
The xanthate route to tetralones, tetralins, and naphthalenes. A brief account†
Béatrice
Quiclet-Sire
and
Samir Z.
Zard
 *
*
Laboratoire de Synthèse Organique UMR 7652, Ecole Polytechnique, 91128 Palaiseau, France. E-mail: samir.zard@polytechnique.edu
First published on 26th December 2022
Abstract
The present account summarises routes to tetralones, tetralines, and naphthalenes based on the chemistry of xanthates developed in the authors’ laboratory. The degenerative reversible transfer of xanthates allows radical addition even to unactivated, electronically unbiased alkenes, and tolerates a broad range of functional groups, in particular common polar groups such as esters, ketones, nitriles, amides, carbamates, etc. Xanthates also allow radical ring closures onto aromatic rings. This feature, in combination with the intermolecular addition to alkenes, can be used to construct tetralones and tetralines. With the appropriate appendages, the former can be converted into napthalenes with a variety of substitution patterns. This translates into a convergent approach to a vast array of building blocks of interest to the pharmaceutical and agrochemical industries, and to material sciences.
 Béatrice Quiclet-Sire | Dr Béatrice Sire (née Quiclet) obtained her MSc in biochemistry in 1976 from Université Paris-Sud, Orsay, France. She then completed her PhD thesis in 1980 at the same university under the supervision of Dr Stéphene Géro, working on the synthesis of pseudo-disaccharides related to the aminoglycoside antibiotics. She spent a post-doctoral year at the Microbiology Department of Sandoz, in Basel, Switzerland, studying biosynthetic modifications of penicillin. In 1981, she obtained a position as a Chargée de Recherches at the CNRS within the research group of Dr Stéphene Géro at the Institut de Chimie des Substances Naturelles (ICSN) in Gif-sur-Yvette. She worked extensively on the chemistry of carbohydrates and on the study of new reactions and total synthesis under the joint supervision of Dr Stéphane Géro and Professor Sir Derek Barton. In 1993, she joined the group of Professor Samir Z. Zard, first at the ICSN, then at Ecole Polytechnique. She retired in 2018. |
 Samir Z. Zard | Dr Samir Z. Zard was born in 1955 in Ife, Nigeria. His training as a chemist started at the American University of Beirut, then at Imperial College, London, and finally at the Université Paris-Sud, Orsay, France, where he received his doctorate under the supervision of Professor Sir Derek Barton in 1983. He is currently an emeritus professor at Ecole Polytechnique and an emeritus Director of Research, Exceptional Class, at the CNRS. His main research concerns the study and development of new reactions and processes, with a special interest in radicals, organosulfur derivatives, alkynes, and nitro compounds. In addition to a number of academic awards, he received in 2007 the Croix de Chevalier de la Légion d'Honneur. |
1 Introduction and background
Tetralones, tetralins, and naphthalenes constitute basic building blocks in organic synthesis. These motifs are present in numerous natural products, pharmaceuticals, agrochemicals, and, in the case of naphthalenes, in chiral ligands for transition metal-based catalysts, such as the widely popular BINAP family, and in material sciences. Examples of natural substances and active ingredients featuring tetralone, tetralins, and naphthalene substructures are displayed in Fig. 1.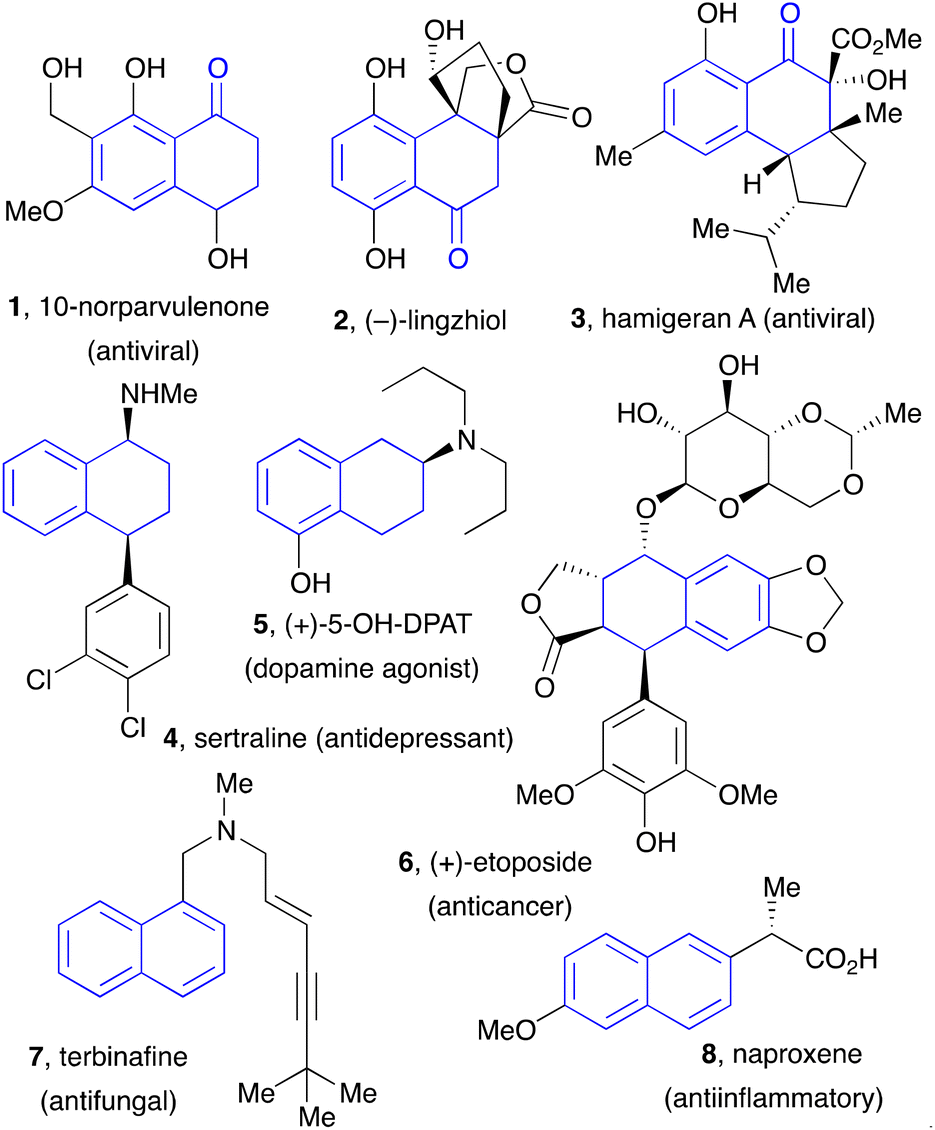 | ||
| Fig. 1 Examples of natural products and biologically active tetralones, tetralins and naphthalenes. | ||
The importance of these compounds in many areas of science has encouraged the development of a variety of synthetic approaches. In the case of 1-tetralones, the traditional route has most often relied on an intramolecular Friedel–Crafts reaction involving an acyl cationic species or a synthetic equivalent.1 The requisite side chain bearing the acid function can be introduced by an intermolecular Friedel–Crafts reaction using succinic anhydride (the Haworth synthesis). A few years ago, Aubé and co-workers have found that the intramolecular Friedel–Crafts can be accomplished by simply dissolving the acid chlorides in hexafluoroisopropanol (HFIP), which acts as both solvent and promoter.2 Organometallic couplings have been used to introduce the side chain bearing the terminal acid moiety and, in some instances, to accomplish also the cyclisation step.3 Radical cyclisation onto the aromatic nucleus to form the desired fused cyclohexanone ring has been reported and represents a complementary approach.4 However, this last strategy has remained anecdotal because of the inherent limitations of the methods used to generate the radical species. In the present overview, we describe the application of the unique radical chemistry of xanthates to the synthesis of 1-tetralones and naphthalenes, which overcomes most of the shortcomings of the previous routes.
The preparation of 2-tetralones and tetralines will also be described, even though much less work has been done on these two families. However, before proceeding with the synthetic aspects, a brief discussion of the general properties of the degenerative addition-transfer of xanthates to alkenes is necessary.5
The mechanism for the peroxide-initiated radical chain reaction of xanthates with alkenes is depicted in a simplified form in Scheme 1. A more comprehensive discussion is provided in references 5e and 6e for readers interested in the actual mechanistic picture, which is far more complex and particularly subtle. Basically, xanthates (and related dithiocarbonyl congeners) have the ability to store active radicals, namely R˙ and 12, in the form of rather unreactive adducts 10 and 13. The latter are bulky, stabilized by three heteroatoms and, since they bear no β-hydrogens, cannot disproportionate. Thus, owing to their steric bulk, their coupling to other radicals is slowed down considerably, leaving β-scission as the main pathway for evolution. This results in the continuous release of radicals R˙ and 12 back into the medium. The effective lifetime of these radicals is hence considerably increased. Furthermore, the equilibrium generally favours stabilized adducts 10 and 13 over active radicals R˙ and 12. The latter therefore spend most of their time sequestered in a non-reactive form and their absolute concentration is significantly lowered. At the same time, the fast equilibrium proceeding via intermediate 13 allows regulating their relative concentration in a manner reflecting their relative thermodynamic stabilities. By biasing this equilibrium so as to favour R˙ over radical 12, it is possible to drive the process towards desired product 14 and simultaneously curtail the unwanted addition of radical 12 to alkene 11 leading ultimately to the formation of oligomers.6 This can be achieved by choosing the reacting partners in such a way that starting R˙ is thermodynamically more stable than adduct radical 12, neglecting possible polar effects which in certain instances can be important.
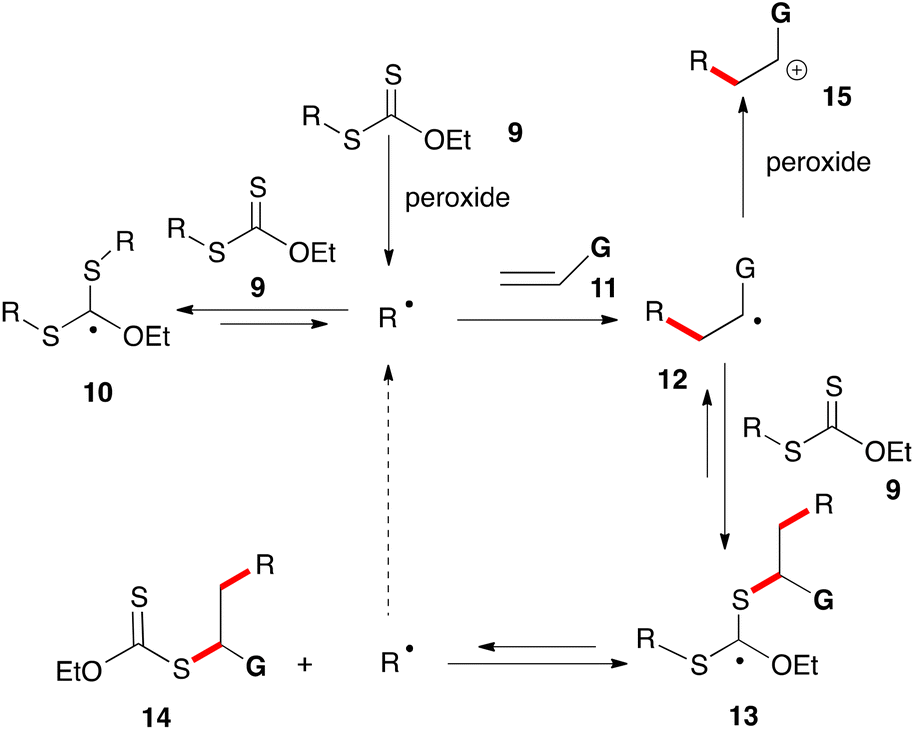 | ||
| Scheme 1 Simplified generic mechanism for the radical addition of xanthates to alkenes. | ||
The extended lifetime of radical R˙ allows it to partake in transformations with relatively slow kinetics, in particular intermolecular additions to unactivated alkenes 11 and cyclizations onto aromatic and heteroaromatic rings, opening up numerous synthetic opportunities not otherwise readily available. In cases where the G substituent is electron-releasing, it is possible to oxidize adduct radical 12 into cationic species 15 by single electron transfer (SET) to the peroxide. This crossover from the radical to the cation manifold provides a pathway for restoring the aromaticity of the (hetero)aromatic ring following the cyclization step, a feature that will be extensively exploited in the present review.
2 Synthesis of 1-tetralones
For the synthesis of 1-tetralones, two routes can be envisaged based on the ability of xanthates to mediate both intermolecular additions to electronically unbiased alkenes and cyclisation onto aromatic rings. The first, depicted in Scheme 2, is to start with phenacyl xanthates 16 derived from aromatic ketones.7 Addition to alkene 11 leads to a new xanthate 17 from which radical 18 can be regenerated and forced, in the absence of an external trap, to react reversibly with the aromatic ring. The resulting cyclohexadienyl radical 19 is more stabilized than initial radical 18 and does not therefore propagate a radical chain. It can, however, irreversibly transfer an electron to the peroxide to furnish corresponding cation 20. Finally, loss of a proton restores aromaticity and completes the synthesis of tetralone 21.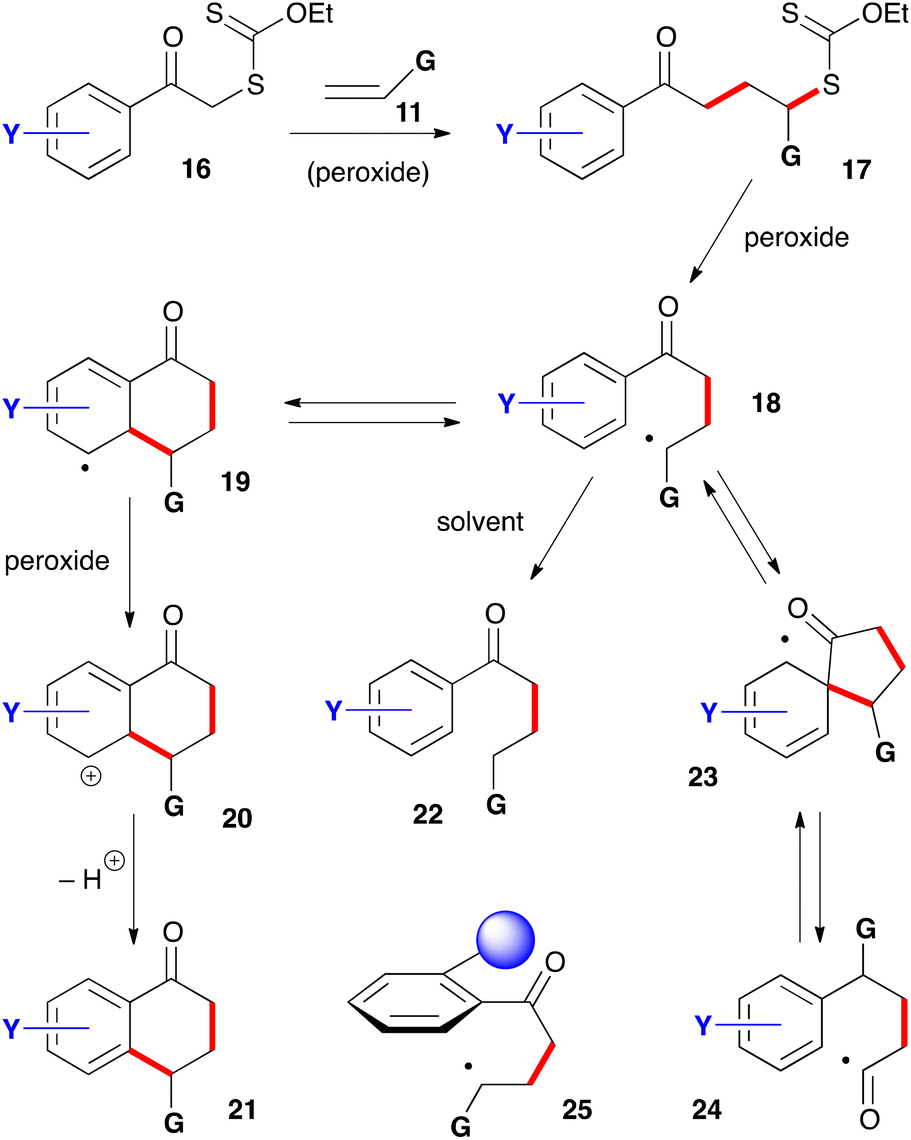 | ||
| Scheme 2 A first route to 1-tetralones. | ||
It is interesting to note that cation 20 is the same Wehland intermediate in typical electrophilic aromatic substitutions epitomized by the Friedel–Crafts reaction. This cationic intermediate is, however, created under very mild, neutral conditions, unlike the classical electrophilic aromatic substitutions which usually require mediation by strong protic or Lewis acids. A second important feature is that the radical cyclization is promoted by both electron-donating and electron-withdrawing groups on the aromatic nucleus. This allows access to a broad range of structures not available by classical Friedel–Crafts type chemistry. A third observation is that no extra functionality is needed on the aromatic ring to construct the tetralone, in contrast to some ionic and organometallic methods which require appropriate substituents in the 1,2-position that can then be manipulated to build the six-membered ketone portion.
There are, however, two main complications that could compete with the desired transformation and lower the yield. The first is the premature reduction of radical 18 by hydrogen atom transfer from the solvent to give compound 22. Factors that slow down the desired cyclisation/rearomatisation will indirectly enhance the extent of premature reduction. The second complication is less obvious and arises from an ipso attack on the aromatic ring leading to intermediate 23 followed by reversible fragmentation into acyl radical 24.8 This radical can then follow various reaction pathways, including cyclization onto the aromatic ring to give a regioisomeric tetralone (not shown). This second side-reaction is encouraged by the presence of a substituent in the ortho position to the ketone that forces the side-chain to adopt a conformation perpendicular to the plane of the aromatic ring, as shown in structure 25. This conformation is propitious for ipso attack on the ring but rather unfavourable for ring closure to the tetralone. The bulkier the ortho substituent the greater the hindrance to tetralone formation.
The second route to 1-tetralones is briefly outlined in Scheme 3. It consists in placing the alkene partner on the ketone as in derivatives 26 and adding xanthate 9 to give adduct 27. Further exposure to the peroxide then induces ring closure into tetralone 28, in the same way as for adduct 17 and with the same possible complications. The two approaches allow placing substituents on almost any position of the tetralone scaffold. These are not shown in the generic structures of Schemes 2 and 3 for the sake of clarity but the numerous possibilities will be evident from the examples presented below.
 | ||
| Scheme 3 An alternative route to 1-tetralones. | ||
Examples of the first route to 1-tetralones are displayed in Scheme 4.7,9–11 The first step is a normal chain process and is initiated by sub-stoichiometric amounts of dilauroyl peroxide (DLP, also sold under lauroyl peroxide). In the second cyclisation step, the peroxide acts as both an initiator and as a stoichiometric oxidant. Throughout this review, when DLP is simply an initiator, it is placed in parenthesis. In most cases, adducts 17 were isolated before being converted into the corresponding tetralones. The first yields after the compound number are for intermediates 17 (structures not shown) and the second is for the cyclisation step leading to tetralones 21. In a few cases, namely 21k–21n, the addition and cyclisation were conducted in the same flask. This one-pot variant could in principle be applied to most other examples since the reagent for the first and second steps is the same. However, a 2- to 5-fold increase in dilution is sometimes needed, especially when the alkene partner is not volatile and an unreacted portion remains in the flask. The bimolecular intermolecular addition to the alkene is disfavoured by a low concentration while the unimolecular cyclisation is unaffected. An increase in the reaction temperature can also be advantageous in the latter case, hence the switch from 1,2-dichloroethane (DCE) or ethyl acetate to chlorobenzene in some of the examples presented in this review.
 | ||
| Scheme 4 Examples of 1-tetralones. | ||
Terminal alkenes are generally the best partners. The radical additions take place at the less hindered unsubstituted terminus. With internal alkenes, problems of regiochemistry arise, except when the alkene is symmetrical or electronically and/or sterically biased. Tetralones 21o and 21p are examples of sterically directed addition–cyclisation. Competing allylic hydrogen atom abstraction can become problematic and explains the low yield of addition to cyclohexene (21s). This difficulty is absent in bridged bicyclic alkenes (21l–n) and less important in strained alkenes, such as cyclobutenes (21o and 21p), and even the slightly strained cyclopentene (21q and 21r). Otherwise, numerous functional groups are tolerated, including pinacolato boronates (21u)10 and heteroaromatics such as triazoles (21v).11 In the latter case, an equivalent of camphorsulfonic acid was added in both steps to subdue the nucleophilicity of the triazole which would otherwise induce an ionic decomposition of the xanthate group and diminish seriously the efficiency of the radical process. Most of the tetralones in Scheme 4 would be quite tedious to make by classical approaches, especially polycyclic derivatives 21m–p.
In the examples in Scheme 4, there is no ambiguity as to the site of cyclisation on the aromatic ring. Meta substituents, however, can, and often do, lead to a mixture of regioisomers. This is illustrated by the examples in Scheme 5.12–14
 | ||
| Scheme 5 Presence of a meta-substituent. | ||
Thus, the cyclisation of adduct 21 derived from xanthate 29 possessing a phthalimide (NPhth) group in the meta position furnishes, in addition to a small amount of prematurely reduced material 34, two regioisomeric tetralones 32 and 33 (Scheme 5a). The former dominates because the steric bulk of the phthalimide hinders somewhat attack at the ortho site (with respect to the phthalimide). In the case of the smaller methoxy substituent in 30, the one-pot addition cyclisation to allyl acetate leads to comparable amounts of both regioisomers 35 and 35 (Scheme 5b). Cyclisation of adduct 37, obtained in 68% yield by a similar addition to allyl acetate as for 31, also furnishes a mixture of two regioisomers 38 and 39 with the former logically dominating, again because of steric factors (Scheme 5c). For reasons not entirely clear, the cyclisation of adducts 40 and 42 to give tetralones 41 and 43 proceeds regioselectively (Scheme 5d).13
When certain substituents, such as halogens, are present in the ortho position, an ipso substitution can intervene and complicate the outcome. For example, difluorophenacyl adduct 45 derived from xanthate 44 and allyl acetate is converted into two tetralones, 46 and 47, upon exposure to stoichiometric amounts of DLP (Scheme 6).15 The latter constitutes a rare example of a fluorine atom departing as a fluorine atom (cf.48).16 Interestingly, heating the “normal” tetralone 46 with hydrazine hydrate leads to indazole 50, with concomitant cleavage of the pendant acetate group. Similarly, acetamide analogue 49 was converted into indazole 51. Such indazoles are of interest to medicinal chemists17 and are easily accessible by the present route.
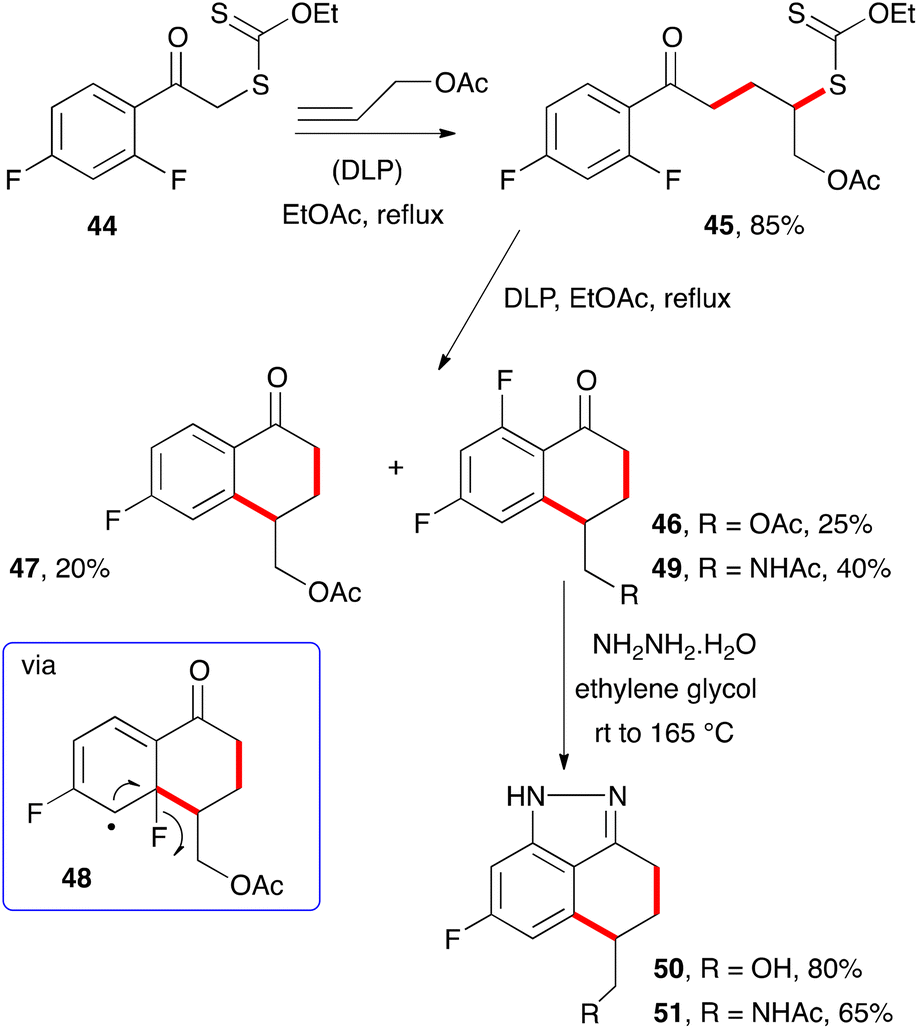 | ||
| Scheme 6 Homolytic ipso substitution of fluorine. | ||
An unusual synthesis of tetracyclic tetralones was accidentally uncovered when an attempt was made to convert tetralone 54a with a pendant benzyl group into naphthylacetamide 56a through the Semmler–Wolff/Schroeter aromatisation, whereby the corresponding oxime 55a is first heated with acetic anhydride to form the acetate and then with acetic acid and a strong acid (Scheme 7).18 Traditionally, it is sulfuric acid, but in this case methanesulfonic acid was used. Under these conditions, naphthylacetamide 56a was indeed produced, but it was the minor component. The major product turned out to be tetracyclic ketone 57a.19 Apparently, strongly electrophilic intermediate 58a reacted in a Friedel–Crafts manner with the phenyl group to furnish acetylimine 59a, in addition to simply losing acetic acid to give naphthylacetamide 56a, as in the normal Semmler–Wolff/Schroeter reaction. Hydrolysis during workup finally provides the observed ketone 57a.
 | ||
| Scheme 7 1-Tetralones by an abnormal Semmler–Wolff/Schroeter reaction. | ||
This transformation was extended to various other analogues, as shown by the examples in Scheme 8.19 Three yields are given. The first is for the intermolecular addition leading to adducts 53. When it is not given (as for 57e, 57g, 57h, and 57l), it is because the radical additions and cyclisations were performed in the same (pot). The second is for the ring closure to tetralones 54; and the third is for the abnormal Semmler–Wolff/Schroeter reaction from corresponding oximes 55 leading to tetracyclic tetralones 57. In the case of 57l, the hydrolytically labile intermediate acetylimine 59l could be isolated in 34% yield thus lending support to the proposed mechanism.19 The yield for the last step is low in structures where the pendant aryl group is substituted with two electron-donating methoxy groups (57c and 57d) because of uncontrolled Friedel-Craft acetylation of the electron-rich aromatic ring in both the starting material and the product. Furthermore, in most of the examples, variable amounts of the corresponding naphthylacetamides 56 were observed but not isolated. Tetracyclic tetralone of general structure 57 are very rare, with only two examples reported prior to this work.20
 | ||
| Scheme 8 Examples of polycyclic 1-tetralones. | ||
As stated in the discussion above in relation with Scheme 2, the presence of an ortho substituent is often detrimental to the cyclisation step because of an unfavourable conformation (cf. structure 25). This is starkly visible in a model study aimed at the synthesis of gilvocarcin M (60, Scheme 9).21 The construction of the AB portion of this molecule could in principle be accomplished by aromatisation of a suitable tetralone precursor. Unfortunately, attempted cyclisation of a variety of xanthate adducts 61 resulted in complex mixtures. In contrast, adducts 62, lacking the ortho methoxy group behaved normally and could be cyclised into corresponding tetralone 63 in a quite reasonable yield. Despite this initial failure, it is noteworthy that such a complex structure as 63 could be obtained in simply two steps.
 | ||
| Scheme 9 Approaches to gilvocarcin M. Effect of an ortho substituent. | ||
A simple solution to this shortcoming emerged later, when it was found that the addition–cyclisation sequence is compatible with the presence of a free ortho hydroxy group.22 The hydrogen bonding between the phenolic hydroxy group and the ketone shown on structure 64 in fact freezes the conformation in a manner inducive to cyclisation to give hydroxytetralones 65. The various examples displayed in Scheme 10 testify to the relative efficiency of this approach.22 Again, the first yield shown is for the intermolecular addition to the alkene, and its absence indicates that the addition–cyclisation sequence was carried in one pot. The second yield is for the cyclisation onto the aromatic ring. Saponification of the acetate in 65f gives shinanolone, 65g, a natural product isolated from various plant sources.23 Example 65h underscores the mildness of the experimental conditions and their compatibility with a fragile trisubstituted epoxide.22
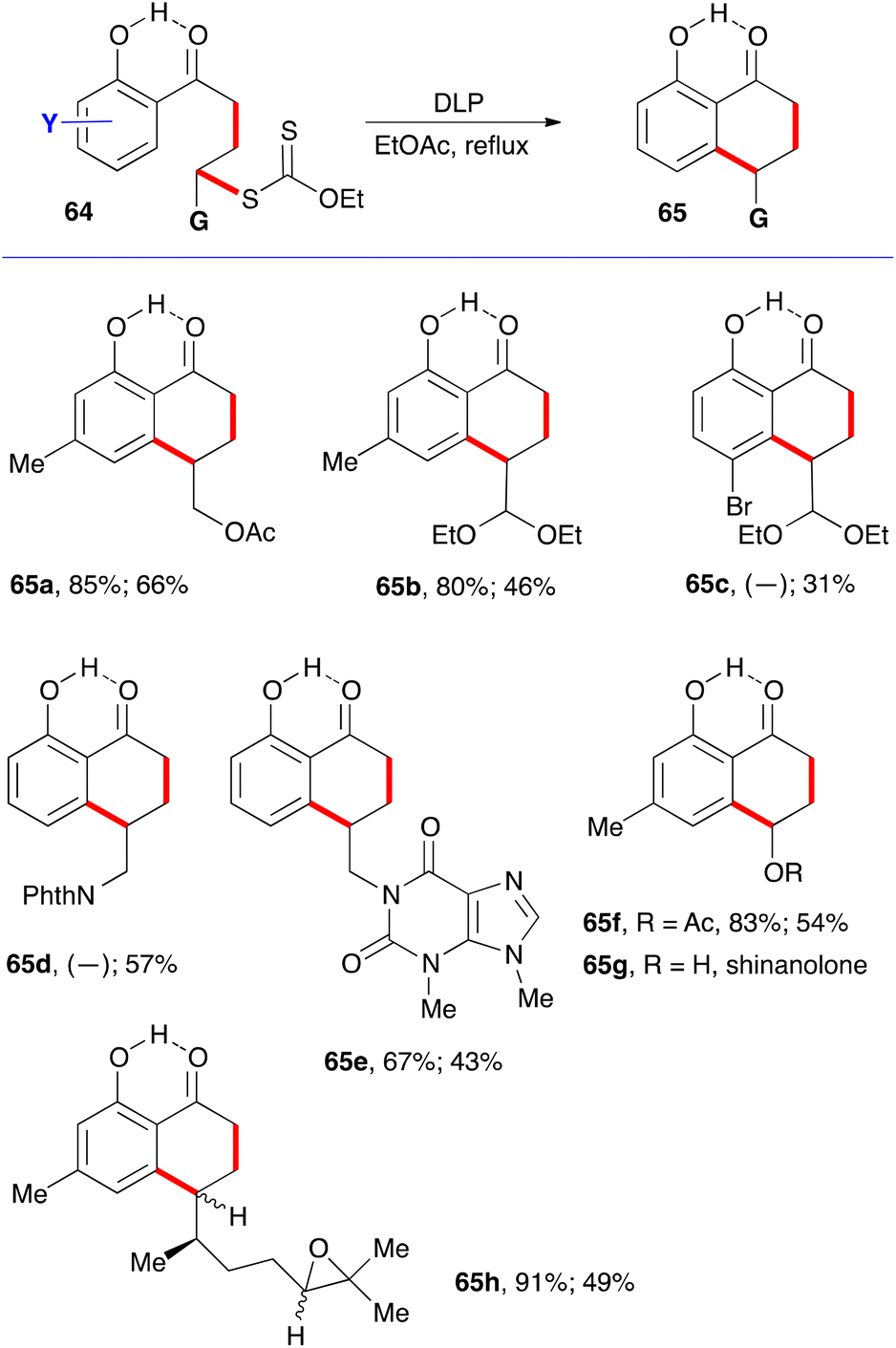 | ||
| Scheme 10 Favourable effect of internal hydrogen bonding. | ||
This finding is of special significance. First is the fact that both intermolecular radical additions and cyclisations can be accomplished in the presence of a free phenol, despite the fact that phenols are commonly used as radical inhibitors.24 In the present case, the hydrogen bonding slows down the abstraction of the phenolic hydrogen to a level that it no longer interferes with the present radical process.25 The second observation is that ortho-hydroxy aromatic ketones and motifs derived therefrom are common structural features in natural products, especially those produced via the polyketide biosynthetic pathway.26 Indeed, the ability to form the tetralone with a free hydroxy group simplified significantly the initial total synthesis of 10-norparvulenone 1 and O-methylasparvenone 72 (Scheme 11).22,27 The former is a promising anti-influenza virus antibiotic isolated from Microsphaeropsis sp.28 and the latter is a nitrogen-free serotonin antagonist with a potential for the treatment of depression and obsessive-compulsive disorders.29,30
 | ||
| Scheme 11 Total synthesis of 10-norparvulenone and O-methylasparvenone. | ||
Xanthate 67 is readily prepared in two steps from inexpensive 3-methoxyphenol 66. Addition to vinyl pivalate [piv = Me3CC(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)-] and ring closure furnishes tetralone 69via adduct 68. Friedel–Crafts formylation according to the procedure of Gross and co-workers31 provides key aldehyde 70 in nearly quantitative yield. Finally, reduction to the corresponding alcohol and saponification of the pivalate completes the synthesis of 10-norparvulenone 1. Wittig olefination of aldehyde 70 affords intermediate 71 in moderate yield. Catalytic hydrogenation of the vinyl group and saponification of the pivalate leads to O-methylasparvenone 72. It remains to be seen whether the use of the free phenol could overcome the difficulties encountered in the model studies aimed at the total synthesis of gilvocarcin M.
O)-] and ring closure furnishes tetralone 69via adduct 68. Friedel–Crafts formylation according to the procedure of Gross and co-workers31 provides key aldehyde 70 in nearly quantitative yield. Finally, reduction to the corresponding alcohol and saponification of the pivalate completes the synthesis of 10-norparvulenone 1. Wittig olefination of aldehyde 70 affords intermediate 71 in moderate yield. Catalytic hydrogenation of the vinyl group and saponification of the pivalate leads to O-methylasparvenone 72. It remains to be seen whether the use of the free phenol could overcome the difficulties encountered in the model studies aimed at the total synthesis of gilvocarcin M.
The second route to tetralones outlined in Scheme 3 has not been extensively studied but exhibits nevertheless a similar scope and limitations to the first and nicely complements it. Two examples are depicted in Scheme 12. Addition of phthalimidomethyl xanthate 74 to alkene 73 gives adduct 75, which can then be cyclised in the usual way into tetralone 76.32 Incidentally, xanthate 74 is more generally an exceedingly useful aminomethylating agent for alkenes and a prototype of a family of reagents for the synthesis of amines.33 In a similar way, reaction of cyanomethyl xanthate 78 with alkene 77 and cyclisation of resulting adduct 79 provides tetralone 80.9 This second example illustrates the possibility of placing a substituent, such as a methyl group, in the β-position to the ketone. This is not usually easy to accomplish with the first route since it requires the use of an internal alkene, with all the attendant complications discussed above (regiochemistry issues and allylic hydrogen abstraction, slow kinetics, etc.).
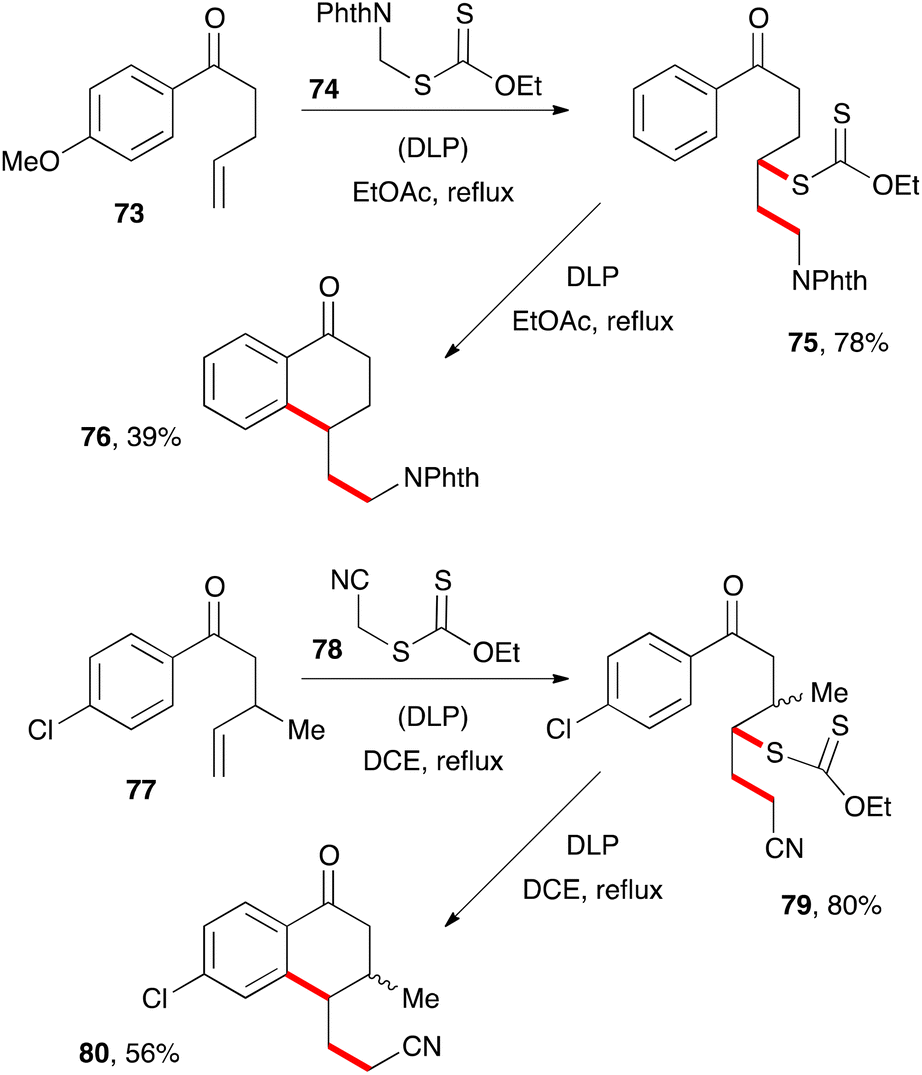 | ||
| Scheme 12 An alternative route to 1-tetralones. | ||
3 Synthesis of 2-tetralones
The synthesis of 2-tetralones using xanthates has hardly been explored, in part because the required precursors are less readily available, unlike the phenacyl xanthates employed in the previous section. One unpublished approach exploits the properties of cyclopropylacyl radicals such as 82.34a These radicals do not readily extrude a molecule of carbon monoxide, nor do they undergo a fast opening of the cyclopropyl ring.34b It is therefore possible to capture them with an external alkene. In the example pictured in Scheme 13, the radical chain process starting from the yellow-coloured S-acyl xanthate 81 can be initiated by irradiation with visible light in the presence of an alkene such as allyl acetate. This furnishes adduct 83, which can then be made to cyclise in the usual manner by treatment with stoichiometric amounts of peroxide to give 2-tetralones 84 and 85. Unexpectedly, partial hydrolysis of the acetate occurred in the second step.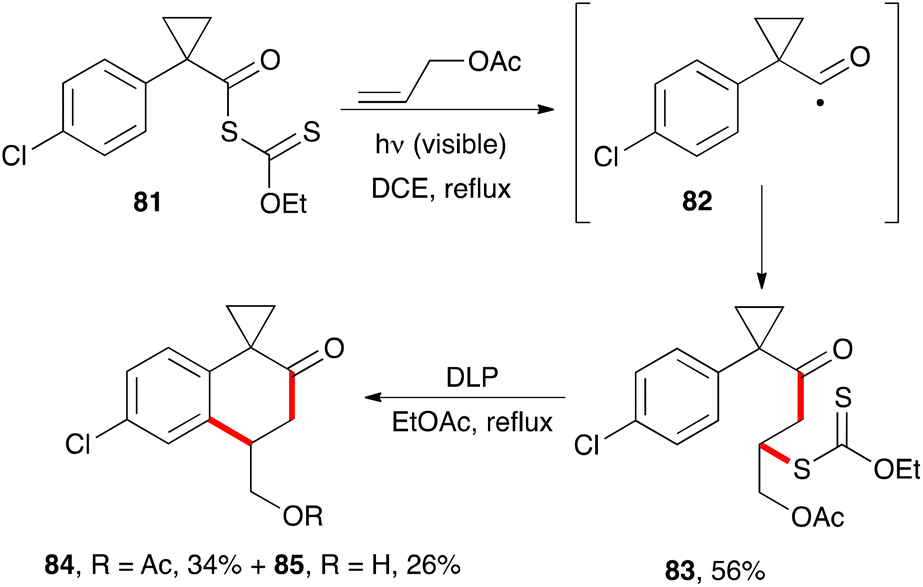 | ||
| Scheme 13 Synthesis of 2-tetralones. | ||
There are various methods that allow the conversion of 1-tetralones into 2-tetralones.35 This would compensate to a certain extent for the limited utility of xanthates for their direct synthesis.
4 Synthesis of tetralins
The synthesis of tetralins has also received only modest attention. Two approaches can be envisaged that parallel those for 1-tetralones, namely the aromatic ring where cyclisation will take place can either be part of the xanthate or the alkene component. The five examples in Scheme 14a illustrate the first possibility.36 The starting material are α-phthalimido xanthates 85 which can undergo a one-pot addition cyclisation to various alkenes to give protected 2-amino tetralins 86a–e. As exemplified by (+)-5-OH-DPAT 5 in Fig. 1, an extensively studied dopamine agonist, aminotetralins are of significant medicinal importance. Their synthesis has hitherto hinged almost solely on the reductive amination of 2-tetralones. The present route opens a rapid access to analogues not readily available by more classical methods.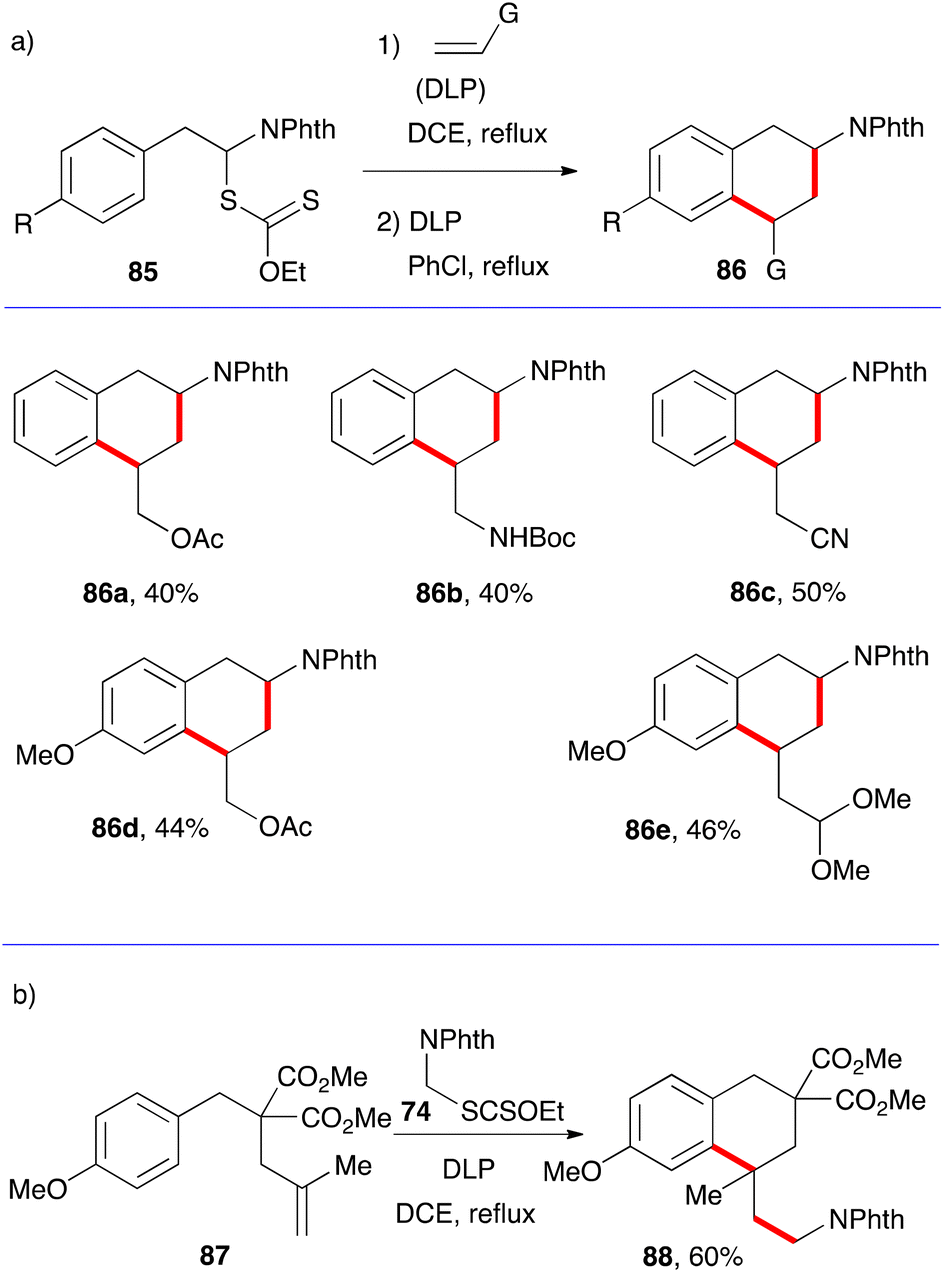 | ||
| Scheme 14 Syntheses of tetralins. | ||
The example displayed in Scheme 14b is a case where the aromatic ring is part of alkene partner 87. Addition–cyclisation with xanthate 74 furnishes tetralin 88 containing a quaternary centre.37
5 Synthesis of naphthalenes
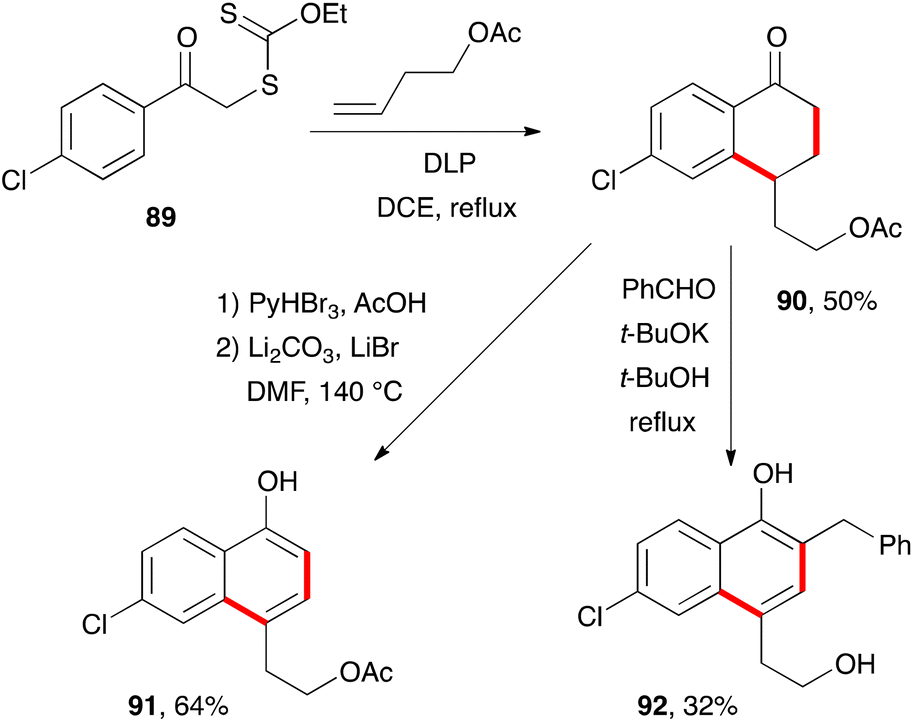
There are countless syntheses of naphthalenes, with the modification and ultimate aromatisation of tetralones constituting one of the main strategies.38 From the standpoint of xanthate chemistry, the multiple possibilities for the construction of tetralones outlined above translate into interesting perspectives for accessing naphthalenes. These will be discussed in the present section.Two classical variants for the aromatisation of tetralone 90, obtained by addition–cyclisation of 4-chlorophenacyl xanthate 89 to butenyl acetate, are shown in Scheme 15.39 The first is a straightforward bromination-elimination sequence leading to naphthol 91. The second involves a condensation with benzaldehyde followed by base induced isomerisation of the intermediate enone into naphthol 92. The acetate in the side-chain is cleaved under the basic reaction conditions.
 | ||
| Scheme 15 Synthesis of naphthalenes. | ||
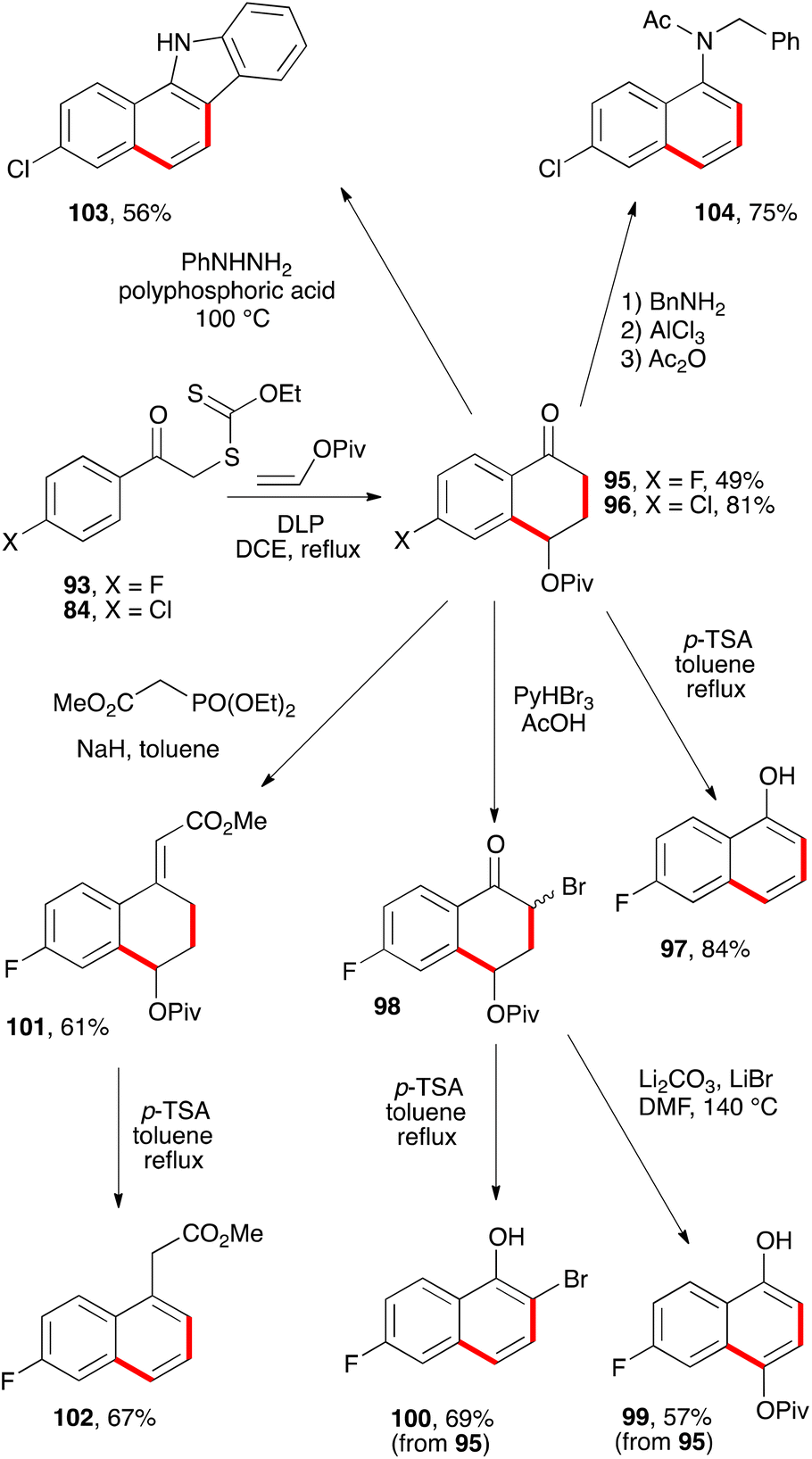
A more interesting route to naphthalene building blocks is to exploit tetralones derived by addition–cyclisation to vinyl pivalate such as 95 and 96 (Scheme 16).39 Simple heating of the former with p-TSA in refluxing toluene causes a clean aromatisation into fluoronaphthol 97. Bromination furnishes bromoketone 98 which can be aromatised with base into naphthol 99, without affecting the pivalate group. Alternatively, acid treatment of bromoketone 98 provides 2-bromo-6-fluoro-1-naphthol 100. The pivalate group in tetralone 95 is sufficiently robust to allow a Horner–Wadsworth–Emmons condensation to give enoate 101, which can then be aromatised by treatment with acid to give fluoronaphthylacetate 102.39
 | ||
| Scheme 16 Divergent syntheses of naphthalenes. | ||
Starting from chlorotetralone 96, it is possible to perform a Fischer indole synthesis which proceeds in this case with concomitant elimination of the pivalate to produce directly carbazole 103. Another transformation concerns condensation first with benzylamine and then forcing the elimination of the pivalate with aluminium trichloride to give the N-benzyl-6-chloro-1-naphthylamine. The latter is finally converted into acetamide 104 to make isolation and handling more convenient.39
Other examples of naphthalenes prepared by similar procedures are presented in Scheme 17. Thus, treatment of tetralone 106, obtained from xanthate 105, with p-TSA in refluxing toluene provides protected 6,7-diamino-1-naphthol 107.39 Such a molecule would be interesting in material sciences and for the synthesis of dyes, since the unprotected parent diaminonaphthol is particularly electron rich and therefore susceptible to various autoxidation and condensation reactions.
 | ||
| Scheme 17 Further syntheses of naphthalenes. | ||
The second series of transformations involves electron rich tetralone 109 which, interestingly, could be obtained despite the presence of an ortho-methoxy group (cf discussion above on the harmful influence of such substituents). In this case, the usual bromination conditions cause spontaneous aromatisation through elimination of the pivalate to give bromonaphthol 110. In contrast, addition of lithium acetylides does not affect the pivalate, as shown by the formation of carbinols 111 and 113. Dehydration then affords naphthalenes 112 and 114.39
The synthesis of phthalimide protected 2-amino-1-naphthols follows the same pathway. It simply involves starting with xanthates 115. Addition–cyclisation with vinyl pivalate to give tetralones 116 and aromatisation by treatment with acid furnishes the corresponding naphthols 117.40 Five examples are collected in Scheme 18, with the two yields given corresponding respectively to the two steps. Evidently, the phthalimido group in starting phenacyl xanthates 115 can be replaced by various other groups to provide a broad variety of naphthalene derivatives.
 | ||
| Scheme 18 Synthesis of aminonaphthols. | ||
This general strategy can be adapted to access other aminonaphthols and aminonaththalenes by exploiting the presence of a favourable hydrogen bonding similar to the phenolic derivatives discussed above (Schemes 10 and 11). This is accomplished by placing an acetamido group in the ortho position as in xanthates 118 (Scheme 19). Again, one-pot addition to vinyl pivalate and cyclisation furnishes tetralones 119 and acid induced aromatisation provides 8-acetamido-1-naphthols 120.40 This variant is illustrated by the five examples assembled in Scheme 19a, with the two yields shown corresponding respectively to the two steps. Prior reduction of the ketone in tetralone 19c before treatment with acid gives rise to N-acetyl-4-trifluoromethyl-1-naphthylamine 121 (Scheme 19b). Such a reduction step could have been applied in essentially all the previous examples in order to obtain the naphthalenes instead of the naphthols.
 | ||
| Scheme 19 Further syntheses of aminonaphthols and aminonaphthalenes. | ||
One more variation concerns the synthesis of protected 1-naphthyl hydrazines, as outlined in Scheme 20.41 Condensation of tetralones 122 with mono Boc-protected hydrazine gives the corresponding hydrazones 123. Prolonged heating in a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of trifluoroacetic anhydride and trifluoroacetic acid then gives the desired hydrazine as the mono- or bis-trifluoroacetylated derivative 124 or 125, depending on the structure and workup as shown by the examples provided. The first yield is for the synthesis of the tetralone and the second is for the combined last two steps. Tetralones of general structure 122 have thus served as versatile springboards to a broad variety of naphthalene derivatives.
:1 mixture of trifluoroacetic anhydride and trifluoroacetic acid then gives the desired hydrazine as the mono- or bis-trifluoroacetylated derivative 124 or 125, depending on the structure and workup as shown by the examples provided. The first yield is for the synthesis of the tetralone and the second is for the combined last two steps. Tetralones of general structure 122 have thus served as versatile springboards to a broad variety of naphthalene derivatives.
 | ||
| Scheme 20 Synthesis of protected naphthylhydrazines. | ||
A final alternative route to naphthols is depicted in Scheme 21.42 It starts with the reaction of ketone 73 with difluorochloro xanthate 126 to give tetralone 127. Gentle exposure to DBU produces difluorovinyl tetralone 128 but a more vigorous and prolonged treatment ultimately results in the formation of 4-difluoroethyl-naphthol 129 by migration of the terminal alkene into the ring. The unusual octadecyl group on xanthate 126 is partly to lower its volatility and make its handling more convenient. The oxygen moiety on the xanthate is just a spectator and is not directly involved in the radical process.
 | ||
| Scheme 21 Synthesis of fluorinated tetralone and naphthol. | ||
6 Concluding remarks
The ability of xanthates to mediate both intermolecular addition to unactivated alkenes and ring closure onto aromatic rings offers many opportunities for the synthesis of tetralones, tetralines, and naphthalenes. It can be further extended to related derivatives, such as the anthracene hydrazide 125c in Scheme 20. Larger polyaromatic derivatives are also accessible. For example, addition–cyclisation of xanthate 130 to bridged alkene 131 gives unusual tetralone 132 (Scheme 22).43 Heating this compound with sulfuric acid in refluxing methanol leads to the formation of dimethoxytetracene 133. Alternatively, addition of methyllithium and phenyllithium to the ketone prior to treatment with acid furnishes analogues 134 and 135. The synthesis of the higher pentacene derivative 138 starts with xanthate 136 and follows a similar sequence to that of tetracene 133.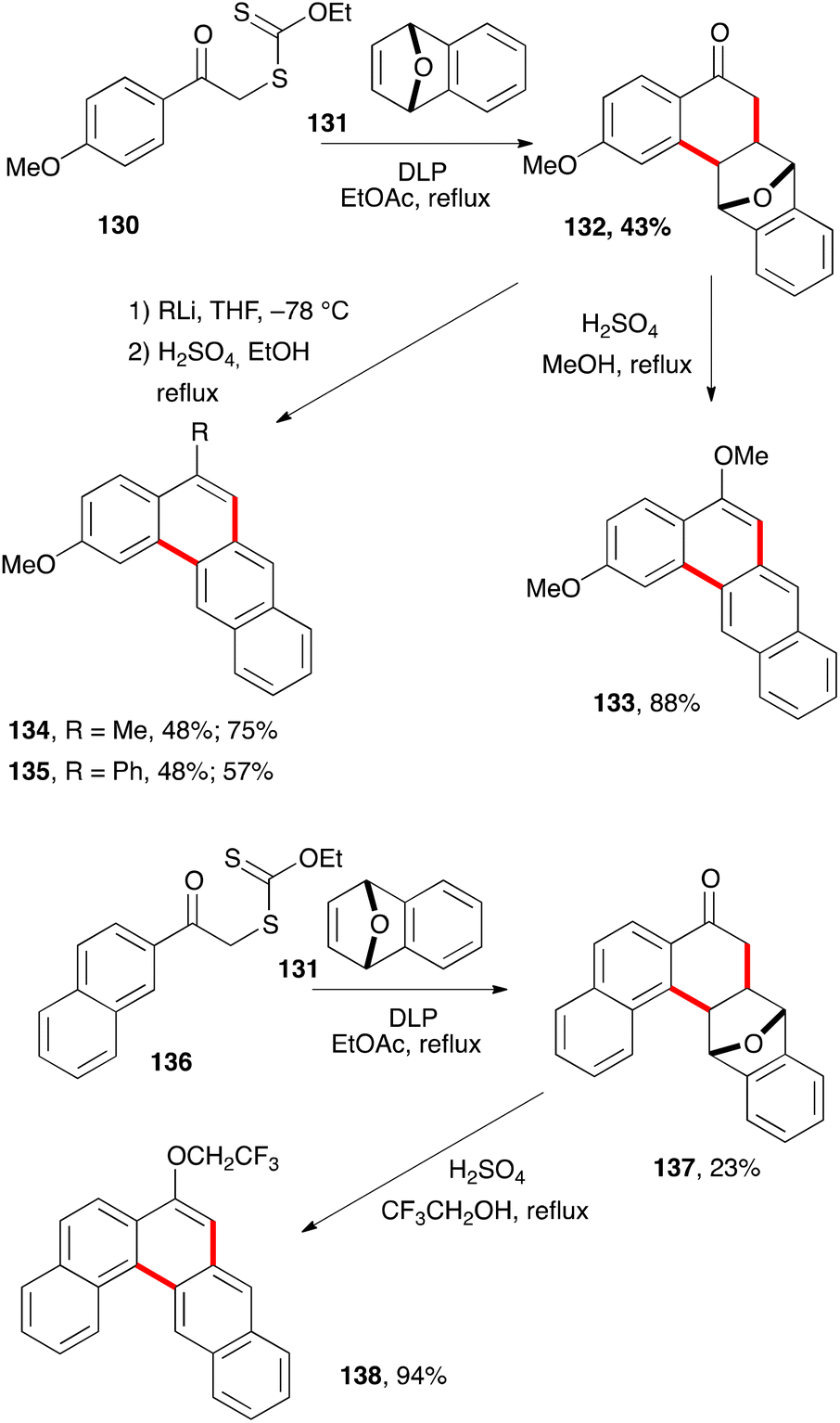 | ||
| Scheme 22 Synthesis of tetracenes and naphthacenes. | ||
The ring closure does not have to be on a benzene type aromatic. Heteroaromatics are frequently more reactive radicophiles and can be employed in the same manner (Scheme 23). Ketones 141 and 143 derived from thiophene bearing xanthate 139via intermediates 140 and 142, and ketone 145 from indole based xanthate 144 are readily prepared by this chemistry.12 They could in principle be converted into the corresponding benzothiophene and carbazole derivatives if so desired. In the case of indole derivative 145, the saponification of the acetate to give the corresponding alcohol was a prelude to its conversion into derivative 147, where X is a leaving group, and ultimately into cyclopropane 148. Such electrophilic cyclopropanes could act as intercalating-alkylating anti-cancer substances by analogy with the important family of the duocarmycins.44
 | ||
| Scheme 23 Thiophene and indole based analogues of tetralones. | ||
The chemistry of tetralones, tetralines, and naphthalenes is intertwined in that they can often be interconverted through appropriate oxidative and reductive transformations. Partial or total reduction of the aromatic ring (or rings in the case of naphthalenes) leads to decalin derivatives. The sequence displayed in Scheme 24 is one interesting illustration.45 Conversion of the acetate in tetralone 151 into a tosylate and treatment with base furnishes tricyclic tetralone 152 by an intramolecular alkylation of the enolate. An alkylative Birch reduction then provides dienone 153. It is remarkable that such a complex structure can be obtained starting with compounds as trivial as phenacyl xanthate 149 and butenyl acetate. This alliance between the powerful alkylative Birch reduction and the radical chemistry of xanthates is indeed a promising strategy for the construction of polycyclic terpene natural products.
 | ||
| Scheme 24 An alliance with the powerful Birch reduction. | ||
In summary, the xanthate based routes to the title compounds described herein nicely complement existing methods. They offer numerous advantages in terms of convergence, flexibility, tolerance for functional groups, mildness of experimental conditions, and cheapness of the reagents. Much has been accomplished, but even more remains to be done. Indeed, the present overview gives but a glimpse of the possibilities.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to express their heartfelt gratitude to all the talented and enthusiastic collaborators who made this chemistry possible and whose names appear in the references. Generous financial support from the following institutions and companies is also acknowledged: Ecole Polytechnique, CNRS, DGA, la Ligue Contre le Cancer, Laboratoires Servier, the Alfred Kastler Foundation, ANR, the China Scholarship Council, Rhodia (now Solvay), and Sanofi.References
- For selected examples, see: (a) T. Venkatesh, P. S. Mainkar and S. Chandrasekhar, J. Org. Chem., 2021, 86, 5412 CrossRef CAS PubMed; (b) L.-M. Mehl and M. E. Maier, J. Org. Chem., 2017, 82, 9844 CrossRef CAS PubMed; (c) J. C. T. Reddel, K. E. Lutz, A. B. Diagne and R. J. Thomson, Angew. Chem., Int. Ed., 2014, 53, 1395 CrossRef CAS PubMed; (d) E. Fillion, D. Fishlock, A. Wilsily and J. M. Goll, J. Org. Chem., 2005, 70, 1316 CrossRef CAS PubMed.
- H. F. Motiwala, R. H. Vekariya and J. Aube, Org. Lett., 2015, 17, 5484 CrossRef CAS PubMed.
- For selected recent examples, see: (a) T. Yumura, T. Nanjo and Y. Takemoto, Eur. J. Org. Chem., 2022, e202200082 CAS; (b) A. Selmani and S. Darses, Org. Lett., 2019, 21, 8122 CrossRef CAS PubMed; (c) H. Zhu, X. Nie, Q. Huang and G. Zhu, Tetrahedron Lett., 2016, 57, 2331 CrossRef CAS; (d) K. Tanaka, D. Hojo, T. Shoji, Y. Hagiwara and M. Hirano, Org. Lett., 2007, 9, 2059 CrossRef CAS PubMed.
- (a) E. I. Heiba and R. M. Dessau, J. Am. Chem. Soc., 1972, 94, 2888 CrossRef CAS; (b) B. B. Snider and L. Han, Synth. Commun., 1995, 25, 2337 CrossRef CAS; (c) F. Z. Yang, M. K. Trost and W. E. Fristad, Tetrahedron Lett., 1987, 28, 1493 CrossRef CAS; (d) Y. Uenoyama, M. Tsukida, T. Doi, I. Ryu and A. Studer, Org. Lett., 2005, 7, 2985 CrossRef CAS PubMed; (e) R. Han, T. H. Han, I. Jeon, B. K. Soh, B. M. Kim, Y. M. Jun, B. M. Lee and B. H. Kim, J. Photochem. Photobiol., A, 2006, 179, 167 CrossRef CAS; (f) J. Fang, L. Li, C. Yang, J. Chen, G.-J. Deng and H. Gong, Org. Lett., 2018, 20, 7308 CrossRef CAS PubMed; (g) M.-J. Jiao, Q. Hu, X.-Q. Hu and P.-F. Xu, J. Org. Chem., 2021, 86, 17156 CrossRef CAS PubMed; (h) A. Petti, P. Natho, K. Lam and P. J. Parsons, Eur. J. Org. Chem., 2021, 854 CrossRef CAS; (i) F. Zhao, X.-W. Gu, R. Franke and X.-F. Wu, Angew. Chem., Int. Ed., 2022, e202214812 CAS.
- For reviews of the xanthate transfer, see: (a) B. Quiclet-Sire and S. Z. Zard, Top. Curr. Chem., 2006, 264, 201 CrossRef CAS; (b) B. Quiclet-Sire and S. Z. Zard, Pure Appl. Chem., 2011, 83, 519 CAS; (c) S. Z. Zard, J. Phys. Org. Chem., 2012, 25, 953 CrossRef CAS; (d) B. Quiclet-Sire and S. Z. Zard, Chem. – Eur. J., 2006, 12, 6002 CrossRef CAS PubMed; (e) B. Quiclet-Sire and S. Z. Zard, Isr. J. Chem., 2017, 57, 202 CrossRef CAS; (f) S. Z. Zard, Helv. Chim. Acta, 2019, 102, e1900134 CrossRef.
- The use of xanthates and related derivatives, such as dithioesters, trithiocabonates and dithiocarbamates, for the formation of oligomers and longer polymers is indeed the basis of the remarkably powerful RAFT/MADIX technology for the manufacture of block copolymers. See: (a) Handbook of RAFT Polymerization, ed. C. Barner-Kowollik, Wiley-VCH, Weinheim, 2008 Search PubMed; (b) K. Matyjaszewski, Macromolecules, 2020, 53, 495 CrossRef CAS; (c) S. Perrier, Macromolecules, 2017, 50, 7433 CrossRef CAS. For an account of the discovery of the basic process, see: (d) S. Z. Zard, Aust. J. Chem., 2006, 59, 663 CrossRef CAS; (e) S. Z. Zard, Macromolecules, 2020, 53, 8144 CrossRef CAS.
- A. Liard, B. Quiclet-Sire, R. N. Saicic and S. Z. Zard, Tetrahedron Lett., 1997, 38, 1759 CrossRef CAS.
- G. A. Russell, P. Chen, B. H. Kim and R. Rajaratnam, J. Am. Chem. Soc., 1997, 119, 8795 CrossRef CAS.
- (a) N. Legrand, PhD Thesis, Ecole Polytechnique, 1999; (b) N. Spassova, PhD Thesis, Ecole Polytechnique, 2000.
- H. Lopez-Ruiz and S. Z. Zard, Chem. Commun., 2001, 2618 RSC.
- F. Gagosz and S. Z. Zard, Org. Lett., 2002, 4, 4345 CrossRef CAS PubMed.
- S. Seguin, PhD Thesis, Université Paris-Sud, 1999.
- D. Dauge, PhD Thesis, Ecole Polytechnique, 2005.
- J. V. Suárez-Meneses, E. Bonilla-Reyes, E. A. Blé-González, M. C. Ortega-Alfaro, R. A. Toscano, A. Cordero-Vargas and J. G. López-Cortés, Tetrahedron, 2014, 70, 1422 CrossRef.
- S. N. Zafar, PhD Thesis, Ecole Polytechnique, 2010.
- (a) Y. Laot, L. Petit and S. Z. Zard, Org. Lett., 2010, 12, 3426 CrossRef CAS PubMed; (b) Y. Laot, L. Petit, N. D. M. Tran and S. Z. Zard, Aust. J. Chem., 2011, 64, 416–425 CrossRef CAS.
- For selected recent examples, see: (a) F. Prati, R. Buonfiglio, G. Furlotti, C. Cavarischia, G. Mangano, R. Picollo, L. Oggianu, A. di Matteo, S. Olivieri, G. Bovi, P. F. Porceddu, A. Reggiani, B. Garrone, F. P. Di Giorgio and R. Ombrato, ACS Med. Chem. Lett., 2020, 11, 825 CrossRef CAS PubMed; (b) G. Luo, L. Chen, A. Easton, A. Newton, C. Bourin, E. Shields, K. Mosure, M. G. Soars, R. J. Knox, M. Matchett, R. L. Pieschl, D. J. Post-Munson, S. Wang, J. Herrington, J. Graef, K. Newberry, D. V. Sivarao, A. Senapati, L. J. Bristow, N. A. Meanwell, L. A. Thompson and C. Dzierba, J. Med. Chem., 2019, 62, 831 CrossRef CAS PubMed; (c) D. Turner, A. J. Summers, L. O. Johnson, M. A. Knowles and C. W. G. Fishwick, ACS Med. Chem. Lett., 2017, 8, 1264 CrossRef PubMed.
- (a) W. Semmler, Ber. Dtsch. Chem. Ges., 1892, 25, 3352 CrossRef; (b) L. Wolff, Justus Liebigs Ann. Chem., 1902, 322, 351 CrossRef CAS; (c) G. Schroeter, A. Gluschke, S. Götzky, J. Huang, G. Irmisch, E. Laves, O. Schrader and G. Stier, Ber. Dtsch. Chem. Ges., 1930, 63, 1308 CrossRef.
- B. Quiclet-Sire, N. Tölle, S. N. Zafar and S. Z. Zard, Org. Lett., 2011, 13, 3266 CrossRef CAS PubMed.
- (a) R. M. Roberts, G. P. Anderson, A. A. Khalaf and C.-E. Low, J. Org. Chem., 1971, 36, 3342 CrossRef CAS; (b) H. Stetter and A. Reischl, Chem. Ber., 1960, 93, 791 CrossRef CAS; (c) M. Makosza, Rocz. Chem., 1967, 41, 1037 CAS.
- (a) A. Cordero Vargas, B. Quiclet-Sire and S. Z. Zard, Tetrahedron Lett., 2004, 45, 7335 CrossRef CAS; (b) A. Cordero Vargas, B. Quiclet-Sire and S. Z. Zard, Org. Biomol. Chem., 2005, 3, 4432 RSC; (c) O. Cortezano-Arellano and A. Cordero-Vargas, Tetrahedron Lett., 2010, 51, 602 CrossRef CAS.
- L. Petit and S. Z. Zard, Chem. Commun., 2010, 46, 5148 RSC.
- (a) M. A. Ferreira, M. A. C. Costa, A. C. Alves and M. H. Lopes, Phytochemistry, 1973, 12, 433 CrossRef CAS; (b) O. Weigenand, A. A. Hussein, N. Lall and J. J. M. Meyer, J. Nat. Prod., 2004, 67, 1936 CrossRef CAS PubMed.
- K. U. Ingold and D. A. Pratt, Chem. Rev., 2014, 114, 9022 CrossRef CAS PubMed.
- D. V. Avila, K. U. Ingold, J. Lusztyk, W. H. Green and D. R. Procopio, J. Am. Chem. Soc., 1995, 117, 2929 CrossRef CAS.
- (a) M. A. Fischbach and C. T. Walsh, Chem. Rev., 2006, 106, 3468 CrossRef CAS PubMed; (b) A. Das and C. Khosla, Acc. Chem. Res., 2009, 42, 631 CrossRef CAS PubMed.
- A. Cordero Vargas, B. Quiclet-Sire and S. Z. Zard, Org. Lett., 2003, 5, 3717 CrossRef PubMed.
- A. Fukami, T. Nakamura, Y.-P. Kim, K. Shiomi, M. Hyashi, T. Nagai, H. Yamada, K. Komiyama and S. Omura, J. Antibiot., 2000, 53, 1215 CrossRef CAS PubMed.
- (a) T. J. Simpson and D. J. Stenzel, J. Chem. Soc., Chem. Commun., 1981, 139 Search PubMed; (b) R. Shan, M. Stadler, H. Anke and O. Sterner, J. Nat. Prod., 1997, 60, 804 CrossRef CAS.
- (a) M. Bös, R. Canesso, N. Inoue-Ohga, A. Nakano, Y. Takehana and A. J. Sleight, Bioorg. Med. Chem., 1997, 5, 2165 CrossRef PubMed; (b) M. Bös, H. Stadler, J. Wichmann, F. Jenck, J. R. Martin, J.-L. Moreau and A. J. Sleight, Helv. Chim. Acta, 1998, 81, 525 CrossRef.
- (a) H. Gross, A. Rieche and G. Mattey, Chem. Ber., 1963, 96, 308 CrossRef CAS; (b) T. M. Cresp, M. V. Sargent, J. A. Elix and D. P. H. Murphy, J. Chem. Soc., Perkin Trans. 1, 1973, 340 RSC.
- M. Boumediene, PhD Thesis, Ecole Polytechnique, 2011.
- (a) B. Quiclet-Sire and S. Z. Zard, Org. Lett., 2008, 10, 3279 CrossRef CAS PubMed; (b) For a review of the field, see: B. Quiclet-Sire and S. Z. Zard, Synlett, 2016, 680 CAS.
- (a) E. Ferrer and S. Z. Zard, unpublished observations; (b) M. Heinrich and S. Z. Zard, Org. Lett., 2004, 6, 4969 CrossRef CAS PubMed. For earlier photochemical work on S-acyl xanthates, see: (c) F. Mestre, C. Tailhan and S. Z. Zard, Heterocycles, 1989, 28, 171 CrossRef CAS; (d) P. Delduc, C. Tailhan and S. Z. Zard, J. Chem. Soc., Chem. Commun., 1988, 308 RSC; (e) D. H. R. Barton, M. V. George and M. Tomoeda, J. Chem. Soc., 1962, 1967 RSC.
- C. C. Silveira, A. L. Braga, T. S. Kaufman and E. J. Lenardaõ, Tetrahedron, 2004, 60, 8295 CrossRef CAS.
- B. Quiclet-Sire, G. Revol and S. Z. Zard, Org. Lett., 2009, 11, 3554 CrossRef CAS PubMed.
- X. Zeng, PhD Thesis, Ecole Polytechnique, 2022.
- For reviews on the synthesis of naphthols and naphthalenes, see: (a) J. A. Mora Vargas, D. P. Day and A. C. B. Burtoloso, Eur. J. Org. Chem., 2021, 741 CrossRef; (b) C. B. de Koning, A. L. Rousseau and W. A. L. van Otterlo, Tetrahedron, 2003, 59, 7 CrossRef CAS; for a review on naphthalenes in medicinal chemistry, see: (c) S. Makar, T. Saha and S. K. Singh, Eur. J. Med. Chem., 2019, 161, 252e276 CrossRef PubMed.
- A. Cordero-Vargas, I. Pérez-Martin, B. Quiclet-Sire and S. Z. Zard, Org. Biomol. Chem., 2004, 2, 3018 RSC.
- N. D. M. Tran and S. Z. Zard, Org. Biomol. Chem., 2014, 12, 3251 RSC.
- N. D. M. Tran, PhD Thesis, Ecole Polytechnique, 2014.
- P. Salomon and S. Z. Zard, Org. Lett., 2014, 16, 2926 CrossRef CAS PubMed.
- R. F. Guignard and S. Z. Zard, Chem. Commun., 2011, 47, 12185 RSC.
- D. Boger, C. Boyce, R. Garbaccio and J. Golberg, Chem. Rev., 1996, 97, 787 CrossRef PubMed.
- M.-G. Braun, PhD Thesis, Ecole Polytechnique, 2011.
Footnote |
| † This article is dedicated to Prof. Shigeru Yamago (Kyoto University) on the occasion of his 60th birthday and to the memory of Prof. Yves Langlois (Université Paris-Saclay). |
| This journal is © The Royal Society of Chemistry 2023 |