
Visible-light-induced cascade reaction: a sustainable approach towards molecular complexity†
Sumit
Ghosh‡
 ,
Pranjal
Pyne‡
,
Anogh
Ghosh‡
,
Swagata
Choudhury‡
and
Alakananda
Hajra
*
,
Pranjal
Pyne‡
,
Anogh
Ghosh‡
,
Swagata
Choudhury‡
and
Alakananda
Hajra
*
Department of Chemistry, Visva-Bharati (A Central University), Santiniketan, 731235, India. E-mail: alakananda.hajra@visva-bharati.ac.in
First published on 18th January 2023
Abstract
Photoredox catalysis has demonstrated rapid evolution in the field of synthetic organic chemistry. On the other hand, the splendour of cascade reactions in providing complex molecular architectures renders them a cutting-edge research area. Therefore, the merging of photocatalysis with cascade synthesis brings out a synthetic paradigm with immense potential. The development of photocascade catalysis for a target molecule with a particular molecular skeleton and stereochemical framework presents certain challenges but provides a robust and environmentally benign synthetic alternative. This comprehensive review assembles all the accomplishments and highlights of visible-light-induced cascade reactions with literature coverage up to October 2022.
 Sumit Ghosh | Sumit Ghosh completed his BSc at Burdwan University in 2014. He achieved his MSc from IIT Kanpur in 2016. He is currently working with Prof. Alakananda Hajra at Visva-Bharati University. He was listed in the Stanford University's single-year top 2% scientists of the world in 2022. His research interest is the functionalization of different heterocycles. |
 Pranjal Pyne | Pranjal Pyne received both his B.Sc. and M.Sc. degrees from Visva-Bharati University in 2018 and 2020, respectively. For his M.Sc. thesis project (2019–2020), he worked under the supervision of Dr Alakananda Hajra at Visva-Bharati University. Currently, he continues his work in the same group. His research interest is in the domain of sustainable catalytic methods via C–H activation and photoredox catalysis. |
 Anogh Ghosh | Anogh Ghosh was born in West Bengal, India (1997). After the completion of his BSc from Visva-Bharati University (2018), he obtained a Post-Graduate degree from the same university (2020). Currently, he is working with Dr Alakananda Hajra at Visva-Bharati University. His research interest lies in organophototredox catalysis and asymmetric C–H functionalization. |
 Swagata Choudhury | Swagata Choudhury was born in 1998 in the state of West Bengal, India. She obtained her BSc degree in Chemistry from Visva Bharati University in 2018. She subsequently completed her MSc in Chemistry from the same university in 2020. Her research interests include C–H activation and photoredox catalysis. |
 Alakananda Hajra | Dr Alakananda Hajra completed his Ph.D. in 2002 under the supervision of Prof. B. C. Ranu from the Indian Association for the Cultivation of Science (IACS), Kolkata, and then joined SUNY in Albany, USA, as a postdoctoral research fellow in Prof. Frank M. Hauser's group (2002–04). He was also a JSPS research fellow at the University of Tokyo and worked with Prof. Eiichi Nakamura and Prof. Masaharu Nakamura from November 2004 to May 2006, and then joined Visva-Bharati University. He also worked with Prof. N. Yoshikai, at NTU, Singapore for one year (2011–2012). His research interests include the development of new synthetic methodologies and green synthetic procedures. |
1. Introduction
Recently, environmental preservation has emerged as a top priority for chemists all over the globe.1 The growing concern for the environment is driving researchers to find new ways to synthesize molecules with medicinal and industrial applications, which produce no waste and minimal pollution using green chemistry principles.2 With this background, after three decades of research and development, the use of visible light to mediate the synthesis and functionalization of a wide range of organic compounds has arrived at a remarkable stage of superiority and efficiency.3 More specifically, after the revolutionary reports by MacMillan,4 Nicewicz,5 Stephenson6 and Yoon,7 the visible-light-induced synthetic methodology has become an important field of research worldwide.8 The increasing fascination with photocatalysis can be attributed to the fact that it is easy to use, uses renewable energy, is cheap, can be easily purified, is biocompatible, and is not toxic.9On the other hand, over the past few years, cascade reaction has gained the attention of organic chemists.10 In organic synthesis, generally, products may be formed through multi-step reactions, but it will be a more efficient process if the product can be formed in one sequence without adding additional reagents, catalysts or solvents. This type of reaction is known as a cascade or domino reaction. Specifically, it can be defined as a one-pot reaction under the same reaction conditions with a single catalytic mechanism deploying a single catalyst, which results in the formation of two or more new bonds.11 To the best of our knowledge, the cascade reaction came into consideration in 1917 when Robinson synthesized tropinone by a one-pot reaction (Scheme 1).12 Since then, a large number of cascade reactions have been reported in chemical literature as they are largely used for the synthesis of various drugs, natural products, and biologically and industrially important compounds; they are also time- and cost-efficient, and atom- and step-economical.13 However, conventional methodologies have several drawbacks and do not meet the twelve principles of green chemistry. Therefore, use of visible-light in cascade reactions may be a game changer and recent years have witnessed a quick surge in the development of various visible-light-induced cascade reactions.
 | ||
| Scheme 1 Robinson's synthesis of tropinone. | ||
Even though the photo-induced cascade reaction is so important and there are a lot of reports on it, there isn't yet a complete, systematic review article in the chemical literature on the proposed topic. However, a recent review by the Xu group only discussed organophotocatalytic cascade reactions.14 Consequently, a complete review is currently needed. Our group has also contributed considerably to both visible-light-induced methodology15 and cascade reactions.16 From our continuous efforts in the exploration of cascade reactions and visible-light-mediated methodology, herein, we delineate the detailed accomplishments of visible-light-induced cascade reactions and highlight their merging as a sustainable tool towards efficient syntheses. For the sake of better understanding, we have categorized this article based on the type of photocatalyst used, for example, (i) Ru-based photocatalyzed cascade reactions, (ii) Ir-based photocatalyzed cascade reactions, (iii) Pd-based photocatalyzed cascade reactions (iv) organic semiconductor-based photocatalyzed cascade reactions and (v) metal-free visible-light-induced cascade reactions. In Fig. 1, we can see the structures of a few photocatalysts that have been employed in cascade reactions.
 | ||
| Fig. 1 Structures of the photocatalysts. | ||
2. Ru-photocatalyzed cascade reactions
The isoxazolidine skeleton is found in various naturally occurring substances, including pyrinodemin A, dactylicapnosinine, and lycojaponicumin A. It is also an important ingredient in pharmaceuticals. Therefore, the synthesis and functionalization of isoxazolidine derivatives are highly important.17 In this context, Zhu and co-workers18 (Jan 2013) reported a methodology for the photoredox-catalyzed synthesis of bicyclic isoxazolidine (2c). Product 2c can be synthesized most effectively when tertiary amine (2a) (0.1 mmol) reacts with α-ketoester (2b) (3.5 equiv.) in the presence of catalyst PC1 (3–5 mol%) under irradiation with 5 W blue LEDs at room temperature in air (Scheme 2). Among all the photocatalysts, PC1 and PC2 were the most efficient. Again, TfOH was shown to increase the yield of the product to 60%. It was observed that yields were higher for solvents like MeOH, EtOH, and i-PrOH than those of MeCN, DMF, and NMP. After achieving the optimal conditions, a variety of substrates were examined. If an electron-withdrawing group was present at the N-substituted aromatic ring, then the reaction became slow. However, in this case, the Ir(III)-photocatalyst PC2 was more fruitful than Ru(III) PC1 because the tertiary amine could transfer a single electron to *Ir(ppy)2(dtbbpy)+. Therefore, its Stokes shift became smaller and the HOMO–LUMO gap increased due to its stronger reductant properties. The yield of this cascade reaction was good when an electron-donating group was attached in the presence of the PC1 photocatalyst. On altering the tert-butyl group to a methyl group on 2-oxobutanoate 3b, diastereoselectivity was obtained, but the yield decreased slightly. | ||
| Scheme 2 Visible-light-induced cascade aerobic C–H/C–N cleavage (Zhu method). | ||
Some control experiments were carried out to demonstrate the mechanism. Very little product was obtained when this cascade reaction was done in the nitrogen atmosphere. Also, BHT addition decreased the yield. This implies that the reaction involved a radical mechanism in air. The isotope labeling study of deuterated N-phenyl-1,2,3,4-tetrahydroisoquinoline with 3b demonstrated that benzylic hydrogen was abstracted and it was 3.5 times faster than deuterium. This proved that benzylic C–H activation was the rate-limiting step of this reaction. Based on the above experiments and earlier literature reports, a plausible mechanism was proposed for the reaction, as shown in Scheme 3. Firstly, Ru(bpy)32+ was excited to Ru(bpy)32+* by the irradiation of visible light, which formed the aminyl radical cation 3I and reductive species Ru(bpy)31+ upon reaction with tertiary amine 3a by a single-electron transfer process. Next, the C–H abstraction of 3I produced the iminium ion 3II. Nucleophilic enol 3III (produced in situ from α-ketoester) on reaction with 3II produced the addition product 3IV. This could isomerize to 3V, which further formed 3VIvia a retro-aza-Michael reaction. Next, 3VI was oxidized to form imine species 3VII, then nitrile oxide 3VIII was formed under oxidative conditions. Finally, the product isoxazolidine 3c was obtained by a [3 + 2] cycloaddition reaction. Therefore, by this method, two C–C, one N–O and one C–O bonds can be constructed along with two new rings and one quaternary carbon center, which is a very good feature of this methodology. Moreover, this mild and environmentally benign method was utilized for the synthesis of dactylicapnosinine derivatives. However, the reaction time was relatively high (60–170 h) with respect to other Ru-catalyzed methodologies.
 | ||
| Scheme 3 Plausible mechanism. | ||
Over the past few years, the incorporation of the trifluoromethyl moiety in various synthetically useful scaffolds has garnered distinct attention.19 In the same vein, Yang & Xia et al.20 developed a difunctionalization methodology using visible-light-induced cascade trifluoromethylation followed by desulfonylation and C–N or C–H bond formation (May, 2015). They exploited carboxyl/tosyl amide (4a, 0.1 mmol), trifluoromethyl agent CF3SO2Cl (2 equiv.), photo redox catalyst Ru(bpy)3Cl2 (0.005 mmol) and base K2HPO4 (0.2 mmol) in acetonitrile as a solvent under blue LED irradiation at room temperature. This strategy produced the desired product (4b/4c/4d) in good yield (Scheme 4). Replacing CF3SO2Cl with different trifluoromethylation agents, viz., Togni's reagent, and CF3I led to inferior yields. Solvent screening disclosed that acetonitrile was preferable. It was observed that an air atmosphere suppressed the desired transformation and the absence of light or photocatalyst abolished the product formation. This protocol was provided with diverse substrate scope. Both electron-donating and electron-withdrawing groups at the para-position in the aromatic ring were suitable substrates and afforded the desired product 4b with good yields, although substitution at the ortho-position led to an undesired complex product. In addition, substituents directly attached to nitrogen atoms, e.g., butyl, isopropyl, and methyl, produced the desired product (4b) in moderate to good yield. If the substituents at the nitrogen atom were replaced with aryl groups, a fascinating change occurred, i.e., a unique 1,4-aryl shift, resulting in a different product 4c without the assistance of base. Substituents at the aryl moiety resulted in diverse electron-donating and electron-withdrawing groups with moderate to good yields. A similar substrate scope at the nitrogen-bound aryl group was observed, albeit with inferior yields. When carboxyl amide was used instead of tosylamide, the desired product 4d was obtained in a moderate to good yield.
 | ||
| Scheme 4 Visible-light-induced trifluoromethylarylation/1,4-aryl shift/desulfonylation cascade reactions for difunctionalization of alkenes (Yang and Xia method). | ||
Based on the results of control experiments and previous literature reports, a plausible mechanism was proposed (Scheme 5). In the beginning, the photocatalyst [Ru(bpy)3]2+ was photo-excited and reduced CF3SO2Cl to generate radical ˙CF3 and liberated sulphur dioxide and chloride ion. This ˙CF3 radical was added to substrate 5a and generated the intermediate 5i with a new C(sp3)–CF3 bond. When tosylamide was incorporated as a substrate (i.e., X = SO2), a consecutive aryl migration/desulfonylation of the intermediate 5I led to a new radical intermediate 5II. Subsequent cyclization of this amidyl radical intermediate 5II produced the desired product 5c. When carboxyl amide (i.e., X = CO) was used, the intramolecular cyclization of the intermediate 5I furnished the intermediate 5III, which engendered the desired isoquinolinedione product (5b/5d) after consecutive oxidation and deprotonation. The important advantages of this methodology are operational simplicity, low catalyst loading and using less additive or no additive (additive-free).
 | ||
| Scheme 5 The most probable mechanism. | ||
Jiang and his group21 in 2015 developed a strategy for the generation of α-aryl esters via the visible-light-driven Meerwein cascade reaction. As per the report, they employed aryl diazonium tetrafluoroborates (6a, 0.5 mmol), acrylonitriles (6c, 3 equiv.) and alcohols (6b, 2 mL) in the presence of Ru(bpy)3Cl2·6H2O (2 mol%) as a catalyst under the irradiation of a blue LED (450 nm) overnight at room temperature in an O2 atmosphere. These conditions are the best suited to produce the desired product 6d with excellent yield (Scheme 6). Trials with other organic dyes such as eosin-Y, and rose bengal as a photocatalyst were unsuccessful. MeOH played both the roles of solvent and reactant very efficiently and suppressed other solvents like DMF, THF, and DMSO. A high yield was obtained when the reaction was done under an oxygen atmosphere rather than air. Both the photocatalyst and visible-light were essential for carrying out this transformation. A broad substrate scope was observed for the reactant with various substituents, which proved its generality. Electron-withdrawing groups like nitro, aryl, cyano, and ester were good substituents for generating good yields. 4-Chlorobenzene diazonium salt was reluctant to generate good yield, whereas a heterocyclic diazonium salt generated good yield under optimized reaction conditions. Electron-donating substituents like para-methylbenzene and para-methoxy benzene diazonium salts were not suitable for making the desired product in good yield. Among some substituted acrylonitriles only crotononitrile showed some tolerance although it produced a lower yield. Substituents on alcohol also showed some influence on product formation. The increment in the chain length of the alkyl group along with the installation of the electron-withdrawing group significantly reduced the yield.
 | ||
| Scheme 6 Preparation of α-aryl esters via visible-light-mediated Meerwein cascade reactions (Jiang method). | ||
Several control studies revealed mechanistic insights. The incorporation of TEMPO confirmed the radical involvement. Based on the results and reports, a plausible mechanism was proposed as depicted in Scheme 6. In the beginning, [Ru(bpy)32+]* was formed from its ground state under the influence of a blue LED, which was further oxidatively quenched by diazonium salt 6a to generate the aryl radical 6I. This aryl radical 6I then attacked the double bond of acrylonitrile (6c) to form the radical intermediate 6II. Next, the generated intermediate 6II underwent a reaction with dioxygen to provide the peroxy radical 6III and the alkoxy radical 6IV. A 1,2-shift of the hydrogen atom in the alkoxy radical 6IV along with SET oxidation generated an aryl acetyl cyanide intermediate 6VI; hence, the catalytic cycle was completed. Finally, the aryl acetyl cyanide intermediate 6VI was engaged in a nucleophilic substitution reaction with alcohol 6b to generate the final product α-aryl ester 6d. This strategy featured a simplistic approach as it was a one-pot strategy, mediated by incorporating molecular oxygen as an oxidant at room temperature to generate higher oxindole analogues.
Two years later in 2017, Zhang and his team22 introduced an efficient and general visible-light-promoted radical cascade reaction strategy using ortho-hydroxyaryl enaminones to synthesize 3-CF2-containing chromones. They initiated with o-hydroxyaryl enaminones (7a, 0.5 mmol) and ethyl bromodifluoroacetate (7b, 2.5 equiv.) in the presence of Ru(bpy)3·Cl2 (1 mol%) as a catalyst, Et3N as an initial reducing agent, and NaHSO3 as a base under the irradiation of a 45 W LED lamp at room temperature for 24 hours, which subsequently generated the desired product 7c with up to 82% yield (Scheme 7). It was observed that bases like Na2SO3 and NaHSO3 were more suitable for producing higher yields with no by-products. In these reaction conditions, DMSO was superior as a solvent. Both the catalyst and visible light were indispensable for the reaction to proceed. Under the optimized conditions, substrate scopes were studied extensively with different o-hydroxyaryl enaminone derivatives attaching both electron-donating and -withdrawing groups. Functional groups like –F, –Cl, –Me, –OMe, and –CN were well tolerated. The position of the substituent played a crucial role in generating products as the ortho- and para-position of the aromatic ring of (E)-3-(dimethylamino)-1-(2-hydroxyphenyl)-prop-2 en-1-one afforded a higher yield in comparison to meta.
 | ||
| Scheme 7 Visible-light-promoted cascade reaction of o-hydroxyaryl enaminones for the synthesis of 3-CF2-containing chromones (Zhang method). | ||
To visualize the mechanistic pathway, some control experiments were performed. The incorporation of TEMPO, which is itself a radical scavenger, scrapped the formation of the desired product and confirmed the involvement of the radical path in the reaction. Based on the literature survey and control experiments, they revealed a mechanism, as shown in Scheme 7. At first, [Ru(bpy)3]2+ got excited to generate [Ru(bpy)3]2+* under the influence of visible-light, which further oxidized Et3N to Et3N+ and was itself reduced to [Ru(bpy)3]1+. The SET process of [Ru(bpy)3]1+ species with C–Br of ethyl bromodifluoroacetate generated a radical 7I and reproduced [Ru(bpy)3]2+. This radical 7I was then attached to the α-position of α,β-unsaturated ketone 7a to generate radical 7II. After that, it participated in the reduction of [Ru(bpy)3]2+* and developed the iminium cation intermediate 7III as well as [Ru(bpy)3]1+. Finally, the iminium cation was attacked by the hydroxyl group on the benzene ring in the presence of the base to form an intermediate 7IV. In the final step, dimethylamine elimination led to the desired product 7c. Mild reaction conditions, broad substrate scope, low catalyst loading, and the construction of biologically active chromone derivatives are important features of this methodology.
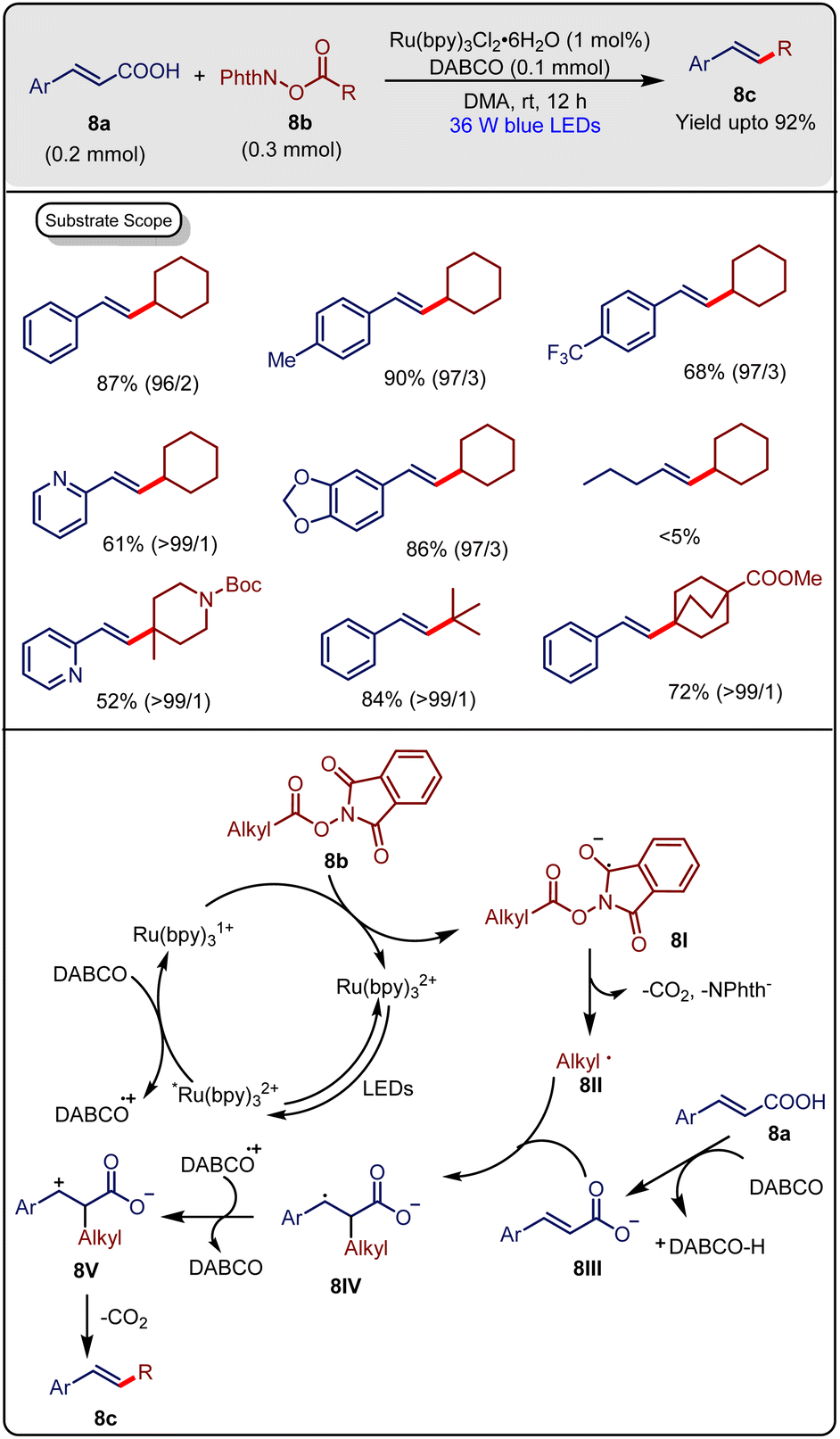
In 2017, Xu & Zhang et al.23 developed a visible-light-induced Ru-photocatalyzed dual decarboxylative coupling between cinnamic acid derivatives and N-(acyloxy)phthalimides to synthesize several α,β-alkylated styrene derivatives. The reaction was done by using 0.2 mmol cinnamic acid 8a and 0.3 mmol of carboxylic acid derivative 8b in the presence of 1 mol% of Ru(bpy)3Cl2·6H2O as photocatalyst and 0.1 mmol of DABCO as an additive under a 36 W blue LED at room temperature. The desired product (E)-(2-cyclohexylvinyl) benzene (8c) was achieved in up to 90% yield with maximum regioselectivity of 98![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 within 12 h (Scheme 8). Another metal photocatalyst [Ir(ppy)3] was not as efficient as Ru(bpy)3Cl2·6H2O. It was observed that both electron-donating and -withdrawing groups containing cinnamic acids produced the desired products with good to excellent yields under optimized reaction conditions. 3,3-Diphenylacrylic acid was also suitable for this reaction. Aliphatic carboxylic acid derivatives having ester, ketone, and terminal alkyne groups were suitable for this reaction. It was observed that secondary, tertiary and quaternary carboxylic acids were also well tolerated. α-Amino acid-containing esters were also able to yield the product under optimized reaction conditions.
:2 within 12 h (Scheme 8). Another metal photocatalyst [Ir(ppy)3] was not as efficient as Ru(bpy)3Cl2·6H2O. It was observed that both electron-donating and -withdrawing groups containing cinnamic acids produced the desired products with good to excellent yields under optimized reaction conditions. 3,3-Diphenylacrylic acid was also suitable for this reaction. Aliphatic carboxylic acid derivatives having ester, ketone, and terminal alkyne groups were suitable for this reaction. It was observed that secondary, tertiary and quaternary carboxylic acids were also well tolerated. α-Amino acid-containing esters were also able to yield the product under optimized reaction conditions.
 | ||
| Scheme 8 Visible-light-induced Ir-photocatalyzed decarboxylative alkylation of alkenyl carboxylic acid (Xu & Zhang method). | ||
Some control experiments were carried out by the authors to propose the mechanism of the reaction. The radical scavenger 1,1-diphenylethylene stopped the reaction. When 2-cyclopropylacetic acid derivative was used, only a radical rearrangement occurred. These results indicated that the aliphatic carboxylic acid ester generated an alkyl radical. Moreover, it was observed by the authors that N-(acyloxy)phthalimide (E½ = −1.26 to −1.37 V vs. SCE) could not be reduced by *Ru(II) catalyst (EIII/II½ = −0.81 V vs. SCE), but could be reduced by Ru(I) (EII/I½ = −1.33 V vs. SCE) and *Ru(II) (EII/I½ = 0.77 V vs. SCE) could oxidize DABCO (E1/2 = 0.6 V vs. SCE). Furthermore, DABCO quenched the excited photoredox catalyst, which could be proved by the Stern–Volmer quenching experiment. Based on these control experiments, a mechanism was proposed, as shown in Scheme 8. The decarboxylation of 8b generated an alkyl radical 8II, which was then added to cinnamate 8III to give benzylic radical 8IV. After that, DABCO oxidized 8IV and the produced benzylic cation 8V and subsequently formed the desired product 8c by decarboxylation. The gram scale applicability and high yield formation with very high regioselectivity are important features of this method.
Like diverse heterocyclic scaffolds, azaspirocycles are found in a wide range of biologically active molecules.24 Tang et al.25 in early 2018, reported the light-mediated general and cascade synthesis of azaspirocyclic cyclohexadienones via the dearomative Meerwein addition/cyclization of alkenes followed by C–F bond cleavage. Despite the chemical stability of C–F bonds, the ready availability of aryl fluorides and scarcity of oxidative C–F cleavage rendered this strategy a good synthetic upgrade. As shown in Scheme 9, N-benzylacrylamides (9a, 0.3 mmol) and 4-nitrophenyldiazonium tetrafluoroborate (9b, 2 equiv.) were reacted in the presence of photocatalyst Ru(bpy)3Cl2 (5 mol%), base K2CO3 (3 equiv.) and water as an external oxygen source (4 equiv.) in DMF as solvent (2 mL) under 5 W blue LED photo-irradiation at room temperature for 24 hours. This reaction led to the intended product 9c with yields of up to 88%. Other photocatalysts and bases were suitable, albeit they provided inferior yields. Moreover, the reaction was abolished in the absence of either light or photocatalyst. This protocol displayed diverse substrate scope for both substrates. Satisfyingly, the optimal condition was fruitful for various aryldiazonium tetrafluoroborates bearing different electron-withdrawing substituents. The other partners, N-benzylacrylamides, showed various scopes in correspondence with phenyl-tethered halides and aliphatic moieties. N-Substituted methyl, benzyl, n-butyl, and i-propyl were also amenable in this protocol. Interestingly, N-benzylacrylamide with a para-methoxyl substituent coupled with aryldiazonium tetrafluoroborate performed excellently in this scheme; it showed an almost similar type of reactivity and yielded the same targeted product. Unlike the previous scheme for this particular method, water was not imperative. Eventually, it afforded a comparably higher yield than the aforementioned reaction involving C–F bond cleavage. Nevertheless, replacing C–F with other components like Cl, Br or H was not effective and provided inferior yields.
 | ||
| Scheme 9 Ru-photocatalyzed synthesis of azaspirocycles via the Meerwein cascade cyclisation of N-benzylacryamides (Tang method). | ||
Based on the mechanistic rationale and literature, a plausible mechanistic pathway was provided (Scheme 9). Photo-excited [Ru(bpy)3]2+* participated in a SET with ArN2+BF4− and was oxidized to [Ru(bpy)3]3+ with the concurrent generation of radical Ar˙. This radical was then added to substrate 9a furnishing a new intermediate radical 9I, which readily transformed into another intermediate 9IIvia the dearomative cyclization process. This intermediate lost a single electron to the Ru(III)-species to afford carbocation 9III, which subsequently reacted with H2O and after C–F cleavage formed a new cationic intermediate 9V. Base-promoted hydrogen abstraction of 9V led to the final product 9c. Readily available aryldiazonium salts were used as the aryl radical source, and environment-friendly water as the external oxygen source, in combination with clean and atomic economy Ru-based photoredox catalysis.
9-H-Pyrrolo[1,2-a]indoles are important heterocycles found in numerous medicinal molecules.26 Liu, Zhou and Xie et al.27 (Nov 2019) employed N-propargylindole (10a) (0.2 mmol) and 4-methoxyphenyldiazonium tetrafluoroborate (10b) (0.4 mmol) under 5 W blue LED light irradiation in the presence of Ru(bpy)3Cl2 (5 mol%) and K2S2O5 (0.4 mmol) in THF (2 mL) and Ar-atmosphere at room temperature for 24 h. The desired product, 2-sulfonyl-substituted 9H-pyrrolo[1,2-a] indole (10c), was afforded in a yield of up to 79% (Scheme 10). Ru(bpy)3Cl2 was the superior catalyst among various catalysts, such as Ir(ppy)3, eosin Y. It was observed that the yield was highest when 5 W blue LED light was used instead of 3 W/12 W blue LED light, 36 W compact fluorescent light, and 5 W green LED light. The solvents were further screened, which resulted in toluene, DMF, DMSO, CH3CN, and 1,4-dioxane being inferior to THF. Next, N-propargylindoles containing substituted aryl groups were investigated. The results demonstrated that the electron-donating groups at the para-position of the aryl ring delivered higher yields than the electron-withdrawing groups. For example, when a NO2 group was present, the yield was 0%. If the terminal position of the triple bond contained a fused ring/heterocyclic ring, the were products formed in 61%–57% yields. However, the desired product could not be obtained for 1-(prop-2-yn-1-yl)-1H-indole. Next, aryldiazonium tetrafluoroborates (10b) were examined. Both electron-donating, as well as electron-withdrawing groups at the para-position of 10b provided the product with moderate to good yields.
 | ||
| Scheme 10 Visible-light-induced cascade sulfonylation/cyclization of N-propargylindoles with aryldiazonium tetrafluoroborates via the insertion of sulfur dioxide (Liu, Zhou and Xie method). | ||
The control experiments with TEMPO, BHT, 1,1-diphenylethene revealed that this cascade reaction proceeded through a radical pathway. Based on other investigations, a plausible mechanism was suggested. Firstly, the strong reductant *Ru(bpy)32+ was generated by absorbing visible-light irradiation. A single-electron transfer process occurred to give aryl radical 10I along with Ru(bpy)33+ from 4-methoxyphenyl-diazonium tetrafluoroborate 10b. On reaction with K2S2O5, radical 10I produced the arylsulfonyl radical 10II and K2SO3. The attack of radical 10II on the carbon–carbon triple bond of 10a generated radical 10III, which led to radical 10IV by intermolecular cyclization. After that, the single-electron oxidation of radical 10IV by Ru(bpy)33+ furnished cation intermediate 10V and Ru(bpy)32+ was regenerated. Finally, 10V immediately underwent isomerization and deprotonation to produce the final product 10c (Scheme 10). This method has excellent functional group tolerance, applicable for gram-scale synthesis, and provides an easy way to assemble a new C–C bond and two new C–S bonds in a single step.
3. Ir-photocatalyzed cascade reaction
Iridium is one of the most used metal-based photocatalysts in organic synthesis.28 The Ir-based photocatalyzed cascade is also familiar in the literature. In this chapter, we will chronologically discuss cascade reactions that are catalyzed by Ir-catalysts.Oxindole is an important heterocycle that exists in many bioactive drugs.29 In 2013, Zhu and co-workers30 developed a synthetic strategy for the synthesis of 3,3-disubstituted oxindoles (11c) by the decarboxylative coupling of phenyliodine(III) dicarboxylate (11b) (3 equiv.) with N-methyl-N-phenylmethacrylamide (11a) (1 equiv.) in the presence of fac-Ir(ppy)3 (1 mol%) photocatalyst under 35 W fluorescent light irradiation at room temperature (Scheme 11). Other photocatalysts, such as Ru(bpy)3Cl2 and Ir(ppy)2(dtbbpy)BF4 were inferior to fac-Ir(ppy)3. The reaction could not occur without a photocatalyst and visible-light. By exploration of the substrate scope, it was seen that the strong electron-withdrawing group (–NO2, –F) (at the benzene ring) containing substrate could not afford the product in good yields. The yield was high if any double bond was present in the substrate. Various aliphatic carboxylic acids (primary, secondary, and tertiary) with various functional groups were suitable for this reaction. The presence of the –CF3 group in the carboxylic acid could enhance the biological activity of the product. PhI(OCOPh)2, produced by ligand metathesis of benzoic acid and 11b, did not function because the decarboxylation process was slower and the nucleophilicity of the aryl radical was low.
 | ||
| Scheme 11 Visible-light-mediated photoredox-catalysed decarboxylation/C–H functionalization cascade (Zhu method). | ||
The proposed mechanism was as follows. Initially, under visible-light irradiation *IrIII(ppy)3 was formed, from which an electron was imparted to PhI(OAc)2 to generate radical 11I and IrIV(ppy)3. Next, radical 11II was formed by the reaction of 11I with 11a, followed by decarboxylation, which produced the radical 11III. After that, a single-electron transfer occurred from 11III to IrIV(ppy)3 to convert the corresponding cation 11IV. Finally, deprotonation of 11IV by the acetate anion generated the desired product 11c (Scheme 11). Mild reaction conditions, broad substrate scope, a wide range of functional group tolerance and low catalyst loading are important advantages of this process.
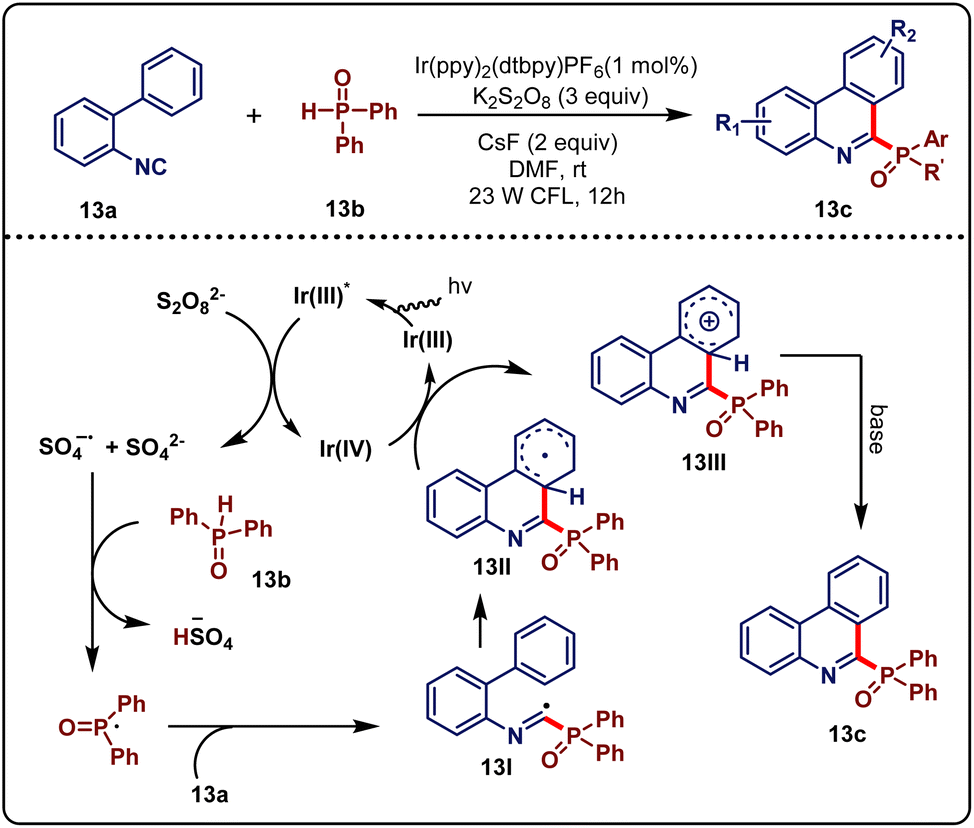
Organophosphorus compounds have many applications in organic synthesis.31 The traditional approach to forming a C–P bond has faced several limitations. In this regard, Yan & Lu et al.32 developed a new methodology of radical C–P and C–C bond formation via the reaction of N-arylacrylamides and isocyanides with diphenylphosphine oxide under the visible-light cascade reaction. As per the literature, they showed the reaction of biphenyl isocyanides, vinyl isocyanates, and N-arylacrylamides with diaryl phosphinesphine oxide changing the reaction condition. At first, biphenyl isocyanides (12a, 0.3 mmol) and diaryl phosphinesosphine oxide (12b, 0.9 mmol) were reacted in the presence of photocatalyst [Ir(ppy)2(dtbpy)PF6] (1 mol%), K2S2O8, CsF as a base and DMF as a solvent under the influence of 23 W CFL for 12 hours, which led to the formation of the desired product 12c in good to moderate yield (Scheme 12a). CsF as a base was the best suited over others. Other photocatalysts except [Ir(ppy)2(dtbpy)PF6] did not perform well and K2S2O8 was the most suitable persulfate. Reaction in a dark environment and in the absence of photocatalyst did not form any products, which proved their necessity in the reaction. Keeping the optimized conditions, the scope of biaryl isocyanides was investigated. The substrate containing substituents such as Me, H, OMe, F, Cl, and CF3 at the para position of the phenyl ring afforded the desired products with yields ranging from 60 to 85%. Whereas, ortho substituents producing some lower yields might be due to steric reasons. An electron-deficient heteroarene, with a pyridyl unit was compatible under standard conditions. However, reactions were unsuccessful when there was a strong electron-withdrawing group at the para-position. Several phosphoryl sources were also investigated. Di-p-tolylphosphine oxide and tert-butyl(phenyl)phosphine oxides produced an ideal yield, whereas diethyl phosphonate generated no desired product. Next, vinyl isocyanides (12d, 0.3 mmol) were also tested with diarylphosphine oxide (12b, 0.9 mmol) imposing a similar condition to biaryl isocyanides, just changing the base from CsF to Cs2CO3 with the time increased to 20 hours. The desired product 12e in 48–70% yield was obtained (Scheme 12b). N-Arylacrylamides (12f, 0.3 mmol), which are good radical receptors, were also tested with diarylphosphine oxide (12b, 0.9 mmol) in a similar condition to vinyl isocyanides. Up to 72% yield of the desired product (12g) was formed within 20 h (Scheme 12c). All of the substrates bearing a para-substituent of arylacrylamides reacted well. Electron-withdrawing group attachment was found to accelerate the reaction. The substrate with the N-benzyl group produced a satisfactory yield but N-free arylacrylamide was not suitable even after more reaction time. A reduced yield was observed for the ortho-substituent as it generated some steric hindrance.
 | ||
| Scheme 12 C–P and C–C bond formation cascade reaction of isocyanides and N-arylacrylamides with diphenylphosphine oxide in the presence of visible light (Yan and Lu method). | ||
Control experiments like TEMPO incorporation and the intermolecular kinetic isotopic effect were executed. A tentative mechanism was proposed based on literature studies and control experiments, as sketched in Scheme 13. Initially, photocatalyst *Ir(III) was formed from its ground state Ir(III) by absorbing visible-light irradiation. Next, the persulfate anion was reduced by *Ir(III) and formed Ir(IV), sulfate dianion, and sulfate radical anion, respectively. Thereafter, the P-centred radical intermediate was generated through hydrogen atom transfer (HAT) between diarylphosphine oxide 13b and the active sulfate radical anion, which further underwent intermolecular addition with 13a to generate intermediate 13I. A cyclohexadienyl-type radical 13II was then formed via the intramolecular cyclization of 13I. Thereafter, radical intermediate 13II could be oxidized by Ir(IV) to reproduce Ir(III) along with the formation of the cationic intermediate 13III. Finally, 13III underwent deprotonation to generate the desired product 13c with the assistance of the base. This process allows direct and convenient access to three types of phosphorylated N-heterocycles using a green energy source, visible light.
 | ||
| Scheme 13 Proposed mechanism. | ||
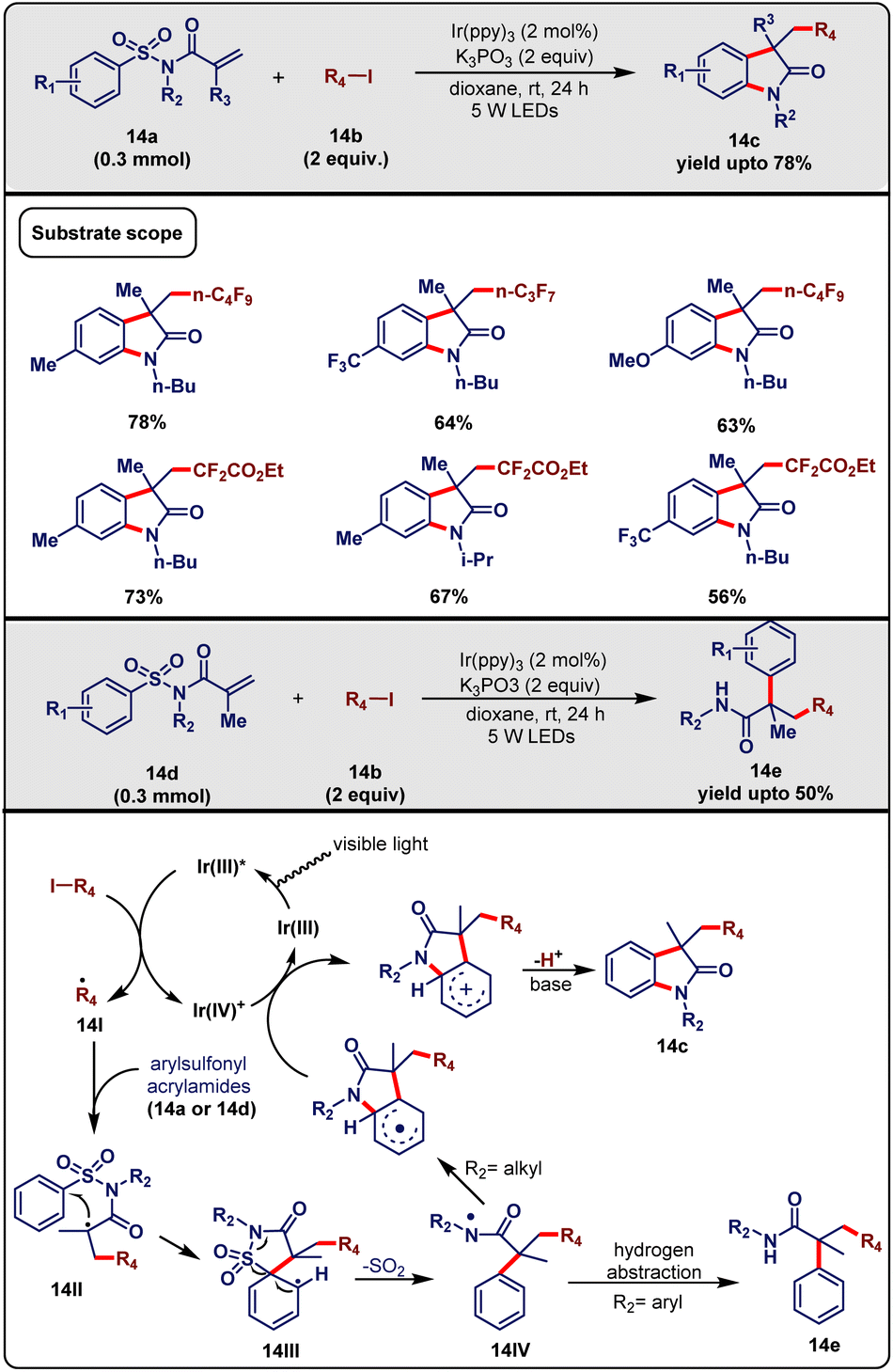
Tang and Sheng et al.33 in 2017 presented an efficient visible-light-driven perfluoroalkylation cascade strategy of conjugated tosyl amides. This strategy introduced an efficient route to synthesize α-aryl-β-fluoroalkyl amides containing several perfluorinated moieties. They exploited N-butyl-N-(tolysulfonyl)methylacrylamides (14a, 0.3 mmol) with perfluoroalkyl iodide (14b, 2 equiv.) in the presence of fac-Ir(ppy)3 (2 mol%) as a photocatalyst, K3PO3 as a base in 1,4-dioxane solvent under the irradiation from 5 W blue LEDs at room temperature. This condition promoted the formation of the desired product 14c with up to 78% yield within 24 h (Scheme 14). Other photocatalysts, such as Ru(bpy)3Cl2, Ru(bpy)3Cl2·6H2O and eosin Y failed when other factors remained the same. The desulfonylation cyclization step required a base and, in this regard, K3PO4 successfully played that role, defeating other inorganic bases, such as K2CO3, CS2CO3, and Na2CO3. Both the light and photocatalyst were essential for carrying out the reaction successfully. Under optimized conditions, further investigation was continued to determine the substrate scope. At first, compatibility was checked for N-substituents of arylsulfonyl acrylamide and it was observed that various alkyl groups (e.g., Bu, i-pr) on the N-atom were well tolerated. Moreover, –OMe, –CF3 and –NO2 groups at the 4-position of the aromatic ring of arylsulfonyl moiety showed good compatibility. Varieties of perfluoroalkyl moieties, such as C3F7I, i-C3F7I, and C6F13I afforded the desired products under optimized reaction conditions.
 | ||
| Scheme 14 Visible-light-mediated cascade reaction of conjugated tosyl amides by perfluoroalkylation/arylmigration/desulfonylation (Tang and Sheng method). | ||
Based on previous literature reports and control experimental results, a reaction mechanism was proposed by the authors (Scheme 14). Initially, the photo-irradiation of photocatalyst led to the excited *[fac-Ir(III)(ppy)3], which in turn reacted with the alkyl iodide and was oxidatively converted to Ir(IV)+ with the simultaneous generation of the alkyl radical species 14I. Subsequent radical addition across the double bond of the substrate (14a/14d) engendered the alkyl radical 14II, which immediately underwent intramolecular 5-ipso-cyclization and was transformed into radical intermediate 14III. Due to instability, this intermediate was readily converted to the amidyl radical species 14IVvia desulfonylation. They propounded that due to a lack of stability in the case of the alkyl group attached to the N-atom of the species 14IV, it was transformed into the perfluorinated oxindole 14cvia the consecutive oxidation/H-atom abstraction. Otherwise, in the case of the aryl substituent, due to substantial stability, the intermediate 14IV was converted to α-aryl-β-di/monofluoromethyl amide 14evia hydrogen abstraction. The use of low-cost fluorine sources, moderate reaction conditions (e.g., room temperature), and broad substrate scope make this protocol attractive.
Benzothiophene and its derivatives have received significant interest due to their widespread application in pharmaceuticals, catalysis, and biology.34 Song et al.35 (Mar 2018) illustrated a metal-free, environment-friendly, photoredox-catalyzed cascade annulation reaction for the synthesis of a series of 3-sulphonyl benzothiophenes (15c). In this methodology, methyl(2-(phenylethynyl)phenyl)sulphane (15a) reacted with sulphonyl chloride (15b) in the presence of 2 mol% [IrdF(CF3)(ppy)2(dtbbpy)]PF6 as a photoredox catalyst, and K2CO3 as a base in CH3CN solvent under 5 W blue LED light at room temperature for 24 h (Scheme 15). A wide variety of sulphonyl chlorides, including those with electron-donating and electron-withdrawing groups, were found to exhibit good performance when kept in their optimum conditions. The aromatic ring of the 2-phenylethynyl group, containing both the electron-donating group and electron-withdrawing group (such as –OMe, –Me, –CF3) was compatible with this reaction. Also, substituents on the aromatic ring of o-methylthio-aryl alkyne did not affect the product formation. Pleasingly, benzoselenophenes (15e) were formed with good yields by replacing substrate 15a with methyl(2-(phenylethynyl)phenyl)selane (15d) (Scheme 15).
 | ||
| Scheme 15 Synthesis of benzothiophenes and benzoselenophenes from methyl(2-(phenylethynyl)phenyl)sulfanes and methyl(2-(phenylethynyl)phenyl)selanes via the photoredox catalyzed cascade reaction (Song method). | ||
Some control experiments were performed to gain insight into the mechanism. An important factor should be noted that if this reaction was carried out in the absence of a base, no reaction occurred. It was observed that the photoredox catalyst and visible-light were both imperative for this scheme. In the presence of radical scavengers like TEMPO or BHT, only a trace amount of the desired product was obtained. Hence, it can be inferred that the reaction proceeded through a radical pathway. Next, in the presence of ethene-1,1-diyldibenzene, a radical addition elimination product was obtained, which suggested that the reaction proceeded through the SET pathway. With the help of the control experiments, a mechanism was designed (Scheme 16). In the initial step, with the irradiation of visible light, photoredox catalyst Ir(III) was converted to its excited state *Ir(III). The sulfonyl radical 16I was subsequently formed in situ from sulfonyl chloride 16bvia a single electron transfer pathway. Simultaneously, Ir(III) was oxidized to Ir(IV). After that, sulfonyl radical 16I was added to an alkynyl moiety of 16a and vinyl radical intermediate 16II was formed. Finally, product 16c was obtained via intramolecular cyclization along with methyl radical elimination. The eliminated methyl radical was oxidized into methyl cation by Ir(IV) and then methyl cation on reaction with Cl− produced CH3Cl as a side product. This procedure has no additive involvement, a simple operation, mild conditions, and good functional group tolerance. However, the use of a very precious catalyst is an important disadvantage of this method.
 | ||
| Scheme 16 Probable mechanism. | ||
Wang et al.36 in May 2018 designed a visible-light-promoted radical dearomatization/cyanation cascade reaction of various indoles. This protocol gave access to gem-difluorinated and cyanated 3,3′-spirocyclic indolines. They reacted a substituted indole (17a, 0.2 mmol) with TMSCN (1.5 equiv.) in the presence of 1 mol% of Ir(ppy)3 as a photocatalyst, and K2HPO4 as a base in DMF solvent under the irradiation of blue LED (450 nm) at room temperature for 24 h, which produced the desired product 17b with up to 86% yield (Scheme 17). However, HFIP was required to achieve the highest yield of the desired product and worked as an additive. Other bases like Na2CO3 and K2CO3 did not work well. Replacement of solvent DMF with MeCN, THF, and MeOH was not fruitful in terms of yield. The generality of this reaction was investigated via the installation of various substituents in the indole moiety. Electron-donating to -withdrawing groups at the position of R1 were feasible for providing a good yield. Both the benzyl and allyl groups were well tolerated at the R3 position. Boc in the position of R3 was not able to produce a yield. Various chemical functionalities R2 at the C-2 position were tested. Phenyl and substituted phenyl groups at the C-2 position of indole afforded a single diastereomer with a good yield. Gram-scale synthesis was also studied by the authors to demonstrate the utility of this protocol. Moreover, this strategy showed a useful pathway to prepare various biologically relevant compounds.
 | ||
| Scheme 17 Synthesis of spiro-γ-lactam indolines with two adjacent stereocenters via the visible-light-promoted dearomatization/cyanation cascade reaction of indoles (Wang method). | ||
For mechanistic insight, control experiments were performed. The addition of TEMPO abolished the formation of the desired product which supported the radically involved pathway. The photocatalyst and light were both necessary to conduct the reaction. Based on the control experiments and earlier literature reports, the following mechanistic steps were proposed. At first, light excited [Ir(III)(ppy)3], which was then oxidised by bromodifluoroacetamide (17a) to make [Ir(IV)(ppy)3] through the SET process, along with the formation of radicals 17I. The radical 17I further underwent regioselective 5-exo cyclization to generate the indole C-2 radical intermediate 17II. The photoredox catalytic cycle was terminated via the oxidation of 17II to the carbocation intermediate 17III by [Ir(IV)(ppy)3] which regenerated [Ir(III)(ppy)3]. In the final step, the nucleophilic addition of the –CN group to that cationic carbon took place from the side with less hindrance to afford the final product 17b (Scheme 17).
Among the nitrogen-containing heterocycles, quinoxalines are widely studied in chemical synthetic literature.37 Xu et al.38 in April 2019 introduced a methodology for the synthesis of the indolo[2,1-a]isoquinoline core structure via a radical cascade reaction driven by visible light. As per the reported methodology, the authors employed 2-aryl indoles (18a, 0.2 mmol), aroyl chloride (18b, 0.4 mmol) along with fac-Ir(ppy)3 (1 mol%) as an active catalyst, 2,4,6-collidine as a base, CH3CN as a solvent under the irradiation of blue LEDs at 25 °C in an N2 atmosphere. The desired product 18c was obtained with up to 94% yield within 24 h (Scheme 18). Under optimized conditions, further investigations were extended to examine the tolerance of various substituents. A wide range of reactivity was observed for aroyl chlorides containing both electron-withdrawing and electron-donating groups. ortho- and meta-methyl substituents showed very good tolerance under the same optimized reaction conditions. Alkyl acid chloride and cinnamoyl chloride failed to meet the expectations due to their high reduction potentials. In the next scope, 2-aryl groups and indoles were verified. Halo-substituted derivatives with F, Cl, or Br at the place of the indole ring or 2-aryl ring were well tolerated. Groups like trifluoromethyl, cyano, and sulfonyl, which are very common in drug molecules, were compatible. Phenyl, n-propyl, and ester substituents at the C-3 position of indole performed well. Attempts with carbazole-related substrates failed miserably. For the demonstration purpose of this strategy, a scale-up reaction was performed, which resulted in the formation of a satisfactory yield.
 | ||
| Scheme 18 Visible-light-mediated cascade cyclization for the synthesis of indolo[2,1-a]isoquinoline derivatives (Xu method). | ||
For a clear idea regarding the mechanism, TEMPO was incorporated into the reaction medium, which resulted in the termination of the reaction. It was a clear indication of the radically involved pathway. Based on previous work and experimental results, a reaction mechanism was outlined (Scheme 18). Photocatalyst fac-Ir(III)(ppy)3 (18I) was promoted to the excited state *fac-Ir(ppy)3 (18II) under visible-light irradiation, which further reduced benzoyl chloride (18b) via SET to generate acyl radical 18IV. The generated acyl radical 18IV then underwent an addition reaction with 18a to produce a new α-acyl radical 18V. Further cyclization of the species 18V produced another radical intermediate 18VI and its oxidation occurred via SET with oxidized fac-Ir(IV)(ppy)318III to generate the corresponding radical cation intermediate 18VII along with the regeneration of fac-Ir(ppy)318I. Alternatively, 18VI could be oxidized with the help of benzoyl chloride to generate cation 18VII along with acyl radical 18IV formation. In the final step, the desired product 18c was obtained via the rapid deprotonation of the intermediate 18VII.
Zhang and his group39 on Sep 2019 devised a visible-light-driven radical cascade cyclization reaction employing cyclobutanone oxime esters and aryl isonitriles for the preparation of a bunch of cyclopenta[b]quinolines. They exploited aryl isonitriles (19b, 0.2 mmol) and cycloketone oxime esters (19a, 0.4 mmol) in the presence of photocatalyst (2 mol% fac-Ir(ppy)3), base (Na2CO3) and solvent DMA with irradiation of 7 W blue LEDs at room temperature for 24 h under an atmosphere of N2. These conditions were convenient for observing the desired product 19c with good to moderate yields (Scheme 19). The presence of an inorganic base was essential and only Na2CO3 was efficient. To observe the generality of this protocol, several substrate scopes were checked via the installation of the substituent. Electron-withdrawing (–Br, –F) and -donating (–OMe, –Me) groups at the para position of the phenyl ring of aryl isonitriles showed well tolerance conferring good yields. –CF3 and acetyl substituents at the para-position of aryl isonitriles were poor groups for producing yields. Naphthalene-derived isocyanides were successful in this transformation. Various cycloketone oxime esters were tested by employing ethyl, methyl, and allyl groups at the 2-position, and all of them were successful. Ester and phenyl groups at the 3-position gave lower yields.
 | ||
| Scheme 19 Synthesis of cyclopenta[b]quinoxalines via the visible-light-promoted cascade cyclization of oxime esters and aryl isonitriles (Zhang method). | ||
Based on relevant literature and control experiments, a probable mechanism was outlined as shown in Scheme 19. Initially, fac-[Ir(III)(ppy)3]* was generated from its ground state fac-[Ir(III)(ppy)3] under the irradiation of visible light. A SET from fac-[Ir(III)(ppy)3]* to cyclobutanone oxime esters (19b) resulted in the reductive cleavage of the N–O bond of 19b and the formation of [fac-Ir(IV)(ppy)3]+ and iminyl radical 19I took place. Next, this iminyl radical 19I underwent C–C bond β-scission in a regioselective manner to generate cyanoalkyl radical 19II. It was further captured by aryl isonitriles 19a to form imidoyl radical 19III. The attachment of the newly formed imidoyl radical 19III with the cyano group occurred in an intramolecular radical addition fashion, resulting in the formation of iminyl radical 19IV, which further cyclized in an intramolecular manner to provide radical intermediate 19V. Single-electron oxidation of intermediate 19V by [fac-Ir(IV)(ppy)3]+ generated a carbocation intermediate 19VI along with ground state [fac-Ir(III)(ppy)3]. Hence, the photocatalytic cycle was completed. Finally, base-assisted deprotonation led to the formation of the desired product 19c. This reaction is a convenient method for obtaining quinoxaline derivatives because of its mild conditions, high functional group tolerance, and wide substrate scope; however, it does not work in the open air.
Zhang et al.40 again in Dec 2019 developed a visible-light-promoted intramolecular radical cascade strategy of α-bromo-N-benzyl-alkylamides for the synthesis of a group of tetracyclic N-fused indolo[2,1-a]isoquinolin-6(5H)-ones. This strategy exhibited a variety of substrate scopes and opened up a milder route to access tetracyclic N-fused indoles. As per the report, when the authors irradiated 20a (0.3 mmol) in the presence of 2 mol% of [fac-Ir(ppy)3] as photocatalyst and Na2HPO4 as the base in DMF solvent with 7 W blue LED light at room temperature for 24 h, the desired product 20b was afforded with up to 88% yield (Scheme 20). Every photocatalyst was unable to produce a good yield except [fac-Ir(ppy)3]. Na2HPO4 beat other inorganic and organic bases in terms of higher yield formation. Both light and photocatalyst were essential for this transformation. After achieving the optimized reaction conditions, the authors evaluated the substrate scope for the reaction. Electron-donating groups (–Me, –t-Bu) and electron-withdrawing groups (–CF3, –COOMe) as substituents at the para-position of the aniline unit were well tolerated to provide cyclized products with good yields. –F, –Cl, and –Br groups were well tolerated under standard reaction conditions. Substituents like ortho-Me, and 3,5-di-Me on the aniline ring showed satisfactory yield. The substituent at the terminal alkyne was also observed to react properly. However, it was found that the chlorinated substrate produced a lower yield. Hence, the protocol was mild and accessible to a wide range of tetracyclic N-fused indoles, demonstrating a broad substrate scope.
 | ||
| Scheme 20 Synthesis of tetracyclic N-fused indolo[2,1-a]isoquinolin-6(5H)-ones by the visible-light-mediated cascade reaction of α-bromo-N-benzyl-alkylamides (Zhang method). | ||
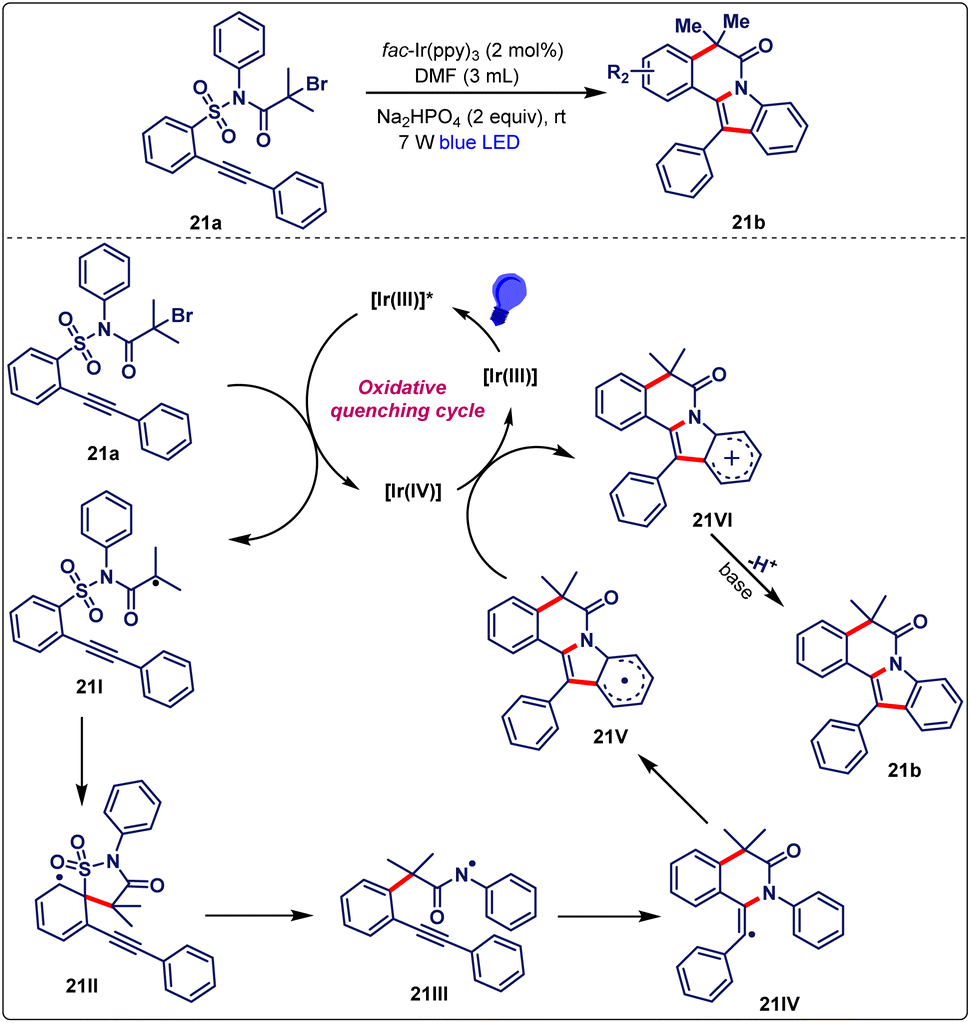
The authors then moved toward the establishment of the reaction mechanism. The incorporation of TEMPO in the reaction medium diminished the formation of the desired product, indicating the involvement of the radical pathway. As per the previous literature reports and several control experiments, a mechanistic pathway was designed (Scheme 21). Initially, visible-light promoted [fac-Ir(ppy)3] to an excited state [fac-Ir(ppy)3]*, which further underwent SET with 21a to form radical intermediate 21I and an Ir(IV) complex. A subsequent 5-ipso cyclization of 21I generated a spirocyclic intermediate 21II, which further underwent desulfonylation to form amidyl radical 21III. After that, this radical 21III cyclized with C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C to afford the intermediate 21IV. Thereafter, the intermediate 21IV was involved in intramolecular cyclization with the aryl ring to produce intermediate 21V, which was next oxidized via SET to form carbocation 21VI along with the regeneration of the photocatalyst. Finally, the deprotonation of 21VI in the presence of a base afforded the targeted product 21b.
C to afford the intermediate 21IV. Thereafter, the intermediate 21IV was involved in intramolecular cyclization with the aryl ring to produce intermediate 21V, which was next oxidized via SET to form carbocation 21VI along with the regeneration of the photocatalyst. Finally, the deprotonation of 21VI in the presence of a base afforded the targeted product 21b.
 | ||
| Scheme 21 Probable mechanism. | ||
In 2019, Studer et al.41 reported a three-component cascade carboamination reaction for unactivated alkenes, exploiting the Giese-type radical addition. They employed a protected N-radical precursor (22a), unactivated alkene (22b), and Michael acceptor (22c) in combination with [Ir(dFCF3ppy)2(dtbbpy)]PF6 as the photocatalyst, 1.5 equiv. of CsCO3 and 2 equiv. of H2O as additives in DCM solvent under blue light radiation to obtain the desired carboaminated product 22d (Scheme 22). The catalyst was so active that only 0.5–1.0 mol% of it afforded up to 92% yield within 24 h at room temperature. Upon optimization, they found that water played a crucial role during transformation; even base, light, and adequate loading of the photocatalyst were necessary for this reaction. Regarding N-protecting groups, Boc, Fmoc, Alloc, Teoc, and Cbz were fruitful and provided the desired conversion. They found that both geminally di-substituted, tri-substituted and tetra-substituted alkenes participated in the reaction quite well under optimized reaction conditions. In the case of the other coupling partners, electron-withdrawing alkenes like 2-methyl acrylate, acrylamides, phenyl vinyl sulfone, dimethyl vinylphosphonate, acrylonitrile, and methacrolein were competent Michael acceptors for this particular transformation.
 | ||
| Scheme 22 Visible-light-induced Ir-catalyzed three-component cascade carboamination reaction (Studer method). | ||
Based on the outcomes of control experiments and previous reports, they propounded the following mechanistic rationale. At first, Ir(III) photocatalyst was excited by photo-irradiation and subsequently underwent reductive SET with 22a, generating the carboxyl radical 22I along with the Ir(II) intermediate. The sequential liberation of CO2 and fragmentation of acetone from 22I furnished the electrophilic N-radical 22II. This radical species chemoselectively added to the electron-rich alkene species 22b and produced the radical adduct 22III. Subsequently, the Michael acceptor 22c trapped the radical species 22III and converted it to radical intermediate 22IV. Finally, the newly generated intermediate 22IV underwent reductive SET with the Ir(II) intermediate and protonation to deliver the desired product 22d (Scheme 22).
In 2020, Murakami and his group42 reported a synthetic methodology for organophosphine oxide in the presence of photoredox catalysis using a three-component cascade reaction. In this methodology, triphenylphosphine (23a), alkene (23b) and water were dearomatized and coupled at room temperature to produce the desired molecule (23c) with up to 87% yield within 20 h (Scheme 23). The authors examined different types of alkenes, such as mono-, di-, tri-, and tetra-substituted alkenes and found that only disubstituted alkenes took part in this particular reaction. Bulky substituents like the tert-butyl group containing disubstituted alkenes underwent this coupling reaction quite well. Additionally, cycloalkanes containing olefins ranging from 4- to 6-membered rings provided the desired yield. They also showed that geminally disubstituted alkenes with hydroxy, amide, ester, ketonic carbonyl, and acetal functionalities performed well under the reaction condition. In the case of the other reaction partner, substituted triarylphosphine reacted well, although the lack of π-electron cloud in trialkylphosphine made it unfit for this scheme.
 | ||
| Scheme 23 Visible-light-induced three-component cascade reaction for the synthesis of organophosphine oxide (Murakami method). | ||
From different control experiments with D2O and H2O18 they realized that water molecules were separated discretely and incorporated into their target molecule. They also accounted that reactants did not participate in the reaction without the water molecules. Based on a few control experiments and previous literature reports, they proposed a suitable mechanism for the reaction as sketched in Scheme 23. At first, the iridium(III) photocatalyst was excited by photo-irradiation and participated in a reductive SET process with triphenylphosphine (23a), generating a triphenylphosphine radical cation 23I. Immediately, this radical cation captured a water molecule in a deprotonated fashion and furnished phosphoranyl radical 23II. Later, this radical was added to the less hindered side of the alkene 23b and produced thermodynamically stable tertiary alkyl radical 23III, which subsequently underwent 5-trig cyclization onto the phenyl group forming a more stable cyclohexadienyl radical 23IV. After that, this species underwent a reductive SET with iridium(II), forming anionic intermediate 23V, and regenerated the catalytic cycle. Simultaneously, the sequential protonation of 23V by water and rearrangement led to the desired product 23c.
Chroman-4-ones and dihydroquinoline-4-ones have several biological and pharmaceutical activities, for example, antitumor, antimicrobial, and antioxidant properties. Ma and co-workers43 in February 2020 investigated the reaction of 2-(allyloxy)arylaldehydes (24a, 0.2 mmol) with α-bromo diethyl malonate (24b, 2 equiv.) in the presence of fac-Ir(ppy)3 (0.005 mmol) as catalyst and 2,6-lutidine (2 equiv.) as a base under irradiation of 5 W blue LED under a N2 atmosphere. They successfully obtained a series of desired chroman-4-ones (24c) in up to 71% yield (Scheme 24). Several solvents, such as THF, DMF, DMSO. 1,4-Dioxane were used in the reaction and the results indicated that DMSO was the most suitable solvent. When K2HPO4 or Et3N replaced 2,6-lutidine, the reaction became less effective. Also, when the base was absent, the reaction was inhibited completely. Due to the lower oxidizing potential, if Ru(bpy)3Cl2 or eosin Y were used in this reaction, the product did not form. On screening the electronic effect of the substituent at the aromatic ring, it was noted that the aryl groups containing electron-withdrawing substituents had higher reactivity than aryl groups having electron-rich substituents.
 | ||
| Scheme 24 Synthesis of chroman-4-ones and dihydroquinolin-4-ones by visible-light-induced cascade reaction (Ma method). | ||
Control experiments ensured that in the absence of fac-Ir(ppy)3 or light, the reaction was stopped. Also, the yield of the product decreased under air. The radical scavenger TEMPO, BHT inhibited the product formation, which indicated that the reaction proceeded via a radical process. Based on these control reactions and previous literature reports, they proposed a probable mechanism as depicted in Scheme 24. Firstly, fac-Ir(ppy)3, via blue LED irradiation, generated fac-*Ir(ppy)3, by which the electron-deficient radical 24I was formed on reduction of 24b. After that, the C–C double bond in 24a captured the radical intermediate 24I to give alkyl radical 24II, which, by intramolecular cyclization, produced an alkoxy radical 24III. Next, radical 24IV was formed by the 1,2-hydrogen atom transfer of newly formed intermediate 24III. Finally, fac-IrIV(ppy)3 oxidized radical 24IV in the presence of 2,6-lutidine to give the desired product 24c and the proton was lost to regenerate fac-IrIII(ppy)3, completing the catalytic cycle.
Sulfonyl functionality plays a viable role in organic synthesis,44 merging it with the dihydrobenzofuran (DHB) skeleton. Various natural products and synthetic medicines contain this component, which unlocks new potential for further synthetic applicability.45 In Nov 2020, Sun and co-workers46 developed a strategy for synthesizing sulfonyl-bearing DHB scaffolds via radical cascade annulation under visible-light irradiation. As shown in Scheme 25, the exploitation of 2-alkynylarylethers (25a, 0.2 mmol) and sodium sulfinates (25b, 0.4 mmol) in conjunction with metal photocatalyst Ir[(ppy)2(dtbbpy)]PF6 (2 mol%), additive AcOH & water and solvent DMF under 30 W blue LED irradiation and N2 atmosphere at room temperature for 24 h provided the intended product 25c with up to 78% yield. PA photocatalyst was mandatory for this transformation and the presence of water and acetic acid promoted the reaction for better outcomes. The optimization of photocatalysts and solvents revealed that other metal-mediated or metal-free catalysts and other organic solvents like acetone, acetonitrile and ethanol were detrimental to the productivity. A broad substrate scope for alkynylaryl ethers was evaluated. Substrates bearing both electron-donating and electron-withdrawing groups at the para-position with respect to the alkoxy group were well tolerated and delivered the targeted product with moderate to good yields. Interestingly, the presence of CN or COOMe moieties reduced the conversion. Different alkyl and aromatic substitutions at the alkoxy chain (R2 and R3) afforded the desired product quite well. Similar or different substituents did not provide any significant difference. Gratifyingly, substrates with cycloalkyl ether, spiro ether and natural product skeletons, viz., menthol and epiandrosterone motifs, were competent substrates for this protocol. Unfortunately, substrates with dimethyl substitutions and no substitutions did not afford the desired product. On the other hand, both electron-rich and -deficient functionalities-tethered sodium phenylsulfinates showed very good amenability in this transformation. Besides, sodium naphthalene-1-sulfinate, sodium pyridine-3-sulfinate and sodium thiophene-2-sulfinate successfully participated. Eventually, sodium ethyl sulfinate also yielded the targeted compound, albeit replacing methyl with ethyl led to no fruition.
 | ||
| Scheme 25 Synthesis of phenylsulfonyl-functionalized dihydrobenzo furans via visible-light-mediated cascade annulation (Sun method). | ||
Upon investigation of the reaction rationale, it was found that the transformation became inhibited in the presence of TEMPO. No targeted product was found in presence of 1,1-diphenylethylene. These findings supported that the reaction might follow a radical pathway. Replacing the model substrate with the deuterated analogue provided the D-substituted benzyl hydrogen, which ascertained the involvement of the 1,5-HAT process. Besides, introducing AcOD and D2O led to a D-containing product, which proved that the α-hydrogen of the sulfonyl group came from acetic acid or water. Based on these control experiments and literature reviews, they proposed a credible mechanistic pathway, as shown in Scheme 25. Initially, sodium benzene sulfinate 25b was oxidized by photoexcited metal catalyst Ir(III) to sulfonyl radical 25I. Subsequently, this radical added to the CC of substrate 25a and generated the vinyl radical 25II. A concomitant intramolecular 1,5-HAT of 25II led to α-oxoalkyl radical species 25III, which transformed into radical 25IVvia 5-exo-trig radical cyclization. Later, this radical 25IV was reduced by Ir(II) and furnished the corresponding anion 25V, which gave the final product 25c after sequential proton abstraction from AcOH/H2O.
Chromones are very important scaffolds because of their pharmaceutical importance.47 Yang and his group48 in January 2022 reported an amination cascade reaction of o-hydroxyarylenaminone 26a (0.2 mmol) with perfluoropyridin-4-yl 26b (0.3 mmol) in the presence of a photocatalyst (5 mol%) under 30 W blue LEDs irradiation. In this reaction, 3-aminochromone 26c was formed within 24 h at room temperature with up to 88% yield (Scheme 26). Among many solvents, toluene was the best choice when other factors remained the same. Moreover, fac-Ir(ppy)3 gave the highest yield among all other photocatalysts, such as eosin Y and rhodamine B. The photocatalyst and light were essential for this reaction. Next, the substrate scope was screened by reacting a variety of o-hydroxyarylenaminones 26a under optimized reaction conditions. The substituents at the meta-position of the phenol resulted in lower yields than the substituent at the para-position. A strong electron-donating methoxy group was inappropriate, whereas halogens were suitable for this reaction under standard reaction conditions. The disubstituted substrates produced a small amount of by-product and unsubstituted chromone. If trifluoroacetic acid (TFA) was used, it was easily possible to remove the Boc group. Carboxybenzyl (Cbz) and 9-fluorenylmethyloxycarbonyl (Fmoc) could not be used as substitutes for the Boc group. The 54% yield in the gram scale reaction proved the practicality of this reaction.
 | ||
| Scheme 26 Accessing 3-aminated chromones via photocascade catalysis (Yang method). | ||
Some control experiments were performed to check the mechanism. The reaction was stopped in the presence of radical scavengers, which indicated that the reaction proceeded through a radical pathway. From the Stern–Volmer studies, it was proved that o-hydroxyarylenaminone 26a quenched the excited fac-Ir(ppy)3 instead of 26b. A plausible mechanism was delineated based on the above experiments and previous literature reports. The excited fac-*Ir(ppy)3 was produced from the photocatalyst fac-Ir(ppy)3 under blue light irradiation. The radical 26I and IrII were formed by the reductive quenching of fac-*Ir(ppy)3 by 26a. Oxidation of IrII occurred by 26b to form amidyl radical 26III along with the reproduction of the photocatalyst. The radical 26I then transformed to 26II, which then coupled with 26III, forming intermediate 26V (path a). In path b, cyclic radical 26IV was afforded from the intramolecular cyclization of 26II. Next, the intermediate 26VI originated from the radical cross-coupling of 26IV and 26III. Finally, the desired product, 3-aminochromone 26c, was obtained by the dimethylamine elimination from 26VI (Scheme 26). This designed protocol is notable for its mild reaction conditions, ease of synthesis, ability to be scaled up and late-stage functionalization. However, this methodology does not work in aerial conditions.
4. Pd-based photocatalyzed cascade reactions
Visible-light-promoted Pd-catalyzed cascade reaction via etherification/C–C cyclization incorporating phenols and α-bromoacetophenone was introduced by Sun and Chu et al.49 in March 2019, which opened up a new route for the synthesis of a bunch of dibenzo[b,d]oxepin-7(6H)-ones. This method is advantageous due to its extensive substrate scope, mild reaction conditions along with wide tolerance of functional groups. According to the reported literature, they utilized phenol 27a with alpha-bromoacetophenone 27b in the presence of Pd(OAc)2 (10 mol%) as a catalyst, DDQ as an oxidant, Cs2CO3 as a base in CH3CN solvent under 25 W blue LED at 80 °C. The targeted product 27c was achieved with a satisfactory yield (Scheme 27). Screening studies revealed that the inorganic base Cs2CO3 was more suitable than organic bases. CH3CN as a solvent showed its superiority and suppressed others like DMSO, DMF, toluene, etc. Knowledge of the generality of this scheme was obtained by installing several substituents in the substrate ranging from electron-donating to -withdrawing groups for both the phenols and α-bromoacetophenone. Installation of –Me at the meta- and para-position of phenols showed a significant yield of 80–88%. Electron-withdrawing groups such as –F, –Cl at the para-position produced lower yields. –OMe was successful at both the meta- and para-positions to afford good yields. Next, electron-withdrawing groups like –OMe, –Me at the meta-position of α-bromoacetophenone (R1) showed high activity in cyclization and generated yields ranging between 79–89%. A slight decrease in yield was observed for –Ph at the meta-position. It is noteworthy that the –F at the meta-position showed a significantly reduced yield. Some control experiments were executed to get an overview of the mechanism involved in incorporating TEMPO. Still, the exact mechanism was not confirmed by the authors at that time and hence is not discussed here. To make this strategy versatile and to reveal its synthetic utility, they applied this methodology in the synthesis of protosappanin A. The technique has simple reaction conditions, easily accessible starting materials, a broad substrate scope, and excellent functional group tolerance. | ||
| Scheme 27 Synthesis of dibenzo[b,d]oxepin-7(6H)-ones via visible-light induced cascade reaction (Sun and Chu method). | ||
One of the important classes of radical ring expansion reaction is the Dowd–Beckwith reaction, which produces cyclic ketones.50 In Nov 2020, Duan et al.51 reported an effective strategy for the synthesis of intermolecular, Pd-catalyzed, visible-light-induced Dowd–Beckwith ring-expansion/C–C bond formation. For this reaction, α-bromomethyl β-keto ester (28a) (0.2 mmol) reacted with styrene (28b) (0.4 mmol) in the presence of Pd(OAc)2 (10 mol%) as the catalyst, 4,5-bis(diphenylphosphino)-9,9-dimethylxanthene (Xantphos) (20 mol%) as ligand and K2CO3 (0.4 mmol) as the base under the irradiation of blue LEDs in toluene solvent. The desired ring-expansion/alkenylation product 28c was obtained with up to 80% yield within 24 h (Scheme 28). Pd(OAc)2 was the best choice among Pd(TFA)2, Pd(PPh3)4 and Pd(OAc)2. The reaction was stopped when Ni(OAc)2 was used instead of Pd(OAc)2. No other phosphine ligands (monodentate/bidentate) were useful for this reaction except Xantphos. Only K2CO3 was effective among other organic and inorganic bases. The visible-light irradiation, catalyst, base, and ligands are vital for this reaction. In this reaction, the direct alkyl-Heck coupling product 28d and β-H elimination product with ring expansion 28e were obtained as the by-products with 10% and 5% yields, respectively. On screening substrate scope, it was noticed that the electron-donating group on styrene was more effective than the electron-withdrawing group. One-carbon enlarged five to eight-membered ring could be obtained from various α-bromomethyl substrates.
 | ||
| Scheme 28 Visible-light-induced Dowd–Beckwith ring expansion/C–C bond formation cascade reaction (Duan method). | ||
Control experiments revealed that the reaction could not proceed in the presence of TEMPO and BHT, which proved that a radical intermediate was involved in this reaction. The photo-absorbing species was none other than the Pd0 complex formed in the reaction, which could be proved by UV-vis quenching study analysis. A mechanism was proposed based on the previous literature and control experiments. In the first step, LnPd0 was irradiated to the excited state LnPd0*. Next, PdI and a primary alkyl radical 29I were obtained by a single-electron-transfer process. The carbon-centre radical 29I underwent cyclization and then C–C bond cleavage took place to expand the ring, producing intermediate 29III. In the last step, styrene 29b underwent radical addition with 29III, followed by β-H elimination to synthesize the desired product 29c and Pd0 was regenerated. In addition, 29e could be formed from intermediate 29IIIvia the formation of 29V (Scheme 29).
 | ||
| Scheme 29 Plausible mechanism. | ||
5. Organic and inorganic semiconductor-based photocatalyzed cascade reactions
Pursuance of sustainable approaches remains an illimitable peregrination.2d,52 In this regard, cascade transformations in correspondence with photocatalytic attributes have achieved huge success towards green syntheses. Many Ir-, Ru-, Cu-based complexes, organic dyes, and inorganic semiconductors have garnished the photocatalytic-synthesis-gallery, but the quest for inorganic nanomaterial as photo-induced aiding agents in syntheses has spurred special interest among the chemists.In the year 2015, the Zhang group53 reported the cascade synthesis of α-aryl-β-trifluoromethyl amides from conjugated tosyl amides by using BiOBr nanosheets as the photocatalyst via consecutive trifluoromethylation/desulfonylation/N–H bond formation processes. They exploited N-aryl-N-tosylmethacrylamide (30a, 0.2 mmol), CF3SO2Cl (30b, 1.0 mmol) in conjunction with BiOBr nanosheets and K2HPO4 under photoirradiation (280–780 nm) in dimethylacetamide (DMAc) as solvent (1.0 mL) at room temperature, which resulted in the desired product 30c with up to 76% yield (Scheme 30). This one-pot protocol showed a dissatisfactory outcome when combined with CdS nanosheets or Ru(bpy)3Cl2·6H2O instead of BiOBr nanosheets as the photocatalyst. Solvent played a significant role and replacing DMAC with others led to inferior yield. Both the nanosheets and light were indispensable for the transformation. Exploration of the generality of this protocol led to good tolerance of different electron-efficient and -deficient groups at the ortho-position of the nitrogen-attached aryl moiety. The meta- and para-bearing halogens, a few alkyls, and alkoxy substituents on the same aryl ring were amenable in this given transformation. On the contrary, the other sulfonamide tethered aryl moiety did not show such variety and displayed very few selective substitutions with synthetically useful yields.
 | ||
| Scheme 30 Synthesis of α-aryl-β-trifluoromethyl amides via a light-induced cascade reaction using BiOBr nanosheets as the photocatalyst (Zhang method). | ||
Control experiments with TEMPO revealed a radical pathway. Depending on the control investigations and literature survey, they propounded a credible mechanistic pathway as sketched in Scheme 31. Initially, photoirradiation facilitated the promotion of electron transfer from the valence band (VB) to the conduction band (CB) of BiOBr nanosheets. Later, CF3SO2Cl was reduced by these excited nanosheets to the trifluoromethyl radical, chloride anion, and sulfur dioxide. The ˙CF3 radical then interacted with the alkene part of substrate 31a and generated the activated radical intermediate 31I. Subsequent dearomatization/5-ipso cyclization converted it to a new aryl radical intermediate 31II. Consecutive desulfonylation led to the amidyl radical 31III, which encountered the solvent engendered amidyl radical cation 31IV, generated via oxidation by the photogenerated hole of the nanosheets. Finally, the intermediate 31III abstracted a hydrogen radical from the amidyl radical cation 31IV and was transformed into product 31c. This low-cost, environmentally friendly, and efficient photocatalytic method may be regarded as a viable alternative to the methods previously mentioned.
 | ||
| Scheme 31 Probable mechanism. | ||
In March 2020, Yao and co-workers54 discovered a methodology for synthesizing 2-arylquinolines through a visible-light-promoted photooxidation-Povarov cascade reaction utilizing alcohol and N-benzylanilines, and using the Ag/g-C3N4 nanometric semiconductor as a photocatalyst. As per the reported literature, they employed N-benzylanilines (32a, 0.1 mmol) and ethanol (2 mL) in the presence of Ag/g-C3N4 as a nanometric photocatalyst under the irradiation of a 3 W blue LED at room temperature for 24 h, which successfully formed the desired product 32b with up to 85% yield (Scheme 32). To get the optimized reaction conditions, several reactions were performed. It was found that both light and photocatalyst were indispensable for the reaction. g-C3N4 and Ag nanoparticles alone were not suitable for generating a desirable yield. A combination of both g-C3N4 and Ag-nanoparticles was found to be the most efficient as a catalyst when other factors remained the same. Metals like Cu were also tried but they failed to generate a good yield. Moreover, 20 mol% of Ag/g-C3N4 was the best loading for the catalyst. The increment in the intensity of light up to 15 W showed a negative impact in terms of yield formation. To widen the generality of this protocol, several substrate scopes were monitored via the installation of the different substituents. It was observed that para-substituted electron-donating groups (methyl, methoxy) were not suitable for generating decent yields. The 4-chloro aniline-derived substrate gave yields in the range of 80%; whereas 4-methoxy-aniline derivatives produced yields of 50–60%. To widen the diversity of the reaction, ethanol was substituted by n-propanol as substrate as well as reagent, which resulted in a reasonable yield.
 | ||
| Scheme 32 The Ag/g-C3N4 nanometric semiconductor-catalysed visible-light-driven cascade reaction for the synthesis of 2-arylquinoline through alcohol and N-benzylanilines (Yao method). | ||
Moreover, the Ag/g-C3N4 nanocomposite was beneficial as it is reusable and easily separable, which is a very important feature of this method. However, the reaction time was a bit high.
Some control studies were performed to gain insight into the mechanism. Based on the results and literature reports, a mechanism was designed as described in Scheme 32. g-C3N4 was excited by visible-light irradiation, which resulted in the formation of conduction band electrons (e−) along with valence band holes (h+). Next, the N-radical cation intermediate 32I was formed from 32avia the release of a single electron on valence band holes. On the other side, photo-generated electrons that were promoted to the conduction band were trapped by Ag nanoparticles and reduced O2 to the superoxide radical anion. Then, N-arylaldimine 32II was generated with the help of the superoxide radical anion. After that, intermediate 32II reacted with acetaldehyde derived from the in situ photo-oxidation of ethanol to form intermediate 32III. Finally, the reaction was terminated with the formation of product 32b by the oxidative dehydroaromatization of intermediate 32III.
The Chen and Yu group,55 in August 2020, reported a methodology for the synthesis of tetrahydro-imidazo[1,5-a]quinoxaline-4(5H)-ones (33c) by the annulation reaction of N-methyl quinoxaline-2(1H)-one (33a) (0.2 mmol) with N-phenylglycine (33b) (0.6 mmol) under 25 W LED irradiation in the presence of CsPbBr3 (5 mol%) as the catalyst at room temperature for 10 h (Scheme 33). The substrate scope was screened and it showed that several N-substituents (methyl, ethyl, propargyl, benzyl etc.) at quinoxaline-2(1H)-ones were suitable for this reaction. The benzene ring of 33a, containing two electron-donating groups (–CH3), afforded the desired products in low yields, whereas, electron-withdrawing groups (–F, –Cl) increased the yield. Moreover, the –Cl group at the meta-position of the N-phenylglycine afforded the product in only a 30% yield. In addition to that, two different electron-withdrawing groups at the para-position generated the desired product in good yields. Next, a gram-scale reaction was carried out to see the applicability of this reaction. This strategy has numerous advantages, including step economy, a broad substrate scope, green solvent, and so on. Moreover, the catalyst is recyclable.
 | ||
| Scheme 33 Visible-light-promoted cascade annulation of quinoxalin-2(1H) ones using photocatalyst perovskite (Chen and Yu method). | ||
Some control experiments were performed to illustrate the mechanism. TEMPO and BHT inhibited the reaction, indicating the radical pathway of this reaction. The addition of a hole-scavenger ammonium oxalate and an electron-scavenger K2S2O8 suppressed the yield, which proved the involvement of electrons and holes in this photocatalytic process. The yield was decreased if the reaction was executed in anaerobic conditions. Based on these results and previous literature reports, the most probable mechanism was introduced by the authors (Scheme 34). Firstly, CsPbBr3 absorbed photons to create electrons in the conduction band (CB) and holes in the valence band (VB). The single-electron transfer process produced the phenylaminomethyl radical 34I along with CO2 and a proton from 34b. The released proton, on reaction with O2 in air, produced a water molecule. The addition of 34I at the C-3 position of 34a produced the radical 34II, which further added to 34I to form the intermediate 34III. Protonation of 34III, followed by the loss of the aniline molecule, generated the iminium cation intermediate 34VI. Next, the generated intermediate 34VI underwent intramolecular nucleophilic attack by the phenylamino group to synthesize intermediate 34VII. In the final step, 34VII was deprotonated to form the desired product 34c.
 | ||
| Scheme 34 Probable mechanism. | ||
6. Organophotoredox-catalyzed cascade reactions
In 2017, Kumar et al.56 developed a light-induced oxidant- and metal-free dehydrogenative cascade methodology for the synthesis of trifluoromethylated C-3 aryloylated heterocycles. They reacted 1-(phenylethynyl)-2-(vinyloxy)benzene/phenolic 1,6-enynes/thiophenolic 1,6-enynes/anilinic 1,6-enynes (35a, 0.1 mmol) and Langlois’ reagent CF3SO2Na (35b, 0.3 mmol) in the presence of photocatalyst phenanthrene-9,10-dione (PQ, 0.01 mmol, 420–505 nm) under an argon atmosphere, which engendered the desired product 35c in up to 75% yield (Scheme 35). A mixture of CH3CN:H2O (9:1) was the most efficient solvent for this reaction. This strategy did not afford the desired product in the absence of a photocatalyst; rather, in the presence of heat, it produced a different outcome, 2-(phenylethynyl)phenol, which was generated after the cleavage of the vinylic carbon–oxygen bond. Replacing the photocatalyst PQ, with other reactants, viz., ketone, α-diketone, ortho-quinones, or 1,10-phenanthroline-5,6-dione was detrimental; instead of the desired product, the above-mentioned byproduct was obtained. This protocol displayed a broad list of diverse substrate scopes. For phenolic 1,6-enynes, the ethynyl phenyl ring containing electron-donating and electron-withdrawing groups afforded the desired product C-3 aryloylated benzofurans quite smoothly in good yield. Instead of benzene and naphthyl substituents, heteroaromatics like pyridyl, thiophenyl, and aliphatic n- and sec-alkyl substituents were also amenable in this protocol. Moreover, thiophenolic 1,6-enynes and anilinic 1,6-enynes have almost similar reactivities.
 | ||
| Scheme 35 Trifluoromethylation and oxidation of 1,6-enynes with water in metal-free conditions in the presence of visible light (Kumar method). | ||
Based on a series of control experiments and other investigations, they proposed the following mechanistic rationale. The photo-excited PQ* oxidized CF3SO2Na to the trifluoromethyl radical and SO2 by single-electron transfer. The generated trifluoromethyl radical was added to the vinylic carbon–carbon double bond in substrate 35a and furnished the radical intermediate 35I. It immediately rearranged to the vinylic radical 35IIvia 5-exo-dig radical translocation. Subsequently, this radical intermediate was converted to the vinylic cation intermediate 35IVvia SET to PQH˙. In an alternative pathway, the radical intermediate 35I might undergo single electron oxidation by PQH˙ to furnish carbocation 35III and after cationic cyclization, it formed the same vinylic intermediate 35IV. The gradual addition of water to the cation 35IV afforded the intermediate 35V. Subsequent proton abstraction from intermediate 35V by PQH− led to an enol intermediate 35VI. Finally, photoaromatization of the enol engendered the desired product 35c with the simultaneous liberation of hydrogen gas from 35VI (Scheme 35). Transition metal-free, mild reaction conditions and large functional group tolerance are important advantages of this method. However, the highest yield was lower than the other related methods.
In June 2018, Zhao & Jiang et al.57 disclosed a transition-metal-free, light-induced cascade enantioselective aerobic oxidation/semipinacol rearrangement reaction of 2-aryl-3-alkylsubstituted indoles. They reacted 2,3-disubstitued indole (36a, 0.10 mmol) in the presence of the chiral organophotocatalyst dicyanopyrazine-based chromophore (DPZ, 0.5 mol%), SPINOL-based spirocyclic CPA (C1) as an extrinsic chiral H-bonding catalyst (10 mol%), 4 Å molecular sieves as an additive in tBuPh solvent in air, and 3 W blue LED irradiation at 8 °C for 40 h (Scheme 36). The reaction was successful in producing the desired chiral indolin-3-one 36b with up to 90% yield and excellent enantioselectivity. It was found that except for C1, other SPINOL-derived spirocyclic CPAs were futile, in fact incorporating BINOL-CPA reversed the absolute configuration of the targeted molecule. Replacing DPZ with other conventional photoredox catalysts decreased the yield, albeit both DPZ and C1 were necessary for an optimized condition. Interestingly, in the absence of both light and air, the reaction was completely inhibited. Further generalization of this protocol led to a wide substrate scope. It was observed that substituents bearing aromatic rings, 2-aryls, and 3-benzyls afforded inferior yields with reduced enantioselectivity. On the other hand, the 3-benzyl-containing meta-substituent showed superior enantioselectivity over para- and ortho-substituents. Moreover, bulkier tBu and iPr moieties at the para-position of the same aryl ring demonstrated better enantioselectivity than the methyl group. Amazingly, replacing this 3-benzyl with thiophenyl-2-methyl afforded the highest selectivity of 94% ee; the cyclohexylmethyl was also a competent alternative for the same in this transformation. Substitution on the 2-aryl moiety showed good amenability for both electron-deficient and efficient groups, albeit with deteriorated enantioselectivity. The reaction occurred at 8 °C, which was a disadvantage as most of the methods worked at room temperature.
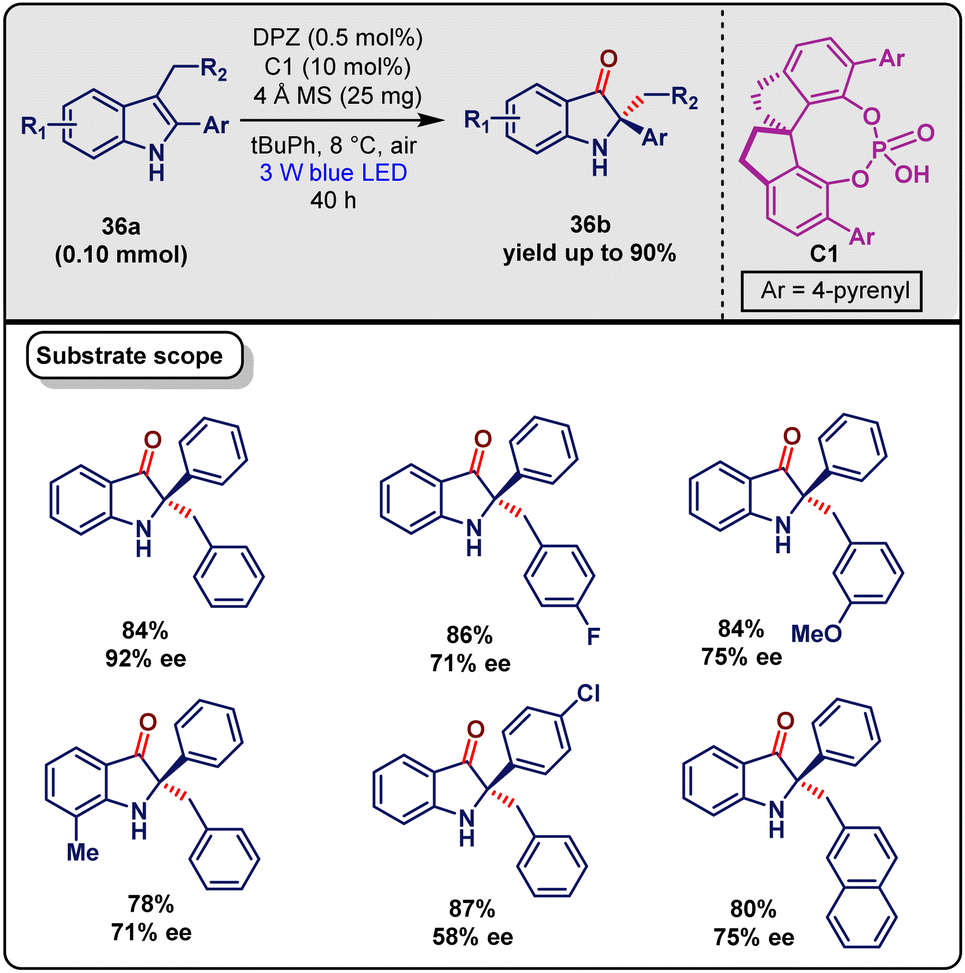 | ||
| Scheme 36 Accessing chiral 2,2-disubstituted indolin-3-ones via organo-photocatalytic cascade reaction (Zhao & Jiang method). | ||
Visible-light-promoted reactions are always desirable due to their greener conditions. Keeping this in mind, Hong et al.58 in March 2019, reported a visible-light-promoted cascade radical cyclization and pyridylation for the sake of the synthesis of tetrahydrofuran. They employed N-alkenyloxypyridinium salts as both heteroaryl sources and radical precursors and furnished a metal-free tool for the functionalization of biorelevant molecules. They investigated N-alkenyloxypyridinium (37a, 0.1 mmol) with Q1 (3-phosphonate quinolinones) (2.5 mol%) as a photocatalyst, and NaHCO3 (0.12 mmol) as the base in CH3CN solvent under the irradiation of blue LEDs at room temperature in a N2 atmosphere for 10 h. The corresponding desired product (37b) was achieved with up to 89% yield (Scheme 37). Compared to several Ru- and Ir-based catalysts, 3-phosphonate quinolinones (Q1) showed the best activity. Investigations revealed that CH3CN was the most suitable among the screened solvents and blue LEDs acted the best in terms of light sources to improve yield. The absence of any one of the photocatalysts Q1 or visible light failed to generate the product, which was a clear indication of their necessity. To explore the generality of this scheme, various substrates were employed under the optimized conditions. The selective functionalization of tertiary, secondary, and primary C–H bonds was viable in this scheme. Quinolines, isoquinolines, and pyridyl-substituted substrates were able to generate products with moderate to good yields. They further extended this strategy for the generation of the pyridine-tethered bicyclic oxaspiro ring. To study the applicability of this approach in late-stage functionalization, the tetrahydrofuran moiety was installed in biologically active molecules like roflumilast, pyriproxyfen, etc., resulting in good yields.
 | ||
| Scheme 37 Synthesis of tetrahydrofurans via visible-light-promoted cascade radical reaction (Hong method). | ||
The reaction was reluctant to proceed in the forward direction to form the desired product in the presence of radical scavenger TEMPO, which confirmed that the reaction involved the generation of radicals inhibited by the incorporation of TEMPO. This result revealed a radical-driven pathway. With the help of experimental evidence and previously reported results, they proposed a plausible mechanistic pathway as depicted in Scheme 37. In the beginning, Q1 was activated under irradiation and promoted to the photoexcited state Q1*, which further underwent SET reduction to generate Q1˙+ and alkoxy radical 37I by N-alkoxypyridinium salt 37a. Subsequently, alkoxy radical 37I was added to an olefin to generate tetrahydrofuran by 5-exo-trig alkoxy-radical cyclization along with carbon-centred radical 37II. This newly formed alkyl radical 37II further engaged with the pyridyl group of substrate 37a to generate a radical cation intermediate 37III. Later, this radical cation 37III underwent consecutive deprotonation followed by SET to give 37IV, whose N–O bond was homolytically cleaved to furnish the final product 37b and alkoxy radical 37I. The generated radical 37I participated in a new reaction cycle. The measured quantum yield (Φ = 9.8) of this strategy suggested that the radical chain pathway was quite effective. This method was found to have good functional group compatibility, broad substrate scope, and metal-free mild conditions, making it a powerful synthetic tool for assembling diverse pyridine-tethered tetrahydrofurans as well as the late-stage functionalization of complex biorelevant molecules.
In 2019, the Zhang and Xu group59 developed a visible-light-induced organophotocatalyzed hydrotrifluoromethylation method for benzyl-protected homoallylic alcohol (38a) and amine derivatives (38d) using various electrophilic trifluoromethylating reagents, such as trifluoromethyl sulfonyl chloride, Togni's reagent, and 2-bromo-2,2difluoroacetate. Also, mild irradiation conditions deprotected the benzyl protecting group to form δ-fluoromethylated alcohols (38c) and amines (38e) as the final products (Scheme 38). Because of the larger band gap (2.4 eV) and suitable HOMO/LUMO gap, 4CzIPN was able to do trifluoromethylation by oxidative or reductive quenching. Togni's reagent, as the CF3 source was more effective than CF3SO2Cl; whereas, Umemoto's reagent (S-(trifluoromethyl)dibenzothiophenium triflate) decreased the yield of the desired product when other factors remained same. The best protecting group was 4-cyanobenzyl because the cyano group could increase the rate of 1,5-H transfer. When α-deuterated benzyl was taken as the protecting group, deuterium was inserted at the γ-position of the product, which was the proof of 1,5-hydrogen-transfer. Next, the substrate scope of alcohol was screened by the authors. β- and γ-substituted homoallylic alcohols were capable of forming the product along with α-substituted homoallylic alcohols. Tertiary and alkyl-substituted alcohols were not suitable for this reaction. If the α- and β-carbons of the homoallylic alcohol contained chiral centers, they would be retained after the reaction. The homoallylic amine derivatives, containing chloro, amide, ester groups, etc., were also capable of this reaction under optimized reaction conditions.
 | ||
| Scheme 38 Visible-light-induced organophotocatalyzed synthesis of δ-fluoromethylated alcohols via cascade reaction (Zhang and Xu method). | ||
The inhibition of the reaction by TEMPO proved that a radical intermediate was formed in the mechanism. The most suitable mechanism proposed by the authors is shown in Scheme 38. At first, the photoexcited 4CzIPN [E½(PC˙+/PC*) = −1.18 V] generated the trifluoromethyl radical, which was then added to the benzyl-protected homoallylic alcohol or amine (38a/38d) to form the radical intermediate 38I. After that, a more stable benzylic radical 38II was formed through the 1,5-H shift. Then, the photoredox catalyst [E½(PC˙+/PC) = +1.49 V], oxidized this benzylic radical 38II to form the cationic intermediate 38III. Next, acetal 38IV was generated by the quenching of 38III by the alcohol solvent. Finally, aqueous workup was done to get the desired product 38c/38e.
Yu et al.60 in Jan 2020 introduced a protocol for the synthesis of functionalized chroman-4-ones via a visible-light-promoted cascade radical cyclization of o-(allyloxy)arylaldehydes along with sulfinic acids. The authors utilized o-(allyloxy) arylaldehydes (39a, 0.2 mmol) and sulfinic acids (39b, 0.6 mmol) in the presence of a photocatalyst (Na2-eosin-Y, 4 mol%), oxidant (K2S2O8, 3 equiv.) and solvent (acetone:H2O) under the irradiation of 8 W blue LED for 24 h at room temperature. This condition led to the formation of the desired product 39c with good to excellent yield (Scheme 39). Acetone:H2O at a 1:1 ratio was perfect as a solvent. Dark conditions did not afford the desired product, indicating the necessity of light in the reaction. To widen the diversity of this protocol, several substrate scopes were observed via the installation of various substituents. Electron-donating, -withdrawing, and sterically hindered groups showed good tolerance in the phenyl ring of sulfinic acids. The compatibility of several electron-donating and -withdrawing groups were also observed for o-(allyloxy) arylaldehydes. To verify the practicality of this strategy, a gram-scale reaction was carried out, which provided excellent yield.
 | ||
| Scheme 39 The synthesis of chroman-4-one derivatives via the visible-light-promoted cascade cyclization of sulfinic acids and o-(allyloxy)arylaldehydes (Yu method). | ||
A series of control experiments and previous literature gave insight into the mechanism. In the beginning, Na2-eosin-Y* was formed from Na2-eosin-Y under photoexcitation. Next, the sulfonyl radical cation 39I and radical anion Na2-eosin-Y˙− were generated from sulfinic acid after oxidization by Na2-eosin-Y*. The successive deprotonation of the sulfonyl radical cation 39I by SO4˙− resulted in the formation of sulfonyl radical 39II, which might be transformed into the resonance structure 39III. At the same time, o-(allyloxy) arylaldehydes 39a were oxidized by K2S2O8 through the SET process to form radical 39IV, which could undergo intramolecular cyclization, generating intermediate 39V. In the last step, the desired product 39c was obtained via the coupling of intermediate 39V with 39II (Scheme 39).
In Feb 2020, Su, Huo, Hu et al.61 illustrated a strategy for the cascade cyclization of N-aryl-N-acryl benzamides with α-keto acids by irradiation with visible light to synthesize acyl-containing isoquinoline-1,3(2H,4H)-diones. In this method, N-methylbenzamide (40a) (0.3 mmol) reacted with 2-oxo-2-phenylacetic acid (40b) (0.4 mmol) in the presence of (NH4)2S2O8 (oxidant) (0.9 mmol) and eosin B (photoredox catalyst) (2 mol%) under irradiation with 2 × 3 W blue LED light and open air in DMSO (3 mL) for the synthesis of 2,4-dimethyl-4-(2-oxo-2-phenylethyl)isoquinoline-1,3(2H,4H)-dione (40c) with up to 92% yield (Scheme 40). The absence of visible light, oxidant, or photocatalyst inhibited the reaction. Eosin B was superior among all other photoredox catalysts. The yield was decreased when K2S2O8, TBHP was used as the oxidant instead of (NH4)2S2O8. DMSO was the best choice among the other solvents, such as CH3CN, DMF, etc. Next, the substrate scope was investigated. The aromatic ring of 40a, bearing different substituents, such as methyl, fluorine, methoxy, and acetyl, at different positions furnished the desired products in good yields. It was noticed that the steric effect negatively influenced the synthesis of bulky products. Moreover, the authors mentioned that electron-withdrawing groups at the α-keto acid were more reactive than electron-donating groups.
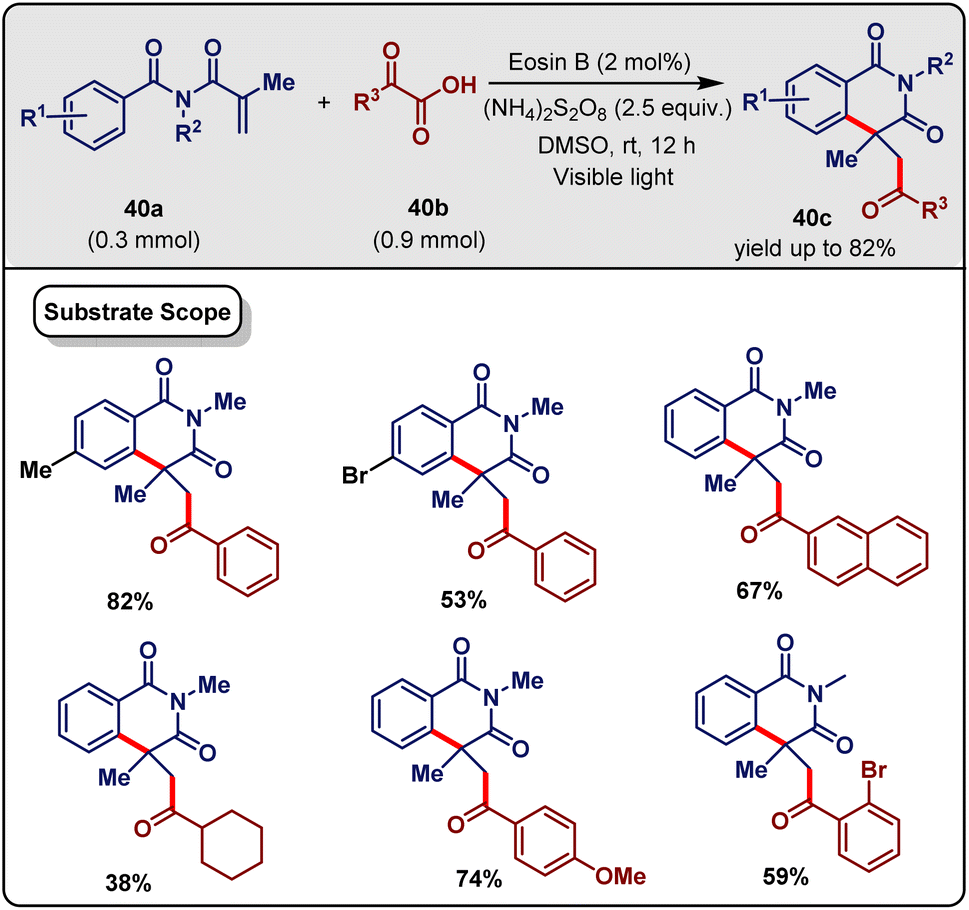 | ||
| Scheme 40 The synthesis of acylated isoquinoline-1,3(2H,4H)-dione via the cascade radical reaction in the presence of visible light (Su, Huo and Hu method). | ||
The reaction was stopped in the presence of TEMPO or BHT, which indicated that this cascade reaction proceeded through a radical mechanism. A mechanism was proposed by the authors based on control experiments and previous literature reports (Scheme 41). In the initial step, eosin B was excited to eosin B* by irradiation with a blue LED. The eosin B radical cation, sulfate dianion, and sulfate radical anion were formed by a single electron transfer from eosin B to the persulfate anion. Next, in path I, 41b and the sulfate radical anion underwent hydrogen atom transfer (HAT) and SET to form a benzoyl radical 41I by decarboxylation. The addition of 41I to the C–C double bond of 41a, followed by intramolecular cyclization resulted in the generation of intermediate 41III. After that, the single-electron oxidation of 41III occurred by the eosin B radical cation to form the carbocation intermediate 41IV. In the final step, the desired product 41c was obtained by the deprotonation of 41IV. This method has mild reaction metal-free conditions, is operationally practical, and has a wide substrate scope.
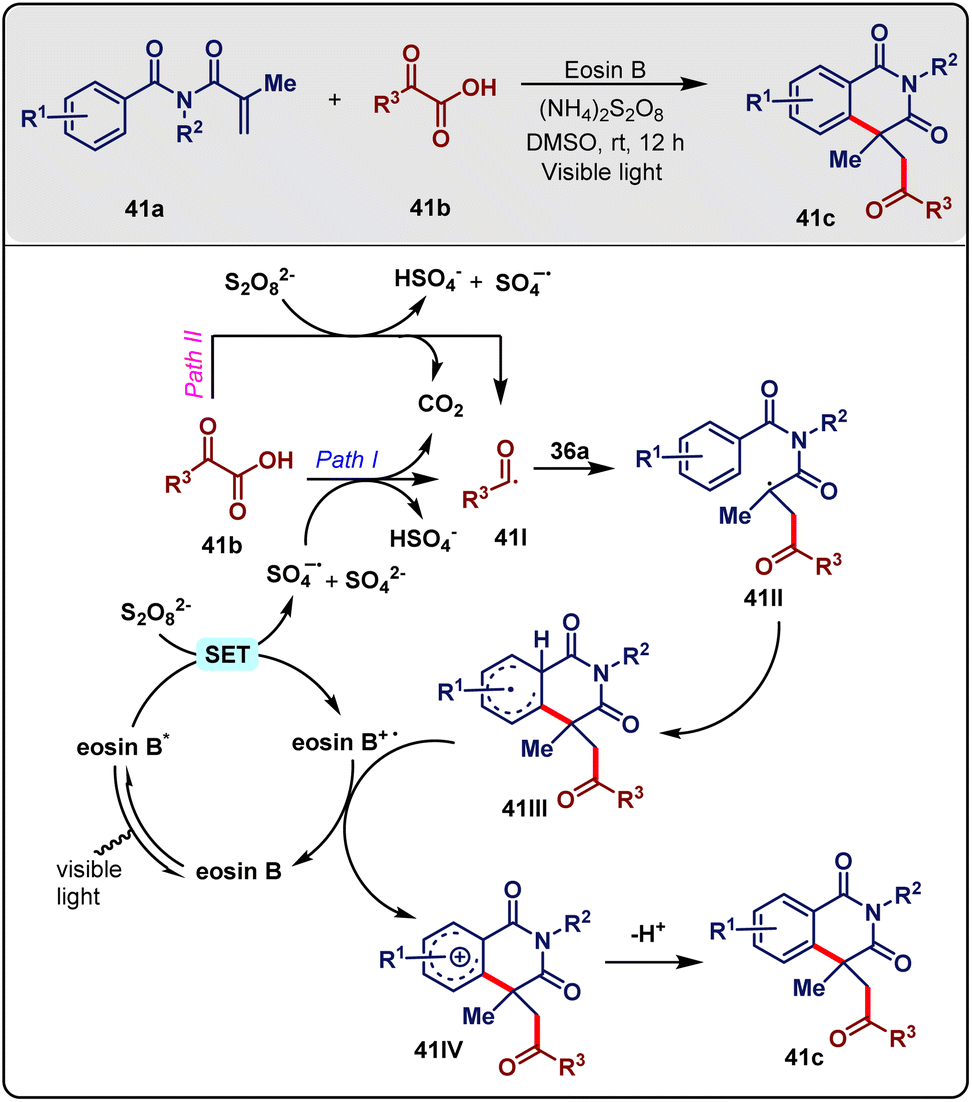 | ||
| Scheme 41 The proposed mechanism. | ||
Xu and Ji et al.62 in June 2020 developed a metal-free methodology for the synthesis of 2-azidoindolin-3-one in the presence of a photocatalyst. According to this methodology, 1H-indol-1-yl(phenyl)methanone 42a (0.2 mmol) reacted with TMSN342b (0.2 mmol) in the presence of 9-mesityl-10-methylacridinium perchlorate (Mes-Acr-Me+) as a photocatalyst (10 mol%) and diphenyl diselenide (PhSeSePh) as an additive (1 equiv.) under irradiation with 40 W blue LED in acetonitrile solvent. The desired product 2-azido-1-benzoylindolin-3-yl-2-benzamidobenzoate (42c) was achieved in up to 75% yield within 3–4 h (Scheme 42). However, an O2 atmosphere was required to achieve the highest yield of the desired product. The reaction was unsuccessful when PhSeSePh, the photocatalyst or irradiation were absent. Among all the photocatalysts, such as rose bengal, fac-Ir(bpy)3, eosin Y, etc., acridine red was the most effective for this reaction. Next, the substrate scope was examined by the authors based on the optimized reaction conditions. Electron-donating groups (–CH3, –OCH3) at benzene ring of indole and at the N-benzoyl group were suitable, whereas electron-withdrawing groups, such as –NO2 were incompatible for this cascade reaction. The gram-scale reaction was studied to bring out the practical applicability of this reaction. They observed that 51% yield of the desired product was achieved under standard conditions in the gram-scale synthesis.
 | ||
| Scheme 42 Transformation of indoles into 2-azidoindolin-3-yl 2-aminobenzoates by the visible-light-induced cascade reaction (Xu and Ji method). | ||
To propose the mechanism, some control experiments were carried out. No product was obtained if the reaction was carried out in an argon atmosphere, so it was noted that O-atoms in the products came from O2. Moreover, 18O-43c was not produced when the experiment was done with H218O. This indicated that the only source of oxygen was molecular oxygen. The reaction proceeded through a radical pathway that could be proved by the fact that by adding radical scavenger TEMPO, no desired product was detected. These experiments and previous studies revealed the mechanism of this ring-opening cascade reaction (Scheme 43). Firstly, TMSN3 was oxidised by visible-light irradiation, producing an azido radical, which was added to the C-2 atom of 43a to form radical intermediate 43I. Next, O2 trapping, followed by protonation resulted in 2-azido-3-hydroperoxyindoline 43III. The reaction proceeded through two pathways. In the first pathway, azidonol 43IV was produced by the reduction of 43III in presence of PhSeSePh. The second path involved the conversion of 43III into intermediate 43V and then to azidonone 43VI. After that, intermediate 43VII was formed by the attack of the hydroxyl oxygen of 43IV on the carbonyl carbon of 43VI. Thereafter, intermediate 43VIII was obtained from 43VII by a ring-opening reaction. Finally, C–N bond cleavage of 43VIII took place to afford the final product 43c. Significant advantages include metal-free environmentally benign conditions and a controllable multi-step reaction.
 | ||
| Scheme 43 The plausible mechanism. | ||
In the same year (Oct 2020), Li and his group63 reported a mild, metal-free, visible-light-mediated oxidative radical cascade synthesis of sulfonated chromanes and sulfonated 1,2,3,4-tetrahydroquinolines compounds. They employed 1-(allyloxy)-2-(arylethynyl)benzenes (44a, 0.10 mmol) or N-allyl-2-(arylethynyl)anilines (44b, 0.10 mmol), a 1,7-enyne system, with benzene sulfinic acids (44c, 0.15 mmol) in conjunction with photocatalyst eosin Y (3 mol%), oxidant TBHP (7.5 mol%) in DCE as solvent (3.0 mL) under green LED irradiation (3 W), N2 atmosphere at room temperature for 12 h. The desired product 44d was obtained with up to 86% yield (Scheme 44). Moreover, the N2 atmosphere was crucial as compared to air for higher yield. Without any oxidants, the reaction afforded the desired product with very poor yield and other oxidants e.g., H2O2, K2S2O8 and DTBP produced inferior yields when other factors remained the same. The absence of either eosin Y or light inhibited the reaction and other photocatalysts were unfit for this reaction protocol. Furthermore, the potential of this scheme was substantiated by the gram-scale (up to 5 mmol scale) synthesis, which obtained 63% of the desired product. Various substitutions in 1-(allyloxy)-2-(arylethynyl)benzenes afforded the desired product with good yield. Substitution in phenylacetylene moiety viz., electron-donating and electron-withdrawing substituents at ortho-, meta- and para-positions provided good results despite steric encumbrance. On the other hand, varieties of substituents on aryl moiety e.g., –Me, –Cl and –Br afforded the anticipated products with good yields. Substitution on the alkene part did not have any negative influence and resulted in moderate output. The substrate scope of N-allyl-2-(arylethynyl)anilines showed good diversity for both phenylacetylene and aryl moieties with moderate to good results. Different substituents such as methyl, chloride and other halides showed good amenability in the case of sulfinic acids. Moreover, the aliphatic analogue of aryl sulfinic acid performed well with a synthetically useful yield.
 | ||
| Scheme 44 Metal-free cascade transformation of 1,7-enynes to sulfonated chromanes and sulfonated tetrahydroquinolines using the sulfinic acid (Li method). | ||
A control experiment with TEMPO revealed the total abolition of the transformation, suggesting a radical path might be involved. Based on the control experiment investigations, a plausible mechanism was proposed. The photo-excited eosin Y* participated in a SET with TBHP and generated the hydroxyl anion, tert-butyloxy radical 44I along with eosin Y˙+. Subsequent abstraction of a hydrogen atom from arylsulfinic acid 44c by 44I furnished sulfonyl radical 44II, which immediately underwent an addition reaction to the carbon–carbon double bond of 44a to generate alkyl radical 44III. The concomitant intramolecular radical cyclization with the carbon–carbon triple bond of the alkyne via a 6-exo-dig formed the vinyl intermediate 44IV. Finally, this newly formed radical 44IV was converted into the desired product 44d after the abstraction of a hydrogen atom from arylsulfinic acid 44b (Scheme 44). This transformation has a low TBHP loading, mild reaction conditions, a simple operation, a wide functional-group tolerance, and high efficiency.
Indazole is one the most important imperative N-containing compounds having several biological and industrial applications and hence is studied largely in synthetic chemistry.64 In March 2021, Liu, Cao and co-workers65 disclosed a photo-induced three-component cascade cyclization strategy to synthesize seleno-functionalized pyrimido[1,2-b]-ioles. The exploitation of 3-aminoindazoles (45a, 0.3 mmol), ynals (45b, 0.3 mmol) and diphenyldiselenide (45c, 0.3 mmol) in the presence of 3 mol% of rose bengal as a photosensitizer, FeCl3 (0.3 mmol) as an additive and MeCN as a solvent (3 mL) under 20 W blue irradiation at room temperature for 12 h generously led them to the desired product 45c with up to 80% yield (Scheme 45). Replacing rose bengal with other photosensitizers, viz., eosin Y, rhodamine, 6G, fluorescein, gave inferior yields in this current scheme. More importantly, light was inevitable for product formation. Solvent screening demonstrated methyl cyanide as the most suitable in this protocol. The substrate scope of this strategy showed that various substituted 3-aminoindazoles, pyrazolo[3,4-b]-pyridines participated and smoothly afforded the desired product. Ynals showed broad amenability. para-Substituted (e.g. –Me, –phenyl, –F, –Cl, –Br, –CN and –OCH3), meta- and ortho-substituted aryl ynals performed quite well and yielded moderate to good outcomes. Along with these di-substituted ynals, 3-(3,4-dimethylphenyl)-propiolaldehyde and aliphatic ynals were substantiated as viable substrates in this scheme. A series of diphenylselenides participated, and meta- and para-phenyl substitutes performed well in this reaction. 1,2-Dimethylselane, 1,2-diethylselane and 1,2-dibenzylselane provided good yields. In addition, biphenyl ditellurium afforded the desired product with 68% yield.
 | ||
| Scheme 45 The visible-light-enabled cascade reaction for the synthesis of chalcogen-containing pyrimido[1,2-b]indazoles (Cao method). | ||
Based on the mechanistic rationale from literature and results from control experiments, two possible mechanistic pathways are shown in Scheme 46. In pathway-I, photo-excited RB* converted diphenyldislenide into the radical cation (PhSe)2˙+ (46I) via SET. The intermolecular condensation adduct 46II was generated from 46a and 46b. 46II subsequently reacted with the radical cation 46I and liberated 0.5 equiv. of (PhSe)2 along with seleniranium cation 46III, which furnished the final product 46d after deprotonation. Molecular oxygen oxidized the RB˙− and closed the catalytic cycle. In the case of path-II, excited RB* transferred energy to the ground-state oxygen and converted it into singlet oxygen, which concomitantly underwent a SET with diselenide (46c) and produced the radical cation 46I, which in turn was converted into the PhSe+ (46IV) intermediate. This intermediate reacted with intermediate 46II and the subsequent intermediate 46III generated the final product 46d.
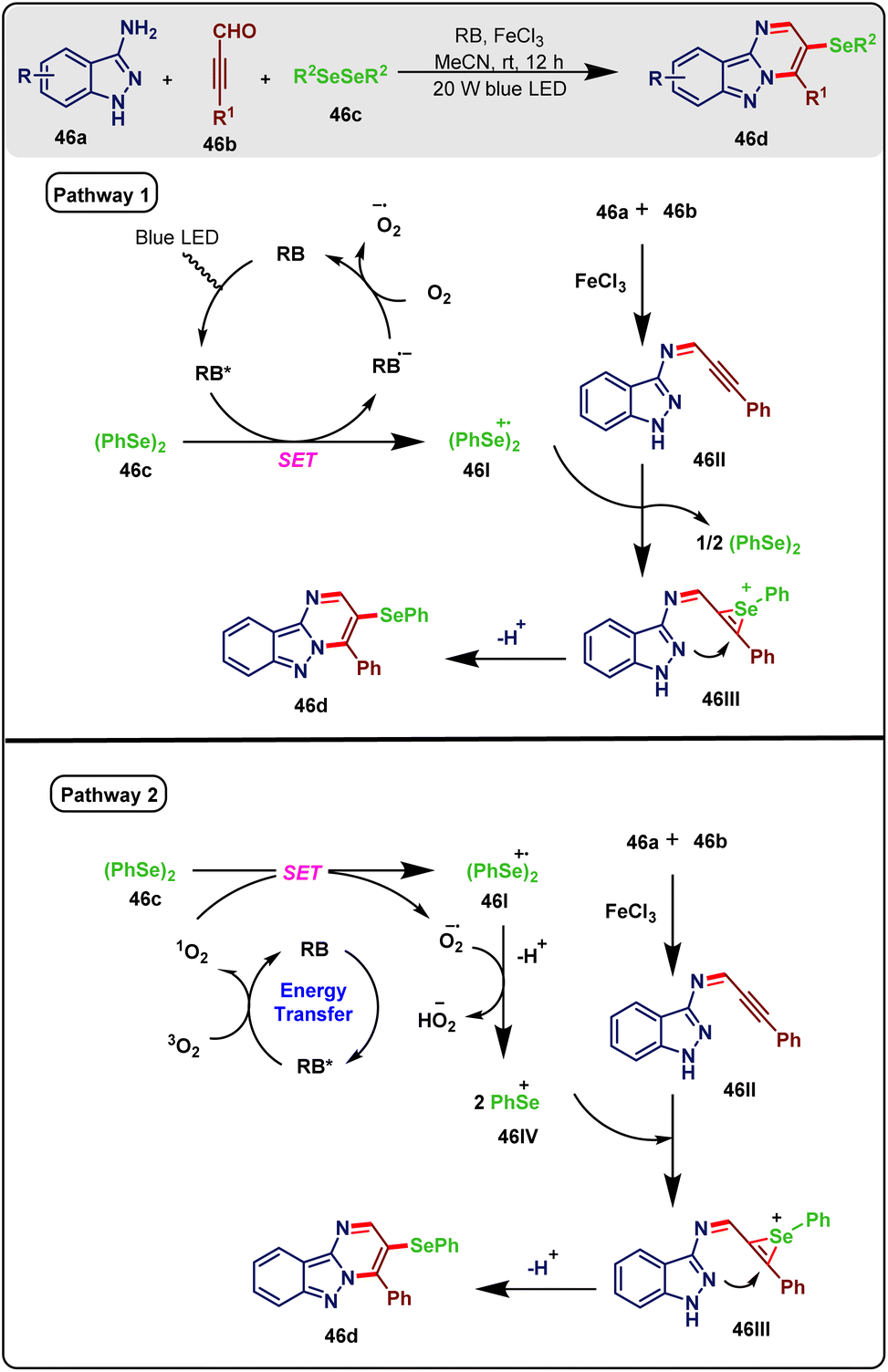 | ||
| Scheme 46 Mechanistic pathway. | ||
Kühn & Guo et al.66 in March 2021 reported a visible-light-induced cascade pericyclic reaction to construct polycyclic compounds with oxabicyclo[4.2.0]octene skeletons. They reacted (E)-4-(((4-bromo-3-methylbut-3-en-1-yl)oxy)methyl)-1-methylquinolin-2(1H)-one or quinolin-2(1H)-one (47a, 0.1 mmol) with thioxanthone (10 mol%) as a photocatalyst and tBuONa (2 equiv.) as a base in anhydrous acetonitrile solvent under violet LED irradiation (410–420 nm) and an Ar-atmosphere at room temperature for 12 h. The desired oxabicyclo[4.2.0]octene product 47b was obtained with the highest yield of 88% (Scheme 47). It was observed that other bases like MeONa, KOH, Cs2CO3 and MeOK provided inferior yields. The absence of light or thioxanthone was detrimental to this transformation, and lowering the catalyst loading decreased the product formation. Without the base, the reaction did not yield the desired product. The amenability of different N-substituted reactants was then examined by the authors. N-Methyl, n-butyl, allyl, propargyl, and benzyl groups afforded the desired product in good yields. Experimentation to measure functional group tolerance showed many compatible moieties, viz., 4-methoxybenzyl, 4-methylbenzyl, 4-phenylbenzyl, 4-bromobenzyl, 4-methoxycarbonylbenzyl, 4-cyanobenzyl and 4-trifluoromethylbenzyl. The installation of different functionalities in the benzene ring demonstrated the high tolerance of strong electron-donating groups in comparison with weak analogues, and halogens also led to moderate to good yields of the desired product under optimized reaction conditions.
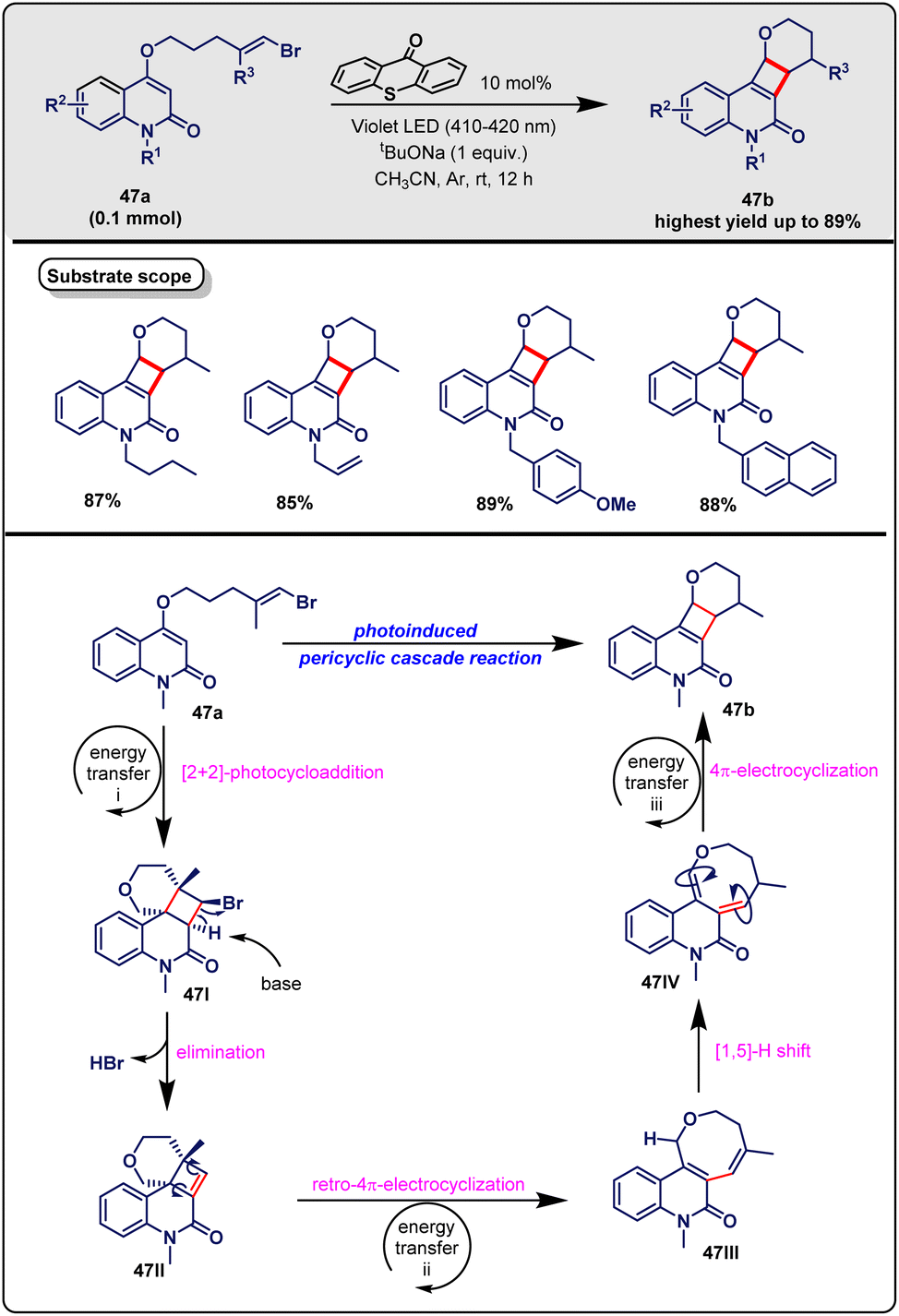 | ||
| Scheme 47 Pericyclic cascade reaction in visible-light to access the oxabicyclo[4.2.0]octene skeleton (Kühn and Guo method). | ||
A series of control experiments suggested that the transformation of 47a was propagated through a thioxanthone-catalyzed photochemical pathway. Moreover, a comparatively higher triplet-state energy of thioxanthone (265 kJ mol−1) than 47a disclosed that an energy-transfer process between thioxanthone (T1) and the substrate (S0) might be possible. Based on these results and previous reports, they proposed a plausible mechanistic pathway as shown in Scheme 47. At the beginning, 47a converted into an intermediate brominated cyclobutene 47I under visible-light irradiation via energy-transfer based [2 + 2]-photocycloaddition. Concomitant base-promoted hydrogen bromide elimination generated the cyclobutene intermediate 47II. Subsequent retro-4π-electrocyclization of this new intermediate 47II furnished the eight-membered ring product 47III. Later, the 1,5-hydrogen shift converted 47III into the new intermediate 47IV and followed with the photoinduced disrotatory 4π-electrocyclization to furnish the final product 47b.
In May 2021, Zhou et al.67 developed a photo-induced cascade sulfonylation/cyclization of 1-(allyloxy)-2-(vinyl)benzene with sulfonyl chloride for the synthesis of sulfonated benzoxepines. The exploitation of 1-(allyloxy)-2-(arylvinyl)benzene (48a, 0.1 mmol) with p-tosyl chloride (48b, 0.2 mmol) in the presence of photocatalyst eosin Y (2 mol%), base Na2CO3 (0.25 mmol) and solvent acetonitrile (1 mL) under 10 W blue LED irradiation at 100 °C for 18 h in an Ar-atmosphere resulted in the desired sulfonated bezoxepine product 48c in up to 92% yield (Scheme 48). Replacing eosin Y with other photoredox catalysts, viz., Ru(bpy)3(PF6)2 and fac-Ir(ppy)3 led to an inferior yield. Solvent screening suggested that acetonitrile was the most suitable as compared to others, e.g., DCM, THF, DMF and DMSO. Of the various bases such as NaHCO3, NaOAc, Na2HPO4, Na2CO3, Et3N and 2,6-luidine, only sodium carbonate afforded the desired product with superior yield. Without a photocatalyst or light source, the transformation was inhibited and the absence of a base led to a very poor yield of the desired product. They found diverse substrate scopes for both substrates. Electron-donating and electron-withdrawing groups in the ortho- or para-position of the phenyl moiety tethered alkene in 1-(allyloxy)-2-(arylvinyl)benzene afforded the desired product with good to excellent yields. Interestingly, no desired product was obtained when the methyl group was tethered in lieu of the phenyl moiety in the alkene part. The reaction went smoothly for substituents like MeO, Me, F, Cl and Br in the 4- or 5-positions of the phenol moiety. Furthermore, disubstitution in the same ring did not hinder the transformation. ((2-Methylallyl)oxy)-2-(1-phenylvinyl)benzene, N-allyl-4-methyl-N-(2-(1-phenylvinyl)phenyl)benzene and 1-(but-3-en-1-yl)-2-(1-phenylvinyl)benzene sulfonamide also displayed good amenability for this protocol. Many aromatic and aliphatic sulfonyl chlorides were found to be viable sulfonylating agents in this strategy. Aryl sulfonyl chlorides with para-substitution of both electron-donating groups (–MeO and –tBu) and electron-withdrawing groups (–F, –Cl and –Br) obtained the desired product in good to excellent yields. Moreover, meta- and ortho-substituted aryl sulfonyl chlorides were compatible partners in this reaction. Besides, naphthalene-1-sulfonyl chloride, naphthalene-2-sulfonyl chloride and thiophene-2-sulfonyl chloride were suitable sulfonating agents, and provided the corresponding products with good yields.
 | ||
| Scheme 48 Synthesis of sulfonated benzoxepines from 1-(allyloxy)-2-(1-arylvinyl)benzenes in the visible light photocatalyzed cascade reaction (Zhou method). | ||
Control experiments with TEMPO and 1,4-dinitrobenzene led to the complete abolition of the transformation, which suggested that a radical path might be involved. Based on these investigations and reports, they propounded the most plausible mechanism as shown in Scheme 48. Initially, the photo-induced excited eosin Y* was oxidized by p-tosyl chloride and converted into eosin Y˙+ and furnished radical anion 48I. The generation of sulfonyl radical 48II was possible by the successive/sequential liberation of chloride ions from 48I. This radical was then added to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond of substrate 48a and engendered the alkyl radical 48III. The concomitant intramolecular cyclization of this radical with another CC bond produced the radical intermediate 48IV. The subsequent oxidation of this radical by eosin Y˙+ led to another cationic intermediate 48V, which was converted into the desired product 48c after base-assisted proton abstraction. Finally, the protocol provided a general approach to sulfonated benzoxepines, with a broad substrate scope, excellent functional group tolerance, and high product yields. However, when R2 was a methyl group, no expected product was achieved due to the lower stability of the benzyl radical intermediate.
C bond of substrate 48a and engendered the alkyl radical 48III. The concomitant intramolecular cyclization of this radical with another CC bond produced the radical intermediate 48IV. The subsequent oxidation of this radical by eosin Y˙+ led to another cationic intermediate 48V, which was converted into the desired product 48c after base-assisted proton abstraction. Finally, the protocol provided a general approach to sulfonated benzoxepines, with a broad substrate scope, excellent functional group tolerance, and high product yields. However, when R2 was a methyl group, no expected product was achieved due to the lower stability of the benzyl radical intermediate.
Very recently, Singh and co-workers68 developed a metal-free visible-light-induced three-component cascade sulfonylative annulation reaction. As per the report, they initially started with benzimidazole (49a) and diazonium salt (49b) in the presence of eosin Y acting as a photocatalyst, DCE as a solvent, and Na2S2O5 as an SO2 precursor under the irradiation of blue LED under an inert N2 atmosphere, which readily delivered the desired product 49c in good yield after 4 h (Scheme 49). These optimized conditions were obtained under several screening experiments. Experiments with several solvents like DCM, DMF, MeCN, and MeOH were carried out but DCE was the best for generating the highest yields. To determine the necessity of the photocatalyst in this scheme, the reaction was carried out in both dark and photocatalyst-free conditions but those attempts were not successful in delivering the desired product. After establishing the optimized conditions, they evaluated the substrate scope to verify its generality. At first, several substituted phenyl diazonium salts were tested. Electron-donating substituents like p-methyl, p-methoxy, and m-methyl groups produced good yields. Electron-withdrawing groups like the para-acylated group also afforded a 60% yield. A diminished yield was observed when ortho –CF3 and ortho –F substituted diazonium salts were used. Similarly, the substrate scope of benzimidazole was also evaluated by the authors. Several electronically distinct groups like –CN, –F, –OMe, –Br were well suited to afford good yield. The nitro-substituted indolyl moiety gave some extent of reduced yield up to 67%. It is worth mentioning that the precursors with internal olefins, acrylate derivatives, and free phenolic –OH failed to generate any yield of the desired product.
 | ||
| Scheme 49 Three-component light-mediated cascade sulfonylative annulation (Singh method). | ||
Thereafter, they studied some control experiments, which helped them to design a plausible pathway for the reaction (Scheme 49). At first, phenyl radical 49I was generated from diazonium salt 49bvia oxidative quenching of the excited state of the photocatalyst. Then, 49I was trapped by Na2S2O5, which resulted in the formation of the phenylsulfonyl radical 49II. The generated radical 49II then reacted with 49a to generate an alkyl radical 49III. In the next step, 49III underwent intra-molecular cyclization and formed an intermediate 49IV. After that, the photocatalytic cycle ended with the generation of 49Vvia the oxidation of 49IV with the help of EY˙+. Alternatively, it can also be said that 49V was generated from 49IV by the single-electron-oxidation of diazonium salt 49b, which finally formed the desired product 49c. Therefore, this strategy simply generates one C–C bond and two C–S bonds via a single-step photochemical cascade cyclization reaction. Moreover, this technique uses a readily available SO2 surrogate and does not necessitate any transition metals, oxidants, or additives.
7. Photocatalyst-free visible-light-induced cascade reactions
Studer and his team69 in 2019 developed a visible-light-promoted electron-catalyzed cascade α-perfluoroalkyl-β-heteroarylation reaction employing several alkenes with perfluoroalkyl iodides and quinoxalin-2(1H)-ones. As per the report, they utilized 1 equiv. of quinoxalin-2(1H)-one (50a) acting as the electrophilic C-radical acceptor, 2.5 equiv. of alkene (50b) as nucleophilic acceptor, and 2 equiv. of perfluorobutyl iodide (50c) as the C-radical precursor along with the presence of 3 equiv. of 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) as base in NMP solvent under the irradiation of blue LED. These conditions successfully converted the expected product (50d) at normal room temperature within 16 h (Scheme 50). These optimized conditions were revealed after several control experiments. A series of different solvents and bases were screened, but none were up to the mark. Next, to evaluate the productivity of this strategy, a series of compounds were tested by changing the substituents at various positions. For the alkenes, non-functionalized alkenes were smooth under this protocol, along with functional groups like phosphonate, alcohol, halo substituents, and nitriles that were well tolerated. Alkenes bearing hetero substituents like carbazole, and coumarin were also well-suited under the same optimized reaction conditions. In the case of quinoxalin-2(1-H)-one, various substituents from electron-donating and -withdrawing groups readily formed the product without any constraints. | ||
| Scheme 50 Photocatalyst-free visible-light-induced α-perfluoroalkyl-β-heteroarylation of various alkenes using the cascade reaction. | ||
The authors performed some control experiments to get a clear picture of the formation of products. As per the results of the control reactions and previous literature reports, they proposed a plausible reaction mechanism as shown in Scheme 50. Initiation of the process occurred via C–I bond homolysis of Rf-I after absorbing visible light, and the radical intermediate 50I was formed. This radical was then attached to alkene 50b to afford another radical intermediate 50II. Thereafter, the addition of radical 50II occurred selectively at the electron-deficient CN bond of 50a to form the N-center radical intermediate 50III. In the next step, the acidic radical 50III was deprotonated by DBU to generate the radical anion 50IV, which acted as a potent reducing agent. It reduced Rf-I via SET to generate 50d along with Rf radical and completed the catalytic cycle. Therefore, this strategy proved to be an efficient approach because of the synthesis of structurally privileged heteroarene moieties. Moreover, this is a rare example of metal-free electron-catalyzed methodology. However, the reaction only works in an inert atmosphere, which is a disadvantage.
In the same year (2019), Yoshino, Kojima and Matsunaga et al.70 showcased a photocatalyst-free visible-light-induced C–H γ,γ,γ-trifluoromethylation of quinolines using Umemoto's reagent II. According to this methodology, quinoline (51a) was reacted with alkene (51b), and Umemoto's reagent II (51c) in MeCN solvent underwent blue LED irradiation leading to the expected γ,γ,γ-trifluoromethylated product 51d with up to 84% yield. The authors observed that Togni's reagents I or II were less effective than Umemoto's reagent II. Moreover, various additives resulted in inferior yields when other factors remained the same. After obtaining the optimized reaction conditions, they evaluated the potential of this strategy by applying it to several substituted alkenes and quinolines. In the case of alkenes, it was found that allylsilanes, chloroalkyl groups and internal alkenes were the perfect sources for obtaining high yields. Apart from this, alkenes with polar functional groups afforded good yields. Simultaneously, the substrate scope of quinolines was also tested. C-4 substituted quinoline with an amide group afforded the C-2 alkylated products. Moreover, Weinreb amide and phenanthridine produced reasonably well-alkylated products. The authors also depicted a plausible mechanistic pathway for a better understanding of the reaction. At first, visible light cleaved the electrophilic trifluoromethyl radical source 51cvia homolysis of the C–F bond. The generated trifluoromethyl radical 51I was then attached to alkene 51b and initiated the first step for the radical propagation step and formed intermediate 51II. The generated trifluoroalkyl radical 51II was nucleophilic and its addition with protonated quinoline 51a formed the α-aminoalkyl radical, which under deprotonation generated the radical anion 51III. The final step terminated with electron transfer from intermediate 51III with a trifluoromethylating reagent (51c), delivering the desired product γ,γ,γ-trifluoro alkylated quinoline 51d (Scheme 51). Thus, this method generated a series of highly value-added biologically relevant trifluoroalkylated products. Metal-free, additive-free and room temperature conditions, high-yield product formation and late-stage functionalization are important features of this method.
 | ||
| Scheme 51 Photocatalyst-free visible-light-induced cascade C–H γ,γ,γ-trifluoroalkylation of quinolines (Yoshino, Kojima and Matsunaga method). | ||
Pan and Xu et al.71 in Jun 2019 enlisted a photocatalyst-free, visible-light-promoted cascade reaction of diselenides and alkyne-tethered cyclohexadienones to generate 5-hydroxy-3-selenyl-4a,8a-dihydro-2H-chromen-6(5H)-ones. This strategy showed great importance as it led to products that work as potential cancer cell growth-inhibiting agents. They initiated the reaction with alkyne-tethered cyclohexadienones (52a, 0.3 mmol) and diaryldiselenides (52b, 0.6 mmol) in the presence of PhCl as a solvent, CsOAc as a base, along with H2O under the influence of 25 W white LEDs. These conditions led to the formation of the major product 52c along with 52d in a trace amount (Scheme 52). When the base CsOAc was not present in the medium, 52d was the major product. PhCl was the best suited among several solvents and CsOAc was the best base, which increased the production of 52c. The increase in temperature resulted in a good yield of 52d, and light was essential to carrying out the reaction. Under optimized conditions, several substrate scopes were observed for this three-component reaction. Alkyl, alkoxy, aldehyde, halide, nitro, ester, and nitrile substituents were easily tolerable at the alkyne-substituent R1. An attempt with 2-methylphenyl substituted alkyne was a failure. 2-Thienyl, and 2-naphthalenyl at the alkyne were also compatible. An exchange of substituent R2 with ethyl, methyl, and 4-bromophenyl was successful. Both electron-withdrawing (–F) and -donating substituents (–OMe, –Me) at the diaryldiselenide took part smoothly in the reaction. Moreover, this catalyst-free, mild and environmentally-benign methodology was used to synthesize two compounds that have in vitro potential for anticancer activity, which is a very important advantage of this method.
 | ||
| Scheme 52 Metal-free synthesis of 5-hydroxy-3-selenyl-4a,8a-dihydro-2H-chromen-6(5H)-ones via visible-light-driven cascade cyclization (Xu method). | ||
Several control experiments were performed to gain insight into the mechanism. Based on literature reports and experiments, the most probable mechanism was proposed by the authors as shown in Scheme 52. In the beginning, phenylselenyl radical 52I was formed from diphenyl diselenide 52b under white light irradiation. Then, this species 52I was added to substrate 52a to generate an alkenyl radical 52II. Next, the participation of alkenyl radical 52II in an intramolecular radical cyclization reaction generated an intermediate 52III, which was further captured by the phenylselenyl free radical to form the side-product 52c. Finally, nucleophilic substitution of 52c with water in the presence of base CsOAc afforded the desired product 52d.
In March 2020, Li and his colleagues72 demonstrated a simple, metal-free, and convenient method for synthesising sulfone-functionalized chroman-4-ones (53c) (Scheme 53). They took o-(allyloxy)benzaldehyde (53a) (0.2 mmol) and 4-methyl benzene sulfonic acid (53b) (0.4 mmol) as the model substrates in the presence of the photocatalyst eosin Y (1 mol%), an oxidant TBHP (1.5 equivalent) and a mixed solvent DMSO/H2O (DMSO: H2O = 1:1, v/v). They stirred the mixture under FSL irradiation (18 W) at room temperature for 12 h and afforded the product 53c in 61% yield. The reaction could not take place without visible light or TBHP. When the wavelength of the light was 410–415 nm, the yield was the highest. The yield again increased to 84% when (NH4)2S2O8 was used as the oxidant instead of TBHP. The use of oxygen or nitrogen balloons afforded the same result as the air atmosphere. Next, the substrate scope was screened. Both electron-donating and electron-withdrawing groups at the substituted 2-(allyloxy)benzaldehydes smoothly underwent the reaction. Again, both types of substituents at the ortho and para-positions of aryl sulfinic acid could produce the desired products under optimized reaction conditions. 2-(Allyl(methyl(amino)benzaldehyde and 2-(allylthio)benzaldehyde could not deliver the desired product even with a longer reaction time.
 | ||
| Scheme 53 Visible-light-enabled metal-free radical cascade cyclisation for accessing sulfone-functionalized chroman-4-ones (Li method). | ||
Some control experiments were performed to reveal the mechanism. The radical scavenger TEMPO inhibited the reaction. Also, 1,1-diphenylethylene reduced the yield of the desired product and the coupling product was also formed. These results proved the radical pathway of the reaction. The proposed mechanism is shown in Scheme 53. Initially, the SO4˙− radical anion formation occurred from (NH4)2S2O8 in the presence of blue light (410–415 nm), which then abstracted a hydrogen radical from 4-methylbenzenesulfinic acid 53b to generate radical 53I or 53II. Next, the generation of intermediate 53III occurred when 53II attacked the C–C double bond of 53a. The radical attacked the aldehyde compound, followed by a 1,2-H shift, resulting in the benzyl radical 53V. In the final step, a hydrogen abstraction by the remaining SO4˙− led to the desired product 53c. Metal-free conditions, broad substrate scope, easily available substrate, good to excellent yields and gram-scale synthesis are important advantages of this methodology. However, when allyl phenyl ether was used as a substrate, the reaction did not respond.
Imidazothiazole, an important heterocyclic compound, displays plenty of photophysical and biological activities.73 Wang and his group74 (in Jan 2021) reported a metal catalyst-free method that required inexpensive substrates and mild reaction conditions for the synthesis of benzo[d]imidazo[5,1-b]thiazoles. As per the reported methodology, 1-iodo-2-isothiocyanatobenzene (54a) (0.2 mmol) and ethyl-2-isocyanoacetate (54b) reacted in the presence of Cs2CO3 (2 equiv.) at room temperature under irradiation by a 23 W compact fluorescence light (CFL) for 15 h in air. These conditions led to the product ethyl benzo[d]imidazo[5,1-b]thiazole-3-carboxylate 54c in good yields (Scheme 54). Among the various solvents, DMSO was excellent. The reaction ceased in the absence of irradiation. The reactant 1-iodo-2-aryl isothiocyanates, containing both electron-donating and electron-withdrawing groups, formed the desired products in 72–73% yield. Again, the products were obtained in 70–84% yields when isocyanides with different carboxylates were examined. However, it was seen that nitro- and phenyl-substituted isonitrile substrates failed to give the desired product under optimized reaction conditions. The two important advantages of this methodology were gram-scale synthesis and accessibility in natural light irradiation.
 | ||
| Scheme 54 Metal-free cascade cyclization to access benzo[d]imidazo[5,1-b]thiazoles in the presence of visible light (Wang method). | ||
To develop a mechanistic pathway, some control experiments were performed. When the reaction was carried out in dark conditions for 15 h and then hydrochloric acid was added for acidification to pH = 5, then the ethyl-1-(2-iodophenyl)-5-mercapto-1H-imidazole-4-carboxylate intermediate was formed, which could be changed to the desired product under standard conditions. This indicated that the above-mentioned intermediate was the active intermediate for this reaction. The product was obtained in low yields when the radical inhibitor TEMPO or BHT was added, suggesting a radical mechanism. Considering these controlled experiments, a plausible mechanism was provided by the authors, which is shown in Scheme 54. In the primary step, in the presence of CsCO3, two reactants, 54a and 54b underwent the [3 + 2] cycloaddition reaction and the intermediate 54I was formed. Then, an EDA complex 54II was produced by the isomerisation of 54I. After being activated by light irradiation, the EDA complex formed an aryl radical, a sulfur radical and an iodine anion by an intramolecular single electron transfer process. Finally, an intramolecular free radical coupling reaction occurred to give the desired product 54c.
In January 2022, Yang et al.75 developed a visible-light-promoted catalyst-free mild cascade iodination reaction for the synthesis of CF2-containing pyrrolidines and tetrahydrofuran with high Z/E stereoselectivity. The authors explored the 1,6-enyne substrate (55a) with ICF2COOEt 55b as the difluoroalkylation reagent in acetone solvent under blue-light irradiation. Here, 1 equiv. of N,N′-dimethylpiperazine (DMP) was required and worked as a base. The desired difluoroalkylated and vinyl C–I bonds containing pyrrolidines and tetrahydrofuran products were achieved with up to 86% yield at room temperature within 24 h (Scheme 55). It was noticed that the organic bases (e.g. – pyridine, TMEDA, DMP) were more efficient than inorganic bases (e.g. – NaHCO3, K2CO3) and among them, DMP was the most efficient. The major products have Z-selectivity. Reaction in dark conditions or in air produced lower yields. Both the electron-donating and electron-withdrawing groups on the aromatic ring were suitable for both pyrrolidine and tetrahydrofuran. The substrate, containing two or three substituents on the aromatic ring afforded the desired products in moderate to good yields under optimized reaction conditions.
 | ||
| Scheme 55 Visible-light-induced radical cascade difluoroalkylation–cyclization–iodination of 1,6-enynes with ethyl difluoroiodoacetate (Yang method). | ||
A mechanism for the reaction was proposed by the authors based on the previous literature (Scheme 55). At first, complex 55I was generated by the reaction of ICF2COOEt 55b with DMP. Then, complex 55I was irradiated by visible light and converted to the radical intermediate 55II. Next, a 5-exo-dig cyclization reaction occurred for the formation of radical intermediate 55III by the attack of 55II on the CC of 1,6-enyne 55a. Finally, ICF2COOEt was used for the iodination of intermediate 55III to form the desired product 55c along with the regeneration of 55II. Advantages such as metal-free mild conditions, high configurational selectivity and good functional group tolerance make this methodology attractive. However, the method worked only in an argon atmosphere.
8. Conclusion
In this article, we have provided a concise summary of the recent progress toward the chemoselective and sequential formation of carbon–carbon and carbon–heteroatom bonds through the effective integration of photocatalysis and cascade syntheses. The cascade reaction, being an eco-friendly approach, is an emerging topic in organic chemistry. In addition, visible-light-mediated photocatalysis is an effective synthetic tool for driving a chemical reaction. The generation of varieties of radicals via SET upon photo-irradiation and promoting multiple sequential radical addition–cyclization reactions in a synergistic fashion is the main essence of photocascade catalysis. Intriguingly, conceptualizing or designing a cascade manifold with a suitable photocatalytic cycle is undoubtedly difficult. However, it is quite reassuring that it can render multiple outcomes with multiple possibilities because a particular cascade sequence may fit into more than one category of cascades as the attacking species are subject to change in the forthcoming steps of a sequence. That is why we have focused on visible-light-induced photocatalyzed cascade reactions. A large number of natural products and drugs can be synthesised by cascade reactions using photocatalysts. Photocatalysts such as Ru(bpy)3, fac-Ir(ppy)3, rose bengal, eosin Y, etc. are very reactive with good regioselectivity. Merging these highly efficient tools resulted in significant progress towards sustainability, as well as broad applicability and scalability, albeit this field requires extensive research exploration. Despite the extremely fast evolution of this field, numerous disadvantages, including the use of costly catalysts, the formation of side-products, and a lack of proper mechanistic information, impede the demonstration of this method's maximum capacity. In this scenario, the creation of a new photocatalyst that can be recycled and its use in cascade reactions is welcome. Using an organophotocatalyst, or even nothing at all, instead of a metallaphotocatalyst in the cascade reaction could be a way to stop more pollution of the environment. Also, the use of modern technologies, such as the continuous flow process and batch reactor process could be a solution for delayed reactions. The exploration of 3d-metal photocatalysts will gain significant attention in the near future. Visible-light-induced I2-catalyzed methodology has not yet been reported. Finally, we hope that gradual interest in photocatalytic synthesis and its vast mechanistic information could enable synthetic practitioners to plan more complex and interesting photocascade catalyses.Conflicts of interest
There are no conflicts to declare.Acknowledgements
A. H. acknowledges the financial support from SERB, New Delhi (grant no. CRG/2020/000362).References
- W. Brack, D. B. Culleres, A. B. A. Boxall, H. Budzinski, S. Castiglioni, A. Covaci, V. Dulio, B. I. Escher, P. Fantke, F. Kandie, D. Fatta-Kassinos, F. J. Hernández, K. Hilscherová, J. Hollender, H. Hollert, A. Jahnke, B. Kasprzyk-Hordern, S. J. Khan, A. Kortenkamp, K. Kümmerer, B. Lalonde, M. H. Lamoree, Y. Levi, P. A. L. Martín, C. C. Montagner, C. Mougin, T. Msagati, J. Oehlmann, L. Posthuma, M. Reid, M. Reinhard, S. D. Richardson, P. Rostkowski, E. Schymanski, F. Schneider, J. Slobodnik, Y. Shibata, S. A. Snyder, F. F. Sodré, I. Teodorovic, K. V. Thomas, G. A. Umbuzeiro, P. H. Viet, K. G. Yew-Hoong, X. Zhang and E. Zuccato, Environ. Sci. Eur., 2022, 34, 21 CrossRef CAS PubMed
.
-
(a) A. DeVierno Kreuder, T. House-Knight, J. Whitford, E. Ponnusamy, P. Miller, N. Jesse, R. Rodenborn, S. Sayag, M. Gebel, I. Aped, I. Sharfstein, E. Manaster, I. Ergaz, A. Harris and L. N. Grice, ACS Sustainable Chem. Eng., 2017, 5, 2927–2935 CrossRef CAS
-
(a) C. Sambiagio and T. Noël, Trends Chem., 2020, 2, 92–106 CrossRef CAS
-
(a) C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS PubMed
-
(a) N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075–10166 CrossRef CAS PubMed
-
(a) J. M. R. Narayanam and C. R. J. Stephenson, Chem. Soc. Rev., 2011, 40, 102–113 RSC
-
(a) M. J. Genzink, J. B. Kidd, W. B. Swords and T. P. Yoon, Chem. Rev., 2022, 122, 1654–1716 CrossRef CAS PubMed
-
(a)
S. Ray, P. K. Samanta and P. Biswas, in Visible Light–Active Photocatalysis, 2018, pp. 393–419 Search PubMed
- G. E. M. Crisenza and P. Melchiorre, Nat. Commun., 2020, 11, 803 CrossRef PubMed
-
(a) C. Grondal, M. Jeanty and D. Enders, Nat. Chem., 2010, 2, 167–178 CrossRef CAS PubMed
- L. F. Tietze and U. Beifuss, Angew. Chem., Int. Ed. Engl., 1993, 32, 131–163 CrossRef
- R. Robinson, J. Chem. Soc., Trans., 1917, 111, 762–768 RSC
-
(a) M. G. Ciulla, S. Zimmermann and K. Kumar, Org. Biomol. Chem., 2019, 17, 413–431 RSC
- G.-Q. Xu and P.-F. Xu, Chem. Commun., 2021, 57, 12914–12935 RSC
-
(a) A. Ghosh, P. Pyne, S. Ghosh, D. Ghosh, S. Majumder and A. Hajra, Green Chem., 2022, 24, 3056–3080 RSC
-
(a) S. Samanta and A. Hajra, Chem. Commun., 2018, 54, 3379–3382 RSC
- M. Berthet, T. Cheviet, G. Dujardin, I. Parrot and J. Martinez, Chem. Rev., 2016, 116, 15235–15283 CrossRef CAS PubMed
- J. Xie, Q. Xue, H. Jin, H. Li, Y. Cheng and C. Zhu, Chem. Sci., 2013, 4, 1281–1286 RSC
- H. Xiao, Z. Zhang, Y. Fang, L. Zhu and C. Li, Chem. Soc. Rev., 2021, 50, 6308–6319 RSC
- L. Zheng, C. Yang, Z. Xu, F. Gao and W. Xia, J. Org. Chem., 2015, 80, 5730–5736 CrossRef CAS PubMed
- T.-f. Niu, L. Li, B.-q. Ni, M.-j. Bu, C. Cai and H.-l. Jiang, Eur. J. Org. Chem., 2015, 5775–5780 CrossRef CAS
- H. Gao, B. Hu, W. Dong, X. Gao, L. Jiang, X. Xie and Z. Zhang, ACS Omega, 2017, 2, 3168–3174 CrossRef CAS PubMed
- K. Xu, Z. Tan, H. Zhang, J. Liu, S. Zhang and Z. Wang, Chem. Commun., 2017, 53, 10719–10722 RSC
- J. A. Burkhard, B. Wagner, H. Fischer, F. Schuler, K. Müller and E. M. Carreira, Angew. Chem., Int. Ed., 2010, 49, 3524–3527 CrossRef CAS PubMed
- L. Yuan, S.-M. Jiang, Z.-Z. Li, Y. Zhu, J. Yu, L. Li, M.-Z. Li, S. Tang and R.-R. Sheng, Org. Biomol. Chem., 2018, 16, 2406–2410 RSC
- C. Lorton and A. Voituriez, Eur. J. Org. Chem., 2019, 5133–5150 CrossRef CAS
- Y. Liu, Q.-L. Wang, Z. Chen, P. Chen, K.-W. Tang, Q. Zhou and J. Xie, Org. Biomol. Chem., 2019, 17, 10020–10029 RSC
-
(a) T. Koike and M. Akita, Inorg. Chem. Front., 2014, 1, 562–576 RSC
- Y. M. Khetmalis, M. Shivani, S. Murugesan and K. V. G. Chandra Sekhar, Biomed. Pharmacother., 2021, 141, 111842 CrossRef CAS PubMed
- J. Xie, P. Xu, H. Li, Q. Xue, H. Jin, Y. Cheng and C. Zhu, Chem. Commun., 2013, 49, 5672–5674 RSC
- S. Demkowicz, J. Rachon, M. Daśko and W. Kozak, RSC Adv., 2016, 6, 7101–7112 RSC
- C.-X. Li, D.-S. Tu, R. Yao, H. Yan and C.-S. Lu, Org. Lett., 2016, 18, 4928–4931 CrossRef CAS PubMed
- S. Tang, L. Yuan, Y.-L. Deng, Z.-Z. Li, L.-N. Wang, G.-X. Huang and R.-L. Sheng, Tetrahedron Lett., 2017, 58, 329–332 CrossRef CAS
- R. S. Keri, K. Chand, S. Budagumpi, S. B. Somappa, S. A. Patil and B. M. Nagaraja, Eur. J. Med. Chem., 2017, 138, 1002–1033 CrossRef CAS PubMed
- J. Yan, J. Xu, Y. Zhou, J. Chen and Q. Song, Org. Chem. Front., 2018, 5, 1483–1487 RSC
- Q. Wang, Y. Qu, Q. Xia, H. Song, H. Song, Y. Liu and Q. Wang, Adv. Synth. Catal., 2018, 360, 2879–2884 CrossRef CAS
-
(a) T. P. Singh and O. M. Singh, Mini-Rev. Med. Chem., 2018, 18, 9–25 CrossRef CAS PubMed
- Y.-L. Wei, J.-Q. Chen, B. Sun and P.-F. Xu, Chem. Commun., 2019, 55, 5922–5925 RSC
- Y. Yuan, W.-H. Dong, X.-S. Gao, X.-M. Xie and Z.-G. Zhang, Chem. Commun., 2019, 55, 11900–11903 RSC
- X. Gao, C. Li, Y. Yuan, X. Xie and Z. Zhang, Org. Biomol. Chem., 2020, 18, 263–271 RSC
- H. Jiang, G. Seidler and A. Studer, Angew. Chem., Int. Ed., 2019, 58, 16528–16532 CrossRef CAS PubMed
- Y. Masuda, H. Tsuda and M. Murakami, Angew. Chem., Int. Ed., 2021, 60, 3551–3555 CrossRef CAS PubMed
- H.-L. Huang, J.-Y. Du, Q.-L. Li, F. Gao and C.-L. Ma, J. Org. Chem., 2020, 85, 3963–3972 CrossRef CAS PubMed
- S. Ghosh, S. Samanta, A. K. Ghosh, S. Neogi and A. Hajra, Adv. Synth. Catal., 2020, 362, 4552–4578 CrossRef CAS
-
(a) Z. Chen, M. Pitchakuntla and Y. Jia, Nat. Prod. Rep., 2019, 36, 666–690 RSC
- S. Xie, Y. Li, P. Liu and P. Sun, Org. Lett., 2020, 22, 8774–8779 CrossRef CAS PubMed
- R. S. Keri, S. Budagumpi, R. K. Pai and R. G. Balakrishna, Eur. J. Med. Chem., 2014, 78, 340–374 CrossRef CAS PubMed
- Z.-W. Wang, Y. Zheng, Y.-E. Qian, J.-P. Guan, W.-D. Lu, C.-P. Yuan, J.-A. Xiao, K. Chen, H.-Y. Xiang and H. Yang, J. Org. Chem., 2022, 87, 1477–1484 CrossRef CAS PubMed
- W. Fu, A. Yu, H. Jiang, M. Zuo, H. Wu, Z. Yang, Q. An, Z. Sun and W. Chu, Org. Biomol. Chem., 2019, 17, 3324–3327 RSC
- T. Singha, A. R. S. Mondal, S. Midya and D. P. Hari, Chem. – Eur. J., 2022, 28, e202202025 CAS
- L. Chen, L.-N. Guo, S. Liu, L. Liu and X.-H. Duan, Chem. Sci., 2021, 12, 1791–1795 RSC
-
(a) M. Rahman, S. Ghosh, D. Bhattacherjee, G. V. Zyryanov, A. K. Bagdi and A. Hajra, Asian J. Org. Chem., 2022, 11, e202200179 CrossRef CAS
- C. Liu and B. Zhang, RSC Adv., 2015, 5, 61199–61203 RSC
- P. Wang, X. Wang, X. Niu, L. Zhu and X. Yao, Chem. Commun., 2020, 56, 4840–4843 RSC
- Y.-F. Si, K. Sun, X.-L. Chen, X.-Y. Fu, Y. Liu, F.-L. Zeng, T. Shi, L.-B. Qu and B. Yu, Org. Lett., 2020, 22, 6960–6965 CrossRef CAS PubMed
- S. Jana, A. Verma, R. Kadu and S. Kumar, Chem. Sci., 2017, 8, 6633–6644 RSC
- L. Bu, J. Li, Y. Yin, B. Qiao, G. Chai, X. Zhao and Z. Jiang, Chem. – Asian J., 2018, 13, 2382–2387 CrossRef CAS PubMed
- Y. Kim, K. Lee, G. R. Mathi, I. Kim and S. Hong, Green Chem., 2019, 21, 2082–2087 RSC
- H. Wang, J. Zhang, J. Shi, F. Li, S. Zhang and K. Xu, Org. Lett., 2019, 21, 5116–5120 CrossRef CAS PubMed
- G.-H. Li, Q.-Q. Han, Y.-Y. Sun, D.-M. Chen, Z.-L. Wang, X.-M. Xu and X.-Y. Yu, Chin. Chem. Lett., 2020, 31, 3255–3258 CrossRef CAS
- Y. Su, R. Zhang, W. Xue, X. Liu, Y. Zhao, K.-H. Wang, D. Huang, C. Huo and Y. Hu, Org. Biomol. Chem., 2020, 18, 1940–1948 RSC
- L.-L. Zhang, M.-M. Xu, W.-B. Cao, X.-P. Xu and S.-J. Ji, Adv. Synth. Catal., 2020, 362, 3131–3136 CrossRef CAS
- Q. Liu, Y. Mei, L. Wang, Y. Ma and P. Li, Adv. Synth. Catal., 2020, 362, 5669–5680 CrossRef CAS
-
(a) S. Ghosh, S. Mondal and A. Hajra, Adv. Synth. Catal., 2020, 362, 3768–3794 CrossRef CAS
- J. Zhou, W. Li, H. Zheng, Y. Pei, X. Liu and H. Cao, Org. Lett., 2021, 23, 2754–2759 CrossRef CAS PubMed
- G. Pan, S. Qin, D. Xu, F. E. Kühn and H. Guo, Org. Lett., 2021, 23, 2959–2963 CrossRef CAS PubMed
- N. Zhou, K. Kuang, M. Wu, S. Wu, Z. Xia, Q. Xu and M. Zhang, Org. Chem. Front., 2021, 8, 4095–4100 RSC
- G. C. Upreti, T. Singh, S. Ranjan, R. K. Gupta and A. Singh, ACS Omega, 2022, 7, 29728–29733 CrossRef CAS PubMed
- D. Zheng and A. Studer, Org. Lett., 2019, 21, 325–329 CrossRef CAS PubMed
- Y. Kumagai, N. Murakami, F. Kamiyama, R. Tanaka, T. Yoshino, M. Kojima and S. Matsunaga, Org. Lett., 2019, 21, 3600–3605 CrossRef CAS PubMed
- X.-L. Ma, Q. Wang, X.-Y. Feng, Z.-Y. Mo, Y.-M. Pan, Y.-Y. Chen, M. Xin and Y.-L. Xu, Green Chem., 2019, 21, 3547–3551 RSC
- Y. Mei, L. Zhao, Q. Liu, S. Ruan, L. Wang and P. Li, Green Chem., 2020, 22, 2270–2278 RSC
- M. L. Fascio, M. I. Errea and N. B. D'Accorso, Eur. J. Med. Chem., 2015, 90, 666–683 CrossRef CAS PubMed
- K. Yan, M. Liu, J. Wen, X. Liu, X. Wang, X. Chen, J. Li, S. Wang, X. Wang and H. Wang, Green Chem., 2021, 23, 1286–1291 RSC
- Y. Wang, R. Liu, P. Zhou, J. Wu, W. Li, C. Wang, H. Li, D. Li and J. Yang, Eur. J. Org. Chem., 2022, e202101395 CAS
Footnotes |
| † This review article is dedicated to Professor Shigeru Yamago on the occasion of his 60th birthday. |
| ‡ These authors have equal contribution and equal authorship. |
| This journal is © The Royal Society of Chemistry 2023 |