
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Catalyst-free thiophosphorylation of in situ formed ortho-quinone methides†
Jeffrey
Ash
and
Jun Yong
Kang
 *
*
Department of Chemistry and Biochemistry, University of Nevada Las Vegas, 4505 S. Maryland Parkway, Las Vegas, Nevada 89154-4003, USA. E-mail: junyong.kang@unlv.edu
First published on 23rd February 2023
Abstract
A metal-, chloride reagent and base-free thiophosphorylation reaction of in situ formed ortho-quinone methide (o-QM) to synthesize functionalized thiophosphates has been developed. The reaction is an atom-economical process, producing water as the sole byproduct. (EtO)2P(O)SH functions as both a Brønsted acid and nucleophilic thiolate to produce the o-QM intermediate and the thiophosphate product, respectively. The aza o-QMs were also successfully thiophosphorylated in the presence of catalytic TsOH to form sulfonamido thiophosphates.
Introduction
Over the past few decades, the organophosphorus chemistry field has witnessed a growing research interest since organophosphorus compounds have found widespread applications in medicinal chemistry, organometallic chemistry, agricultural chemistry, and materials chemistry.1 Specifically, phosphorodithioate compounds bearing the thiophosphoryl bond have useful properties such as insecticides (Fig. 1, A and B) and neurotoxins (Fig. 1, C).2 They also display important biological activity for the treatment of cancer (Fig. 1, D) and glaucoma (Fig. 1, E).3 In addition, thiophosphates have been known for significant antibacterial activities against common strains of bacteria (Fig. 1, F).4 | ||
| Fig. 1 Applications of phosphorodithioate and thiophosphate. | ||
With a plethora of applications, various methods have been developed to access thiophosphoryl bond motifs. A conventional method to synthesize thiophosphate uses electrophilic P(V) or P(III) compounds such as chlorophosphine oxides and chlorophosphines which undergo a nucleophilic substitution reaction with thiols to produce thiophosphates.5 Another approach to thiophosphate synthesis employs cross dehydrogenative coupling (CDC) reactions which couple a thiol and pentavalent phosphorous reagent in the presence of a metal catalyst. This CDC synthetic methodology has been extensively explored and various catalysts/activators such as Ni, Cu, Fe, Cs, Pd, NCS, peroxides, Bunte salts, and quaternary ammonium salts have been demonstrated.6 Photocatalysis and electrochemical process have also been applied to the synthesis of thiophosphates via coupling reactions.7 Nevertheless, catalyst-free methods of thiophosphate synthesis are underdeveloped with a few precedents: a reaction of disulfides with secondary phosphine oxides in the presence of silica gel via a radical pathway and the direct substitution reaction between N-chalcogenoimides and diethyl phosphites.8 These methods, however, are limited to a trivalent phosphorus tautomer as an active nucleophile to react with electrophilic sulfur reagents.9
An alternative method uses (EtO)2P(O)SH as a pentavalent nucleophile and a synthon for the thiophosphate group (Scheme 1). (EtO)2P(O)SH has been utilized in a Michael reaction of activated alkenes to synthesize functionalized thiophosphates at elevated temperatures (Scheme 1a).10 In addition, the Wu group demonstrated a Ga(OTf)3-catalyzed sulfur substitution reaction on activated alcohols (benzylic or allylic alcohols) with hydrogen phosphorothioates (EtO)2P(O)SH to generate the thiophosphate compounds (Scheme 1b).11 They also reported a photochemical method for the synthesis of thiophosphate compounds.12 The Xiao group also explored the utility of (EtO)2P(O)SH by coupling with a propargylic alcohol partner to construct S-(2H-chromen-4-yl) phosphorothioates via a cascade reaction and the synthesis of allenyl thiophosphates under elevated thermal conditions (Scheme 1c and d).13
 | ||
| Scheme 1 S–C bond formation using (EtO)2P(O)SH. | ||
Reactive o-QM intermediates can be generated under thermal, photochemical, acidic, or basic conditions and they have been employed in various addition reactions.14 For example, phosphorylation of o-QM and p-QM using secondary phosphine oxides and H-phosphonates was recently reported.15 In addition, enantioselective phosphorylation reactions of o-QM and aza o-QM using bifunctional cinchona catalysts were released.16 In 2017, we also disclosed the phospha-Michael addition reaction of trialkylphosphites to in situ generated o-QMs using N-heterocyclic phosphorodiamidic acid (NHPA) catalyst. This transformation demonstrated the formation of carbon–phosphorous bonds employing trivalent phosphorus nucleophiles.17 Sulfa-Michael addition reaction of thiophosphates to o-QMs for the construction of carbon–sulfur bond, however, has remained unexplored. This thiophosphorylation reaction of o-QM revealed a dual role of phosphorothioic acid, (EtO)2P(O)SH; a Brønsted acid and thiolate P(V) nucleophile, unrealized yet in o-QM chemistry.
Results and discussion
To develop a mild, atom-efficient thiophosphorylation of o-QM that avoids a toxic metal, moisture-sensitive chloride reagents, and base, we hypothesized that the phosphorothioic acid, (EtO)2P(O)SH, can serve as both a Brønsted acid to generate o-QMs and a phosphorothioate nucleophile to react with the o-QMs for the synthesis of functionalized diaryl thiophosphates. To test our hypothesis, we used 2-(hydroxy(phenyl)methyl)phenol 1a and (EtO)2P(O)SH 2a as a model substrate for the reaction optimization (Table 1). The reaction was first tested using a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar equivalent of 1a:2a and product 3a was generated in 37% yield (Table 1, entry 1). Next, an increment in the molar ratio of 1a:2a to 1:1.5 afforded the desired product 3a in 91% yield (Table 1, entry 2). Solvent effects were then examined and other solvents (THF, DCE, toluene, ACN, and ether) were inferior to DCM (Table 1, entries 3–7). Finally, the reaction can be performed under neat conditions but a lower yield of 44% was observed (Table 1, entry 8).
:1 molar equivalent of 1a:2a and product 3a was generated in 37% yield (Table 1, entry 1). Next, an increment in the molar ratio of 1a:2a to 1:1.5 afforded the desired product 3a in 91% yield (Table 1, entry 2). Solvent effects were then examined and other solvents (THF, DCE, toluene, ACN, and ether) were inferior to DCM (Table 1, entries 3–7). Finally, the reaction can be performed under neat conditions but a lower yield of 44% was observed (Table 1, entry 8).

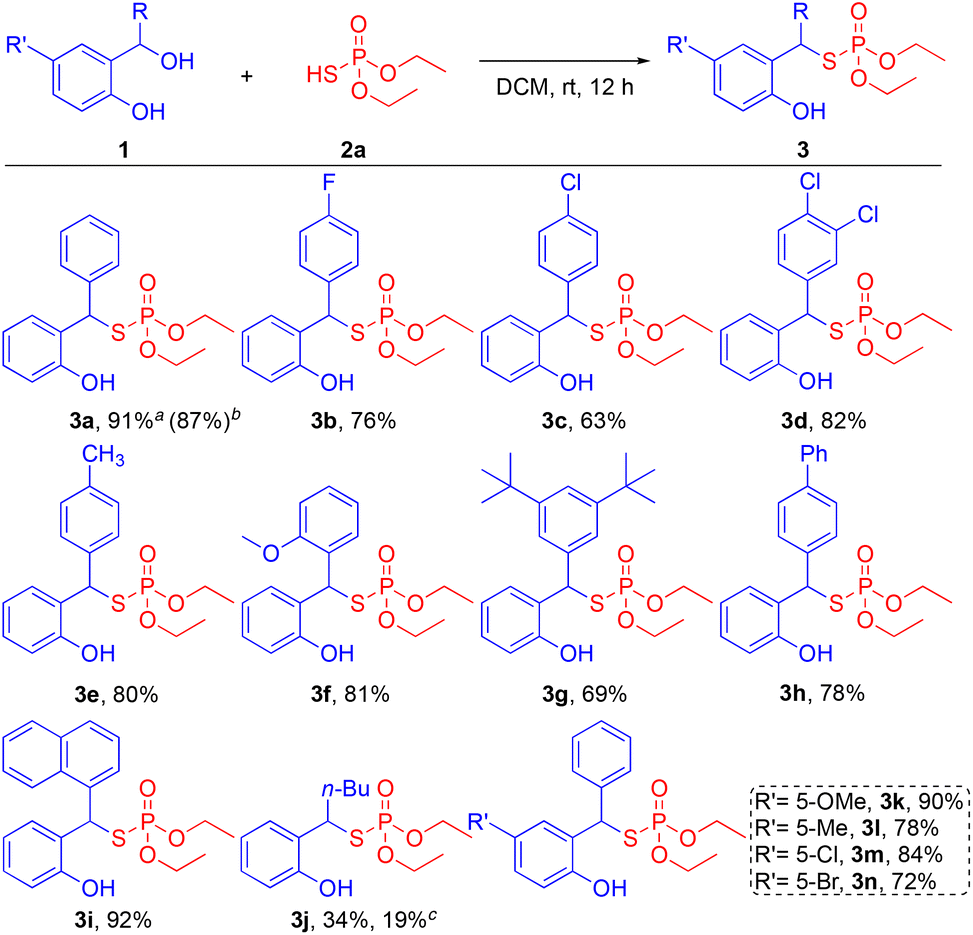
With the optimized conditions in hand, the scope of diaryl alcohols was examined for the steric and electronic effects on reaction outcomes (Scheme 2). First, various substituents on the benzylic carbon of the diaryl alcohol substrates were screened. Halogenated diaryl alcohols 1b–1d (4-F, 4-Cl, and 3,4-diCl) were well tolerated to give the desired products 3b–3d in moderate to high yields (63–82%). Diaryl alcohols containing electron-donating groups 1e–1g (4-Me, 2-MeO, and 3,5-tert-Bu) furnished the target products 3e–3g in high yields (69–81%). Poly aromatic groups on diaryl alcohols 1h and 1i (biphenyl and naphthyl) also smoothly generated the desired products 3h and 3i with 78% and 92% yields, respectively. Aliphatic-substituted phenol 1j (n-Bu), however, provided the corresponding product 3j in a low yield of 34%, presumably due to the reduced stabilization of the o-QM intermediate. To inquire about the effect of water molecules in the reaction, a reaction with molecular sieves was performed. The use of molecular sieves, however, did not improve the yield (19%) of 3j. This result suggests that the water in fact may help to stabilize the o-QM intermediate through intermolecular hydrogen bonding.18 The positive effect of in situ generated water molecule on the stabilization of o-QM intermediate and the product yield is aligned with our observation on the phospha-Michael reaction.17 In addition, various substituents on the phenol motif were examined for the substrate scope of the reaction. Electron donating groups on the phenols 1k and 1l (5-OMe and 5-Me) were smoothly tolerated to provide the corresponding products 3k and 3l in high yields of 90% and 78%, respectively. Phenols with halogenated substrates 1m and 1n (5-Cl and 5-Br) also afforded the desired products 3m and 3n in 84% and 72% yields, respectively. Furthermore, to demonstrate the scalability of this reaction and its applicability in pharmaceutical processes, a scaled-up experiment with 1a (4.5 mmol) was demonstrated without sacrificing the reactivity by providing 3a in 87% yield.
 | ||
| Scheme 2 Substrate scope of thiophosphorylation reaction. Reaction conditions: 1 (0.1 mmol) and 2a (0.15 mmol) in DCM (0.5 mL) for 12 h. aIsolated yield. bA gram-scale experiment with 1a (4.5 mmol).cAddition of molecular sieves. | ||
Next, thioacids with different alkoxy substituents were evaluated to test their effects on reaction outcomes (Scheme 3). Various alkoxy-substituted thioacids 2b and 2c (n-Bu and i-Pr) smoothly provided the target compounds 4a and 4b in high yields of 82% and 93%, respectively. In addition, diphenyl thiophosphinic acid 2d also gave the desired product 4c in a synthetically useful yield of 51%. These results indicate that the structural variation of thioacids is tolerable.
 | ||
| Scheme 3 Substrate scope of thioacid nucleophiles. Reaction conditions: 1a (0.1 mmol) and 2 (0.15 mmol) in DCM (0.5 mL) for 12 h. aIsolated yield. | ||
With promising results of sulfa-Michael addition reaction of thiophosphates to o-QMs, we were interested in applying this method to aza o-QM intermediates (Scheme 4). In our preliminary screening, it was found that (EtO)2P(O)SH 2a was not a strong enough acid to dehydrate sulfonamido alcohol 5a. With the addition of a 10 mol% TsOH, the reaction proceeded efficiently giving 6a in 90% yield. Having the optimized reaction conditions, the substrate scope was tested with various benzylic aryl substituents. Sulfonamindo alcohols bearing electron-donating groups 5b and 5c (4-Me and 4-OMe) furnished the target products 6b and 6c in 90% and 82% yields, respectively. Next, halogenated sulfonamido alcohols 5d and 5e (4-F and 4-Cl) were examined and they provided the desired products 6d and 6e with 81% and 78% yields, respectively. In addition, polyaryl sulfonamido alcohol 5f generated the naphthyl sulfonamido thiophosphate product 6f with 74% yield. Furthermore, a different protecting group on the nitrogen atom (Bz) 5g was investigated, but it provided 6g in a low yield (34%), presumably due to the weak polarizability of the o-QM intermediate compared to a tosyl group on the nitrogen atom.19 Finally, alkyl-protecting groups on the amine 5h–5j (benzyl, methyl, and allyl) were screened. It was, however, revealed that the final products 6h–6j were unstable and rapidly decomposed after isolation. It is noteworthy to mention that attempts to deprotect the tosyl group on the amine moiety 6a under both basic conditions (4-OMePhSH/DIPEA)20 and acidic conditions (TFA)21 were unsuccessful, leaving the decomposition of the substrates.
 | ||
| Scheme 4 Substrate Scope of sulfonamido thiophosphate synthesis. Reaction conditions: 5 (0.1 mmol), 2a (0.2 mmol), and TsOH (10 mol%) in DCM (0.5 mL) for 12 h. aIsolated yield. bND (not determined due to instability). | ||
On the basis of our experimental data and previous work,17 a plausible mechanistic pathway is proposed (Scheme 5). Diaryl alcohol 1a is protonated by (EtO)2P(O)SH 2a and then water is released to form o-QM intermediate and O,O-diethyl phosphorothioate. Subsequently, sulfa-Michael addition reaction of o-QM with O,O-diethyl phosphorothioate provides the addition product 3a.
 | ||
| Scheme 5 Proposed mechanism. | ||
To rationalize the proposed mechanism of this thiophosphorylation reaction, control experiments were performed to gain a mechanistic perspective on this synthetic transformation (Scheme 6). Diaryl alcohol 1a was treated with (EtO)2P(O)SH 2a and (PhO)2P(O)OH 2aa, and the reaction provided the thiophosphorylation product 3a in 83% yield (Scheme 6, eqn (1)). This competition reaction generated only the phosphorothioate product. In another control experiment, the reaction of diaryl alcohol 1a with diphenylphosphoric acid 2aa did not generate the desired product which suggests that diphenylphosphoric acid is not acidic enough to generate the o-QM intermediate under the reaction conditions (Scheme 6, eqn (2)). These reaction outcomes support the dual role of phosphorothioic acid 2a as a Brønsted acid and thioate nucleophile; these results also suggest that phosphorothioic acid 2a is a better nucleophile than the diphenylphosphoric acid 2aa. It is noteworthy that phosphorothioic acid (pKa = 1.0) is more acidic than phosphoric acid is (pKa = 3.88).22 Additionally, the reaction of benzyl alcohol 1aa or diphenyl methanol 1ab with phosphorothioic acid 2a did not afford the target thiophosphate product (Scheme 6, eqn (3) and (4)). Therefore, these results indicate that a reaction mechanism involving carbocation intermediates is an unlikely pathway.
 | ||
| Scheme 6 Control experiments. | ||
Conclusions
We have developed a metal-, catalyst-, chloride reagent-, and base-free thiophosphorylation reaction of o-QM to synthesize functionalized thiophosphates. The reaction tolerates a wide range of functional groups, proceeds under environmentally benign conditions, and fulfills an atom-economical process. It also demonstrated the dual role of phosphorothioic acid as a Brønsted acid and a thiolate nucleophile in o-QM chemistry. Future experiments involving cascade and multicomponent reactions to harness the dual role of phosphorothioic acid, (EtO)2P(O)SH, are underway and will be reported in due course.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Knowledge Fund which is administered by the Nevada Governor's Office of Economic Development (GOED) and University of Nevada Las Vegas. Liangqiao Bian at SCAAC is acknowledged for mass spectra data.References
- (a) S. Demkowicz, J. Rachon, M. Daśko and W. Kozak, RSC Adv., 2016, 6, 7101–7112 RSC; (b) T. Baumgartner and R. Réau, Chem. Rev., 2006, 106, 4681–4727 CrossRef CAS PubMed; (c) M. Eto, Organophosphorous pesticides, CRC Press, Cleveland, 1974 Search PubMed; (d) C. Xie, A. J. Smaligo, X.-R. Song and O. Kwon, ACS Cent. Sci., 2021, 7, 536–558 CrossRef CAS PubMed; (e) P.-H. Leung, Acc. Chem. Res., 2004, 37, 169–177 CrossRef CAS PubMed; (f) V. Iaroshenko, Organophosphorus Chemistry: From Molecules to Applications, John Wiley & Sons, 2019 CrossRef; (g) F. Palacios, C. Alonso and J. M. de los Santos, Chem. Rev., 2005, 105, 899–932 CrossRef CAS PubMed; (h) M. Wehbi, A. Mehdi, C. Negrell, G. David, A. Alaaeddine and B. Améduri, ACS Appl. Mater. Interfaces, 2020, 12, 38–59 CrossRef CAS PubMed.
- (a) Y. Fan, K. Lai, B. A. Rasco and Y. Huang, Food Control, 2014, 37, 153–157 CrossRef CAS; (b) H. Shapiro and S. Micucci, Can. Med. Assoc. J., 2003, 168, 1427–1430 Search PubMed; (c) E. R. White, K. M. Al-Adil, W. L. Winterlin and W. W. Kilgore, J. Agric. Food Chem., 1972, 20, 1184–1186 CrossRef CAS PubMed; (d) K. Sinderhauf and W. Schwack, J. Labelled Compd. Radiopharm., 2004, 47, 509–512 CrossRef CAS; (e) C. E. Berkman, C. M. Thompson and S. R. Perrin, Chem. Res. Toxicol., 1993, 6, 718–723 Search PubMed.
- (a) B. A. T. Gabelt, E. A. Hennes, J. L. Seeman, B. Tian and P. L. Kaufman, Invest. Ophthalmol. Visual Sci., 2004, 45, 2732–2736 CrossRef PubMed; (b) J. R. Kouvaris, V. E. Kouloulias and L. J. Vlahos, Oncologist, 2007, 12, 738–747 CrossRef CAS PubMed; (c) S. Song, Y. Zhang, A. Yeerlan, B. Zhu, J. Liu and N. Jiao, Angew. Chem., Int. Ed., 2017, 56, 2487–2491 CrossRef CAS PubMed; (d) C. L. Kraus, R. H. Trivedi and M. E. Wilson, J. AAPOS, 2015, 19, 116–118 CrossRef PubMed.
- (a) S. Mitra, S. Mukherjee, S. K. Sen and A. Hajra, Bioorg. Med. Chem. Lett., 2014, 24, 2198–2201 CrossRef CAS PubMed; (b) H. Huang, J. Denne, C.-H. Yang, H. Wang and J. Y. Kang, Angew. Chem., Int. Ed., 2018, 57, 6624–6628 CrossRef CAS PubMed.
- X. Bi, J. Li, F. Meng, H. Wang and J. Xiao, Tetrahedron, 2016, 72, 706–711 CrossRef CAS.
- (a) X.-Y. Chen, M. Pu, H.-G. Cheng, T. Sperger and F. Schoenebeck, Angew. Chem., Int. Ed., 2019, 58, 11395–11399 CrossRef CAS PubMed; (b) H. Huang, J. Ash and J. Y. Kang, Org. Biomol. Chem., 2018, 16, 4236–4242 RSC; (c) B. Kaboudin, Y. Abedi, J.-y. Kato and T. Yokomatsu, Synthesis, 2013, 2323–2327 CrossRef CAS; (d) T. Liu, Y. Zhang, R. Yu, J. Liu and F. Cheng, Synthesis, 2020, 253–262 CAS; (e) Y.-C. Liu and C.-F. Lee, Green Chem., 2014, 16, 357–364 RSC; (f) C. Min, R. Zhang, Q. Liu, S. Lin and Z. Yan, Synlett, 2018, 2027–2030 CAS; (g) S. Song, Y. Zhang, A. Yeerlan, B. Zhu, J. Liu and N. Jiao, Angew. Chem., Int. Ed., 2017, 56, 2487–2491 CrossRef CAS PubMed; (h) J. Wang, X. Huang, Z. Ni, S. Wang, J. Wu and Y. Pan, Green Chem., 2015, 17, 314–319 RSC; (i) C. Wen, Q. Chen, Y. Huang, X. Wang, X. Yan, J. Zeng, Y. Huo and K. Zhang, RSC Adv., 2017, 7, 45416–45419 RSC; (j) J. Xu, L. Zhang, X. Li, Y. Gao, G. Tang and Y. Zhao, Org. Lett., 2016, 18, 1266–1269 CrossRef CAS PubMed; (k) J.-W. Xue, M. Zeng, S. Zhang, Z. Chen and G. Yin, J. Org. Chem., 2019, 84, 4179–4190 CrossRef CAS PubMed; (l) L. Zhang, P. Zhang, X. Li, J. Xu, G. Tang and Y. Zhao, J. Org. Chem., 2016, 81, 5588–5594 CrossRef CAS PubMed; (m) P. Zhang, G. Yu, W. Li, Z. Shu, L. Wang, Z. Li and X. Gao, Org. Lett., 2021, 23, 5848–5852 CrossRef CAS PubMed; (n) X. Zhang, D. Wang, D. An, B. Han, X. Song, L. Li, G. Zhang and L. Wang, J. Org. Chem., 2018, 83, 1532–1537 CrossRef CAS PubMed; (o) Y. Zhu, T. Chen, S. Li, S. Shimada and L.-B. Han, J. Am. Chem. Soc., 2016, 138, 5825–5828 CrossRef CAS PubMed.
- (a) X. Gong, J. Chen, J. Liu and J. Wu, Org. Chem. Front., 2017, 4, 2221–2225 RSC; (b) Y. Guo, Y. Luo, S. Mu, J. Xu and Q. Song, Org. Lett., 2021, 23, 6729–6734 CrossRef CAS PubMed; (c) C.-Y. Li, Y.-C. Liu, Y.-X. Li, D. M. Reddy and C.-F. Lee, Org. Lett., 2019, 21, 7833–7836 CrossRef CAS PubMed; (d) J.-G. Sun, H. Yang, P. Li and B. Zhang, Org. Lett., 2016, 18, 5114–5117 CrossRef CAS PubMed; (e) H. Zhang, Z. Zhan, Y. Lin, Y. Shi, G. Li, Q. Wang, Y. Deng, L. Hai and Y. Wu, Org. Chem. Front., 2018, 5, 1416–1422 RSC.
- (a) R. Choudhary, P. Singh, R. Bai, M. C. Sharma and S. S. Badsara, Org. Biomol. Chem., 2019, 17, 9757–9765 RSC; (b) M. Mondal and A. Saha, Tetrahedron Lett., 2019, 60, 150965 CrossRef CAS.
- W. J. Pietro and W. J. Hehre, J. Am. Chem. Soc., 1982, 104, 3594–3595 CrossRef CAS.
- (a) A. Bacsó, M. Szigeti, S. Varga and T. Soós, Synthesis, 2017, 429–439 Search PubMed; (b) E. Desforges, A. Grysan, N. Oget, M. Sindt and J.-L. Mieloszynski, Tetrahedron Lett., 2003, 44, 6273–6276 CrossRef CAS.
- X. Han and J. Wu, Org. Lett., 2010, 12, 5780–5782 CrossRef CAS PubMed.
- X. Han, Y. Zhang and J. Wu, J. Am. Chem. Soc., 2010, 132, 4104–4106 CrossRef CAS PubMed.
- (a) X.-R. Song, T. Yang, H. Ding and Q. Xiao, Synthesis, 2020, 208–218 Search PubMed; (b) Y. Zhang, S. Du, T. Yang, F. Jin, J. Zhou, B. Cao, Z.-J. Mao, X.-R. Song and Q. Xiao, Org. Chem. Front., 2022, 9, 3156–3162 RSC.
- (a) W.-J. Bai, J. G. David, Z.-G. Feng, M. G. Weaver, K.-L. Wu and T. R. R. Pettus, Acc. Chem. Res., 2014, 47, 3655–3664 CrossRef CAS PubMed; (b) M. S. Singh, A. Nagaraju, N. Anand and S. Chowdhury, RSC Adv., 2014, 4, 55924–55959 RSC; (c) R. M. Jones, R. W. Van De Water, C. C. Lindsey, C. Hoarau, T. Ung and T. R. R. Pettus, J. Org. Chem., 2001, 66, 3435–3441 CrossRef CAS PubMed; (d) P. Batsomboon, W. Phakhodee, S. Ruchirawat and P. Ploypradith, J. Org. Chem., 2009, 74, 4009–4012 CrossRef CAS PubMed; (e) D. Liao, H. Li and X. Lei, Org. Lett., 2012, 14, 18–21 CrossRef CAS PubMed; (f) R. W. Van De Water and T. R. R. Pettus, Tetrahedron, 2002, 58, 5367–5405 CrossRef CAS; (g) H. Sugimoto, S. Nakamura and T. Ohwada, Adv. Synth. Catal., 2007, 349, 669–679 CrossRef CAS; (h) B. J. Nachtsheim, Nat. Chem., 2020, 12, 326–328 CrossRef CAS PubMed.
- (a) Z. Chen, Q. Shi, G. Wang, S. Chen and J. Hu, Molecules, 2018, 23, 1240 CrossRef PubMed; (b) B. Zhang, L. Liu, S. Mao, M.-D. Zhou, H. Wang and L. Li, Chem. – Eur. J., 2019, 2019, 3898–3907 CrossRef CAS; (c) H. Yuan, J. A. H. Kowah and J. Jiang, Tetrahedron Lett., 2020, 61, 151748 CrossRef CAS; (d) Y. N. Aher and A. B. Pawar, Org. Biomol. Chem., 2019, 17, 7536–7546 RSC; (e) P. Arde and R. V. Anand, Org. Biomol. Chem., 2016, 14, 5550–5554 RSC; (f) B. Xiong, G. Wang, C. Zhou, Y. Liu, W. Xu, W.-Y. Xu, C.-A. Yang and K.-W. Tang, Eur. J. Org. Chem., 2019, 3273–3282 CrossRef CAS; (g) B. Yang, W. Yao, X.-F. Xia and D. Wang, Org. Biomol. Chem., 2018, 16, 4547–4557 RSC.
- (a) X. Gu, H. Yuan, J. Jiang, Y. Wu and W.-J. Bai, Org. Lett., 2018, 20, 7229–7233 CrossRef CAS PubMed; (b) F. Yang, X. Zhou, Y. Wei, L. Wang and J. Jiang, Org. Chem. Front., 2021, 8, 5064–5070 RSC.
- H. Huang and J. Y. Kang, Org. Lett., 2017, 19, 5988–5991 CrossRef CAS PubMed.
- S. González-Pelayo and L. A. López, Eur. J. Org. Chem., 2017, 2017, 6003–6007 CrossRef.
- M. M. Toteva and J. P. Richard, in Advances in Physical Organic Chemistry, ed. J. P. Richard, Academic Press, 2011, vol. 45, pp. 39–91 Search PubMed.
- Y. Ge, H. Wang, H.-N. Wang, S.-S. Yu, R. Yang, X. Chen, Q. Zhao and G. Chen, Org. Lett., 2021, 23, 370–375 CrossRef CAS PubMed.
- S. Nakamura, M. Hayashi, Y. Hiramatsu, N. Shibata, Y. Funahashi and T. Toru, J. Am. Chem. Soc., 2009, 131, 18240–18241 CrossRef CAS PubMed.
- (a) G. Cote and D. Bauer, Anal. Chem., 1984, 56, 2153–2157 CrossRef CAS; (b) D. M. Rubush, in Encyclopedia of Reagents for Organic Synthesis, 2014, pp. 1–6, DOI:10.1002/047084289X.rn01742.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ob02169b |
| This journal is © The Royal Society of Chemistry 2023 |