
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
π-Facial selectivity in the Diels–Alder reaction of glucosamine-based chiral furans and maleimides†
Cornelis H. M.
van der Loo
 a,
Rutger
Schim van der Loeff
a,
Avelino
Martín
c,
Pilar
Gomez-Sal
c,
Mark L. G.
Borst
b,
Kees
Pouwer
b and
Adriaan J.
Minnaard
*a
a,
Rutger
Schim van der Loeff
a,
Avelino
Martín
c,
Pilar
Gomez-Sal
c,
Mark L. G.
Borst
b,
Kees
Pouwer
b and
Adriaan J.
Minnaard
*a
aDepartment of Chemical Biology, Stratingh Institute for Chemistry, University of Groningen, Groningen, The Netherlands. E-mail: A.J.Minnaard@RUG.nl
bSymeres B.V., Kadijk 3, 9747 AT Groningen, The Netherlands
cDepartamento de Química Orgánica y Química Inorgánica, Instituto de Investigación Química “Andres M. Del Rio” (IQAR), Universidad de Alcalá. Alcalá de Henares, 28805, Madrid, Spain
First published on 30th December 2022
Abstract
Furans derived from carbohydrate feedstocks are a versatile class of bio-renewable building blocks and have been used extensively to access 7-oxanorbornenes via Diels–Alder reactions. Due to their substitution patterns these furans typically have two different π-faces and therefore furnish racemates in [4 + 2]-cycloadditions. We report the use of an enantiopure glucosamine derived furan that under kinetic conditions predominantly affords the exo-product with a high π-face selectivity of 6.5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :1. The structure of the product has been resolved unequivocally by X-ray crystallography, and a multi-gram synthesis (2.8 g, 58% yield) confirms the facile accessibility of this multifunctional enantiopure building block.
:1. The structure of the product has been resolved unequivocally by X-ray crystallography, and a multi-gram synthesis (2.8 g, 58% yield) confirms the facile accessibility of this multifunctional enantiopure building block.
Introduction
With the emergence of carbohydrate-based furans, a readily available class of versatile building blocks is now at the disposal of the synthetic chemist.1,2 Dehydration of carbohydrates not only opens the door to a bio-renewable source of furan derivatives, but also provides access to heterocyclic building blocks with a substitution pattern that is not easily accessible otherwise. Prime examples of such heterocycles are the 3-amido-furans derived from N-acetyl glucosamine: dihydroxyethyl acetamidofuran (Di-HAF (1)) and 3-acetamido-5-acetylfuran (3A5AF (2)). Since practical methods for their preparation have become available in the last decade,3–6 these building blocks have found several synthetic applications.7–9The use of furans in the Diels–Alder (DA) cycloaddition is as old as the named reaction itself.10,11 The reaction generates a well-defined three-dimensional 7-oxanorbornene structure, with up to four stereocenters, in a single step. Since their discovery, 7-oxanorbornenes have found diverse synthetic utility,12 such as in natural product synthesis,13 in the field of therapeutics,14 as well as in polymer chemistry and material science.15–17 Furans make for atypical Diels–Alder reactions as these not always adhere to the well-known “endo-rule”.18 This rule states that under kinetic conditions the product with endo topology is favored.19 Although the selectivity with furans is strongly substrate and solvent dependent, typically a small difference in ΔG‡ between the endo- and exo-products results in low selectivity for one or the other isomer.20,21 In addition, due to the regeneration of aromaticity combined with the relief of ring strain, 7-oxanorbornenes readily undergo a retro-cycloaddition, at a rate depending on their substituents.22
The DA reaction has been applied extensively to carbohydrate-derived furans.23–26 This normally requires that these typically electron poor dienes are first converted into more electron rich derivatives.27,28 However, very recently examples of the direct Diels–Alder reaction with carbohydrate-based furans have been reported. The group of Bruijnincx29 overcame the thermodynamic challenges by trapping the Diels–Alder products of, among others, furfural (3) and 5-HMF (4) as hydrates. And Gomes et al.30 reported the use of the more electron rich 3A5AF (2) and attained high yields and exo-selectivities (Scheme 1).
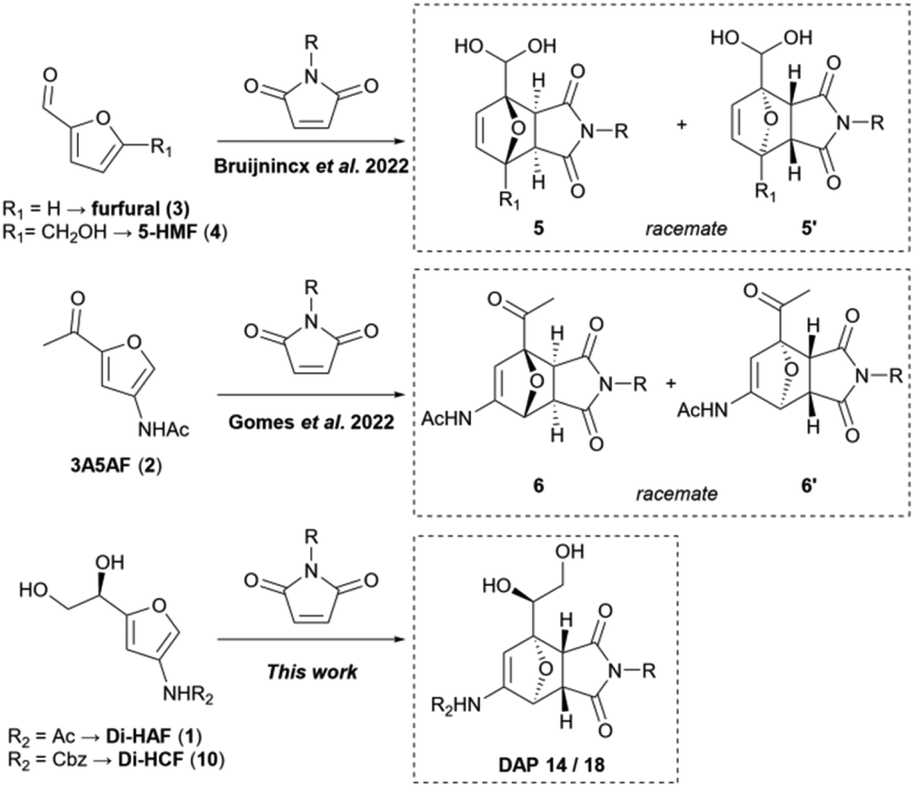 | ||
| Scheme 1 Recent advances in the application of carbohydrate derived furans in direct Diels–Alder cycloaddition reactions with maleimides. | ||
In these examples, the products are formed as racemates since the furans have nonequivalent π-faces. This characteristic diminishes the application potential of their cycloaddition products as building blocks for optically active compounds, such as pharmaceuticals and natural products. Research into π-face selective Diels–Alder reactions with carbohydrate-based furans is still in its infancy, and only a few examples have been described in literature. Sakamoto reported the synthesis of the DA adduct of 2-methyl furan and N-phenylmaleimide, in 90% ee by an elegant dynamic stereoselective crystallization.31 A more common approach to induce enantioselectivity however, is via π-facial differentiation. To achieve this for non-chiral substrates, a source of chirality is required, typically in the form of a temporary auxiliary group or a chiral catalyst.32,33 Alternatively, if an appropriate enantiopure reactant is used, stereochemical induction can in principle be achieved without the need for external aids.
Recently, our group reported on the synthesis of enantiopure dihydroxyethyl acetamidofuran (Di-HAF (1)),3 an unique enantiopure carbohydrate derived furan (Scheme 2). Application of this chiral pool building block is part of our ongoing research and we expected this electron rich diene to be well suited for Diels–Alder reactions. With its stereogenic center favorably positioned to induce π-facial selectivity, we were interested in its ability to transfer this chirality to the stereocenters formed in a Diels–Alder reaction. After this chirality transfer, the directing stereogenic center can be readily removed if desired, by oxidative vicinal diol cleavage, producing an aldehyde function.
 | ||
| Scheme 2 The synthesis of Di-HAF (1) from GlcNAc (7).3 | ||
Herein, we report the effective chirality transfer in glucosamine-derived furans via stereocenter induced π-facial selectivity in a Diels–Alder reaction.
Results and discussion
As the aim of our research included the preparation of versatile enantiopure amido-oxanorbornene based building blocks, we looked for an amine protecting group complementing the acetamide found in Di-HAF (1). The benzyloxycarbonyl (-Cbz) protecting group, removable via hydrogenolysis, was chosen. Glucosamine was reacted with benzyl chloroformate under standard conditions to afford N-benzyloxycarbonyl glucosamine (9)34 (Scheme 3). The dehydration procedure of N-acetyl glucosamine (7) developed by our group, was applied to this substrate. After adaptation of the work-up and purification protocol, Di-HCF (10) was isolated in high chemical- and enantiopurity (97.4% ee). Compared to an acyl group, a Cbz group can be cleaved under mild conditions, with a broad functional group tolerance. This characteristic makes the cycloaddition products of Di-HCF (10) more versatile as building blocks than their acetamide analogues, and therefore the Cbz-derivative was used for the Diels–Alder study discussed below. | ||
| Scheme 3 Synthesis of Di-HCF (10), the Cbz-analogue of Di-HAF (1). | ||
endo/exo-selectivity
We started our study with the examination of the endo-exo selectivity of the Diels–Alder reaction of Di-HCF (10) with substituted maleimides, which are reactive dienophiles. Because of the stereocenter in the starting material, both π-face products are formed as a set of diastereomers. Four different diastereomers can be formed, two with the endo-configuration and two with the exo-configuration (Scheme 4, left side). Our aim was to monitor the endo/exo-ratio by NMR, and to differentiate between the endo and exo products in the 1H-NMR spectrum, we resorted to the Karplus equation.35 The difference in dihedral angle between the ether bridgehead proton (red colored) and its adjacent proton (blue colored) gives rise to a distinct difference in coupling constant. This feature has been reported in literature for structurally similar compounds.29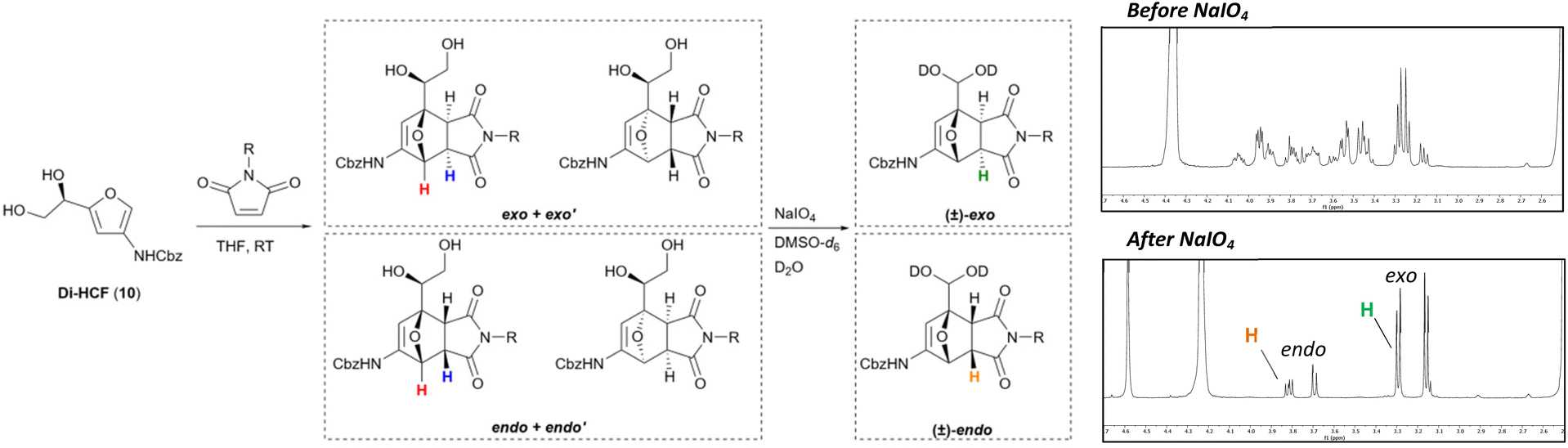 | ||
| Scheme 4 Diels–Alder reactions of Di-HCF (10) with substituted maleimides afford a mixture of four different diastereomers. Periodate oxidation simplifies the structure to facilitate NMR analysis. Top 1H-NMR spectrum: before periodate treatment, bottom 1H-NMR spectrum: after periodate treatment. | ||
The endo- and exo-isomers were separated by flash column chromatography, furnishing two sets of diastereomers with matching topology. 1H-NMR analysis was hampered by signal overlap, and to resolve this issue, the vicinal diol-moiety was oxidized with periodate, prior to analysis (Scheme 4, right side). The oxidation removed the interfering protons, along with the diastereogenic center, to furnish a set of racemates with significantly simplified spectra. In case of the exo-configuration, the dihedral angle between the bridgehead proton and the adjacent proton approximates 90°, therefore the Karplus equation predicts the 3J-coupling to be minimal, and the protons appear as a singlet and doublet (green colored, 3.3 ppm), respectively. In the endo-configuration, the dihedral angle leads to a larger 3J-coupling and a doublet (J = 5.4 Hz) and double doublet (J = 7.6, 5.4 Hz, orange colored, 3.8 ppm) are observed (all spectra can be found in the ESI†). After identification of the isomers, diagnostic signals in the spectra were assigned to the products before periodate treatment. We were fortunate to find that the proton of the primary alcohol of each diastereomer appeared as a triplet with a different chemical shift (Fig. 1). This proved to be a highly beneficial characteristic for both endo/exo- and facial selectivity determination. Since the absolute stereochemistry of the newly formed chiral centers could not be elucidated based on the NMR data alone, the endo and exo diastereomers were provisionally assigned the labels endo & endo′ and exo & exo′.
 | ||
| Fig. 1 The 1H-NMR signal used to determine the product distribution. The proton of the primary alcohol (highlighted in purple) has a different chemical shift for each of the four diastereomeric products. NMR solvent: DMSO-d6. | ||
Aided by this convenient analysis, a dienophile screening was performed and the effect of maleimide substitution on the product distribution was recorded (Scheme 4 and Table 1). The reactions were stirred for 24 h at room temperature, although reaction monitoring revealed that steady state was typically reached within the first hour. All dienophiles were modestly exo-selective, except for methylmaleimide (entry 2) that showed a ratio of 6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 in favor of the endo-isomer.
:4 in favor of the endo-isomer.
| No. | R |
endo:exob |
endo’:endob |
exo’:exob |
|---|---|---|---|---|
| Conditions: THF, 1 M of 10, RT, 24 h.a 66 °C.b Determined by 1H-NMR. | ||||
| 1 | H | 4:6 |
1.0:1 |
3.0:1 |
| 2 | Me | 6:4 |
1.1:1 |
2.8:1 |
| 3a | Me | 3:7 |
1.4:1 |
2.6:1 |
| 4 | t-Bu | 3:7 |
1.0:1 |
3.0:1 |
| 5a | t-Bu | 3:7 |
0.8:1 |
2.3:1 |
| 6 | Bn | 4:6 |
1.2:1 |
3.5:1 |
| 7a | Bn | 2:8 |
1.0:1 |
3.1:1 |
| 8 | Ph | 3:7 |
1.2:1 |
3.0:1 |
| 9a | Ph | 3:7 |
1.3:1 |
2.4:1 |
| 10 | p-OMe-Ph | 3:7 |
1.1:1 |
2.7:1 |
| 11 | p-NO2-Ph | 4:6 |
1.5:1 |
2.7:1 |
To obtain insight into the thermodynamics of the reaction, we probed its reversibility at different temperatures by 1H-NMR. Under thermodynamic control, e.g. when the reaction is reversible, the product mixture will eventually reach an equilibrium. Pure samples of the endo- and exo-isomers of the DA reactions with N-methylmaleimide and N-phenylmaleimide were dissolved in DMSO-d6 and kept at room temperature, 40 °C, 50 °C, 60 °C and 70 °C. After 24 h, the degree of retro reaction was determined, based on the amount of isomerization and formation of starting materials (Fig. 2) (for more details see ESI†). At room temperature, none of the isomers decreased in concentration. In other words, there is no equilibrium, and therefore the corresponding Diels–Alder reaction is under kinetic control. Above room temperature, the relative isomer concentrations decreased due to the backward reaction. In line with expectation, the retro-reaction accelerated as the temperature increased and was faster with endo-products than with exo-products. Based on these results we concluded that a thermodynamic equilibrium is in operation when the reaction is conducted above 50 °C.
 | ||
| Fig. 2 Reversibility experiments with the DA products of N-phenylmaleimide (left) and N-methylmaleimide (right) at different temperatures. The remaining isomer concentration was determined after 24 h. At room temperature the reaction is under kinetic control and above 40 °C a thermodynamic equilibrium sets in. | ||
The experiments with methyl-, tert-butyl-, benzyl- and phenylmaleimide were repeated in THF at reflux (66 °C). While the endo-exo ratio in the reaction with phenylmaleimide and tert-butylmaleimide remained unchanged (3:7, entries 5 and 9), methylmaleimide now also showed the same 3:7 product distribution (entry 3). The reaction with benzylmaleimide showed an increased exo-selectivity of 2:8 (entry 7). In all experiments, prolonged heating was accompanied with decomposition and after 48 h, accurate analysis was severely hampered by the interference of degradation products.
Next, we focused our attention on π-face selectivity (Scheme 5).
 | ||
| Scheme 5 π-facial selectivity in Di-HCF (10). | ||
π-Face selectivity
A clear difference in π-face selectivity was observed between the endo- and exo-products (Table 1). For the endo-products, π-facial differentiation was minimal. In contrast, a pronounced facial selectivity was found for the exo-products. At room temperature the exo′/exo-ratios were approximately 3:1. Notably, the selectivity slightly decreased at higher temperature. For further optimization of the π-facial selectivity, N-phenylmaleimide was used as dienophile. It is reported in literature that π-facial selectivity can be solvent dependent,36 and therefore a comprehensive solvent screening was performed (Table 2). With many common solvents, the π-face selectivity remained approximately 3:1, hardly influenced by solvent polarity and the protic or non-protic nature of the solvent. In chlorinated solvents (DCE, DCM, CHCl3, entries 9–11), however, a jump in selectivity was observed, with the highest selectivity found in chloroform (4.3:1, entry 11). In other chlorinated solvents (CCl4, trichlorotoluene, chlorobenzene) no reaction occurred, likely due to the poor solubility of the starting materials.
| No. | Solvent | Temp. | Ratio exo′:exo |
|---|---|---|---|
| Conditions: 0.75 M, 24 h.a Di-HCF (10) was sparingly soluble. | |||
| 1 | Tetrahydrofuran | RT | 3.0:1 |
| 2 | Water | RT | 3.0:1 |
| 3 | Acetonitrile | RT | 2.9:1 |
| 4 | 1,4-Dioxane | RT | 2.9:1 |
| 5 | Ethyl acetate | RT | 3.1:1 |
| 6 | 2-Methyltetrahydrofuran | RT | No reactiona |
| 7 | t-Butyl methyl ether | RT | 2.8:1 |
| 8 | Toluene | RT | No reactiona |
| 9 | 1,2-Dichloroethane | RT | 3.7:1 |
| 10 | Dichloromethane | RT | 4.1:1 |
| 11 | Chloroform | RT | 4.3:1 |
| 12 | Tetrachloromethane | RT | No reactiona |
| 13 | Chlorobenzene | RT | No reactiona |
| 14 | α,α,α-Trichlorotoluene | RT | No reactiona |
| 15 | 2,2,2-Trifluoro ethanol | RT | 3.2:1 |
| 16 | Chloroform | 0 °C | 5.7:1 |
| 17 | Chloroform | −10 °C |
6.5:1
|
| 18 | Chloroform | −20 °C | No reaction |
As earlier experiments at elevated temperatures showed reduced π-facial selectivity, the temperature of the reaction in chloroform was stepwise lowered, which ultimately led to a high π-face selectivity of 6.5:1 at −10 °C (entry 17). This corresponds to an overall NMR-yield of the major diastereomer (exo′) of 65% (Fig. 3).
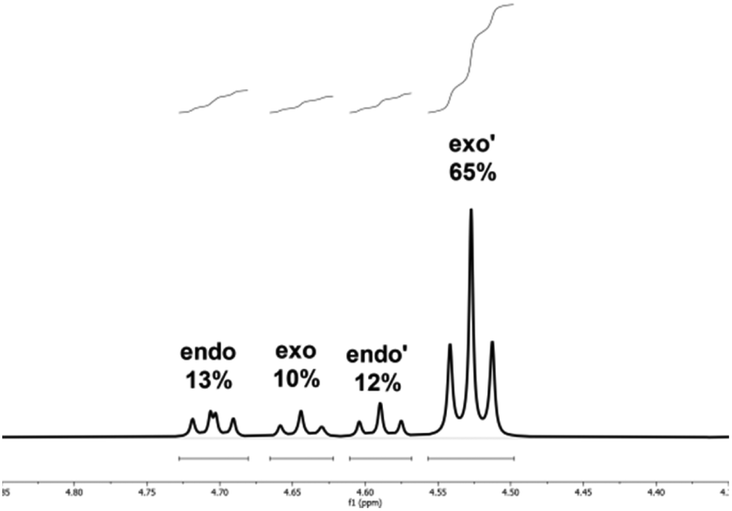 | ||
| Fig. 3 1H-NMR spectrum of the highest π-face selectivity observed in chloroform at −10 °C. With a 65% overall NMR yield of the major exo-isomer. | ||
With π-face selectivity achieved, we set out to synthesize the major product on a preparative scale and to resolve the absolute stereochemistry by X-ray diffraction. Facile synthetic availability and full structure elucidation are key to enable the future application of the DA product as chiral synthon. Furthermore, the absolute configuration could provide clues to get an understanding of the driving force behind the chiral induction.
The major product of the Diels–Alder reaction with phenylmaleimide could easily be isolated by chromatography. A mobile phase of dichloromethane and methanol was crucial for peak separation. However, during manipulation and concentration of the column fractions, methanol addition to the enamide function in 11 occurred (Scheme 6). A similar side reaction of alcohol nucleophiles on amido-oxanorbornenes has been reported by Bridson et al.37 Removing the methanol from the collected column eluent by washing with water, completely suppressed this unwanted reaction. The major exo′-diastereomer was isolated on preparative scale in 58% yield (2.8 g). Because all attempts to crystallize the exo′-product were unsuccessful, a crystalline derivative was synthesized as shown in Scheme 7.
 | ||
| Scheme 6 Methanol addition to amino-oxanorbornene 11. | ||
 | ||
| Scheme 7 Synthesis of crystalline derivative 16 to enable X-ray diffraction. | ||
The oxanorbornene (14) was hydrogenated to afford oxanorbornane 15. This modification both blocked the retro-cycloaddition pathway and the addition of nucleophiles. Hydrogenation with platinum selectively reduced the olefin and a new stereocenter was formed exclusively in the endo-configuration, bringing the chirality-transfer-sum to a total of five new stereocenters. To obtain suitable crystals for X-ray diffraction, different forms of derivatization were explored. Hydrazone synthesis, via the corresponding aldehyde, and salt formation with the deprotected amine were fruitless. Ultimately, benzoylation of the primary alcohol afforded derivative 16, that was successfully crystallized from ethanol. The obtained crystals were fine needles and diffracted relatively weak. Therefore, data collection was only performed up to theta 25 degrees and, ultimately the structure was successfully solved (Fig. 4). Details about the crystal structure determination and crystallographic data for 16 can be found in the ESI†.
 | ||
| Fig. 4 X-Ray structure of compound 16. | ||
Based on the resolved structure, we deduced that addition of the dienophile takes place preferentially at the alpha face of the furan (Scheme 8). This result is consistent with the trend reported in literature of Diels–Alder reactions with dienes that have stereogenic groups on the allylic position. An oxygen bearing allylic chiral center with R-configuration, typically directs dienophile attack to the Re-face of the diene,38 equivalent to the alpha face of our furan. Although π-facial selectivity has been the subject of numerous studies,39 there is no consensus regarding the driving force behind facial selectivity.40,41 Often the effect is rationalized with a combination of electrostatic-, secondary orbital- and steric interactions. In this specific case, the model proposed by Cieplak offers an adequate explanation. It postulates that each diastereomeric transition state is stabilized to varying degrees by electron donation from the antiperiplanar σ-bond into the σ*‡ of the newly formed bond. The stabilization is governed by the hyperconjugative capabilities of the substituents on the furanylic position (σC–H > σC–C > σC–O) in combination with the conformation of the transition state.42,43 Due to free rotation, the stereocenter can adopt various conformations with the σ-donor in different orientations relative to the π-plane. For optimal stabilization the strongest donor (σC–H) needs to be aligned parallel with the neighboring σ*‡-orbital, which is likely one of the major contributing conformers in alpha-face attack (Fig. 5A). In this conformation the transition state can be optimally stabilized by electron donation from σC–H, assisted by lone-pair back-donation from the oxygen. Based on the experimental results we can conclude that the transition states of the other diastereomeric products are not stabilized to the same extent. If the energy penalty of a combination of steric interactions between the reactants and torsional strain within the diene is larger than the possible hyperconjugative effect, the transition state may be forced into a conformer less suitable for hyperconjugation (Scheme 5B).
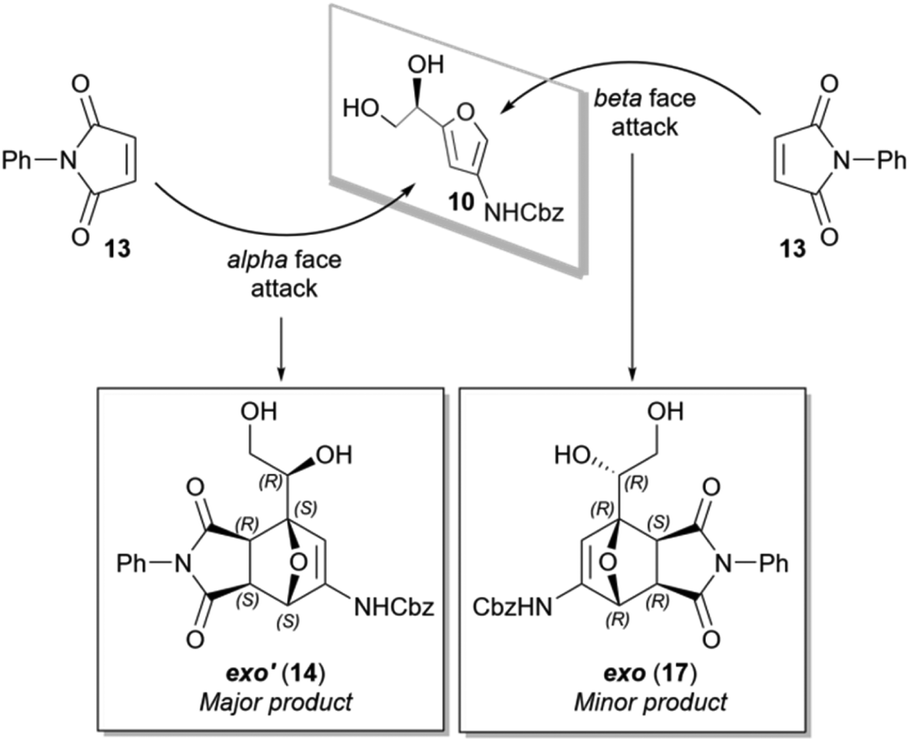 | ||
| Scheme 8 π-facial diastereomeric induction results in preferred alpha face addition of the dienophile. | ||
 | ||
| Fig. 5 (A) Transition state stabilization via electron donation of the σ-bond (σC–H) into the σ*‡ of the newly formed bond (red arrow) and lone-pair back donation (blue arrow). (B) Steric interactions and torsional strain force the transition state in a conformer with less favourable orbital overlap for hyperconjugation. | ||
Temperature dependence of the selectivity is easily explained by basic chemical kinetics, at lower temperature the transition state with the lower activation energy is favored. Meanwhile, we can only speculate on the exact role of chlorinated solvents. Although, as such solvents possess a unique set of polarity descriptors44 they may also play a role in stabilizing the contributing conformer for π-facial selectivity.
To complete the study, we applied the optimized conditions to N-acetyl glucosamine derived Di-HAF (1). Because of the low solubility of Di-HAF (1), DMSO was added as co-solvent. Compared to Di-HCF (10), a slightly lower facial selectivity was observed; 4:1 instead of 6.5:1. However, this was compensated by a higher exo-selectivity (2:8) which ultimately led to a similar isolated yield of 59% of 18 (Scheme 9).
 | ||
| Scheme 9 Diels–Alder cycloaddition of Di-HAF (1) and N-phenylmaleimide in chloroform-DMSO at −10 °C. endo/exo-ratio: 2:8 and exo-facial selectivity: 4:1, isolated yield of major isomer: 59%. | ||
Conclusions
In this study, we demonstrate efficient chirality-transfer in a Diels–Alder reaction with carbohydrate-based furan Di-HCF (10) and maleimides. A solvent and temperature dependence was found, and after optimization an exo′/exo-selectivity of 6.5:1 was obtained in combination with an endo/exo-selectivity of 3:7. Ultimately the multifunctional enantiopure amido-oxanorbornene (14) building block was synthesized in 58% yield. Subsequent stereoselective hydrogenation brought the chirality-transfer-sum to a total of five new stereocenters and afforded a chemically stable 7-oxanorbornane derivative (15). Based on the structure resolved by X-ray crystallography, the major product from the Diels–Alder reaction is the result of dienophile addition to the alpha face of the diene. In addition, the optimized Diels–Alder reaction conditions were also applied to Di-HAF (1) to afford the acyl analogue 18 in 59% yield.
The results show that Diels–Alder reactions with chiral carbohydrate-based furans, which up until now provided mixtures of stereoisomers, can be steered to favor one product. This is a conditio sine qua non for application in fine chemicals and medicinal chemistry. The next step will be to access also other stereoisomers, produced in this reaction. The current results provide a good starting point for such a study.
Author contributions
All authors took part in the conceptualization of this study. C. van der Loo and R. Schim van der Loeff carried out the experimental work and A. Martín and P. Gomez-Sal the crystallography. C. van der Loo and A. Minnaard wrote the original draft of the manuscript. All authors contributed to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The CCC Carbobased program is acknowledged for funding. We thank the analytical department of Symeres Groningen for analytical support, in particular chiral HLPC, and R. Sneep (University of Groningen) for HRMS analysis.References
- P. Gallezot, Chem. Soc. Rev., 2012, 41, 1538–1558 RSC.
- F. M. Kerton, Y. Liu, K. W. Omari and K. Hawboldt, Green Chem., 2013, 15, 860–871 RSC.
- C. H. M. Van Der Loo, M. L. G. Borst, K. Pouwer and A. J. Minnaard, Org. Biomol. Chem., 2021, 19, 10105–10111 RSC.
- X. Chen, S. L. Chew, F. M. Kerton and N. Yan, Green Chem., 2014, 16, 2204–2212 RSC.
- K. W. Omari, L. Dodot and F. M. Kerton, ChemSusChem, 2012, 5, 1767–1772 CrossRef CAS PubMed.
- D. Padovan, H. Kobayashi and A. Fukuoka, ChemSusChem, 2020, 13, 3594–3598 CrossRef CAS PubMed.
- A. D. Sadiq, X. Chen, N. Yan and J. Sperry, ChemSusChem, 2018, 11, 532–535 CrossRef CAS PubMed.
- T. T. Pham, X. Chen, T. Söhnel, N. Yan and J. Sperry, Green Chem., 2020, 22, 1978–1984 RSC.
- J. C. Neville, M. Y. Lau, T. Söhnel and J. Sperry, Org. Biomol. Chem., 2022, 20, 6562–6565 RSC.
- O. Diels and K. Alder, Justus Liebigs Ann. Chem., 1928, 460, 98–122 CrossRef CAS.
- O. Diels and K. Alder, Ber. Dtsch. Chem. Ges., 1929, 62, 554–562 CrossRef.
- C. O. Kappe, S. S. Murphree and A. Padwa, Tetrahedron, 1997, 53, 14179–14233 CrossRef CAS.
- P. Vogel, J. Cossy, J. Plumet and O. Arjona, Tetrahedron, 1999, 55, 13521–13642 CrossRef CAS.
- C. E. Puerto Galvis, L. Y. Vargas Méndez and V. V. Kouznetsov, Chem. Biol. Drug Des., 2013, 82, 477–499 CrossRef CAS PubMed.
- A. D. Pehere, S. Xu, S. K. Thompson, M. A. Hillmyer and T. R. Hoye, Org. Lett., 2016, 18, 2584–2587 CrossRef CAS PubMed.
- M. Shiramizu and F. D. Toste, Chem. – Eur. J., 2011, 17, 12452–12457 CrossRef CAS PubMed.
- C. L. Williams, C. C. Chang, P. Do, N. Nikbin, S. Caratzoulas, D. G. Vlachos, R. F. Lobo, W. Fan and P. J. Dauenhauer, ACS Catal., 2012, 2, 935–939 CrossRef CAS.
- S. Calvo-Losada and D. Suárez, J. Am. Chem. Soc., 2000, 122, 390–391 CrossRef CAS.
- J. G. Martin and R. K. Hill, Chem. Rev., 1961, 61, 537–562 CrossRef CAS.
- V. K. Yadav, D. L. V. K. Prasad, A. Yadav and K. Yadav, J. Phys. Org. Chem., 2021, 34, 1–8 Search PubMed.
- R. C. Cioc, M. Crockatt, J. C. van der Waal and P. C. A. Bruijnincx, Angew. Chem., Int. Ed., 2022, 61, e202114720 CrossRef CAS PubMed.
- R. C. Boutelle and B. H. Northrop, J. Org. Chem., 2011, 76, 7994–8002 CrossRef CAS PubMed.
- F. A. Kucherov, L. V. Romashov, G. M. Averochkin and V. P. Ananikov, ACS Sustainable Chem. Eng., 2021, 9, 3011–3042 CrossRef CAS.
- K. I. Galkin and V. P. Ananikov, Int. J. Mol. Sci., 2021, 22, 11856 CrossRef CAS PubMed.
- J. M. J. M. Ravasco and R. F. A. Gomes, ChemSusChem, 2021, 14, 3047–3053 CrossRef CAS PubMed.
- C. S. Santos, R. R. Mattioli, J. S. Baptista, V. H. M. Da Silva and J. C. Pastre, ChemRxiv, 2022, DOI:10.26434/chemrxiv-2022-9ld3l.
- I. Scodeller, S. Mansouri, D. Morvan, E. Muller, K. de Oliveira Vigier, R. Wischert and F. Jérôme, Angew. Chem., Int. Ed., 2018, 57, 10510–10514 CrossRef CAS PubMed.
- I. van Scodeller, K. De Oliveira Vigier, E. Muller, C. Ma, F. Guégan, R. Wischert and F. Jérôme, ChemSusChem, 2021, 14, 313–323 CrossRef CAS PubMed.
- R. C. Cioc, M. Lutz, E. A. Pidko, M. Crockatt, J. C. Van Der Waal and P. C. A. Bruijnincx, Green Chem., 2021, 23, 367–373 RSC.
- J. G. Pereira, J. M. J. M. Ravasco, J. R. Vale, F. Queda and R. F. A. Gomes, Green Chem., 2022, 2022, 7131–7136 RSC.
- N. Uemura, S. Toyoda, H. Ishikawa, Y. Yoshida, T. Mino, Y. Kasashima and M. Sakamoto, J. Org. Chem., 2018, 83, 9300–9304 CrossRef CAS PubMed.
- K. Shibatomi, F. Kobayashi, A. Narayama, I. Fujisawa and S. Iwasa, Chem. Commun., 2012, 48, 413–415 RSC.
- D. H. Ryu, K. H. Kim, J. Y. Sim and E. J. Corey, Tetrahedron Lett., 2007, 48, 5735–5737 CrossRef CAS.
- E. Kamst, K. Zegelaar-Jaarsveld, G. A. Van Der Marel, J. H. Van Boom, B. J. J. Lugtenberg and H. P. Spaink, Carbohydr. Res., 1999, 321, 176–189 CrossRef CAS.
- M. Karplus, J. Am. Chem. Soc., 1963, 85, 2870–2871 CrossRef CAS.
- W. H. Watson, J. Galloy, P. D. Bartlett and A. A. M. Roof, J. Am. Chem. Soc., 1981, 103, 2022–2031 CrossRef CAS.
- J. N. Bridson, Can. J. Chem., 1979, 57, 314–317 CrossRef CAS.
- R. Tripathy, R. W. Franck and K. D. Onan, J. Am. Chem. Soc., 1988, 110, 3257–3262 CrossRef CAS.
- J. B. Macaulay and A. G. Fallis, J. Am. Chem. Soc., 1990, 112, 1136–1144 CrossRef CAS.
- M. Ishida and S. Inagaki, Orbitals Chem., 2009, 289, 183–218 CAS.
- M. Ishida, H. Kobayashi, S. Tomohiro and S. Inagaki, J. Chem. Soc., Perkin Trans. 2, 2000, 1625–1630 RSC.
- A. S. Cieplak, Chem. Rev., 1999, 99, 1265–1336 CrossRef CAS PubMed.
- A. S. Cieplak, B. D. Tait and C. R. Johnson, J. Am. Chem. Soc., 1989, 111, 8447–8462 CrossRef CAS.
- A. Jordan, P. Stoy and H. F. Sneddon, Chem. Rev., 2021, 121, 1582–1622 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2223346. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2ob02221d |
| This journal is © The Royal Society of Chemistry 2023 |