
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGenerating water/MeOH-soluble and luminescent polymers by grafting 2,6-bis(1,2,3-triazol-4-yl)pyridine (btp) ligands onto a poly(ethylene-alt-maleic anhydride) polymer and cross-linking with terbium(III)†
Isabel N.
Hegarty
a,
Adam F.
Henwood
ab,
Samuel J.
Bradberry
*a and
Thorfinnur
Gunnlaugsson
 *ab
*ab
aSchool of Chemistry and Trinity Biomedical Sciences Institute (TBSI), Trinity College Dublin, The University of Dublin, Dublin 2, Ireland. E-mail: gunnlaut@tcd.ie; bradberry.samuel@gmail.com
bSynthesis and Solid-State Pharmaceutical Centre (SSPC), School of Chemistry, Trinity College Dublin, The University of Dublin, Dublin 2, Ireland
First published on 25th January 2023
Abstract
The synthesis of two new polymers made from P(E-alt-MA) (poly(ethylene-alt-maleic anhydride) and possessing 2,6-bis(1,2,3-triazol-4-yl)pyridine (btp) ligand side chains in 3 and 6 mol%, respectively (P1 and P2, respectively) is described. These polymers were shown to be soluble in MeOH solution and, in the case of P1, also in water, while P2 needed prolonged heating to enable water dissolution. Btp ligands are known for coordinating both d- and f-metal ions and so, herein, we demonstrate by using both UV-Vis absorption, fluorescence emission, as well as time-gated phosphorescence spectroscopies, that both P1 and P2 can bind to Tb(III) ions to give rise to luminescent polymers. From the analysis of the titration data, which demonstrated large changes in the emission intensity properties of the polymer upon Tb(III) binding (ground state changes were also clearly observed, with the absorption being red-shifted at lower energy), we show that the dominant stoichiometry in solution is 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 (M:L; Tb(III):btp ratio) which implies that two btp ligands from the polymer background are able to crosslink through lanthanide coordination and that the backbone of the polymer is very likely to aid in coordinating the ions.
:2 (M:L; Tb(III):btp ratio) which implies that two btp ligands from the polymer background are able to crosslink through lanthanide coordination and that the backbone of the polymer is very likely to aid in coordinating the ions.
Introduction
The development of functional and robust materials with unique properties is highly topical within the areas of supramolecular and materials chemistries.1–3 As part of such developments the formation and study of responsive luminescent and/or supramolecular polymers is gaining significant interest.4–6 For over a decade now we have been reporting examples of both all-organic and lanthanide-based polymers and soft-materials (such as self-assembled gels and cross-linked supramolecular polymers) formed by using lanthanide coordinating ligands such as cyclens (1,4,7,10-tetraazacyclododecanes) and self-recognising, hydrogen bond-driven self-assembling BTAs (benzene-1,3,5-tricarboxamides).7,8 In particular, we have been interested in probing the evolution of the photophysics of such systems upon coordination to various lanthanide ions,9 especially as many lanthanides possess unique and desirable luminescence properties such as long-lived excited states and emissions occurring at long wavelengths with line-like features.10 Such properties have applications in various fields including optoelectronics,11 medicine,12 chemosensing,13 counterfeiting,14etc. Furthermore, depending on the coordination properties of the ligands employed, they can also be used as cross-linking agents within supramolecular polymers, rendering such polymers with additional properties.15The 2,6-bis(1,2,3-triazol-4-yl)pyridine (btp) structure is an attractive ligand that has been used in various areas of research.16 In particular, the btp has been employed in coordination chemistry, due to its similarity to the well-known terpy ligand.17 Recently, Byrne et al. utilised this ligand upon complexation to Ru(II) in antifouling18 as well as in the formation of chiral luminescent lanthanide complexes that give rise to circularly polarised luminescence (CPL) from the lanthanide centre.19 In addition to its favourable metal ion coordination properties, the btp motif has been shown to form dimers via self-templation. This was recently demonstrated by our research group where we exploited the self-templating features of both chiral as well as achiral olefin btp precursors to synthesise homocircuit [2]catenanes and pseudo-rotaxanes using ring closing metathesis (RCM) reactions.20,21 We have also been interested in the use of btp complexes in the formation of responsive polymers, and demonstrated that such complexes can be incorporated into HEMA polymers.22 However, since these complexes are doped non-covalently into the polymer hosts, they are prone to leaching from the material during solvent swelling processes. With this in mind, we recently developed btp monomers possessing a methacrylamide unit which could be co-polymerised with 2-hydroxyethyl methacrylate (HEMA), methylmethacrylate (MMA) and ethylene glycol dimethacrylate (EGDMA) to give polymers containing covalently attached btp units.23 This gave rise to a hard ‘plastic’ like material that was luminescent upon treatment with lanthanide ions such as Eu(III) and Tb(III). While being highly successful, unfortunately this method sometimes resulted in the formation of films that lacked homogeneity upon treatment with the lanthanides. With this in mind, we set out to develop poly(ethylene-alt-maleic anhydride) [P(E-alt-MA)]24,25 polymers as the basis for new, alternative, btp-containing polymers that were soluble in competitive media and would give more consistent homogeneity within their structures upon doping with lanthanides.
Results and discussion
Design, synthesis and NMR characterisations of P1 and P2
The P(E-alt-MA) polymer is water soluble in moderate alkaline environments as, upon polymerisation, each anhydride moiety is converted into two carboxylate groups.26 An important application of this polymer is in rendering water-soluble compounds insoluble for extraction from aqueous environments.27 The anhydride functionality of this polymer has been shown to react readily with even moderate nucleophiles, undergoing a variety of reactions including amidation, esterification, hydrolysis, thioesterification, and imidization, generating a functionalised water-insoluble polymer.28,29 In our previous work within this area of research, we developed the btp ligand 1 as an intermediate in the formation of the methacryloyl amide based btp ligands.23 This final structure was then incorporated by co-polymerisation into a hydrogel polymer using HEMA, MMA and EGDMA.23 Here, however, we generated the polymers P1 and P2 in a single step, with a low loading of compound 1, from P(E-alt-MA), Fig. 1.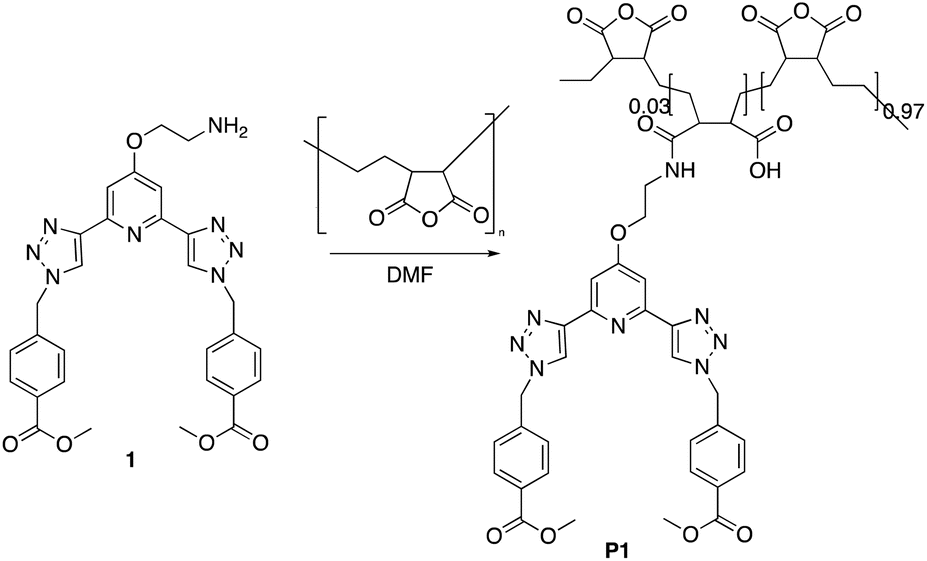 | ||
| Fig. 1 The polymer P1 formed by grafting the btp ligand 1 onto poly(ethylene-alt-maleic anhydride) [P(E-alt-MA)] (P2 was formed in a similar manner). | ||
The synthesis of P1 was achieved by first dissolving P(E-alt-MA) in anhydrous DMF and, to this solution, 3 mol% of ligand 1 in DMF was added dropwise. The reaction was complete after one hour, and the P1 polymer was isolated by dropping the reaction mixture into 0.5 M HCl. This caused the formation of a white precipitate, which was isolated by filtration, followed by washing with water to yield the pure product. This procedure was repeated using 6 mol% of ligand 1 to generate polymer P2. The resulting polymers were characterised by NMR spectroscopies. The 1H NMR of P1 is shown in Fig. 2, while the 13C NMR of 1 (top) and P1 (bottom) are shown in Fig. 3.
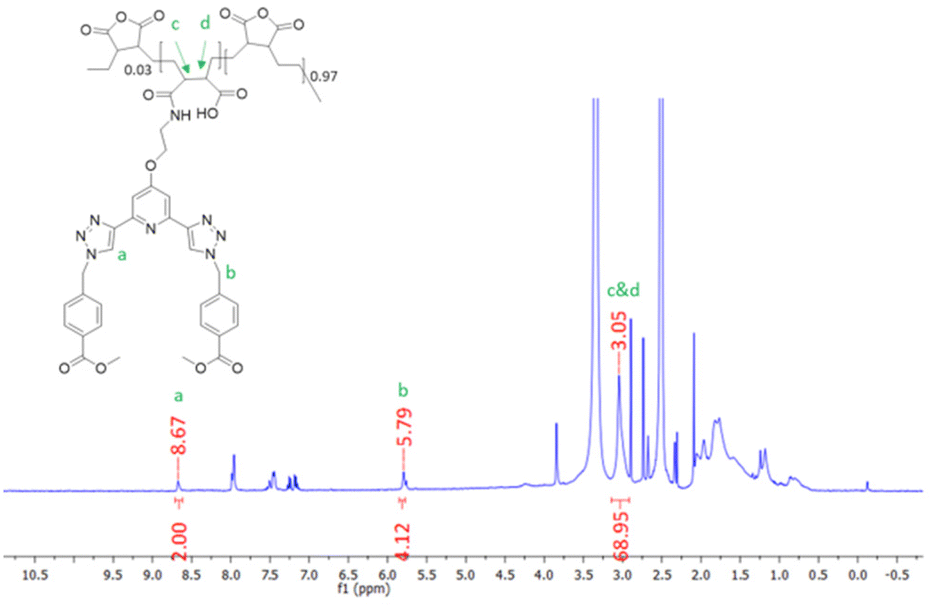 | ||
| Fig. 2 The 1H NMR spectrum (600 MHz, DMSO-d6) of P1. | ||
 | ||
| Fig. 3 The 13C NMR spectra (600 MHz, DMSO-d6) of 1 (top) and P1 (bottom). The resonances assigned to the ligand are also clearly visible. | ||
As demonstrated in Fig. 2, 1H NMR (600 MHz, DMSO-d6) studies were used to confirm the formation of the grafted polymer P1. The resonance corresponding to the triazole protons occurs at 8.67 ppm, which is at a similar resonance to the analogous proton in the free ligand 1. The signal at 5.79 ppm can be assigned to the CH2 protons of ligand 1. The signal upfield at 3.05 ppm corresponds to protons c and d of the polymer backbone, while the remaining protons of the polymer chain resonate as a broad signal at approximately 0.85–1.96 ppm. Integration of the 1H NMR peaks confirmed an approximately 3% loading of compound 1 onto the polymer backbone.
As shown in Fig. 3, 13C NMR spectroscopy also confirmed the formation of the desired product P1. The appearance of two signals at 175.1 and 173.6 ppm in the polymer 13C NMR are indicative of the carbonyl carbons of the polymer backbone while the carbonyl carbon associated with the arms of ligand 1 is located at 165.9 ppm. Additional resonances are also visible in the base line, that are more deshielded within the polymer structure. The formation of P2 was also confirmed by NMR spectroscopies (see ESI†).
Solution studies of P1 and P2
As mentioned above, the objective herein was to develop simple, water-soluble, lanthanide-based polymers. Although P2, which was soluble in CH3OH, could only dissolve in water after prolonged heating at 60 °C for several days, it was gratifying to find that, upon solvation, polymer P1 was found to readily dissolve in both CH3OH and water (cf. as demonstrated with a picture shown as insert in Fig. 4). Addition of the polymers to water gave a colourless solution in the case of P1 and a more viscous, and white suspension in the case of P2. By contrast, both polymers readily dissolved in CH3OH, generated a colourless solutions (see inset in Fig. S2 ESI†).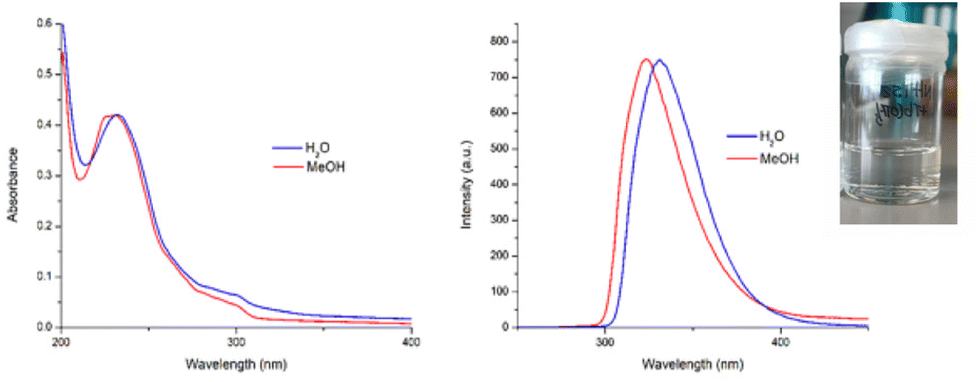 | ||
| Fig. 4 (Left) Overlaid absorption spectra of P1 in H2O and CH3OH and (right) fluorescence spectra of P1 in H2O and CH3OH recorded at a concentration of approximately c ∼ 6 × 10−6 M. Inset: A photograph of P1 dissolved in H2O demonstrating the transparent nature of the solution. | ||
The UV-visible absorption and fluorescence emission spectra of P1 and P2 were recorded in both H2O and CH3OH and are shown in Fig. 4. In all cases there were two main bands observed in the UV-visible absorption spectra at λmax = 231 nm and λ = 298 nm. As would be expected, the features of the absorption spectra of both polymers resemble that of the btp ligands with only a slight loss of resolution in the band at 298 nm, confirming that the P(E-alt-MA) polymer does not significantly interfere with the absorption profile of compound 1. This provides a useful spectroscopic handle for monitoring the evolution of the self-assembly of our polymers by UV-visible absorption spectroscopy (vide infra). Upon excitation of the btp within the polymers (λexc = 240 nm) ligand-centred fluorescence was observed (λmax = 323 nm) which is characteristic of the photophysical behaviour of many btp ligands.30
Solution studies of P1 and P2 in the presence of Tb(III)
In our previous works we have demonstrated the efficacy of btp ligands in binding lanthanide ions and giving rise to highly luminescent complexes.31 This is particularly the case with Tb(III) ions whose resultant complexes exhibit photoluminescent quantum yields that are almost an order of magnitude higher than those measured for analogous Eu(III) complexes in organic solutions. This has been ascribed to well-matched energy levels between the triplet state of the btp and Tb(III). Thus, with this in mind, we next investigated the use of Tb(III) ions for cross-linking polymer chains of P1 and P2: by titrating CH3OH solutions of these polymers with Tb(III), we could monitor changes in both the ground and the excited states of these polymers as they undergo cross-linking. CH3OH was chosen to minimise any quenching of the Tb(III) excited state from water oscillators. The solutions of P1 and P2 were prepared so that the overall concentrations of the btp units within the solutions were 1 × 10−5 M. These solutions were then titrated against specific aliquots of Tb(CF3SO3)3, and the changes to the UV-visible absorption, ligand fluorescence, and delayed lanthanide luminescence were analysed.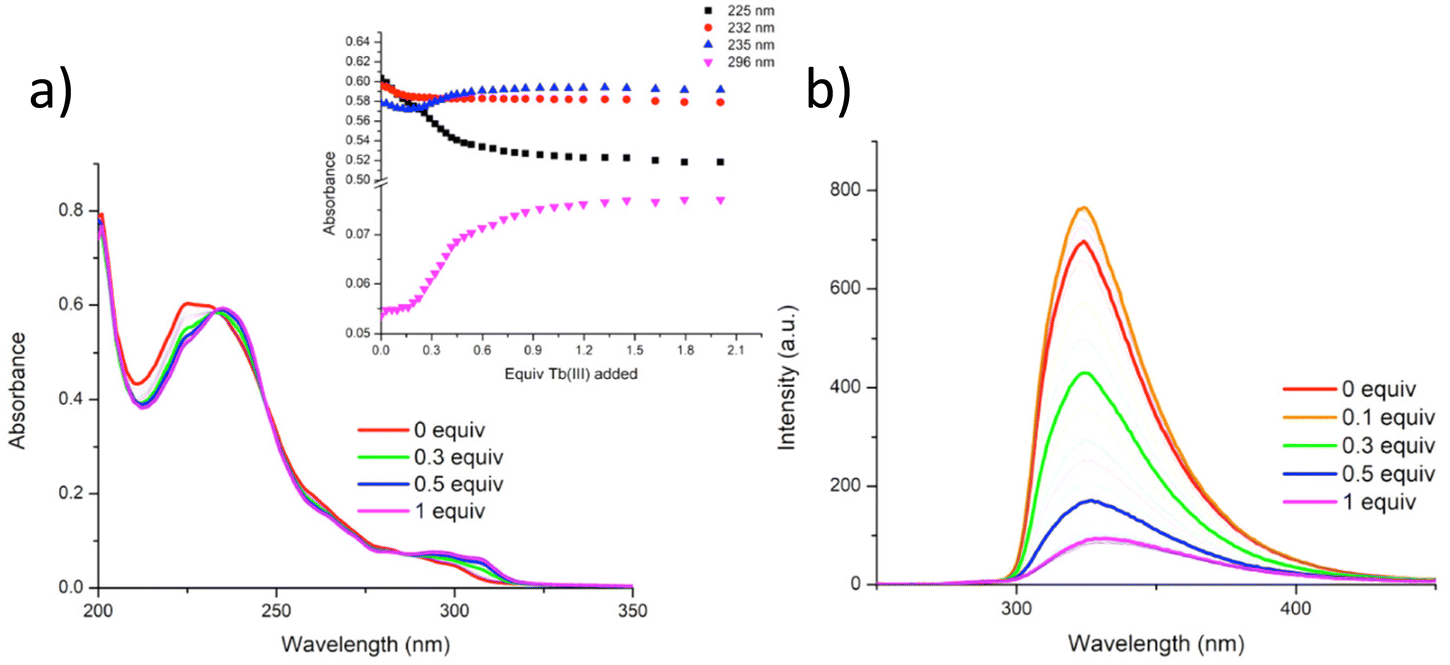 | ||
| Fig. 5 (a) The overall changes in the UV-vis absorption spectra, and (b) fluorescence spectra (λexc = 237 nm) upon titrating P1 (1 1 × 10−5 M within P1) against Tb(CF3SO3)3 (0 → 3 equiv.) in CH3OH at RT. Inset (a): corresponding experimental binding isotherms of absorbance at λ = 325, 232, 235 and 295 nm. | ||
In a similar manner the changes in the absorption spectra of P2 upon addition of Tb(CF3SO3)3 were also monitored. As for P1, the absorption spectrum of P2 consisted of two main bands located at 231 nm and 296 nm, respectively (see ESI†), and three isosbestic points were observed at 233 nm, 246 nm and 287 nm, respectively. Upon addition of Tb(III), the higher energy band experienced a redshift and concomitant decrease in absorbance up to the addition of 0.5 equivalents of Tb(III). Subsequently, this band increased upon addition of further 0.5 equivalents of Tb(III). There were also changes at longer energy with the band centred at 296 nm experiencing a hyperchromic shift (see ESI†). These changes also occur up to the addition of 0.5 equivalents of Tb(III). From the changes to the UV-visible absorption and luminescence spectra, it can be concluded that multiple species coexist in solution. As the most significant changes to the spectra occur up to the addition of 0.5 equivalents of Tb(III), it is likely that the more emissive M:L3 and/or M:L2 species are the ones generated. The speciation above 0.5 equivalents does not appear to undergo changes upon the addition of excess Tb(III), and thus suggests there is unlikely to be any dissociation to the less emissive ML species.‡
The fluorescence emission properties of P2 were also probed. Upon excitation of P2 at 237 nm, analogous ligand-centred fluorescence emission to P1 was observed, localised at 335 nm (see ESI†) and, as in the case of P1, addition of Tb(III) efficiently quenched the ligand fluorescence due to metal coordination to the btp ligand, ultimately being reduced to 87% of the original emission intensity upon the addition of 0.5 equivalents of Tb(III).
 | ||
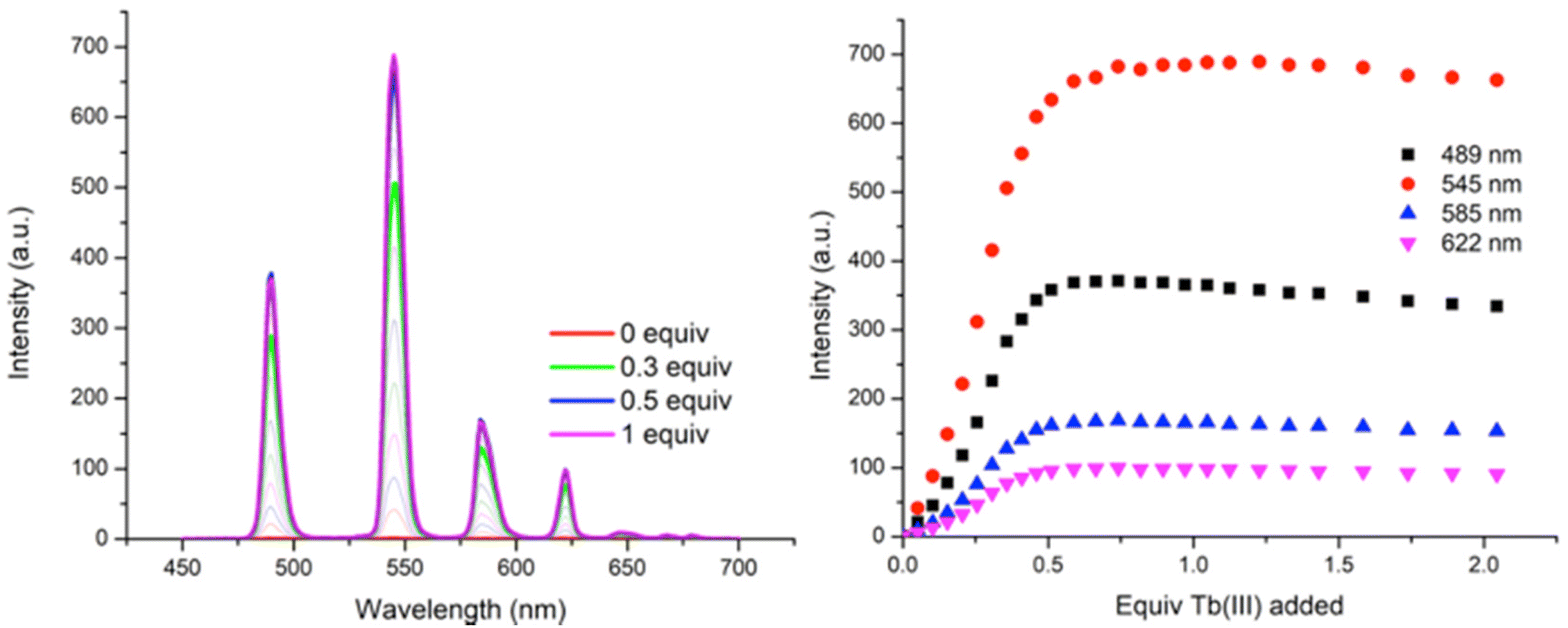
| Fig. 6 The overall changes in the (left) Tb(III)-centred phosphorescence spectra (λexc = 237 nm) and (right) binding isotherms at λ = 490, 545, 585 and 622 nm upon titrating P1 (1 × 10−5 M) against Tb(CF3SO3)3 (0 → 3 equiv.) in CH3OH at RT. The equivalents added are in respect to the ligand concentration. | ||
 | ||
| Fig. 7 The overall changes in the (left) Tb(III)-centred phosphorescence spectra and (right) binding isotherms at λ = 490, 545, 585, 622, 647, 667, and 656 nm upon titrating P2 (1 × 10−5 M) against Tb(CF3SO3)3 (0 → 3 equiv.) in CH3OH at RT. | ||
As can be seen in Fig. 6, titrating P1 with Tb(CF3SO3)3 gives rise to Tb(III) luminescence that is progressively enhanced with increasing equivalents of Tb(III). A gradual enhancement was observed in this luminescence in going from 0 → 0.3 equivalents of Tb(III), after which a more rapid increase was observed up to approximately 0.5 equivalents. The characteristic Tb(III)-centred emission bands were observed at λ = 490, 545, 585, 622, 647, 667, and 675 nm, corresponding to radiative deactivation from the 5D4 → 7FJ states. Subsequent additions of Tb(III), from 0.5 to 3.0 equivalents, resulted in no noticeable changes in the luminescence, indicating the formation of a stable Tb(III) complex with a 1:2 (M:L) stoichiometry. This species is also the most emissive species in solution as, upon further addition of Tb(III), no noticeable changes in the emission intensity were observed. However, btp ligands are known to give rise to complexes with 1:3 (M:L) stoichiometries and analysis of the titration isotherm in Fig. 6 shows that at that point, the emission is ca. 60% switched on, being fully switched on at the 1:2 stoichiometry, with no changes in the emission intensity at higher stochiometries. Analysis of the overall changes show that the initially slow onset of the emission can be explained by the presence of Tb(III) species whose coordination spheres are made up primarily of bound solvent molecules which can deactivate the excited state through their O–H oscillations. This phenomenon is possibly more pronounced for polymer P1 –with its lower loading of btp ligands – as it is possible that, in the absence of btp moieties close to the metal ion, coordination to the Tb(III) ion could occur via other coordination moieties competing with coordinating solvent molecules, such as the carbonyl groups contained within the p(E-alt-MA) polymer itself. Therefore, the speciation in solution is necessarily complex, and random, with coordination to the metal ions during the titrations arising from a potential combination of btp units, polymer backbone and solvent molecules. However, it is important to note that at higher Tb(III) loadings the emission is not being appreciably enhanced or quenched. This feature means that cannot conclusively determine if additional ions are binding to the polymer backbone alone as such species are, in all likelihood, non-emissive as these sites do not possess an antenna that can effectively populate the Tb(III) excited state.
This effect of higher loading of btp ligands within the polymer does indeed seem to be important in affecting the emission properties, as upon measuring the delayed Tb(III)-centred luminescence arising from P2 upon titrating with Tb(CF3SO3)3, the aforementioned ‘slow onset’ of the Tb(III) emission was absent. The overall changes in the Tb(III) emission of P2 upon excitation at 237 nm is shown in Fig. 7. A gradual enhancement in the Tb(III) luminescence was observed up to the addition of 0.5 equivalents of the metal, with characteristic Tb(III)-centred emission transitions appearing at λ = 490, 545, 585, 622, 647, 667, and 656 nm. Subsequent additions of Tb(III) did not result in any change in the luminescence intensity, which indicates the formation of stable emissive species in solution.
While the concentration of compound 1 was the same within these two samples (1 × 10−5 M), and hence, for each titration, the higher number of grafted btp ligands at the backbone of the polymer P(E-alt-MA) in P2 would increasing the probability of forming cross-linked moieties (e.g. more of bridged ML2 points within the polymer). Hence, the likelihood of the existence of great numbers of crosslinking structures exists within P2 than P1. This indicates that subtle changes to the loading levels of btp units within the p(E-alt-MA) polymers can have profound effects on the subsequent self-assembly of the polymer with metal ions. With this in mind, and to gain further insights into the M:L stoichiometries of the polymer assemblies, we carried out studies on mixtures of Tb(III) and ligand 1. The results (see ESI†) indeed showed that the 2:1 complex was the only observed signal in the HRMS, with the calculated and experimental isotopic distribution patterns matching a molecular species of the formula [Tb(1)2](CF3SO3)2+. While this does not exclude the formation of the 1:3 stoichiometry, as discussed above, it does seem to indicate that in the case of 1 the 1:2 stoichiometry is the most likely and/or the most stable structure to form upon self-assembly with Tb(III), supporting our observations from the ground and excited state measurements for P1 and P2. Although traditionally the most emissive species possess the 1:3 stoichiometry, we have nevertheless observed efficient Tb(III)-based luminescence from both polymers upon titrating Tb(III). In addition, this 1:2 M:L stoichiometry is more than enough to structurally modify the polymer materials with the lanthanide ions acting to crosslink neighbouring polymer chains while the coordination sphere about the Tb(III) ions being completed by solvent molecules and/or coordination by carbonyl moieties on the polymer backbones. Accordingly, the presence of multiple different, competing coordinating species means that such crosslinking is likely to be random, but nevertheless would be expected to modulate the mechanical properties of such systems. Such studies are currently on going in our laboratory.
Conclusions
In this study, we have developed two new CH3OH and water-soluble polymers P1 and P2, generated by modifying the polymer backbone of P(E-alt-MA) with the btp ligand 1. By using UV-visible absorption and emission spectroscopies, we showed that these polymers could complex Tb(III) and form highly luminescent, cross-linked polymer structures. Analysis of the spectroscopic changes clearly demonstrate the formation of stable complexes exhibiting 1:2 (M:L) stoichiometries between the btp-based polymers (P1 and P2) and Tb(III). However, given the high coordination requirements of the lanthanides, other stoichiometries can also occur. Furthermore, it cannot be ruled out that the backbone of the P(E-alt-MA) polymer itself takes part in the coordination of the ions. However, as no changes in the Tb(III)-centered emission were seen at high ion loadings, if such coordination is occurring, it does not affect the photophysical properties of the polymers. With the view of depicting the coordination environment around the bpt units alone within the polymer, and with the view of showing the three dimensional nature of the crosslinking, we have drawn the three different M:L coordination environments in Fig. 8 in a schematic manner. It is important to note that we show these in the stepwise manner (1:1, 1:2 and 1:3 M:L). However, upon higher loading of the Tb(III) ions within the polymer, the reverse effect can also occur where the ligands in the 1:3 (which is commonly the most stable stoichiometry, with the ligands fulfilling the 9-coordiantion environment of the ion) are stripped off due to increasing Tb(III) concentrations. This we have seen in many of our lanthanide titrations in the past. This normally leads to quenching in the lanthanide luminescence if the displaced ligands are exchanged for O–H oscillators as we discussed above. However, the fact that the emission is not quenched above 0.5 equivalents, would indicated that the lanthanide centre is shielded from such coordination, possibly by the polymer backbone. We are in the process of developing other examples of lanthanide-based polymers and supramolecular polymers that utilise the btp motif.
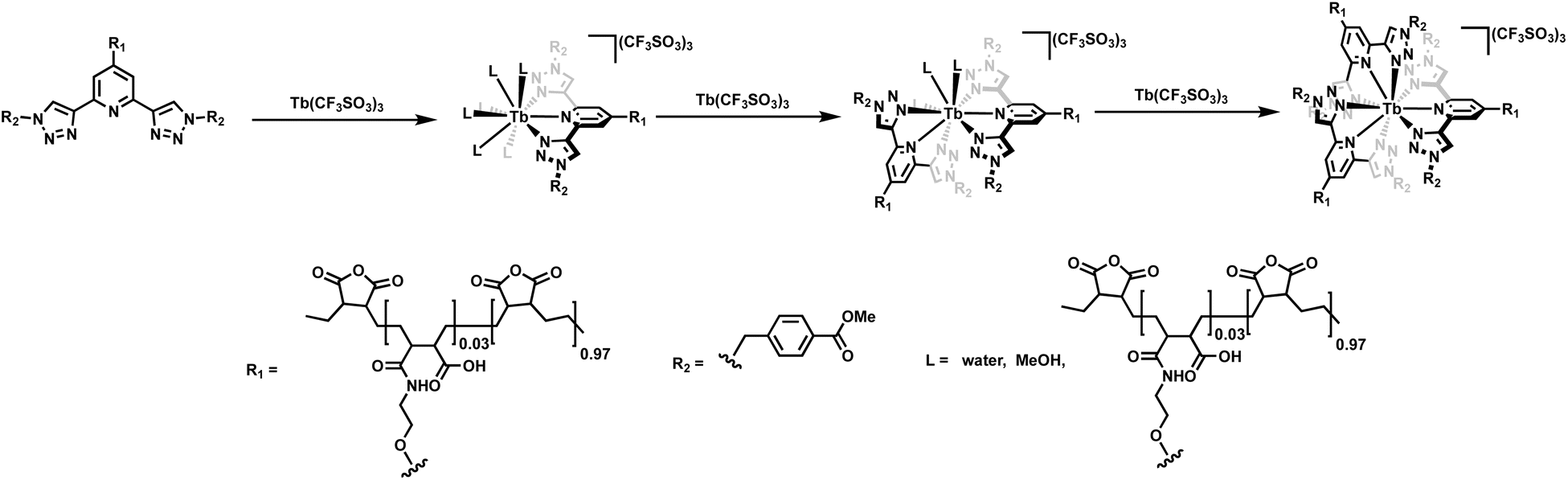 | ||
| Fig. 8 The stepwise formation of the 1:1, 1:2 and 1:3 (M:L) stoichiometries possible with the btp ligands alone within the polymer. | ||
Experimental
Materials and methods
All chemicals and solvents were obtained commercially and used without further purification. All NMR spectra were recorded using a Bruker Avance III 600 NMR spectrometer operating at 400 MHz for 1H NMR and 101 MHz for 13C NMR. Chemical shifts (δ) were referenced relative to the internal solvent signals. The synthesis and characterisation of 1 has been previously reported and is only given here for a reference.23Synthesis of 1
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to thank Science Foundation Ireland (SFI) (SFI PI Award 13/IA/1865 to T. G.) and (the SFI funded) Solid-State Pharmaceutical Centre (SSPC, Research Centres Phase 2: 12/RC/2275_P2) for financial support. We would especially like to thank Drs Gary Hessman, John E. O'Brien and Manuel Ruether for the help with HRMS and NMR studies.References
- (a) P. K. Hashim, J. Bergueiro, E. W. Meijer and T. Aida, Prog. Polym. Sci., 2020, 105, 101250 CrossRef CAS; (b) G. J. Scerri, M. Caruana, N. Agius, G. Agius, T. J. Farrugia, J. C. Spiteri, A. D. Johnson and D. C. Magri, Molecules, 2022, 27, 5939 CrossRef CAS PubMed; (c) T. Gorai, J. I. Lovitt, D. Umadevi, G. McManus and T. Gunnlaugsson, Chem. Sci., 2022, 13, 7805 RSC; (d) Y. Li, D. J. Young and X. J. Loh, Mater. Chem. Front., 2019, 3, 1489 RSC.
- (a) A. J. Savyasachi, O. Kotova, S. Shanmugaraju, S. J. Bradberry, G. M. Ó. Máille and T. Gunnlaugsson, Chem, 2017, 3, 764 CrossRef CAS; (b) M. Coste, E. Suárez-Picado and S. Ulrich, Chem. Sci., 2022, 13, 909 RSC; (c) G. Olivo, G. Capocasa, D. Del Giudice, O. Lanzalunga and S. Di Stefano, Chem. Soc. Rev., 2021, 50, 7681 RSC; (d) E. R. Draper and D. J. Adams, Chem, 2017, 3, 390 CrossRef CAS; (e) E. R. Draper and D. J. Adams, Langmuir, 2019, 35, 6506 CrossRef CAS PubMed.
- (a) J.-F. Lutz, J.-M. Lehn, E. W. Meijer and K. Matyjaszewski, Nat. Rev. Mater., 2016, 1, 16024 CrossRef CAS; (b) Y. Liu, Z. Wang and X. Zhang, Chem. Soc. Rev., 2012, 41, 5922 RSC; (c) A. B. Aletti, S. Blasco, S. J. Aramballi, P. E. Kruger and T. Gunnlaugsson, Chem, 2019, 5, 2617 CrossRef CAS; (d) T. Gunnlaugsson, Nat. Chem., 2016, 8, 6 CrossRef CAS PubMed; (e) Y. T. Shen, C. H. Li, K. C. Chang, S. Y. Chin, H. A. Lin, Y. M. Liu, C. Y. Hung, H. F. Hsu and S. S. Sun, Langmuir, 2009, 25, 8714 CrossRef CAS PubMed; (f) A. D. Lynes, C. S. Hawes, K. Byrne, W. Schmitt and T. Gunnlaugsson, Dalton Trans., 2018, 47, 5259 RSC; (g) J. Kopeček, Biomaterials, 2007, 56, 1078–1109 Search PubMed.
- (a) D. Zhao, J. Yang, Y. Wang and H. Li, Dyes Pigm., 2022, 197, 109864 CrossRef CAS; (b) O. Kotova, S. J. Bradberry, A. J. Savyasachi and T. Gunnlaugsson, Dalton Trans., 2018, 47, 16377 RSC; (c) I. N. Hegarty, H. L. Dalton, A. D. Lynes, B. Haffner, M. E. Möbius, C. S. Hawes and T. Gunnlaugsson, Dalton Trans., 2020, 49, 7364 RSC; (d) S. Wei, Z. Li, W. Lu, H. Liu, J. Zhang, T. Chen and B. Z. Tang, Angew. Chem., Int. Ed., 2021, 60, 8608 CrossRef CAS PubMed; (e) G. C. Le Goff, R. L. Srinivas, W. A. Hill and P. S. Doyle, Eur. Polym. J., 2015, 72, 386 CrossRef CAS PubMed.
- (a) S. Afzala and U. Maitra, Helv. Chim. Acta, 2022, 105, e202100194 Search PubMed; (b) T. Gorai and U. Maitra, Angew. Chem., Int. Ed., 2017, 56, 10730 CrossRef CAS PubMed; (c) R. Laishram and U. Maitra, Chem. Commun., 2022, 58, 3162 RSC; (d) J. C. Yuan, X. L. Fang, L. X. Zhang, G. N. Hong, Y. J. Lin, Q. F. Zheng, Y. Z. Xu, Y. H. Ruan, W. G. Weng, H. P. Xia and G. H. J. Chen, Mater. Chem., 2012, 22, 11515 RSC; (e) B. Yang, H. Zhang, H. Peng, Y. Xu, B. Wu, W. Weng and L. Li, Polym. Chem., 2014, 5, 1945 RSC.
- (a) S. J. Bradberry, A. J. Savyasachi, R. D. Peacock and T. Gunnlaugsson, Faraday Discuss., 2015, 185, 413 RSC; (b) S. J. Bradberry, G. Dee, O. Kotova, C. P. McCoy and T. Gunnlaugsson, Chem. Commun., 2019, 55, 1754 RSC.
- (a) R. Daly, O. Kotova, M. Boese, T. Gunnlaugsson and J. J. Boland, ACS Nano, 2013, 7, 4838 CrossRef CAS PubMed; (b) O. Kotova, R. Daly, C. L. M. dos Santos, P. E. Kruger, J. J. Boland and T. Gunnlaugsson, Inorg. Chem., 2015, 54, 7735 CrossRef CAS; (c) M. Martínez-Calvo, O. Kotova, M. E. Möbius, A. P. Bell, T. McCabe, J. J. Boland and T. Gunnlaugsson, J. Am. Chem. Soc., 2015, 137, 1983 CrossRef PubMed.
- (a) E. M. Surender, S. J. Bradberry, S. A. Bright, C. P. McCoy, D. C. Williams and T. Gunnlaugsson, J. Am. Chem. Soc., 2017, 139, 381 CrossRef CAS PubMed; (b) C. P. McCoy, F. Stomeo, S. E. Plush and T. Gunnlaugsson, Chem. Mater., 2006, 18, 4336 CrossRef CAS.
- O. Kotova, C. O'Reilly, S. T. Barwich, L. E. Mackenzie, A. D. Lynes, A. J. Savyasachi, M. Ruether, R. Pal, M. E. Möbius and T. Gunnlaugsson, Chem, 2022, 8, 1395 CAS.
- (a) D. E. Barry, D. F. Caffrey and T. Gunnlaugsson, Chem. Soc. Rev., 2016, 45, 3244 RSC; (b) T. Gunnlaugsson and F. Stomeo, Org. Biomol. Chem., 2007, 5, 1999 RSC; (c) T. Gunnlaugsson and J. P. Leonard, Chem. Commun., 2005, 25, 3114 RSC.
- H. Xu, Q. Sun, Z. An, Y. Wei and X. Liu, Coord. Chem. Rev., 2015, 293–294, 228 CrossRef CAS.
- Monographs in Supramolecular Chemistry No. 33, in Supramolecular Chemistry in Biomedical Imaging, ed. S. Faulkner, T. Gunnlaugsson and G. M. Ó. Máille, The Royal Society of Chemistry, 2022 Search PubMed.
- (a) A. B. Aletti, D. M. Gillen and T. Gunnlaugsson, Coord. Chem. Rev., 2018, 354, 98 CrossRef CAS; (b) D. Parker, J. D. Fradgley and K.-L. Wong, Chem. Soc. Rev., 2021, 50, 8193 RSC; (c) S. E. Bodman and S. J. Butler, Chem. Sci., 2021, 12, 2716 RSC; (d) E. Mathieu, A. Sipos, E. Demeyere, D. Phipps, D. Sakaveli and K. E. Borbas, Chem. Commun., 2018, 54, 10021 RSC; (e) T. Gorai, W. Schmitt and T. Gunnlaugsson, Dalton Trans., 2021, 50, 770 RSC.
- (a) L. E. MacKenzie and R. Pal, Nat. Rev. Chem., 2021, 5, 109 CrossRef CAS; (b) J. Andres, R. D. Hersch, J.-E. Moser and A.-S. Chauvin, Adv. Funct. Mater., 2014, 24, 5029 CrossRef CAS.
- (a) E. P. McCarney;, J. P. Byrne;, B. Twamley;, M. Martínez-Calvo, G. Ryan, M. E. Mobius and T. Gunnlaugsson, Chem. Commun., 2015, 51, 14123 RSC; (b) I. N. Hegarty, H. L. Dalton, A. D. Lynes, B. Haffner, M. E. Möbius, C. S. Hawes and T. Gunnlaugsson, Dalton Trans., 2020, 49, 7364 RSC.
- (a) A. F. Henwood, I. N. Hegarty, E. P. McCarney, J. I. Lovitt, S. Donohoe and T. Gunnlaugsson, Coord. Chem. Rev., 2021, 449, 214206 CrossRef CAS; (b) J. P. Byrne, J. A. Kitchen and T. Gunnlaugsson, Chem. Soc. Rev., 2014, 43, 5302 RSC; (c) J. P. Byrne, J. A. Kitchen, O. Kotova, V. Leigh, A. P. Bell, J. J. Boland, M. Albrecht and T. Gunnlaugsson, Dalton Trans., 2014, 43, 196 RSC; (d) D. Preston and P. E. Kruger, Chem. – Eur. J., 2019, 25, 178 CrossRef PubMed; (e) Y. Li, J. C. Huffman and A. H. Flood, Chem. Commun., 2007, 26, 2692 RSC.
- (a) Q. V. C. van Hilst, N. R. Largesse, D. Preston and J. D. Crowley, Dalton Trans., 2018, 47, 997 RSC; (b) H. Hofmeier and U. S. Schubert, Chem. Soc. Rev., 2004, 33, 373 RSC; (c) J. D. Crowley and P. H. Bandeen, Dalton Trans., 2010, 39, 612 RSC; (d) Q. V. C. van Hilst, R. A. S. Vasdev, D. Preston, J. A. Findlay, S. Ø. Scottwell, G. I. Giles, H. J. L. Brooks and J. D. Crowley, Asian J. Org. Chem., 2019, 8, 496 CrossRef CAS; (e) J. E. M. Lewis, F. Modicom and S. M. Goldup, J. Am. Chem. Soc., 2018, 140, 4787 CrossRef CAS PubMed; (f) M. Cirulli, A. Kaur, J. E. M. Lewis, Z. Zhang, J. A. Kitchen, S. M. Goldup and M. M. Roessler, J. Am. Chem. Soc., 2019, 141, 879 CrossRef CAS PubMed; (g) Y. Li, M. Pink, J. A. Karty and A. H. Flood, J. Am. Chem. Soc., 2008, 130, 17293 CrossRef CAS PubMed; (h) E. M. Zahran, Y. Hua, S. Lee, A. H. Flood and L. G. Bachas, Anal. Chem., 2011, 83, 3455 CrossRef CAS PubMed; (i) D. Preston and P. E. Kruger, Chem. – Eur. J., 2019, 25, 1781 CrossRef CAS PubMed; (j) D. A. W. Ross, D. Preston and J. D. Crowley, Molecules, 2017, 22, 1762 CrossRef PubMed; (k) J. D. Crowley, P. H. Bandeen and L. R. Hanton, Polyhedron, 2010, 29, 70 CrossRef CAS.
- C. O'Reilly, S. Blasco, B. Parekh, H. Collins, G. Cooke, T. Gunnlaugsson and J. P. Byrne, RSC Adv., 2021, 11, 16318 RSC.
- J. P. Byrne, M. Martínez-Calvo, R. D. Peacock and T. Gunnlaugsson, Chem. – Eur. J., 2016, 22, 486 CrossRef CAS PubMed.
- J. P. Byrne, S. Blasco, A. B. Aletti, G. Hessman and T. Gunnlaugsson, Angew. Chem., Int. Ed., 2016, 55, 8938 CrossRef CAS PubMed.
- E. P. McCarney, J. I. Lovitt and T. Gunnlaugsson, Chem. – Eur. J., 2021, 27, 12052 CrossRef CAS PubMed.
- S. J. Bradberry, J. P. Byrne, C. P. McCoy and T. Gunnlaugsson, Chem. Commun., 2015, 51, 16565 RSC.
- I. N. Hegarty, S. J. Bradberry, J. I. Lovitt, J. M. Delente, N. Fox, R. Daly and T. Gunnlaugsson, Mater. Chem. Front., 2023 10.1039/D2QM00998F , QM-RES-09-2022-000998 (at revision stage).
- G. H. Hu and J. T. Lindt, Polym. Bull., 1992, 29, 357 CrossRef CAS.
- (a) D. E. Bergbreiter and Y. S. Liu, Tetrahedron Lett., 1997, 38, 3703 CrossRef CAS; (b) C. Ladaviere, T. Delair, A. Domard, C. Pichot and B. Mandrand, J. Appl. Polym. Sci., 1999, 71, 927 CrossRef CAS.
- O. Stoilova, M. Ignatova, N. Manolova, T. Godjevargova, D. G. Mita and I. Rashkov, Eur. Polym. J., 2010, 46 Search PubMed.
- Y. S. Choe, M. H. Yi, J. H. Kim, G. S. Ryu, Y. Y. Noh, Y. H. Kim and K. S. Jang, Org. Electron., 2016, 36, 171 CrossRef CAS.
- G. Panzarasa, A. Osypova, G. Consolati, F. Quasso, G. Soliveri, J. Ribera and F. Schwarze, Nanomaterials, 2018, 8, 54 CrossRef PubMed.
- C. Ladavière, T. Delair, A. Domard, C. Pichot and B. Mandrand, J. Appl. Polym. Sci., 1999, 72, 1565 CrossRef.
- (a) E. P. McCarney, C. S. Hawes, J. A. Kitchen, K. Byrne, W. Schmitt and T. Gunnlaugsson, Inorg. Chem., 2018, 57, 3920 CrossRef CAS PubMed; (b) I. N. Hegarty, H. L. Dalton, A. D. Lynes, B. Haffner, M. E. Möbius, C. S. Hawes and T. Gunnlaugsson, Dalton Trans., 2020, 49, 7364 RSC.
- J. P. Byrne, J. A. Kitchen, J. E. O'Brien, R. D. Peacock and T. Gunnlaugsson, Inorg. Chem., 2015, 54, 1426 CrossRef CAS PubMed.
- D. Parker and J. A. G. Williams, J. Chem. Soc., Dalton Trans., 1996, 3613 RSC.
- J. C. G. Bünzli, Acc. Chem. Res., 2006, 39, 53 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Characterisation and NMR studies. See DOI: https://doi.org/10.1039/d2ob02259a |
| ‡ The fitting of the changes in the acetamide version of 1 (to prevent PET from the free amine) has previously been determined by observing the changes in both the ground and the excited state of the btp ligand.23 From the Tb(III) luminescent changes, the binding constants were determined as being: logβ1:3 = 22.7, logβ1:2 = 14.5 ± 0.1, and logβ1:1 = 7.0. While we attempted to fit the data obtained from the titrations shown in Fig. 6 and 7, then these gave not very reliable fits. |
| This journal is © The Royal Society of Chemistry 2023 |