years following the initial applications, these types of compounds were found to be extremely cysteine selective and have been used for blocking sulfhydryl residues in proteins providing an approach to systematically map the cysteine residues in proteins with the aid of fluorescent maleimides and also for bioconjugation of drugs to macromolecules.86 However, traditional maleimides suffer from limitations (Scheme 24).87
 | ||
| Scheme 24 a) General Michael addition and hydrolytic pathway of thiosuccinimides. (b) Maleimide–thiol conjugate reagents participate in disruptive cleavage by thiol exchange reactions with another thiol, resulting in the formation of a new maleimide–thiol conjugate. | ||
The hydrolysis and oxidation of maleimide-peptide adducts to the isomeric succinic acid thioether adducts can compete significantly with thiol modification, particularly above pH 8 and may also result in decomposition of protein conjugates and unwanted product mixtures (Scheme 24a).88,89 Moreover, irreversible thiol exchange of maleimide-peptide adducts with another thiol is another concern leading to the loss of therapeutic efficacy since premature release of a drug may cause toxicity issues (Scheme 24b).86
The thiol-maleimide linkages are less robust than previously thought. Because of their potential problems, novel conjugation chemistries for drug attachment to cysteine residues as an alternative to maleimide constructs are being pursued.
Recently, Kavianinia and Brimble reported the first examples of thiol-selective heterobifunctional electrophiles, N-vinyl acrylamides 85, that enable the execution of thiol–thiol conjugation in a site-selective manner to afford stable, irreversible conjugate addition adducts when exposed to GSH rendering N-vinyl acrylamides as a valuable alternative to commonly used maleimides (Scheme 25).90 These new classes of thiol-selective scaffolds possess both an α,β-unsaturated Michael acceptor that can readily undergo a thia-Michael addition and a vinyl amide functionality that is amenable to radical induced thiol–ene “click” reaction (Scheme 25a). In the course of this work, it was discovered that the enamide functionality of the N-vinyl acrylamide linker is highly reactive towards nucleophilic addition of thiols and can participate in a rapid, green, and unusual Markovnikov-selective hydrothiolation reaction in less than one minute (Scheme 25b). The formation of an unexpected Markovnikov N,S-acetal hydrothiolation was further explained using computational studies. It was discovered that N-methylation of the N-vinyl acrylamide scaffold leads to new selectivity resulting in complete formation of the thia-Michael adduct (Scheme 25c). The utility of the N-methyl-N-vinyl acrylamide linker for facile construction of fluorophore-labelled peptides and proteins was also demonstrated.90
 | ||
| Scheme 25 a) N-Vinyl acrylamides can participate in thia-Michael additions as well as radical thiol–ene “click” reactions. (b) and (c) N-vinyl acrylamides, versatile heterobifunctional electrophiles for facile access to effective bis-thioether linkages. | ||
3. Conclusions
Covalent derivatization of biomolecules under conditions compatible with proteins, in a chemoselective and high-yielding manner has been the focus of intensive research in last few decades and became a hot area in the nexus of bioorthogonal chemistry, peptide and protein engineering, and drug development, leading to the generation of peptide drugs, antibody–drug conjugates, and probes for molecular imaging.This review showcases the advances made in chemical methodologies that exhibit control over product selectivity with the focus on cysteine selective modification through thia-Michael addition reaction.
Author contributions
Conceptualization, M. A; writing—initial draft, M. A; writing—review & editing, I. K., M. A. B.; supervision, M. A. B.; funding acquisition. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
The authors declare no conflict of interest.References
- L. Pauling and N. A. Campbell, BioScience, 1986, 36, 737–739 CrossRef.
- P. A. Frey, Biochem. Mol. Biol. Educ., 2002, 30, 152–162 CrossRef CAS.
- M. Ahangarpour, I. Kavianinia, P. W. R. Harris and M. A. Brimble, Chem. Soc. Rev., 2021, 50, 898–944 RSC.
- G. E. Means and R. E. Feeney, Bioconjugate Chem., 1990, 1, 2–12 CrossRef CAS PubMed.
- R. J. Collier, Bacteriol. Rev., 1975, 39, 54–85 CrossRef CAS PubMed.
- C. Jones, Ann. Acad. Bras. Cienc., 2005, 77, 293–324 CrossRef CAS PubMed.
- O. T. Avery and W. F. Goebel, J. Exp. Med., 1931, 54, 437–447 CrossRef CAS PubMed.
- V. Verez-Bencomo, V. Fernandez-Santana, E. Hardy, M. E. Toledo, M. C. Rodríguez, L. Heynngnezz, A. Rodriguez, A. Baly, L. Herrera and M. Izquierdo, Science, 2004, 305, 522–525 CrossRef CAS PubMed.
- H. Park, A. Otte and K. Park, J. Controlled Release, 2022, 342, 53–65 CrossRef CAS PubMed.
- M. Ibrahim, E. Ramadan, N. E. Elsadek, S. E. Emam, T. Shimizu, H. Ando, Y. Ishima, O. H. Elgarhy, H. A. Sarhan, A. K. Hussein and T. Ishida, J. Controlled Release, 2022, 351, 215–230 CrossRef CAS PubMed.
- A. Abuchowski, J. R. McCoy, N. C. Palczuk, T. van Es and F. F. Davis, J. Biol. Chem., 1977, 252, 3582–3586 CrossRef CAS PubMed.
- A. Rondon, S. Mahri, F. Morales-Yanez, M. Dumoulin and R. Vanbever, Adv. Funct. Mater., 2021, 31, 2101633 CrossRef CAS.
- S. Panowski, S. Bhakta, H. Raab, P. Polakis and J. R. Junutula, mAbs, 2014, 6, 34–45 CrossRef PubMed.
- J. T. W. Tong, P. W. R. Harris, M. A. Brimble and I. Kavianinia, Molecules, 2021, 26, 5847 CrossRef CAS PubMed.
- E. L. Sievers, Expert Opin. Biol. Ther., 2001, 1, 893–901 CrossRef CAS PubMed.
- J. Katz, J. E. Janik and A. Younes, Clin. Cancer Res., 2011, 17, 6428–6436 CrossRef CAS PubMed.
- H. S. Olcott and H. Fraenkel-Conrat, Chem. Rev., 1947, 41, 151–197 CrossRef CAS PubMed.
- O. Boutureira and G. J. Bernardes, Chem. Rev., 2015, 115, 2174–2195 CrossRef CAS PubMed.
- J. M. Chalker, G. J. Bernardes, Y. A. Lin and B. G. Davis, Chem. – Asian J., 2009, 4, 630–640 CrossRef CAS PubMed.
- C. D. Spicer and B. G. Davis, Nat. Commun., 2014, 1, 4740 CrossRef PubMed.
- E. M. Sletten and C. R. Bertozzi, Angew. Chem., Int. Ed., 2009, 48, 6974–6998 CrossRef CAS PubMed.
- E. A. Hoyt, P. M. Cal, B. L. Oliveira and G. J. Bernardes, Nat. Rev. Chem., 2019, 3, 147–171 CrossRef CAS.
- E. Basle, N. Joubert and M. Pucheault, Chem. Biol., 2010, 17, 213–227 CrossRef CAS PubMed.
- N. Stephanopoulos and M. B. Francis, Nat. Chem. Biol., 2011, 7, 876–884 CrossRef CAS PubMed.
- J. Rademann, Angew. Chem., Int. Ed., 2004, 43, 4554–4556 CrossRef CAS PubMed.
- N. H. Fischer, M. T. Oliveira and F. Diness, Biomater. Sci., 2023, 11, 719–748 RSC.
- S. B. Gunnoo and A. Madder, ChemBioChem, 2016, 17, 529–553 CrossRef CAS PubMed.
- D. R. Goddard and L. Michaelis, J. Biol. Chem., 1935, 112, 361–371 CrossRef CAS.
- N. Lundell and T. Schreitmüller, Anal. Biochem., 1999, 266, 31–47 CrossRef CAS PubMed.
- D. Cavallini, B. Mondovì and C. de Marco, Biochim. Biophys. Acta, 1955, 18, 122–130 CrossRef CAS PubMed.
- C. E. Hopkins, G. Hernandez, J. P. Lee and D. R. Tolan, Arch. Biochem. Biophys., 2005, 443, 1–10 CrossRef CAS PubMed.
- W. Jiskoot and D. J. A. Crommelin, Biophysical and Biochemical Characteristics of Therapeutic Proteins, Springer, 2019, pp. 19–32 Search PubMed.
- A. Chowdhury, V. Gour, B. K. Das, S. Chatterjee and A. Bandyopadhyay, Org. Lett., 2023, 25, 1280–1284 CrossRef CAS PubMed.
- M. V. Trivedi, J. S. Laurence and T. J. Siahaan, Curr. Protein Pept. Sci., 2009, 10, 614–625 CrossRef CAS PubMed.
- A. Fontana, E. Scoffone and C. A. Benassi, Biochemistry, 1968, 7, 980–986 CrossRef CAS PubMed.
- D. P. Gamblin, P. Garnier, S. van Kasteren, N. J. Oldham, A. J. Fairbanks and B. G. Davis, Angew. Chem., Int. Ed., 2004, 43, 828–833 CrossRef CAS PubMed.
- S. I. van Kasteren, H. B. Kramer, H. H. Jensen, S. J. Campbell, J. Kirkpatrick, N. J. Oldham, D. C. Anthony and B. G. Davis, Nature, 2007, 446, 1105–1109 CrossRef CAS PubMed.
- Y. Zhu and W. A. van der Donk, Org. Lett., 2001, 3, 1189–1192 CrossRef CAS PubMed.
- G. J. L. Bernardes, J. M. Chalker, J. C. Errey and B. G. Davis, J. Am. Chem. Soc., 2008, 130, 5052–5053 CrossRef CAS PubMed.
- M. D. Nolan and E. M. Scanlan, Front. Chem., 2020, 8, 583272 CrossRef CAS PubMed.
- M. E. B. Smith, F. F. Schumacher, C. P. Ryan, L. M. Tedaldi, D. Papaioannou, G. Waksman, S. Caddick and J. R. Baker, J. Am. Chem. Soc., 2010, 132, 1960–1965 CrossRef CAS PubMed.
- P. Moody, M. E. Smith, C. P. Ryan, V. Chudasama, J. R. Baker, J. Molloy and S. Caddick, ChemBioChem, 2012, 13, 39–41 CrossRef CAS PubMed.
- L. M. Tedaldi, M. E. B. Smith, R. I. Nathani and J. R. Baker, Chem. Commun., 2009, 6583–6585 RSC.
- Y. A. Lin, J. M. Chalker, N. Floyd, G. J. L. Bernardes and B. G. Davis, J. Am. Chem. Soc., 2008, 130, 9642–9643 CrossRef CAS PubMed.
- A. On-Yee Chan, J. Lui-Lui Tsai, V. Kar-Yan Lo, G.-L. Li, M.-K. Wong and C.-M. Che, Chem. Commun., 2013, 49, 1428 RSC.
- D. Crich, V. Krishnamurthy and T. K. Hutton, J. Am. Chem. Soc., 2006, 128, 2544–2545 CrossRef CAS PubMed.
- T. Schlatzer, J. Kriegesmann, H. Schröder, M. Trobe, C. Lembacher-Fadum, S. Santner, A. V. Kravchuk, C. F. W. Becker and R. Breinbauer, J. Am. Chem. Soc., 2019, 141, 14931–14937 CrossRef CAS PubMed.
- N. C. Reddy, R. Molla, P. N. Joshi, S. T. K., I. Basu, J. Kawadkar, N. Kalra, R. K. Mishra, S. Chakrabarty, S. Shukla and V. Rai, Nat. Commun., 2022, 13, 6038 CrossRef CAS PubMed.
- G. T. Hermanson, Bioconjugate techniques, Academic press, 2013 Search PubMed.
- A. Michael, J. Am. Chem. Soc., 1887, 9, 115 Search PubMed.
- D. P. Nair, M. Podgorski, S. Chatani, T. Gong, W. Xi, C. R. Fenoli and C. N. Bowman, Chem. Mater., 2014, 26, 724–744 CrossRef CAS.
- P. Wadhwa, A. Kharbanda and A. Sharma, Asian J. Org. Chem., 2018, 7, 634–661 CrossRef CAS.
- A. K. Sinha and D. Equbal, Asian J. Org. Chem., 2019, 8, 32–47 CrossRef CAS.
- D. Berne, V. Ladmiral, E. Leclerc and S. Caillol, Polymers, 2022, 14, 4457 CrossRef CAS PubMed.
- A. M. Costa, L. Bosch, E. Petit and J. Vilarrasa, J. Org. Chem., 2021, 86, 7107–7118 CrossRef CAS PubMed.
- G.-Z. Li, R. K. Randev, A. H. Soeriyadi, G. Rees, C. Boyer, Z. Tong, T. P. Davis, C. Remzi Becer and D. M. Haddleton, Polym. Chem., 2010, 1, 1196–1204 RSC.
- S. Chatani, D. P. Nair and C. N. Bowman, Polym. Chem., 2013, 4, 1048–1055 RSC.
- R. B. Roseli, A. B. Keto and E. H. Krenske, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2022, e1636 Search PubMed.
- J. W. Chan, C. E. Hoyle, A. B. Lowe and M. Bowman, Macromolecules, 2010, 43, 6381–6388 CrossRef CAS.
- I. M. Serafimova, M. A. Pufall, S. Krishnan, K. Duda, M. S. Cohen, R. L. Maglathlin, J. M. McFarland, R. M. Miller, M. Frödin and J. Taunton, Nat. Chem. Biol., 2012, 8, 471–476 CrossRef CAS PubMed.
- P. A. Jackson, J. C. Widen, D. A. Harki and K. M. Brummond, J. Med. Chem., 2017, 60, 839–885 CrossRef CAS PubMed.
- J. J. Reddick, J. Cheng and W. R. Roush, Org. Lett., 2003, 5, 1967–1970 CrossRef CAS PubMed.
- E. H. Krenske, R. C. Petter and K. N. Houk, J. Org. Chem., 2016, 81, 11726–11733 CrossRef CAS PubMed.
- J. M. Bradshaw, J. M. McFarland, V. O. Paavilainen, A. Bisconte, D. Tam, V. T. Phan, S. Romanov, D. Finkle, J. Shu, V. Patel, T. Ton, X. Li, D. G. Loughhead, P. A. Nunn, D. E. Karr, M. E. Gerritsen, J. O. Funk, T. D. Owens, E. Verner, K. A. Brameld, R. J. Hill, D. M. Goldstein and J. Taunton, Nat. Chem. Biol., 2015, 11, 525–531 CrossRef CAS PubMed.
- M. H. Johansson, Mini-Rev. Med. Chem., 2012, 12, 1330–1344 CAS.
- S. Krishnan, R. M. Miller, B. Tian, R. D. Mullins, M. P. Jacobson and J. Taunton, J. Am. Chem. Soc., 2014, 136, 12624–12630 CrossRef CAS PubMed.
- L. T. T. Nguyen, M. T. Gokmen and F. E. Du Prez, Polym. Chem., 2013, 4, 5527–5536 RSC.
- F.-J. Chen and J. Gao, Chem. – Eur. J., 2022, 28, e202201843 CAS.
- A. Abbas, B. Xing and T.-P. Loh, Angew. Chem., Int. Ed., 2014, 53, 7491–7494 CrossRef CAS PubMed.
- A. J. Cameron, P. W. R. Harris and M. A. Brimble, Angew. Chem., Int. Ed., 2020, 59, 18054–18061 CrossRef CAS PubMed.
- E. Petit, L. Bosch, A. M. Costa and J. Vilarrasa, J. Org. Chem., 2019, 84, 11170–11176 CrossRef CAS PubMed.
- B. Bernardim, P. M. S. D. Cal, M. J. Matos, B. L. Oliveira, N. Martínez-Sáez, I. S. Albuquerque, E. Perkins, F. Corzana, A. C. B. Burtoloso, G. Jiménez-Osés and G. J. L. Bernardes, Nat. Commun., 2016, 7, 13128 CrossRef CAS PubMed.
- B. Bernardim, M. J. Matos, X. Ferhati, I. Compañón, A. Guerreiro, P. Akkapeddi, A. C. B. Burtoloso, G. Jiménez-Osés, F. Corzana and G. J. L. Bernardes, Nat. Protoc., 2019, 14, 86–99 CrossRef CAS PubMed.
- M. J. Matos, C. D. Navo, T. Hakala, X. Ferhati, A. Guerreiro, D. Hartmann, B. Bernardim, K. L. Saar, I. Compañón, F. Corzana and T. P. Knowles, Angew. Chem., Int. Ed., 2019, 58, 6640–6644 CrossRef CAS PubMed.
- S. Ariyasu, H. Hayashi, B. Xing and S. Chiba, Bioconjugate Chem., 2017, 28, 897–902 CrossRef CAS PubMed.
- N. J. Smith, K. Rohlfing, L. A. Sawicki, P. M. Kharkar, S. J. Boyd, A. M. Kloxin and J. M. Fox, Org. Biomol. Chem., 2018, 16, 2164–2169 RSC.
- J.-R. Deng, S.-F. Chung, A. S.-L. Leung, W.-M. Yip, B. Yang, M.-C. Choi, J.-F. Cui, K. K.-Y. Kung, Z. Zhang, K.-W. Lo, Y.-C. Leung and M.-K. Wong, Commun. Chem., 2019, 2, 1–10 CrossRef CAS.
- B. Bernardim, L. Dunsmore, H. Li, B. Hocking, R. Nuñez-Franco, C. D. Navo, G. Jiménez-Osés, A. C. B. Burtoloso and G. J. L. Bernardes, Bioconjugate Chem., 2020, 31, 1604–1610 CrossRef CAS PubMed.
- E. D. Geyter, E. Antonatou, D. Kalaitzakis, S. Smolen, A. Iyer, L. Tack, E. Ongenae, G. Vassilikogiannakis and A. Madder, Chem. Sci., 2021, 12, 5246–5252 RSC.
- J. Yu, X. Yang, Y. Sun and Z. Yin, Angew. Chem., 2018, 130, 11772–11776 CrossRef.
- Y. Zhang, X. Zhou, Y. Xie, M. M. Greenberg, Z. Xi and C. Zhou, J. Am. Chem. Soc., 2017, 139, 6146–6151 CrossRef CAS PubMed.
- P. Evans and R. J. K. Taylor, J. Sulfur Chem., 2005, 26, 481–497 CrossRef CAS.
- M. S. Masri and M. Friedman, J. Protein Chem., 1988, 7, 49–54 CrossRef CAS PubMed.
- E. Friedmann, Biochim. Biophys. Acta, 1952, 9, 65–75 CrossRef CAS PubMed.
- J. E. Moore and W. H. Ward, J. Am. Chem. Soc., 1956, 78, 2414–2418 CrossRef CAS.
- S. D. Fontaine, R. Reid, L. Robinson, G. W. Ashley and D. V. Santi, Bioconjugate Chem., 2015, 26, 145–152 CrossRef CAS PubMed.
- A. Witter and H. Tuppy, Biochim. Biophys. Acta, 1960, 45, 429–442 CrossRef CAS PubMed.
- R. I. Nathani, V. Chudasama, C. P. Ryan, P. R. Moody, R. E. Morgan, R. J. Fitzmaurice, M. E. B. Smith, J. R. Baker and S. Caddick, Org. Biomol. Chem., 2013, 11, 2408–2411 RSC.
- B.-Q. Shen, K. Xu, L. Liu, H. Raab, S. Bhakta, M. Kenrick, K. L. Parsons-Reponte, J. Tien, S.-F. Yu, E. Mai, D. Li, J. Tibbitts, J. Baudys, O. M. Saad, S. J. Scales, P. J. McDonald, P. E. Hass, C. Eigenbrot, T. Nguyen, W. A. Solis, R. N. Fuji, K. M. Flagella, D. Patel, S. D. Spencer, L. A. Khawli, A. Ebens, W. L. Wong, R. Vandlen, S. Kaur, M. X. Sliwkowski, R. H. Scheller, P. Polakis and J. R. Junutula, Nat. Biotechnol., 2012, 30, 184–189 CrossRef CAS PubMed.
- M. Ahangarpour, I. Kavianinia, P. A. Hume, P. W. R. Harris and M. A. Brimble, J. Am. Chem. Soc., 2022, 144, 13652–13662 CrossRef CAS PubMed.
- M. A. Kasper, M. Glanz, A. Stengl, M. Penkert, S. Klenk, T. Sauer, D. Schumacher, J. Helma, E. Krause, M. C. Cardoso, H. Leonhardt and C. P. R. Hackenberger, Angew. Chem., Int. Ed., 2019, 58(34), 11625–11630 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2023 |