
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCalcium-catalysed synthesis of amines through imine hydrosilylation: an experimental and theoretical study†
Yougourthen
Boumekla
,
Fengjie
Xia
,
Lucas
Vidal
,
Cédric
Totée
,
Christophe
Raynaud
 * and
Armelle
Ouali
*
* and
Armelle
Ouali
*
ICGM, Univ Montpellier, CNRS, ENSCM, Montpellier, France. E-mail: armelle.ouali@enscm.fr; christophe.raynaud1@umontpellier.fr
First published on 23rd December 2022
Abstract
A method to reduce aldimines through hydrosilylation is reported. The catalytic system involves calcium triflimide (Ca(NTf2)2) and potassium hexafluorophosphate (KPF6) which have been shown to act in a synergistic manner. The expected amines are obtained in fair to very high yields (40–99%) under mild conditions (room temperature in most cases). To illustrate the potential of this method, a bioactive molecule with antifungal properties was prepared on the gram scale and in high yield in environmentally friendly 2-methyltetrahydrofuran. Moreover, it is shown in this example that the imine can be prepared in situ from the aldehyde and the amine without isolating the imine. The mechanism involved has been explored experimentally and through DFT calculations, and the results are in accordance with an electrophilic activation of the silane by the calcium catalyst.
Introduction
Amines constitute highly important building blocks in organic synthesis due to their omnipresence in natural products, polymers,1 pharmaceuticals2 and agrochemicals.3,4 Among the synthetic strategies towards these key compounds, catalytic reduction of imines including hydrogenation, hydrogen transfer or hydrosilylation appear to be efficient methodologies. In the last few years, hydrosilylation processes involving silanes as the H source significantly contributed to the preparation of amines.5,6 Hydrosilanes, readily produced on the industrial scale, indeed constitute useful reducing agents due to their air- and water stability, ease of handling and ability to be activated under mild conditions. They thus appear as attractive alternatives to molecular hydrogen often requiring high temperature and pressure conditions as well as a specific set-up. Although ytterbium-, aluminium-, boron7,8- and even ionic liquid9-based or organic10 catalysts have been shown to promote imine hydrosilylation, the most ubiquitous catalysts for these transformations involve transition metals. Historically, the first systems reported involved noble ones (ruthenium, rhodium, iridium, and palladium) and later on, some catalysts based on more Earth abundant metals such as copper,11 zinc,12 nickel,13 and iron14–17 have been developed.5 Some of the complexes required multi-step ligand synthesis and/or are air- or moisture sensitive. Our objective is to provide the actual toolbox for imine hydrosilylation with a catalytic system involving a benign metal, with attractive features for organic chemists, namely ready availability and ease of handling (air- and moisture stable). We then turned our attention to calcium. Generally, alkaline earth metals (Ae = Mg, Ca, Sr, and Ba) attracted much attention over the last decade and catalytic systems have been reported to promote a wide range of processes including ring-opening polymerization,18 C–C or C–heteroatom bond-forming transformation,19,20 dehydrocoupling21–24 or hydroelementation of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C, CO or CN bonds.25–28 Along these lines, alkaline earth complexes were shown to promote hydrosilylation of olefins29–31 or pyridines.32–35 Interestingly, with regard to non-activated CN bonds, Sarazin et al. reported stoichiometric hydrosilylation of imine ligands in barium complexes.36 In addition, a recent breakthrough by Harder et al. demonstrated for the first time the ability of calcium, strontium and barium complexes involving amide and/or benzyl ligands to catalyse the hydrosilylation of aldimines and one ketimine with phenylsilane.37,38
C, CO or CN bonds.25–28 Along these lines, alkaline earth complexes were shown to promote hydrosilylation of olefins29–31 or pyridines.32–35 Interestingly, with regard to non-activated CN bonds, Sarazin et al. reported stoichiometric hydrosilylation of imine ligands in barium complexes.36 In addition, a recent breakthrough by Harder et al. demonstrated for the first time the ability of calcium, strontium and barium complexes involving amide and/or benzyl ligands to catalyse the hydrosilylation of aldimines and one ketimine with phenylsilane.37,38
In this report, a commercial and moisture stable calcium salt Ca(NTf2)2 is shown to act in a synergistic manner with KPF6 and in the absence of any additional ligands to catalyse the hydrosilylation of aldimines under mild conditions. DFT studies have been carried out to get insight into the involved mechanism. To the best of our knowledge, while calcium-based Lewis acids have emerged as important catalysts in green chemistry, Ca(NTf2)2 has not been used yet to promote imine hydrosilylation.20,28 In addition, it is the first time that calcium triflimide is shown to activate Si–H bonds, opening new prospects in the field of hydrosilylation reactions as well.
Results and discussion
Imine 1a, which results from the condensation of 4-bromobenzaldehyde and 4-iodoaniline, was chosen as the model substrate. The performance of several commercial calcium salts in the hydrosilylation of 1 with phenylsilane at room temperature was evaluated. Although calcium hydroxide and alkoxides were inefficient (no conversion of 1a), Ca(NTf2)2, a calcium precursor that has a strongly electron-withdrawing and weakly coordinating counter anion, allowed the formation of the expected amine 2a, however, with low yield (10%) (Table 1, entry 1). The addition of catalytic amounts of KPF6 (20 mol%) enabled a significant increase in the performance (entry 2), with 2a being quantitatively obtained. It is noteworthy that Ca(NTf2)2 and KPF6 acted in a synergistic manner, with KPF6 being inefficient when used alone (entry 3). It has previously been shown that Ca(NTf2)2 and PF6 salts undergo anion metathesis to form KNTf2 and Ca(NTf2)PF6.20 The latter salt, which is more Lewis acidic than Ca(NTf2)2 and has two binding sites available on the calcium center, was reported to activate a wide range of substrates such as cyclopropanes,39,40 carbonyl compounds,41,42 alcohols,43–48 alkenes49,50 and more recently sulfonyl51,52 fluorides.20,28,53 Ca(NTf2)PF6, whose Lewis acidity has been evaluated by the Childs method,41 has also been associated with hexafluoroisopropanol as the solvent and the resulting system has been shown by Gandon and Lebœuf to be highly active in various transformations including aza-Piancatelli,54,55 hydroarylation,56 hydroamidation57 and hydroacyloxylation58 reactions. It is noteworthy that under our conditions, KNTf2 alone was unable to promote imine 1a hydrosilylation (entry 4), suggesting that Ca(NTf2)PF6 is the catalytically active species. Furthermore, triflimic acid was unable to promote hydrosilylation, which ruled out the possibility that traces of Tf2NH were the source of the observed activity and confirmed that the calcium center played a role in the catalytic process.|
|
||||
|---|---|---|---|---|
| Entry | [Ca] (mol%) | Additive (mol%) | Eq. PhSiH3 | Yield 2a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a,b a,b |
| a Typical procedure: Ca(NTf2)2, KPF6, PhSiH3 and THF (1 mL) were stirred at 25 °C for 10 minutes before adding aldimine 1a (0.25 mmol) and THF (0.5 mL). The reaction mixture was stirred at 25 °C for 16 h and next hydrolysed with MeOH (1.5 mL) and NaOH (2 M, 1.5 mL). b Yield determined by NMR using 1,3,5-trimethoxybenzene as the standard (selectivity >99%). c After 6 h instead of 16. | ||||
| 1 | Ca(NTf2)2 (20) | — | 4 | 10 |
| 2 | Ca(NTf2)2 (20) | KPF6 (20) | 4 | 99 |
| 3 | — | KPF6 (20) | 4 | 0 |
| 4 | — | KNTf2 (20) | 4 | 0 |
| 5 | Ca(NTf2)2 (10) | KPF6 (10) | 4 | 99 |
| 6 | Ca(NTf2)2 (10) | KPF6 (10) | 2 | 99 (86c) |
| 7 | Ca(NTf2)2 (5) | KPF6 (5) | 2 | 87 |

The amounts of catalyst and additive could be successfully decreased to 10 mol% without significant change (entry 5). Under these conditions, by decreasing the amount of silane by two equivalents (2 equiv. instead of 4, entry 6), amine 2a could be obtained quantitatively when the reaction was stopped after 16 hours at 25 °C (entry 6). It is worth noting that 2a is formed in 86% yield after only 6 hours, suggesting that the conversion is attained before 16 h. Decreasing the amount of Ca(NTf2)PF6/KPF6 (1:1) from 10 to 5 mol% only led to a slight decrease in the yield of amine 2a from 99 to 87% (entry 7).
With these optimized conditions in hand, phenylsilane was replaced with several cheaper counterparts including diphenylsilane, triphenylsilane, dimethylphenylsilane, triethylsilane and 1,1,3,3-tetramethyldisiloxane. However, none of the tested silanes led to the conversion of imine 1a at 25 °C.
Next, the scope of the method was investigated on aromatic aldimines using Ca(NTf2)2/PF6 (1:1) (10 mol%) as the catalytic system (Fig. 1).
 | ||
| Fig. 1 Scope of the catalytic system (NMR yield/isolated yield). Typical procedure (NMR yields): Ca(NTf2)2 (0.025 mmol), KPF6 (0.025 mmol), PhSiH3 (0.5 mmol), and THF (1 mL) were stirred at 25 °C for 10 minutes before adding aldimine 1x (0.25 mmol) and THF (0.5 mL). The reaction mixture was stirred at 25 °C for 16 h and then hydrolysed with MeOH (1.5 mL) and NaOH (2 M, 1.5 mL). Yield was determined by NMR using 1,3,5-trimethoxybenzene as the standard. Typical procedure (isolated yields): Ca(NTf2)2 (0.1 mmol), KPF6 (0.1 mmol), PhSiH3 (2 mmol), and THF (4 mL) were stirred at 25 °C for 10 minutes before adding aldimine 1x (1 mmol) and THF (2 mL). The reaction mixture was stirred at 25 °C for 16 h and then hydrolysed with MeOH (6 mL) and NaOH (2 M, 6 mL). a65 °C. bQuantitative NMR yield after 6 h. cFormation of N,N-di(4-bromobenzyl)benzylamine 3s (20% isolated yield, see the ESI†). | ||
(Benzylidene)phenylamine 1b was quantitatively transformed into amine 2b. Imines 1c and 1d, both derived from toluidine, underwent hydrosilylation in good yields as well (98 and 82% isolated yields for 2c and 2d, respectively), even in the case of the bulkier ortho-substituted naphthyl derivative 1d. Substrates 1e–g bearing electron-donating methoxy substituents were converted in fair to high yields (66, 82 and 99% for 2f, 2g and 2e, respectively). While imine 1e was fully converted within 16 h at 25 °C, 2f and 2g derived from electron-rich p-methoxyaniline were less reactive and an increase in temperature to reflux THF (65 °C) was required to improve the conversions. The hydrosilylation of imines 1h–j bearing electron-withdrawing trifluoromethyl groups was then investigated. Amines 2h, 2i and 2j could be formed quantitatively and isolated in very high yields (88, 97 and 90%, respectively). Here again, the effect of the substituent was dependent on its position: while 1h and 1j produced from electron-poor p-trifluoromethylaniline were fully converted at 25 °C within 6 and 16 h, respectively, imine 1d bearing the CF3 substituent on the “aldehyde” side required higher temperature to be reduced (refluxing THF instead of 25 °C). As in the case of 1a, the halogenated imines 1k and 1l were quantitatively transformed into the corresponding amines 2k and 2l under the standard conditions. The method was shown to be compatible with several functional groups including esters (90% and 82% isolated yields for 2m and 2n), cyano substituents (88% and 76% isolated yields for 2o and 2p) and ketones (94% yield for 2q). Significantly lower yields were obtained for amines 2r–u bearing alkyl substituents at N in refluxing THF. Increasing the amount of the calcium catalyst from 10 to 20 mol% did not significantly improve the performance. Therefore, this system appears to be complementary to the one reported by Harder, which has been shown to be particularly efficient for imines bearing an alkyl group at N and aryl substituents at C.37 Interestingly, in addition to the expected amines 2r and 2s, tertiary amines were also formed; for example, N,N-di(4-bromobenzyl)benzylamine 3s, isolated in 20% yield (see ESI p. S12†), is an appealing starting compound for application in pharmaceuticals or sensors.59 The synthetic pathway to explain the formation of this by-product is still unclear. The heteroaromatic imines 1v and 1w were quantitatively reduced and the expected amines were isolated in good to very high yields (72 and 90% for 2v and 2w, respectively). While 2v is a ligand with potential for application in catalysis, 2w has potential for application in medicinal chemistry due to its antifungal properties and also as a precursor of antitubercular agents.60,61
At this stage, we were keen to get insights into the processes involved in the previously described hydrosilylation.
From a mechanistic point of view, two main hydrosilane activation pathways have been previously proposed (Scheme 1): (i) the Chalk–Harrod and the modified Chalk–Harrod mechanism occurring for e.g. with rhodium or platinum catalysts and relying on two-electron oxidative addition and reductive elimination steps (Scheme 1A); and (ii) an ionic mechanism in which a Lewis acidic catalyst enhances the electrophilic character of the silicon center through the activation of the Si–H bond (Scheme 1B).
 | ||
| Scheme 1 Main mechanisms of silane activation pathways. | ||
In Ae-catalyzed CC or CN hydrosilylation, metal hydride species play a key role in the mechanism and are proposed to be the active species.29–32,37,62 In some of these reactions, the pre-catalyst involves the metal–hydrogen bond. The calcium hydrido complexes can also be generated from amide/alkyl/benzyl calcium precursors and silanes through σ-bond metathesis (Scheme 1C). This process has been widely described and exploited in catalytic reactions including hydrogenation or hydro-elementation,62 and more recently in the redistribution of hydrosilanes63 or dehydrogenative silylation of aromatic ethers.64
Under our conditions, calcium hydroxide, alkoxides and even calcium triflimidate Ca(NTf2)2 alone are not efficient to promote the hydrosilylation of imine 1a. The fact that hydrosilylation only occurs in the presence of the highly Lewis acidic calcium salt Ca(NTf2)PF6 might suggest that the mechanism involves an electrophilic activation of the Si–H bond (Scheme 1B). Such a pathway was reported for the hydrosilylation of ketones and imines promoted by perfluoroarylboranes such as B(C6F5)3 (Scheme 1B).65–69 In these processes, hydrosilylation proceeded more rapidly in the case of least basic substrates.67 Under our conditions, the electronic properties of the substituents were shown to have an effect on the catalytic performance. For example, electron-poor imine 1h with a CF3 group at the para position of the imine nitrogen was significantly more reactive than imine 1f with an electron-donating methoxy group at the same position. Indeed, while 1h was quantitatively converted into 2h within 6 h at room temperature (Fig. 1, note d), 1f required heating to reach a conversion of 66% after 16 h.
Next the interaction between the Ca(NTf2)PF6 catalyst and each of the substrates, imines and silane, was investigated. As a first attempt, the evolution of a stoichiometric mixture of the calcium catalyst and imine 1h in deuterated THF was then monitored by 1H NMR at different temperatures (25 °C to −65 °C and back to 25 °C). No obvious change could be observed in the imine spectrum irrespective of the temperature, suggesting that the presence of the hydrosilane is required for the two substrates to react together. The same behaviour was observed in the case of more electron-rich imine 1f. The same NMR experiment was then performed in the case of phenylsilane. The addition of one equivalent of Ca(NTf2)PF6 to phenylsilane did not induce any change in the 1H NMR spectrum of the silane at room temperature. Next 1H NMR spectra were collected at lower temperatures, from 25 °C to −65 °C. Interestingly at −25 °C (ESI § 4.1†), the appearance of broad signals at around 6–6.4 ppm was observed and the signals became increasingly sharp when the temperature was decreased to −65 °C (ESI, Fig. S1 p. S14†). The phenomenon was shown to be reversible when the temperature returned to room temperature (ESI, Fig. S2 p. S14†). These observations suggest a weak coordination of the silane to calcium.67,70
From all these experimental observations, we propose a putative catalytic cycle (Scheme 2). The process starts with the generation of the catalytically active species and highly Lewis acidic Ca(NTf2)PF6 (represented as [Ca]), where the PF6− counteranion is not expected to be in the first coordination sphere.20,71 The first step of the catalytic cycle would consist in the coordination of the silane to the calcium center Ca(NTf2)+ (Scheme 2, step a). The subsequent intermolecular nucleophilic attack of the imine on the silicon center may lead to the silyliminium intermediate (Scheme 2, step b), by analogy with the mechanism proposed in the case of B(C6F5)3.69 In the following step c, H-transfer may occur. The last decoordination step d would enable the release of the expected silylamine and the regeneration of the catalytically active calcium species.
 | ||
| Scheme 2 Mechanism of the Ca-catalyzed hydrosilylation of imines proposed by analogy with that of B(C6F5)3. | ||
To gain a better understanding of the mechanism of this catalytic hydrosilylation, DFT studies were carried out with a dispersion corrected hybrid functional and an implicit model for the solvent (THF). Natural bond orbital (NBO) analyses of the stationary points of the potential energy surface have been carried out. Full computational details are given in the ESI.†
The Ca2+ complex with the NTf− ligand, and the substrates (2b imine and PhSiH3) were represented in full. The PF6− counter-anion was supposed to be non-coordinated and was not taken into account for the modelling.
The overall reaction is found to be exergonic (ΔrG° = −16.9 kcal mol−1) and requires a Gibbs activation energy of 26.1 kcal mol−1 relative to that of the separate reagents (Fig. 2). The driving force of the reaction relies on the C–H and Si–N bonds, which are slightly stronger than the broken Si–H and CN bonds. The reaction mechanism is found to be the succession of two elementary steps.
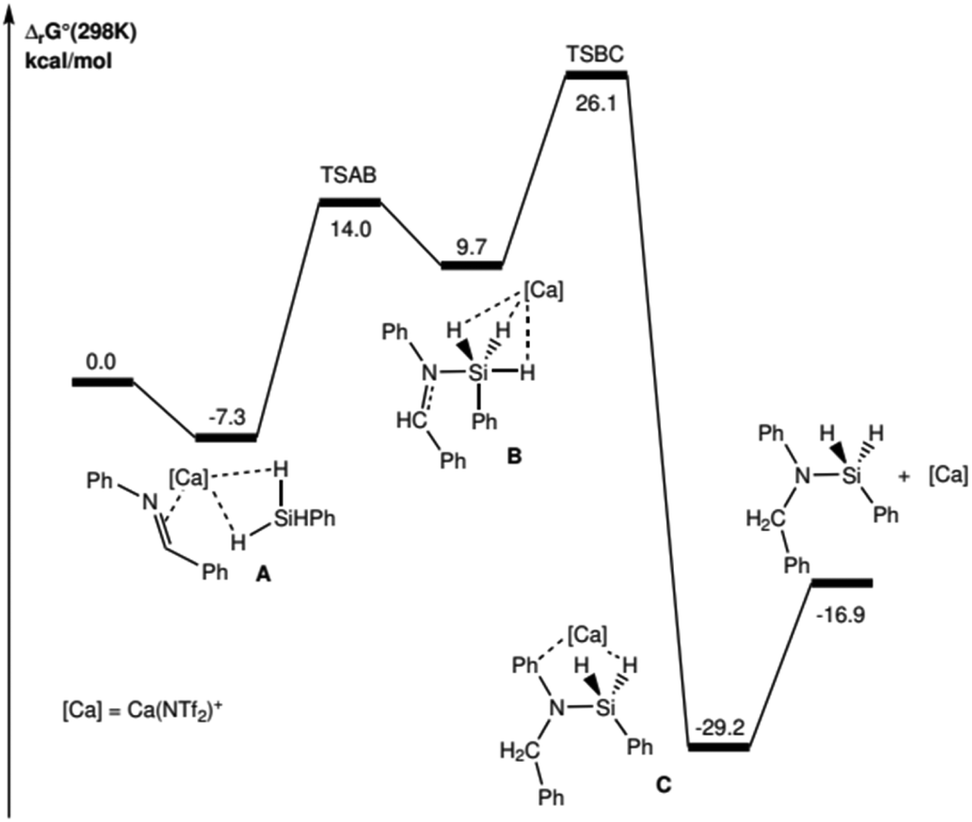 | ||
| Fig. 2 Gibbs energy profile (kcal mol−1) for the hydrosilylation of 2b imine catalysed by Ca(NTf)+. | ||
The reaction sequence begins with the coordination of the imine and PhSiH3 to the metal cation, leading to adduct A. The first step requires a low activation barrier (ΔG‡ = 21.3 kcal mol−1, relative to that of adduct A, i.e. 14.0 kcal mol−1 relative to the energy reference), and corresponds to the step during which the Si–N bond is formed, leading to intermediate B (dSi–N = 2.181 Å and 2.019 Å for TSAB and B, respectively). It is noteworthy that for this intermediate B, the Si atom is found to be pentavalent and at the center of a trigonal bipyramid. The NBO analysis for this intermediate shows that its relative stability is essentially based on favorable donor–acceptor interactions, on the one hand between the N lone-pair and the three σ*Si–H antibonds (mainly with the σ*Si–H trans to N) and on the other hand between the three σSi–H bonds and the vacant 3s orbital of Ca (see ESI § 5.3†). It is also important to note that all attempts to optimize the structure of such a reaction intermediate, where Si is hypervalent, in the absence of the Ca(NTf2)+ catalyst, have been unsuccessful: the stability of this intermediate is a key aspect of the process and can only be ensured with the electrophilic assistance of Ca. The intermediate B also has a characteristic 1H NMR signature: calculations suggest an isotropic chemical shift δ for the H in alpha from the C functional of the imine to 9.0 ppm and shifts for the 3 H bonded to the Si and in interaction with Ca, between 5.0 and 5.8 ppm (ESI § 5.4†); these calculated values are consistent with the transiently experimentally observed values of the chemical shift (see ESI Fig. S3 p. S15†).
The second step corresponds to the transition from the intermediate B to the product C and is the kinetic determining step (ΔG‡ = 26.1 kcal mol−1 with respect to the reference energy). This step corresponds to the β-migration of one of the three H atoms carried by the Si towards the imine C atom. An NBO analysis of the electronic structure of this TSBC transition state shows clearly that the transferred H has a strong hydride character (the corresponding NBO charge is −0.81e). This electrophilic nature allows for better understanding of the greater efficiency of the process for imines substituted with an electron-withdrawing group with respect to imines substituted with electron-donating groups (2hvs.2f, Fig. 2), which enables, respectively, the stabilisation/destabilisation of this transition state (see ESI § 5.3†).
The final step corresponds to the decoordination of the silylamine to the metal (adduct C), leading to the free Ca(NTf2)+ complex and the reaction product without interactions.
The proposed mechanism, consistent with the experimental observations previously presented, favors an electrophilic Si–H activation pathway.
Lastly, to illustrate the potential of the method for application in organic chemistry, the catalytic system reported here was applied to the synthesis of bioactive amine 2w on the gram-scale through reductive amination and by optimizing the conditions (solvent and amounts of metal and silane, Scheme 3). Therefore, in this synthesis, equimolar amounts of 2-furfural, a chemical obtained from biomass,72 and 3,4-dichloroaniline (7 mmol) were allowed to react in 2-methyltetrahydrofuran, a renewable and more eco-friendly solvent.73 After 6 h, 5 mol% of the Ca catalyst and 1.2 equiv. of silane were added and the mixture was stirred for 24 h at 25 °C before achieving basic hydrolysis. 1H NMR of the crude confirmed the complete conversion of both reactants, the absence of the intermediate imine and the quantitative formation of 2w. The latter was isolated in 85% yield after column chromatography, which is in the same order of magnitude as on the 1 mmol scale from imine 1w (90%, Fig. 1). It is worth noting that for this substrate, the amount of the catalyst could be decreased from 10 to 5 mol% and the excess of silane from 2 to 1.2 equivalents, while maintaining good performance. Under these conditions, more than 1.4 g of the expected amine could be obtained with high purity.
 | ||
| Scheme 3 Application of the method for gram-scale synthesis of an amine with anti-fungal activity. | ||
Conclusions
This method reports a simple and efficient method for imine hydrosilylation using a catalytic system based on non-toxic calcium, the fifth most frequent element in the Earth's crust. The proposed catalyst is based on Ca(NTf2)2 and KPF6, which act in a synergistic manner. Both precursors are readily available and easy-to-handle salts that can be stored on shelves, open to air and used for several months without loss of activity. This catalytic system is able to promote the hydrosilylation of a variety of aldimines under mild conditions (25–65 °C, 6–16 h) without requiring an inert atmosphere, and shows tolerance towards functional groups including halides (I, Br, and F), trifluoromethyls, ketones, esters or nitriles. The expected amines are obtained in fair to excellent yields.The value of the method has been shown in the synthesis of a bioactive amine with antifungal properties, which was prepared on the gram scale and in high yield in environmentally friendly 2-methyltetrahydrofuran. It is shown in this example that the imine can be generated in situ from the corresponding aldehyde and amine without isolating the imine, which is interesting from a practical point of view. Overall, due to the nature of the metal (calcium is non-toxic even in large amounts) and the mild reaction conditions, the reported methodology contributes to the search for sustainable methods for the synthesis of amines, which are highly important building blocks in many fields. The resulting amines are free from traces of transition metals, which is of great interest in the field of pharmaceuticals, wherein metal contaminants are strictly regulated. No tedious and costly purification is thus needed. In addition, experimental data and DFT calculations are in accordance with an electrophilic Si–H activation pathway catalyzed by the Ca(NTf2)+ catalyst shown to promote imine hydrosilylation for the first time. To the best of our knowledge, such an electrophilic activation of the Si–H bond, well-known for boron catalysts, is not common in alkaline earth chemistry where an Ae–hydride complex is generally generated from calcium precursors and silanes through σ-bond metathesis (Scheme 1C). For the first time, Ca(NTf2)+ has been shown to activate Si–H bonds and studies to extend the scope of this catalyst to other challenging hydrosilylation reactions are currently underway.
Author contributions
Yougourthen Boumekla: investigation and methodology. Fengxie Xia: investigation. Lucas Vidal: investigation. Cédric Totee: methodology. Christophe Raynaud: methodology, software, formal analysis, and writing – review and editing. Armelle Ouali: conceptualization, methodology, writing – original draft, review and editing, and supervision.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank CNRS, ENSCM and the University of Montpellier for funding.References
- V. Froidevaux, C. Negrell, S. Caillol, J. P. Pascault and B. Boutevin, Biobased Amines: From Synthesis to Polymers; Present and Future, Chem. Rev., 2016, 116, 14181–14224 CrossRef CAS PubMed.
- O. I. Afanasyev, E. Kuchuk, D. L. Usanov and D. Chusov, Reductive Amination in the Synthesis of Pharmaceuticals, Chem. Rev., 2019, 119, 11857–11911 CrossRef CAS.
- S. A. Lawrence, Amines: Synthesis, Properties and Applications, Cambridge University Press, Cambridge, UK, 1st edn, 2004, 450 pp Search PubMed.
- P. F. Vogt and J. J. Gerulis, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, Weinheim, Germany, 2000, p. 699 Search PubMed.
- B. Li, J.-B. Sortais and C. Darcel, Amine synthesis via transition metal homogeneous catalysed hydrosilylation, RSC Adv., 2016, 6, 57603–57625 RSC.
- N. S. Shaikh, Sustainable Amine Synthesis: Iron Catalyzed Reactions of Hydrosilanes with Imines, Amides, Nitroarenes and Nitriles, ChemistrySelect, 2019, 4, 6753–6777 CrossRef.
- X. Zhu and H. Du, A Chiral Borane Catalyzed Asymmetric Hydrosilylation of Imines, Org. Biomol. Chem., 2015, 13, 1013–1016 RSC.
- M. Mewald and M. Oestreich, Illuminating the Mechanism of the Borane-Catalyzed Hydrosilylation of Imines with Both an Axially Chiral Borane and Silane, Chem. – Eur. J., 2012, 18, 14079–14084 CrossRef.
- B. Li, S. Zhang, W. Wu, L. Liang, S. Jiang, L. Chen and Y. Li, Imidazolium-based ionic liquid-catalyzed hydrosilylation of imines and reductive amination of aldehydes using hydrosilane as the reductant, RSC Adv., 2017, 7, 31795–31799 RSC.
- A. V. Malkov, A. J. P. Stewart-Liddon, G. D. McGeoch, P. Ramirez-Lopez and P. Kocovsky, Catalyst development for organocatalytic hydrosilylation of aromatic ketones and ketimines, Org. Biomol. Chem., 2012, 10, 4864–4877 RSC.
- B. H. Lipshutz and H. Shimizu, Copper(I)-Catalyzed Asymmetric Hydrosilylations of Imines at Ambient Temperatures, Angew. Chem., Int. Ed., 2004, 43, 2228–2230 CrossRef PubMed.
- M. Swewczyk, A. Bezlada and J. Mlynarski, Zinc–Catalyzed Enantioselective Hydrosilylation of Ketones and Imines under Solvent–Free Conditions, ChemCatChem, 2016, 8, 3575–3579 CrossRef.
- J. Zheng, T. Roisnel, C. Darcel and J.-B. Sortais, Nickel-Catalysed Reductive Amination with Hydrosilanes, ChemCatChem, 2013, 5, 2861–2864 CrossRef.
- H. Jaafar, L. C. Li, M. Castro, J. Zheng, T. Roisnel, V. Dorcet, J.-B. Sortais and C. Darcel, Phosphane-Pyridine Iron Complexes: Synthesis, Characterization and Application in Reductive Amination through the Hydrosilylation Reaction, Eur. J. Inorg. Chem., 2012, 3546–3550 CrossRef.
- L. C. M. Castro, J.-B. Sortais and C. Darcel, NHC-carbene cyclopentadienyl iron based catalyst for a general and efficient hydrosilylation of imines, Chem. Commun., 2012, 48, 151–153 RSC.
- M. Bhunia, P. K. Hota, G. Vijaykumar, D. Adhikari and S. K. Mandal, A Highly Efficient Base-Metal Catalyst: Chemoselective Reduction of Imines to Amines Using An Abnormal-NHC−Fe(0) Complex, Organometallics, 2016, 35, 2930–2937 CrossRef.
- A. Saini, C. R. Smith, F. S. Weseka, A. K. Helms and M. Findlater, Conversion of aldimines to secondary amines using iron-catalysed hydrosilylation, Org. Biomol. Chem., 2018, 16, 9368–9372 RSC.
- Y. Sarazin and J.-F. Carpentier, Discrete Cationic Complexes for Ring-Opening Polymerization Catalysis of Cyclic Esters and Epoxides, Chem. Rev., 2015, 115, 3864–3614 CrossRef.
- S. Kobayashi and Y. Yamashita, Alkaline Earth Metal Catalysts for Asymmetric Reactions, Acc. Chem. Res., 2011, 44, 58–71 CrossRef.
- D. Lebœuf and V. Gandon, Carbon-Carbon and Carbon–Heteroatom Bond-Forming Transformations Catalyzed by Calcium(II) triflimide, Synthesis, 2017, 49, 1500–1508 CrossRef.
- N. Li and B.-T. Guan, A Dialkyl Calcium Carbene Adduct: Synthesis, Structure, and Catalytic Cross-Dehydrocoupling of Silanes with Amines, Eur. J. Inorg. Chem., 2019, 2231–2235 CrossRef.
- M. Rauch, R. C. Roberts and G. Parkin, Reactivity of [TismPriBenz]MgMe towards Secundary Amines and Terminal Alkynes : catalytic Dehydrocoupling with Hydrosilanes to Afford Si-N and Si-C Bonds, Inorg. Chem. Acta, 2019, 494, 271–279 CrossRef.
- N. V. Forosenko, I. V. Basalov, A. V. Chersakov, G. K. Fukin, E. S. Shubina and A. Trifonov, Amido Ca(II) Complexes Supported by Schiff Base Ligands for Catalytic Cross-Dehydrogenative Coupling of Amines with Silanes, Dalton Trans., 2018, 47, 12570–12581 RSC.
- L. Zhao, X. Shi and J. Cheng, Calcium-Catalyzed Dehydrogenative Silylation of Aromatic Ethers with Hydrosilanes, ACS Catal., 2021, 11, 2041–2046 CrossRef.
- S. Harder, From Limestone to Catalysis: Application of Calcium Compounds as Homogeneous Catalysts, Chem. Rev., 2010, 110, 3852–3876 CrossRef PubMed.
- S. Harder, Molecular early main group metal hydrides: synthetic challenge, structures and applications, Chem. Commun., 2012, 48, 11165–11177 RSC.
- M. S. Hill, D. J. Liptrot and C. Weetman, Alkaline earths as main group reagents in molecular catalysis, Chem. Soc. Rev., 2016, 45, 972–988 RSC.
- L. Yu, R. Qian, X. Deng, F. Wang and Q. Xu, Calcium-Catalyzed Reactions of Element-H bonds, Sci. Bull., 2018, 15, 1010–1016 CrossRef.
- L. Garcia, C. Dinoi, M. F. Mahon, L. Maron and M. S. Hill, Magnesium Hydride Alkene Insertion and catalytic Hydrosilylation, Chem. Sci., 2019, 10, 8108–8118 RSC.
- D. Schuhknecht, T. P. Spaniol, L. Maron and J. Okuda, Regioselective Hydrosilylation of Olefins Catalyzed by a Molecular Calcium Hydride Cation, Angew. Chem., Int. Ed., 2020, 59, 310–314 CrossRef PubMed.
- L. M. Rauch, S. Ruccolo and G. Parkin, Synthesis, Structure, and Reactivity of a Terminal Magnesium Hydride Compound with a Carbatrane Motif, [TismPriBenz]MgH : A Multifunctional Catalyst for Hydrosilylation and Hydroboration, J. Am. Chem. Soc., 2017, 139, 13264–13267 CrossRef.
- L. Fohlmeister and A. Stasch, Ring-Shaped Phosphinoamido-Magnesium-Hydride Complexes : Syntheses, Structures, Reactivity, and Catalysis, Chem. – Eur. J., 2016, 22, 10235–10246 CrossRef.
- D. Mukherjee, D. Schuhknecht and J. Okuda, Hydrido Complexes of Calcium: A new Family of Molecular Alkaline-Earth-Metal Compounds, Angew. Chem., Int. Ed., 2018, 57, 9590–9602 CrossRef PubMed.
- J. Intemann, H. Bauer, J. Phal, L. Maron and S. Harder, Calcium Hydride Catalyzed Highly 1,2-Selective Pyridine Hydrosilylation, Chem. – Eur. J., 2015, 21, 11542–11461 CrossRef.
- For related hydroboration: M. Arrowsmith, M. S. Hill, T. Hadlington, G. Kociok-Köhn and C. Weetman, Magnesium-Catalyzed Hydroboration of Pyridines, Organometallics, 2011, 30, 5556–5559 CrossRef.
- P. M. Chapple, M. Cordier, V. Dorcet, T. Roisnel, J.-F. Carpentier and Y. Sarazin, A versatile nitrogen ligand for alkaline-earth Chemistry, Dalton Trans., 2020, 49, 11878–11889 RSC.
- H. Elsen, C. Fischer, C. Knüpfer, A. Escolana and S. Harder, Early Main Group Metal Catalysts for Imine Hydrosilylation, Chem. – Eur. J., 2019, 25, 16141–16147 CrossRef.
- Similar complexes are able to promote the related imine hydrogenation: H. Bauer, M. Alonso, C. Färber, H. Elsen, J. Pahl, A. Causero, G. Ballmann, F. De Proft and S. Harder, Imine hydrogenation with simple alkaline earth metal catalysts, Nat. Catal., 2018, 1, 40–47 CrossRef.
- T. P. Maloney, K. L. Murphy, T. L. Mainsah and K. A. Nolin, Friedel-Crafts Alkylation of Benzo[b]furan with Activated Cyclopropanes by a Calcium(II) Complex, Tetrahedron Lett., 2018, 59, 18–21 CrossRef.
- C. C. Dulin, K. L. Murphy and K. A. Nolin, Calcium-Catalyzed Friedel-Crafts Addition of 1-methylindole to Activated Cyclopropanes, Tetrahedron Lett., 2014, 55, 5380–5382 CrossRef.
- J. Davies and D. Leonori, The First Calcium-Catalyzed Nazarov Cyclisation, Chem. Commun., 2014, 50, 15171–15174 RSC.
- S. P. Morcillo, M. Presset, S. Floquet, V. Coeffard, C. Greck, C. Bour and V. Gandon, Site-Selective Calcium-Catalyzed/Organocatalyzed Condensation of Propargyl Alcohols Tethered to β-Keto-esters, Eur. J. Org. Chem., 2016, 2688–2694 CrossRef CAS.
- V. J. Meyer and M. Niggemann, Highly Chemoselective Calcium-Catalyzed Propargylic Deoxygenation, Chem. – Eur. J., 2012, 18, 4687–4691 CrossRef CAS.
- A. J. Basson and M. G. McLaughlin, Synthesis of Functionalized Isoindolinones via Calcium Catalyzed Generation and Trapping of N-Acyliminium Ions, J. Org. Chem., 2020, 85, 5615–5628 CrossRef CAS.
- P. Xie, Z. Sun, S. Li, L. Zhang, X. Cai, W. Fu, X. Yang, Y. Liu, X. Wo and T.-P. Loh, Dehydrative Cross-Coupling of Allylic Alcohols with Alkynes, Org. Lett., 2020, 22, 1599–1604 CrossRef CAS PubMed.
- P. Xie, Z. Sun, S. Li, X. Cai, J. Qiu, W. Fu, C. Gao, S. Wu, X. Yang and T.-P. Loh, Reciprocal-Activation Strategy for Allylic Sulfination with Unactivated Allylic Alcohols, Org. Lett., 2020, 22, 4893–4897 CrossRef CAS.
- S. P. Morcillo, D. Lebœuf, C. Bour and V. Gandon, Calcium-Catalyzed Synthesis of Polysubstituted 2-Alkenylfurans from β-Keto Esters Tethered to Propargyl Alcohols, Chem. – Eur. J., 2016, 22, 16974–16978 CrossRef CAS.
- S. Yang, C. Bour, D. Lebœuf and V. Gandon, DFT Analysis into the Calcium(II)-Catalyzed Coupling of Alcohols with Vinylboronic Acides: Cooperativity of Two Different Lewis Acids and Counterion Effects, J. Org. Chem., 2021, 86, 9134–9144 CrossRef CAS PubMed.
- A. K. Diba, J.-M. Begouin and M. Niggemann, Calcium-Catalyzed Hydroalkoxylation, Tetrahedron Lett., 2012, 53, 6629–6632 CrossRef.
- M. Niggemann and N. Bisek, Calcium-Catalyzed Hydroarylation of Alkenes at Room Temperature, Chem. – Eur. J., 2010, 16, 11246–11249 CrossRef CAS PubMed.
- P. Mukherjee, C. P. Woroch, L. Cleary, M. Rusznak, R. W. Franzese, M. R. Reese, J. W. Tucker, J. M. Humphrey, S. M. Etuk, S. C. Kwan, C. W. am Ende and N. D. Ball, Sulfonamide Synthesis via Calcium Triflimide Activation of Sulfonyl Fluorides, Org. Lett., 2018, 20, 3943–3947 CrossRef CAS PubMed.
- P. Mukherjee, C. P. Woroch, T. W. Butler, S. N. Cameiro, S. C. Kwan, S. R. Khasnavis, J. Gu, J. K. Dutra, B. C. Vetelino, J. Bellenger, C. W. am Ende and N. D. Ball, SuFEx Activation with Ca(NTf2)2: A Unified Strategy to Access Sulfamides, Sulfamates, and Sulfonamides from S(VI) Fluorides, Org. Lett., 2020, 22, 4389–4394 CrossRef.
- J.-M. Begouin and M. Niggemann, Calcium-Catalyzed Lewis Acid Catalysts, Chem. – Eur. J., 2013, 19, 8030–8041 CrossRef CAS.
- S. P. Morcillo, D. Lebœuf, C. Bour and V. Gandon, Harnessing the Lewis Acidity of HFIP through its Cooperation with a Calcium(II) Salt: Application to the Aza-Piancatelli Reaction, Chem. – Eur. J., 2016, 22, 16165–16171 CrossRef.
- L. Marin, S. Jerhaoui, E. Kolodziej, R. Guillot, V. Gandon, F. Colobert, E. Schultz, J. Wencel-Delord and D. Lebœuf, Sulfoxide-Controlled Stereoselective Aza-Piancatelli Reaction, ACS Catal., 2021, 363, 4277–4282 CAS.
- C. Qi, V. Gandon and D. Lebœuf, Calcium(II)-Catalyzed Intermolecular Hydroarylation of Deactivated Styrenes in Hexafluoroisopropanol, Angew. Chem., Int. Ed., 2018, 57, 14245–14249 CrossRef CAS PubMed.
- C. Qi, F. Hasenmaile, V. Gandon and D. Lebœuf, Calcium(II)-Catalyzed Intra- and Intermolecular Hydroamidation of Unactivated Alkenes in Hexafluoroisopropanol, ACS Catal., 2018, 8, 1734–1739 CrossRef.
- C. Qi, S. Yang, V. Gandon and D. Lebœuf, Calcium(II)- and Triflimide-Catalyzed Intramolecular Hydroacyloxylation of Unactivated Alkenes in Hexafluoroisopropanol, Org. Lett., 2019, 21, 7405–7409 CrossRef.
- C. Li, K.-F. Wan, F.-Y. Guo, Q.-H. Wu, M.-L. Yuan, R.-X. Li, H.-Y. Fu, X.-L. Zheng and H. Chen, Iridium-Catalyzed Alkylation of Amine and Nitrobenzene with Alcohol to Tertiary Amine under Base- and Solvent-Free Conditions, J. Org. Chem., 2019, 84, 2158–2168 CrossRef.
- V. V. Kouznetsov, L. Y. V. Mendéz, M. Sortino, Y. Vásquez, M. P. Gupta, M. Freile, R. D. Enriz and S. A. Zacchino, Antifungal and Cytotoxic Activities of some N-substituted Aniline Derivatives bearing a Hetaryl Fragment, Bioorg. Med. Chem., 2008, 16, 794–809 CrossRef PubMed.
- M. F. Arshad, S. Kumar, A. H. Al Rohaimi, A. A. Hassan, A. Elkerdasy and N. Upmanyu, Synthesis of 3-chloro-4-fluorophenyl-3,4-dichlorophenyl substituted thiocarbamide derivatives as potential antitubercular agents, Med. Chem. Res., 2005, 24, 334–343 CrossRef.
- D. Mukherjee, D. Schuhknecht and J. Okuda, Hydrido Complexes of Calcium: A new Family of Molecular Alkaline-Earth-Metal Compounds, Angew. Chem., Int. Ed., 2018, 57, 9590–9602 CrossRef PubMed.
- T. Li, K. N. McCabe, L. Maron, X. Leng and Y. Chen, Organocalcium Complex-Catalyzed Selective Redistribution of ArSiH3 or Ar(alkyl)SiH2 to Ar3SiH or Ar2(alkyl)SiH, ACS Catal., 2021, 11, 6348–6356 CrossRef CAS.
- L. Zhao, X. Shi and J. Cheng, Calcium-Catalyzed Dehydrogenative Silylation of Aromatic Ethers with Hydrosilanes, ACS Catal., 2021, 11, 2041–2046 CrossRef CAS.
- D. J. Parks and W. E. Piers, Tris(pentafluorophenyl)boron-Catalyzed Hydrosilylation of Aromatic Aldehydes, Ketones and Esters, J. Am. Chem. Soc., 1996, 118, 9440–9441 CrossRef CAS.
- S. Rendler and M. Oestreich, Conclusive Evidence for an SN2-Si Mechanism in the B(C6F5)3-Catalyzed Hydrosilylation of Carbonyl Compounds: Implications for the Related Hydrogenation, Angew. Chem., Int. Ed., 2008, 47, 5997–6000 CrossRef CAS PubMed.
- M. C. Lipke, A. L. Liberman-Martin and T. Don Tilley, Electrophilic Activation of Silicon-Hydrogen Bonds in catalytic Hydrosilylations, Angew. Chem., Int. Ed., 2017, 56, 2260–2294 CrossRef CAS PubMed.
- W. E. Piers, A. J. V. Marwitz and L. G. Mercier, Mechanistic Aspects of Bond Activation with Perfluoroarylboranes, Inorg. Chem., 2011, 50, 12252–12262 CrossRef CAS PubMed.
- M. Mewald and M. Oestreich, Illuminating the Mechanism of Imines with Both an Axially Chiral Borane and Silane, Chem. – Eur. J., 2012, 18, 14079–14084 CrossRef CAS PubMed.
- M. C. Lipke, M.-N. Poradowski, C. Raynaud, O. Eisenstein and T. D. Tilley, Catalytic Olefin Hydrosilylation Mediated by Ruthenium η3-H2Si σ Complexes of Primary and Secondary Silanes, ACS Catal., 2018, 8, 11513–11523 CrossRef CAS.
- L. Xue, D. D. Desmarteau and W. T. Pennington, Synthesis and Structures of Alkaline Earth Metal Salts of Bis[(trifluoromethyl)sulfonyl]imide, Solid State Sci., 2005, 7, 311–318 CrossRef CAS.
- J. A. Caetano and A. C. Fernandes, One-Pot synthesis of amines from biomass resources catalyzed by HReO4, Green Chem., 2018, 20, 2494–2498 RSC.
- E. Bisz and M. Szostak, 2-Methyltetrahydrofuran: A Green Solvent for Iron-Catalyzed Cross-Coupling Reactions, ChemSusChem, 2018, 11, 1290–1294 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ob02243e |
| This journal is © The Royal Society of Chemistry 2023 |