
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Hydrogen bond templated synthesis of catenanes and rotaxanes from a single isophthalic acid derivative†
Sean R.
Barlow
 ,
Geoffrey R.
Akien
and
Nicholas H.
Evans
*
,
Geoffrey R.
Akien
and
Nicholas H.
Evans
*
Department of Chemistry, Lancaster University, Lancaster, LA1 4YB, UK. E-mail: n.h.evans@lancaster.ac.uk
First published on 7th December 2022
Abstract
Hydrogen bond templated [2]catenanes and [2]rotaxanes have been synthesized using azide precursors derived from a single isophthalic acid derivative precursor. The interlocked molecules were prepared using either stoichiometric or near stoichiometric amounts of macrocycle and CuAAC “click” precursors, with yields of up to 70% for the mechanical bond formation step. Successful preparation of the interlocked structures was confirmed by NMR spectroscopy and mass spectrometry, with detail of co-conformational behaviour being elucidated by a range of 1H NMR spectroscopic experiments.
Introduction
Mechanically interlocked molecules such as catenanes1 (consisting of interlocked macrocyclic rings) and rotaxanes2 (macrocyclic rings(s) trapped on stoppered axles) have unusual properties arising from the mechanical bond such as the potential for the controlled, large amplitude motion of the interlocked components and the 3D cavities and spaces arising from their exotic architectures.3An array of template methodologies have been used to prepare interlocked molecules, including but not limited to, metal cations,4 π–π stacking (or π-donor/π-acceptor interactions),5 hydrogen bonding6 and anions.7 However, the bespoke nature of many syntheses of catenanes and rotaxanes is often a drawback when trying to exploit their properties and potential applications. Development of modular methodologies based on easy to access building blocks is desirable.
While our group has reported upon the rapid hydrogen bond templated synthesis of [2]rotaxanes in good yield,8,9 our previous serendipitous [2]catenane synthesis is low yielding (12%) with regard to the crucial covalent capture step that forms the interlocked compound.10,11 We hypothesized that with design modification to allow for more efficient hydrogen bonding templation, that synthesis of [2]catenanes in higher yields should be possible.
Indeed, here in we report the successful synthesis of three novel [2]catenanes derived from a simple isophthalic acid precursor in good yield. Furthermore, starting from the same isophthalic acid derivative allowed for the synthesis of two novel [2]rotaxanes in reasonable yield. All interlocked molecules were characterized by NMR and IR spectroscopy and mass spectrometry.
Results and discussion
Synthesis and characterization of catenanes
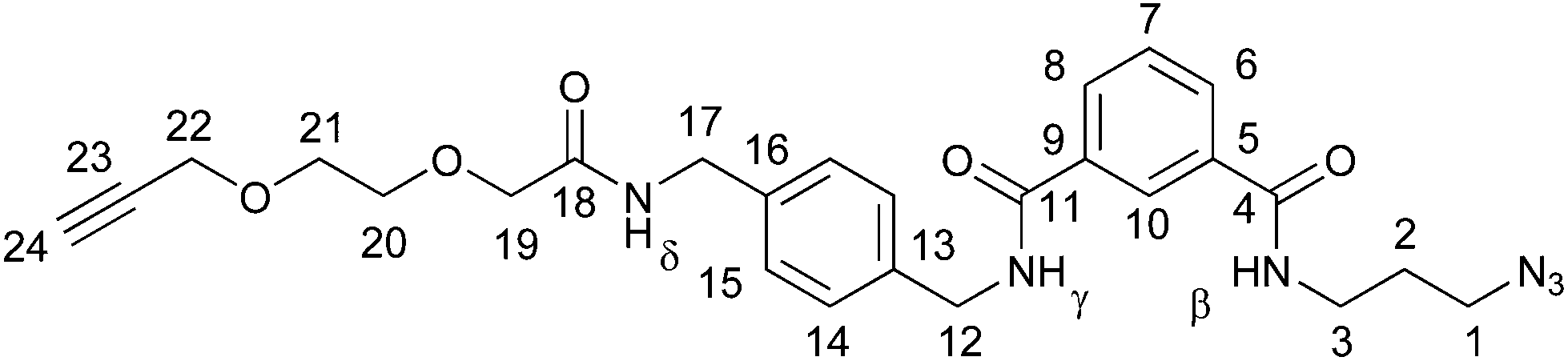
The catenane syntheses all build upon azido-functionalized isophthalic acid derivative 1 (Scheme 1), which may be rapidly prepared from commercially available mono-methyl ester of isophthalic acid.12,13 | ||
| Scheme 1 Synthesis of [2]catenanes 15–17. | ||
Three azide–alkyne coupling partners were prepared. Initially DCC/N-hydroxysuccinimide-mediated amide coupling of alkyne carboxylic acids 2–4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 14–16 with commercially available 4-(Boc-amino)benzylamine afforded alkynes 5–7. These were subsequently deprotected using TFA, with removal of the Boc group in quantitative yield confirmed by 1H NMR spectroscopy.
14–16 with commercially available 4-(Boc-amino)benzylamine afforded alkynes 5–7. These were subsequently deprotected using TFA, with removal of the Boc group in quantitative yield confirmed by 1H NMR spectroscopy.
The alkyne–azide precursor targets 11–13 were synthesized via the coupling of the amine trifluoroacetate salts 8–10 with isophthalic acid derivative 1 using DCC and N-hydroxysuccinimide. Following workup and purification by silica gel column chromatography, 11–13 were isolated in reasonable yields (51–64%). The successful preparation of novel compounds 11–13 was confirmed by NMR spectroscopy and mass spectrometry (see ESI, pp. S9 and S10; S14 and S15; S19 and S20†).
Mechanical bond formation was accomplished via the threading and cyclization of alkyne-azides 11–13 through previously reported macrocycle 14.10 In each case, to a solution of 14 in dry CH2Cl2, 1.0 equivalents of 11–13 was added and allowed to stir for 15 minutes to facilitate pseudorotaxane formation (Scheme 1). Then, catalytic Cu(CH3CN)4BF4 and TBTA (tris((1-benzyl-4-triazolyl)methyl)amine), and 1.2 equivalents of N,N-diisopropylamine were added. The reactions were stirred overnight at room temperature under an inert atmosphere, and then submitted to aqueous workup and purification by silica gel column chromatography. Novel heterocircuit [2]catenanes 15–1717,18 were isolated in good yields (59–70%). The [2]catenanes were characterized by 1H & 13C NMR and IR spectroscopy, with molecular ions being detected by high resolution mass spectrometry (see ESI, pp. S21–S26†).
Catenane formation is evident upon comparison of the 1H NMR spectra of precursor 13, macrocycle 14 and [2]catenane 17 (Fig. 1). The upfield shift and splitting of aromatic protons 14/15 and h/i in catenane 17 compared to 13 and 14 is consistent with the intercalation of aromatic rings within the interlocked structure. The downfield shift of internal isophthalamide proton d is indicative of interactions with a hydrogen bond acceptor on the cyclized thread. Another notable characteristic of the 1H NMR spectra of catenanes 15–17 is the splitting of certain protons (e.g. f and k) due to the two faces of the rotationally symmetric macrocycle becoming inequivalent due to the directionality of the newly formed rotationally asymmetric macrocyclic ring.
 | ||
| Fig. 1
1H NMR spectra of (a) alkyne–azide 13, (b) [2]catenane 17 and (c) macrocycle 14 (1:1 CDCl3/CD3OD, 400 MHz, 298 K). | ||
Further evidence of the interlocked nature of the macrocyclic rings is provided by a molecular ion peak being identifiable by positive ion electrospray mass spectrometry (see ESI, pp. S22, S24 & S26†). In addition, for each catenane there is the appearance of multiple through-space correlations in the 1H–1H ROESY NMR spectra between resonances arising from protons in the two interlocked macrocycles (e.g.Fig. 2 and see ESI, pp. S39–S41†). Inspection of the entire spectrum in each case reveals sufficient intercomponent correlations to indicate the macrocyclic rings of the catenane are switching between multiple co-conformations in 1:1 CDCl3/CD3OD (see ESI, p. S42†).
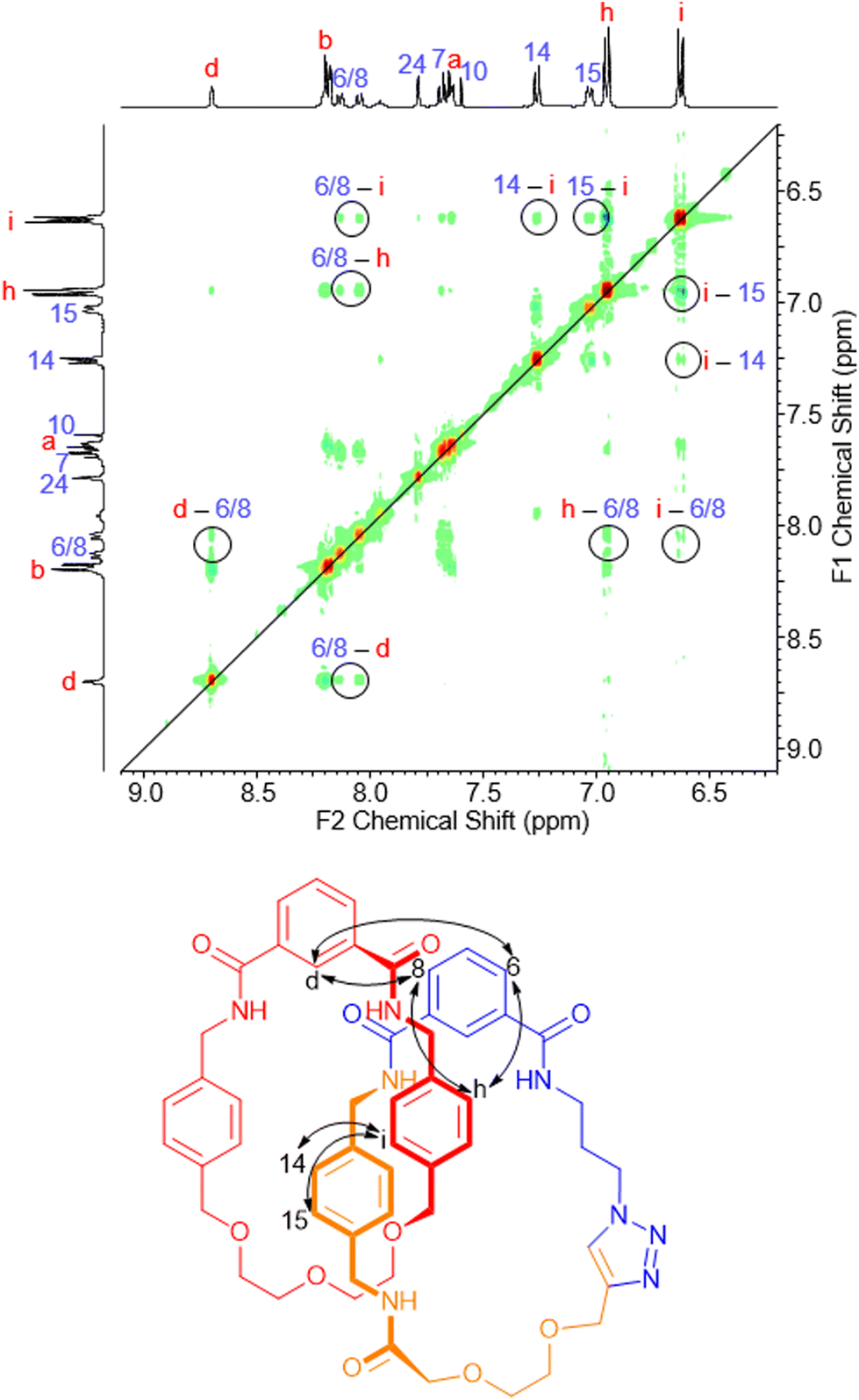 | ||
| Fig. 2 Section of 1H–1H ROESY NMR spectrum of [2]catenane 17 with intercomponent through-space correlations highlighted (1:1 CDCl3/CD3OD, 400 MHz, 298 K). | ||
To probe potential dynamic interactions (e.g. the pirouetting of the interlocked macrocyclic rings) the most soluble [2]catenane, glycol catenane 17, was studied by variable temperature (VT) 1H NMR spectroscopy in C2D2Cl4 (Fig. 3). At (and below) room temperature, several C–H resonances are broad, but these sharpen upon heating. Three of the amide N–H resonances not only sharpen but move upfield, while one (δ) has almost no change in chemical shift, indicating that this amide N–H is participating in intramolecular hydrogen bonding.
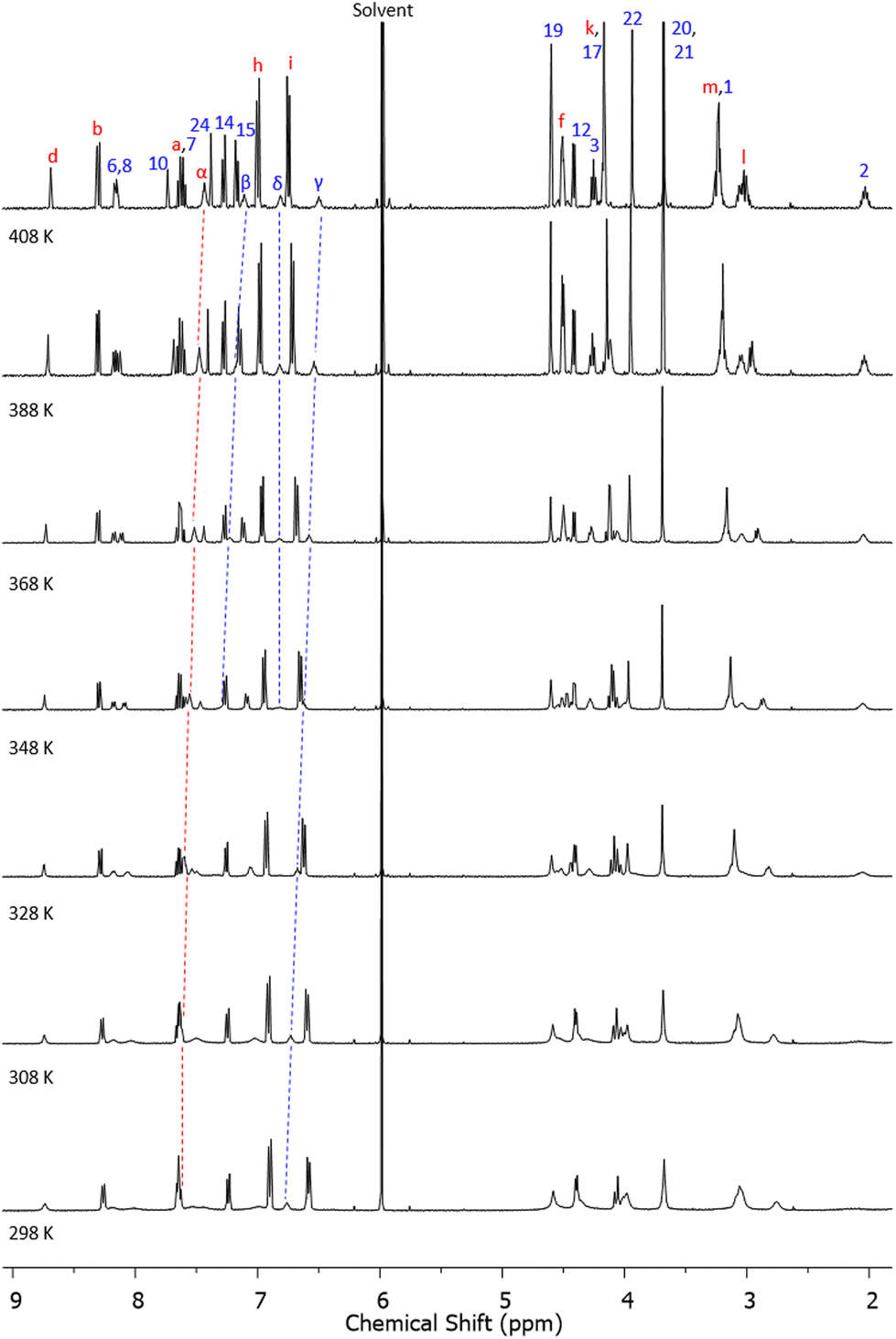 | ||
| Fig. 3 1H NMR spectra of [2]catenane 17 recorded at T = 298 K to 408 K in C2D2Cl4 (400 MHz). See Scheme 1 or Fig. 1 for atom labels. | ||
Synthesis and characterization of rotaxanes
The azido-functionalized isophthalic acid derivative 1 can also be derivatized with bulky stopper groups to allow for the formation of rotaxanes (Scheme 2). Novel half-axle components 18 and 19 were prepared in good yield by reaction of 3,5-bis(trifluoromethyl)benzylamine and 3,5-bis(trifluoromethyl)-aniline with isophthalic acid derivative 1 activated using DCC and N-hydroxysuccinimide. | ||
| Scheme 2 Synthesis of [2]rotaxanes 21 and 23. | ||
To prepare [2]rotaxanes 21 and 23, 1.1 equivalents of 18 or 19 and alkyne 208 were added to a solution of macrocycle 14 in dry CH2Cl2, followed by catalytic Cu(CH3CN)4BF4 and TBTA, and 1.2 equivalents of N,N-diisopropylamine. Following aqueous workup and purification of the crude material by preparative TLC, [2]rotaxanes 21 and 23 were isolated in 29% and 40% yields respectively.19 While the isolated yields are reasonable, it should be noted the actual yields of rotaxane formation are almost certainly higher. Isolation of pure rotaxane was hindered in both cases by incomplete separation of product bands during chromatographic purification.
Successful formation of the [2]rotaxanes was confirmed by analysis of NMR spectra and detection of molecular ion peaks in high resolution mass spectrometry (see ESI, pp. S31–S34†). The 1H NMR spectrum of rotaxane 21, along with that of non-interlocked macrocycle 14 and axle 22 for comparison, is shown in Fig. 4. Once again, the upfield shift and splitting of aromatic protons h and i in the interlocked molecule compared to non-interlocked macrocycle 14 is consistent with presence of the second component (here an axle) between the aromatic rings of the macrocycle. The downfield shift of proton d and the amide protons α of the macrocycle in the rotaxane are indicative of interactions with a hydrogen bond acceptor on the axle. In the 1H–1H ROESY NMR spectra of both rotaxanes there are through-space correlations between resonances arising from protons in the two interlocked components (see ESI, pp. S43 & S44†). For rotaxane 21, the multiple observed intercomponent correlations strongly support the primary location of the macrocyclic ring component as being in the vicinity of the axle isophthalamide in CDCl3 (see ESI, p. S45†). Meanwhile for rotaxane 23 there is evidence the macrocyclic ring preferentially resides over the isophthalamide amide N–Hγ due to the observable correlations being between stopper proton 22 and ring protons i and l (see ESI, p. S45†).
 | ||
| Fig. 4 1H NMR spectra of (a) axle 22, (b) [2]rotaxane 21 and (c) macrocycle 14 (CDCl3, 400 MHz, 298 K). | ||
Conclusions
Using precursors prepared from isophthalic acid derivative 1, we have demonstrated the hydrogen bond templated synthesis of both [2]catenanes and [2]rotaxanes. Despite using only stoichiometric (or near stoichiometric) quantities of precursors, yields of up to 70% for the crucial covalent capture step are possible. This work demonstrates synthetic methodologies that have the potential to incorporate a range of functionality into either the ring of a catenane or the stoppers of a rotaxane in a modular approach in good yields. Investigations into deploying these methodologies, including preparing mechanically chiral analogues,20 are ongoing in our laboratories.Experimental
General information
All reagents and solvents were used as obtained from commercial suppliers, unless otherwise stated. Dry solvents, Et3N and DIPEA were purchased dry and stored under an inert atmosphere. Cu(CH3CN)4BF4 was stored in a desiccator over P4O10. Petrol refers to the fractions of petroleum that boil between 40 °C and 60 °C. Deionized water was used in all cases. All aqueous solutions are saturated unless otherwise stated.Azido-functionalized isophthalic acid derivative 1,12 alkyne benzoic acids 214 and 3,15 alkyne ethylene glycol acid 4,16 macrocycle 14,9,10 alkyne 208 were all synthesized based on previously reported procedures.
Silica gel with a 60 Å particle size was used as the stationary phase for column chromatography. Analytical TLC was used to monitor the progress of column chromatography, with TLC plates examined under short wavelength (254 nm) UV light, or staining with potassium permanganate and phosphomolybdic acid solutions as appropriate. Preparatory TLC was carried out on silica gel possessing a fluorescent indicator to allow for examination with short wavelength UV light.
IR spectra were recorded on an Agilent Technologies Cary 630 FTIR spectrometer. NMR spectra were recorded on a Bruker AVANCE III 400 or a Bruker Fourier 300 spectrometer at 298 K (unless otherwise stated). Mass spectra were recorded on a Shimadzu LCMS IT ToF instrument. Melting points were recorded on a Gallenkamp capillary melting point apparatus and are uncorrected.
Experimental procedures
:petrol 2:1) gave the title product (262 mg, 59%) as a colourless solid.
 :petrol 2:1).
:petrol 2:1).
m.p. 84–85 °C.
ν
max
/cm
−1
(neat): 3315 (N–H), 3274 (N–H), 2928 (C–H), 1677 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 1632 (CO).
O), 1632 (CO).
δ H (400 MHz, CDCl 3 ): 7.77 (2H, d, J = 9.0 Hz, H5), 7.34–7.20 (4H, m, H11 & H12), 7.01 (2H, d, J = 9.0 Hz, H6), 6.28 (1H, bs, NHδ), 4.83 (1H, bs, NHγ), 4.75 (2H, d, J = 2.4 Hz, H3), 4.63 (2H, d, J = 5.7 Hz, H9), 4.31 (2H, d, J = 6.1 Hz, H14), 2.54 (1H, t, J = 2.4 Hz, H1), 1.47 (9H, s, H17).
δ C (100 MHz, CDCl 3 ): 166.6 (C8) 166.5 (C15), 160.0 (C4), 138.4 (C7), 137.4 (C10), 128.7 (C6), 128.2 (C12), 127.8 (C11), 127.4 (C13), 114.6 (C5), 79.5 (C2), 79.1 (C16), 76.0 (C1), 55.6 (C3), 44.3 (C14), 43.7 (C9), 28.3 (C17).
m/z (ESI): 417.1785 ([M + Na]+ C23H26N2NaO4 requires 417.1769).
 :petrol 3:1–4:1) gave the title product (77 mg, 59%) as a colourless solid.
:petrol 3:1–4:1) gave the title product (77 mg, 59%) as a colourless solid.
 :petrol 4:1).
:petrol 4:1).
m.p. 120–122 °C.
ν
max
/cm
−1
(neat): 3281 (C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C–H), 2929 (C–H), 2102 (NNN), 1621 (CO), 1541 (C–N).
C–H), 2929 (C–H), 2102 (NNN), 1621 (CO), 1541 (C–N).
δ
H
(400 MHz, 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :1 CDCl
3
/CD
3
OD): 8.17 (1H, bs, H10), 7.94 (1H, d, J = 9.0 Hz, H6/8) 7.90 (1H, d, J = 9.0 Hz, H6/8), 7.74 (2H, d, J = 8.9 Hz, H21), 7.44 (1H, app. t, H7), 7.20–7.10 (4H, m, H14 & H15), 6.94 (2H, d, J = 8.9 Hz, H20), 4.69 (2H, d, J = 2.4 Hz, H23), 4.48 (2H, d, J = 6.0 Hz, H17), 4.43 (2H, d, J = 5.7 Hz, H12), 3.39–3.35 (2H, m, H3), 3.30 (2H, t, J = 9.0 Hz, H1), 2.55 (1H, t, J = 2.4 Hz, H25), 1.78 (2H, quintet, J = 6.5 Hz, H2).
:1 CDCl
3
/CD
3
OD): 8.17 (1H, bs, H10), 7.94 (1H, d, J = 9.0 Hz, H6/8) 7.90 (1H, d, J = 9.0 Hz, H6/8), 7.74 (2H, d, J = 8.9 Hz, H21), 7.44 (1H, app. t, H7), 7.20–7.10 (4H, m, H14 & H15), 6.94 (2H, d, J = 8.9 Hz, H20), 4.69 (2H, d, J = 2.4 Hz, H23), 4.48 (2H, d, J = 6.0 Hz, H17), 4.43 (2H, d, J = 5.7 Hz, H12), 3.39–3.35 (2H, m, H3), 3.30 (2H, t, J = 9.0 Hz, H1), 2.55 (1H, t, J = 2.4 Hz, H25), 1.78 (2H, quintet, J = 6.5 Hz, H2).
δ
c
(100 MHz, 9:1 CDCl
3
/CD
3
OD): 167.6 (C11), 167.4 (C18), 167.2 (C4), 160.1 (C22), 137.6 (C13), 137.1 (C5/9/19) 137.0 (C16), 134.3 (C5/9/19), 134.2 (C5/9/19), 130.4 (C6/8), 130.4 (C6/8) 129.1 (C21), 129.0 (C7), 127.9 (C15), 127.0 (C14), 125.2 (C10), 114.6 (C20), 77.8 (C24), 76.0 (C25), 55.7 (C23), 49.1 (C1), 43.7 (C17), 43.4 (C12), 37.4 (C3), 28.5 (C2).
m/z (ESI): 525.2245 ([M + H]+ C29H29N6O4 requires 525.2231).
:Hexane 2:3–1:1) gave the title product (306 mg, 61%) as a colourless solid.
 :petrol 1:2).
:petrol 1:2).
m.p. 118–120 °C.
ν
max
/cm
−1
(neat): 3310 (CC–H), 3270 (N–H), 2930 (C–H), 1672 (CO), 1635 (CO).
δ H (400 MHz, CDCl 3 ): 7.48–7.45 (1H, m, H8), 7.38–7.29 (6H, m, H14, H13, H5 & H7), 7.16–7.11 (1H, m, H6), 6.38 (1H, bs, NHδ), 4.88 (1H, bs, NHγ), 4.75 (2H, d, J = 2.4 Hz, H3), 4.64 (2H, d, J = 5.8 Hz, H11), 4.32 (2H, d, J = 5.8 Hz, H16), 2.55 (1H, t, J = 2.4 Hz, H1), 1.48 (9H, s, H19).
δ C (100 MHz, CDCl 3 ): 166.9 (C10) 166.8 (C17), 157.8 (C4), 138.5 (C12), 137.1 (C15), 135.9 (C13), 129.7 (C6), 128.2 (C14), 127.8 (C19), 119.6 (C5), 118.5 (C7), 113.5 (C8), 79.1 (C18), 78.0 (C2), 75.9 (C1), 55.9 (C3), 44.3 (C11), 43.8 (C16), 28.4 (C19).
m/z (ESI): 417.1785 ([M + Na]+ C23H26N2NaO4 requires 417.1771).
 :petrol 5:1) gave the title product (253 mg, 64%) as a colourless solid.
:petrol 5:1) gave the title product (253 mg, 64%) as a colourless solid.
 :petrol 3:1).
:petrol 3:1).
m.p. 122–124 °C.
ν
max
/cm
−1
(neat): 3291 (N–H), 2926 (C–H), 2098 (NNN), 1634 (CO), 1533 (C–N).
δ H (400 MHz, CDCl 3 ): 8.36 (1H, s, H10), 8.08 (1H, d, J = 7.7 Hz, H6/8), 7.98 (1H, d, J = 7.7 Hz, H6/8), 7.53–7.48 (1H, m, H7), 7.49–7.47 (1H, m, H20), 7.46–7.43 (1H, m, H24/22), 7.35 (1H, t, J = 7.7 Hz, H23), 7.21–7.13 (4H, m, H22/24 & NHβ, γ, δ), 7.10 (2H, d, J = 8.9 Hz, H15), 7.01 (2H, d, J = 8.9 Hz, H14), 4.71 (2H, d, J = 2.4 Hz, H25), 4.51 (2H, d, J = 5.8 Hz, H17), 4.36 (2H, d, J = 5.8 Hz, H12), 3.32 (2H, q, J = 6.6 Hz, H3), 3.25 (2H, t, J = 6.6 Hz, H1), 2.53 (1H, t, J = 2.4 Hz, H27), 1.71 (2H, quintet, J = 6.6 Hz, H2).
δ C (100 MHz, CDCl 3 ): 167.6 (C18), 166.8 (C11), 166.3 (C4), 157.7 (C21), 137.4 (C16), 137.0 (C13), 135.2 (C5/9/19), 134.1 (C5/9/19), 134.0 (C5/9/19), 130.7 (C6/8), 130.6 (C6/8), 129.7 (C23), 129.0 (C7), 128.3 (C15), 127.3 (C14), 124.9 (C10), 119.8 (C24), 118.6 (C22), 113.7 (C20), 78.0 (C26), 75.9 (C27), 55.9 (C25), 49.1 (C1), 43.9 (C17), 43.8 (C12), 37.6 (C3), 28.5 (C2).
m/z (ESI): 525.2245 ([M + H]+ C29H29N6O4 requires 525.2261).
:Hexane 1:1–2:1) gave the title product (1.67, 75%) as a clear oil.
 :hexane 1:1).
:hexane 1:1).
ν
max
/cm
−1
(neat): 3330 (CC–H), 2976 (C–H), 1664 (CO), 1515 (C–N), 1095 (C–O–C).
δ H (400 MHz, CDCl 3 ): 7.30–7.23 (5H, m, H10, H11 & NHδ), 4.88 (1H, bs, NHγ), 4.47 (2H, d, J = 5.9 Hz, H8), 4.31 (2H, d, J = 5.7 Hz, H13), 4.06–4.03 (4H, m, H6 & H3), 3.72–3.65 (4H, H4 & H5), 2.41 (1H, t, J = 2.4 Hz, H1), 1.46 (9H, s, H16).
δ C (100 MHz, CDCl 3 ): 169.6 (C7), 155.8 (C14), 138.2 (C9), 137.1 (C12), 128.2 (C10), 127.7 (C11), 79.5 (C2), 79.1 (C15), 74.9 (C1), 70.8 (C5), 70.4 (C4), 68.6 (C6), 58.2 (C3), 44.3 (C13), 42.6 (C8), 28.3 (C16).
m/z (ESI): 399.1890 ([M + Na]+ C20H28N2NaO5 requires 399.1890).
 :Hexane 6:1–8:1) gave the title product (259 mg, 51%) as a colourless solid.
:Hexane 6:1–8:1) gave the title product (259 mg, 51%) as a colourless solid.
 :Hexane 4:1).
:Hexane 4:1).
m.p. 78–80 °C.
ν
max
/cm−1(neat): 3291 (CC–H), 3060 (N–H), 2916 (C–H), 2102 (NNN), 1649 (CO), 1530 (C–N), 1097 (C–O–C).
δ H (400 MHz, CDCl 3 ): 8.22 (1H, s, H10), 8.00–7.92 (2H, m, H6 & H8), 7.53 (1H, app. t, H7), 7.34 (1H, bs, NHδ), 7.31–7.22 (4H, m, H14 & H15), 6.75–6.66 (2H, m, NHβ,γ), 4.61 (2H, d, J = 5.6 Hz, H12), 4.44 (2H, d, J = 5.9 Hz, H17), 4.08 (2H, d, J = 2.4 Hz, H22), 4.03 (2H, s, H19), 3.73–3.66 (4H, m, H21 & H20), 3.52 (2H, q, J = 6.6 Hz, H3), 3.42 (2H, t, J = 6.5 Hz, H1), 2.42 (1H, t, J = 2.4 Hz, H24), 1.88 (2H, quintet, J = 6.6 Hz, H2).
δ C (100 MHz, CDCl 3 ): 169.9 (C18), 166.6 (C4), 166.3 (C11), 137.6 (C16), 137.0 (C13), 134.6 (C5/9), 134.5 (C5/9), 130.1 (C6/8), 130.1 (C6/8), 129.0 (C7), 128.2 (C14), 128.1 (C15), 125.2 (C10), 79.1 (C23), 75.0 (C24), 70.5 (C19), 70.0 (C20), 68.7 (C21), 58.3 (C22), 49.4 (C1), 43.9 (C12), 42.5 (C17), 37.8 (C3), 28.7 (C2).
m/z (ESI): 529.2170 ([M + Na]+ C26H30N6O5 requires 529.2175).
:2) gave the title product (76 mg, 59%) as a colourless solid.
 :2).
:2).
m.p. 165–167 °C.
ν
max
/cm
−1
(neat): 3345 (N–H), 2868 (C–H), 1630 (CO), 1578 (C–N), 1533 (C–N), 1416 (Ar-C).
δ
H
(400 MHz, 50:50 CDCl
3
/CD
3
OD): 8.70 (1H, s, Hd), 8.21–8.16 (2H, m, Hb), 8.11 (1H, d, J = 7.7 Hz, H6/8), 8.02 (1H, d, J = 7.8 Hz, H6/8), 7.82–7.72 (3H, m, H20 & H25), 7.68–7.59 (2H, m, H7 & Ha), 7.52 (1H, s, H10), 7.40 (2H, d, J = 7.9 Hz, H14), 7.07–6.97 (4H, m, H15 &H21), 6.94 (4H, d, J = 7.9 Hz, Hh), 6.56 (4H, d, J = 7.9 Hz, Hi), 5.42 (2H, s, H23), 4.58 (2H, dd, J = 14.2, 5.1 Hz, Hf), 4.47–4.36 (4H, m, H12 & Hf′), 4.25 (2H, bs, H1), 3.97–3.79 (6H, m, Hk, Hk′ & H17), 2.98 (2H, bs, H3), 2.93–2.75 (4H, m, Hl), 2.64–2.48 (4H, m, Hm), 1.84 (2H, bs, H2).
δ
C
(100 MHz, 50:50 CDCl
3
/CD
3
OD): 167.4 (Ce), 167.0 (C4), 166.4 (C18), 166.3 (C11), 159.8 (C22), 143.4 (C24), 139.3 (C13), 139.2 (C16), 135.2 (Cj), 133.9 (Cc), 133.9 (C5/9), 133.8 (C5/9), 133.5 (C6/8), 131.8 (C6/8), 131.6 (Cb), 131.5 (Cg), 129.6 (Ci), 129.3 (C15), 129.3 (C14), 128.9 (Ca), 128.9 (C7), 128.7 (C20), 128.1 (Ch), 126.3 (C19), 124.6 (Cd) 124.6 (C10), 123.4 (C25) 115.3 (C21), 73.4 (Ck), 69.9 (Cm), 68.4 (Cl), 60.5 (C23), 47.9 (C1), 44.3 (Cf), 43.4 (C17), 43.2 (C12), 37.4 (C3), 29.5 (C2).
m/z (ESI): 999.4400 ([M + H]+ C57H59N8O9 requires 999.4426).
:2) gave the title product (79 mg, 60%) as a colourless solid.
 :2).
:2).
m.p. 168–170 °C.
ν
max
/cm
−1
(neat): 3341(N–H), 2868 (C–H), 1630 (CO), 1578 (C–N), 1533 (C–N), 1416 (Ar–C).
δ
H
(400 MHz, 50:50 CDCl
3
/CD
3
OD): 8.71 (1H, s, Hd), 8.19 (2H, dd, J = 7.8 Hz, Hb), 8.12 (1H, d, J = 7.7 Hz, H6/8), 8.05 (1H, d, J = 7.8 Hz, H6/8), 7.80 (1H, s, H27), 7.69–7.63 (2H, m, H7 & Ha), 7.62 (1H, s, H10), 7.51 (1H, d, J = 7.5 Hz, H24), 7.43–7.35 (3H, m, H23 & H14), 7.32 (1H, s, H20), 7.22 (1H, dd, J = 7.2, 2.4 Hz, H22), 7.04 (2H, d, J = 7.7 Hz, H15), 6.94 (4H, d, J = 7.9 Hz, Hh), 6.56 (4H, d, J = 7.9 Hz, Hi), 5.34 (2H, s, H25), 4.59 (2H, d, J = 14.2 Hz, Hf), 4.47–4.35 (4H, m, H12 & Hf′), 4.26 (2H, bs, H1), 3.98–3.90 (4H, m, Hk & Hk′), 3.88 (2H, bs, H17), 3.07–2.92 (8H, m, Hl, Hl′, Hm & H3), 2.78–2.71 (2H, m, Hm′), 1.96 (2H, bs, H2).
δ
C
(100 MHz, 50:50 CDCl
3
/CD
3
OD): 167.8 (Ce), 166.5 (C18), 166.4 (C11), 165.8 (C4), 157.24 (C21), 143.4 (C26), 144.3 (C5/9), 138.4 (C5/9), 137.1 (Cj), 136.7 (C16/19/c), 136.5 (C13), 135.0 (Cg), 134.6 (C16/19/c), 131.6 (C6/8), 131.6 (C6/8), 131.6 (Cb), 130.0 (C23), 129.8 (Ci) 129.5 (C15) 129.3 (C14) 129.1 (Ca), 129.0 (C7), 128.4 (Ch), 124.6 (C10), 124.6 (Cd), 123.1 (C27), 119.3 (C24), 117.3 (C22), 114.7 (C20), 72.9 (Ck), 69.9 (Cl), 68.5 (Cm), 60.9 (C25), 52.8 (C1), 43.6 (Cf), 42.8 (C12), 42.7 (C17), 36.3 (C1), 29.2 (C2).
m/z (ESI): 999.4400 ([M + H]+ C57H59N8O9 requires 999.4427).
:CH3OH 96:4–94:6) gave the title product (89 mg, 70%) as a colourless solid.
 :CH3OH 96:4).
:CH3OH 96:4).
m.p. 118–121 °C.
ν
max
/cm
−1
(neat): 3215 (C–H), 2860 (C–H), 1638 (CO), 1522 (C–N), 1017 (C–O–C).
δ
H
(400 MHz, 50:50 CDCl
3
/CD
3
OD): 8.70 (1H, s, Hd), 8.18 (2H, dd, J = 7.8, 1.8 Hz, Hb), 8.13 (1H, d, J = 7.7 Hz, H6/8), 8.05 (1H, d, J = 7.8 Hz, H6/8), 7.80 (1H, s, H24), 7.70–7.63 (3H, m, H7 & Ha), 7.62 (1H, s, H10), 7.26 (2H, d, J = 8.1 Hz, H14), 7.03 (2H, d, J = 8.1 Hz, H15), 6.96 (4H, d, J = 7.9 Hz, Hh), 6.63 (4H, d, J = 7.9 Hz, Hi), 4.63–4.53 (4H, m, H22 & Hf), 4.42–4.36 (4H, m, Hf′ & H12), 4.32 (2H, bs, H1), 4.11–4.02 (4H, m, Hk), 3.98 (2H, m, H19), 3.90 (2H, bs, H17), 3.71 (4H, bs, H20 & H21), 3.15–3.00 (8H, dt, m, Hl, H3, Hm & Hl′), 2.90–2.85 (2H, m, Hm′), 2.08 (2H, bs, H2).
δ
C
(100 MHz, 50:50 CDCl
3
/CD
3
OD): 170.0 (C18), 167.4 (Ce), 167.0 (C4), 166.4 (C11), 144.3 (C23), 138.7 (C16),138.4 (C5/9/c), 138.3 (C5/9/c), 137.1 (C5/9/c), 136.9 (C13), 135.4 (Cj), 131.7 (Cb) 131.6 (C6/8), 131.4 (C6/8), 131.2 (Cg), 129.5 (Ci) 129.3 (C15), 129.1 (Ca), 128.7 (C7), 128.5 (C14), 128.2 (Ch), 124.8 (C10), 124.6 (Cd), 123.6 (C24), 73.5 (Ck), 70.4 (C19), 70.3 (Cm), 70.2 (C21), 69.7 (C20) 68.7 (Cl), 64.2 (C22), 47.5 (C1), 44.2 (Cf), 43.3 (C17), 42.1 (C12), 36.6 (C3), 29.5 (C2).
m/z (ESI): 1003.4325 ([M + H]+ C54H61N8O10 requires 1003.4372).
:1) to afford the title product as a waxy clear oil (129 mg, 68%).
 :Hexane 3:7).
:Hexane 3:7).
ν
max
/cm
−1
(neat): 3330 (CH2), 2933 (ArCH), 2096 (NNN), 1638 (CO), 1531 (C–N).
δ H (400 MHz, CDCl 3 ): 8.23 (1H, s, H10), 8.01–7.96 (1H, m, H8), 7.93–7.88 (1H, m, H6), 7.80 (3H, s, H14 & H16), 7.54 (1H, app. t, H7), 6.95 (1H, bs, NHγ), 6.54 (1H, bs, NHβ), 4.77 (2H, d, J = 6.1 Hz, H12), 3.56 (2H, t, J = 6.6 Hz, H3), 3.45 (2H, t, J = 6.5 Hz, H1), 1.91 (2H, quintet, J = 6.6 Hz, H2).
δ C (100 MHz, CDCl 3 ): 166.6 (C11), 166.5 (C4), 140.7 (C13), 134.8 (C9), 134.0 (C5), 131.9 (q, 2J = 34 Hz, C15), 130.2 (C8), 130.0 (C6), 129.2 (C7), 128.0 (C14), 125.5 (C10), 124.6 (C16), 121.9 (q, 1J = 271 Hz, C17), 49.5 (C1), 43.4 (C12), 38.0 (C3), 28.7 (C2).
δ F (377 MHz, CDCl3): −62.8
m/z (ESI): 474.1359 ([M + H]+ C20H18N5O2F6 requires 474.1346).
:1) to afford the title product as a brown oil (89 mg, 51%).
 :hexane 3:7).
:hexane 3:7).
ν
max
/cm
−1
(neat): 3330 (N–H), 2935 (ArCH), 2096 (NNN), 1638 (CO), 1531 (C–N).
δ H (400 MHz, D 6 -DMSO): 10.99 (1H, bs, NHγ), 8.72 (1H, bs, NHβ), 8.55 (2H, s, H13), 8.49 (1H, s, H15), 8.15 (1H, d, J = 7.1 Hz, H6), 8.10 (1H, d, J = 7.2 Hz, H8), 7.84 (1H, s, H10), 7.68 (1H, t, J = 7.8 Hz, H7), 3.45 (2H, t, J = 6.5 Hz, H3), 3.38 (2H, t, J = 6.5 Hz, H1), 1.82 (2H, quintet, J = 6.6 Hz, H2).
δ C (100 MHz, D 6 -DMSO): 166.3 (C11), 166.1 (C4), 141.4 (C12), 135.4 (C9), 134.5 (C5), 131.3 (q, 2J = 34 Hz, C14), 131.1 (C7), 130.9 (C15), 129.1 (C6), 127.2 (C8), 122.3 (C13), 120.3 (q, 1J = 271 Hz, C16), 117.3 (C10), 48.9 (C1), 37.2 (C3), 28.8 (C2).
δ F (377 MHz, D6-DMSO): −61.6.
m/z (ESI): 458.1057 ([M − H]− C19H16F6N5O2 requires 458.1065).
:2:3 CH2Cl2/CH3OH/CH3COCH3) which allowed for isolation of the product with contaminated macrocycle 14. Pure rotaxane was isolated after running another preparative TLC (run twice in 95:2:3 CH2Cl2/CH3OH/CH3COCH3) to give the title product as a colourless glassy film (14 mg, 29%).
 :CH3OH 96:4).
:CH3OH 96:4).
ν
max
/cm
−1
(neat): 3332 (N–H), 2929 (C–H), 2862 (C–H),1638 (CO), 1528 (C–N), 1276 (C–O).
δ H (400 MHz, CDCl 3 ): 8.66 (1H, s, Hd), 8.31–8.21 (3H, m, Hb & H15/17), 8.07 (1H, bs, H15/17), 7.84 (1H,s, H25), 7.78 (2H, bs, H23), 7.73 (2H, m, H16 & H19), 7.62 (1H, t, J = 7.8 Hz, Ha), 7.56 (2H, bs, NHα), 7.32–7.16 (11H, m, H1, H2, H3 & H9), 6.84 (4H, d, J = 7.9 Hz, Hh), 6.53 (4H, d, J = 8.0 Hz, Hi), 4.64 (2H,s, H7), 4.59–4.37 (4H, m, Hf), 4.32 (1H, t, J = 7.7 Hz, H5), 4.14–4.02 (8H, m, H6, Hk &H10), 3.97 (2H, bs, H21), 3.70–3.55 (2H, m, Hl), 3.44–3.30 (4H, m, Hm & Hl′), 3.28–3.20 (2H, m, Hm′), 2.78 (2H, bs, H12), 1.78 (2H, bs, H11).
δ C (100 MHz, CDCl 3 ): 166.9 (C13), 166.6 (C20), 166.4 (Ce), 145.2 (C8), 141.9 (C4), 136.4 (Cg), 134.8 (Cj), 133.8 (C14/18/22), 133.7 (C14/18/22), 133.1 (C14/18/22), 132.1 (C24), 132.0 (Cb), 131.8 (C15/17), 131.6 (C15/17), 129.9 (Ci), 129.5 (Ca), 129.2 (C23), 128.8 (C16), 128.4 (C3), 128.3 (Ch), 128.2 (C2), 126.5 (C1), 124.5 (C25), 123.8 (Cd), 122.6 (C9), 121.7 (C19), 73.9 (Ck), 73.7 (C6), 70.7 (Cm), 68.8 (Cl), 64.5 (C7), 50.9 (C5), 47.6 (C10), 44.5 (Cf), 43.3 (C21), 36.8 (C12), 29.6 (C11).
δ F (377 MHz, CDCl3): −62.6.
m/z (ESI): 1184.4715 ([M + H]+ C65H64F6N7O8 requires 1184.4752).
:2 CH2Cl2/CH3OH) which allowed for isolation of the product with contaminated macrocycle 14. Pure rotaxane was isolated after running another preparative TLC (repeated running in 96:2:2 CH2Cl2/CH3OH/CH3COCH3) to give the title product as a colourless glassy film (20 mg, 40%).
 :4).
:4).
ν
max
/cm
−1
(neat): 3319 (N–H), 2918 (C–H), 1645 (CO), 1528 (C–N), 1276 (C–O).
δ H (400 MHz, CDCl 3 ): 8.55 (1H, s, Hd), 8.32–8.22 (3H, m, Hb & H15/17), 8.06–7.87 (3H, m, H15/17 & H19), 7.84 (1H, bs, H22), 7.72–7.62 (2H, m, H16 & Ha), 7.58 (1H, s, H24), 7.43–7.17 (13H, m, H1, H2, H3, H9 & NHα), 6.78 (4H, d, J = 7.9 Hz, Hh), 6.54 (4H, d, J = 8.0 Hz, Hi), 4.67 (2H, s, H7), 4.52–4.36 (4H, m, Hf), 4.34–4.20 (3H, m, H5 & H10), 4.18–4.10 (4H, m, Hk), 4.09 (2H, d, 7.3 Hz, H6), 3.79–3.72 (2H, m, Hm), 3.68–3.60 (2H, m, Hm′), 3.55–3.44 (4H, m, Hl), 3.33 (2H, bs, H12), 2.14 (2H, bs, H11).
δ C (100 MHz, CDCl 3 ): 166.3 (Ce), 166.2 (C20), 165.7 (C13), 145.3 (C8), 141.9 (C18/14), 139.4 (C21/c), 137.4 (Cj), 134.8 (Cg), 133.7 (C14/18), 133.7 (C21/c), 132.8 (C15/17), 132.0 (C15/17), 131.9 (Cb), 129.4 (Ca), 129.4 (C4), 129.3 (Ci), 129.0 (C16), 128.5 (C22), 128.4 (C2), 128.2 (C3), 128.2 (Ch), 126.5 (C1), 124.8 (C22), 123.1 (Cd), 122.7 (C9), 121.6 (C19), 117.0 (C24), 73.9 (Ck), 73.7 (C6), 70.6 (Cm), 68.8 (Cl), 64.6 (C7), 50.9 (C5), 47.8 (C10), 44.4 (Cf), 37.1 (C12), 29.7 (C11).
δ F (377 MHz, CDCl3): −63.0.
m/z (ESI): 1170.4559 ([M + H]+ C64H62F6N7O8 requires 1170.4595).
Author contributions
NHE proposed the study. NHE conducted initial experiments (see ESI†). SRB conducted the synthesis, characterization and analysis of all materials in the main article with assistance from GRA. NHE supervised the work. SRB and NHE wrote the manuscript. All authors discussed and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
SRB acknowledges PhD funding from EPRSC DTP (EP/T518037/1) and a Sydney Andrew Scholarship from the Society of Chemical Industry. NHE acknowledges the prize sponsored by Amigo Chem, that was awarded at the EPSRC Dial-a-Molecule and Directed Assembly Networks ECR “Supporting Synthesis and Self-Assembly” event held at the University of Liverpool in 2017, part of which funded the initial experiments reported in the ESI.† We thank Dr David Rochester (Lancaster University) for the recording of mass spectrometry data.Underlying data for this paper are provided in the Experimental section and ESI.† Electronic copies of NMR spectra (including fid files) will be available upon publication from: https://doi.org/10.17635/lancaster/researchdata/554, https://doi.org/10.17635/lancaster/researchdata/569, https://doi.org/10.17635/lancaster/researchdata/570, https://doi.org/10.17635/lancaster/researchdata/574.
References
- G. Gil-Ramírez, D. A. Leigh and A. J. Stephens, Angew. Chem., Int. Ed., 2015, 54, 6100–6150 CrossRef PubMed.
- M. Xue, Y. Yang, X. Chi, X. Yan and F. Huang, Chem. Rev., 2015, 115, 7398–7501 CrossRef CAS PubMed.
- C. J. Bruns and J. F. Stoddart, The Nature of the Mechanical Bond: From Molecules to Machines, Wiley & Sons, 2017 Search PubMed.
- Review: (a) J. E. M. Lewis, P. D. Beer, S. J. Loeb and S. M. Goldup, Chem. Soc. Rev., 2017, 46, 2577–2591 RSC Seminal examples: (b) C.-O. Dietrich-Buchecker, J.-P. Sauvage and J.-P. Kintzinger, Tetrahedron Lett., 1983, 34, 5095–5098 CrossRef; (c) V. Aucagne, K. D. Hänni, D. A. Leigh, P. J. Lusby and D. B. Walker, J. Am. Chem. Soc., 2006, 128, 2186–2187 CrossRef CAS PubMed Recent examples: (d) A. W. Heard and S. M. Goldup, Chem, 2020, 6, 994–1006 CrossRef CAS PubMed; (e) R. C. Knighton and P. D. Beer, Org. Chem. Front., 2021, 8, 2468–2472 RSC.
- Review: (a) G. Barin, A. Coskin, M. M. G. Fouda and J. F. Stoddart, ChemPlusChem, 2012, 77, 159–185 CrossRef CAS Seminal examples: (b) P. R. Ashton, T. T. Goodnow, A. E. Kaifer, M. V. Reddington, A. M. Z. Slawin, N. Spencer, J. F. Stoddart, C. Vicent and D. J. Williams, Angew. Chem., Int. Ed. Engl., 1989, 28, 1396–1399 CrossRef; (c) D. G. Hamilton, J. K. M. Sanders, J. E. Davies, W. Clegg and S. J. Teat, Chem. Commun., 1997, 897–898 RSC Recent examples: (d) T.-M. Gianga, E. Audibert, A. Trandafir, G. Kociok-Köhn and G. D. Pantoş, Chem. Sci., 2020, 11, 9685–9690 RSC; (e) Y. Jiao, L. Đorđević, H. Mao, R. M. Young, T. Jaynes, H. Chen, Y. Qiu, K. Cai, L. Zhang, X.-Y. Chen, Y. Feng, M. R. Waseielewski, S. I. Stupp and J. F. Stoddart, J. Am. Chem. Soc., 2021, 143, 8000–8100 CrossRef CAS PubMed.
- Review: (a) N. H. Evans, Eur. J. Org. Chem., 2019, 3320–3343 CrossRef CAS Seminal examples: (b) C. A. Hunter, J. Am. Chem. Soc., 1992, 114, 5303–5311 CrossRef CAS; (c) F. Vögtle, S. Meier and R. Hoss, Angew. Chem., Int. Ed. Engl., 1992, 31, 1619–1622 CrossRef; (d) P. R. Ashton, P. T. Glink, J. F. Stoddart, P. A. Tasker, A. J. P. White and D. J. Williams, Chem. – Eur. J., 1996, 2, 729–726 CrossRef CAS; (e) F. G. Gatti, D. A. Leigh, S. A. Nepogodiev, A. M. Z. Slawin, S. J. Teat and J. K. Y. Wong, J. Am. Chem. Soc., 2001, 123, 5983–5989 CrossRef CAS PubMed Recent examples: (f) S. D. P. Fielden, D. A. Leigh, C. T. McTernan, B. Pérez-Saavdra and I. J. Vitorica-Yrezabal, J. Am. Chem. Soc., 2018, 140, 6409–6052 CrossRef PubMed; (g) S. Amano, S. D. P. Fielden and D. A. Leigh, Nature, 2021, 594, 529–534 CrossRef CAS PubMed.
- Relevant reviews: (a) G. T. Spence and P. D. Beer, Acc. Chem. Res., 2013, 46, 571–586 CrossRef CAS PubMed; (b) K. M. Bąk, K. Porfyrakis, J. J. Davis and P. D. Beer, Mater. Chem. Front., 2020, 4, 1052–1073 RSC Seminal examples: (c) G. M. Hübner, J. Gläser, C. Seel and F. Vögtle, Angew. Chem., Int. Ed., 1999, 38, 383–386 CrossRef; (d) J. A. Wisner, P. D. Beer, M. G. B. Drew and M. R. Sambrook, J. Am. Chem. Soc., 2002, 124, 12469–12476 CrossRef CAS PubMed; (e) N. L. Kilah, M. D. Wise, C. J. Serpell, A. L. Thompson, N. G. White, K. E. Christensen and P. D. Beer, J. Am. Chem. Soc., 2010, 132, 11893–11895 CrossRef CAS PubMed; (f) S. Lee, C. H. Chen and A. H. Flood, Nat. Chem., 2013, 5, 704–710 CrossRef CAS PubMed.
- N. H. Evans, C. E. Gell and M. J. G. Peach, Org. Biomol. Chem., 2016, 14, 792–7981 RSC.
- B. E. Fletcher, M. J. G. Peach and N. H. Evans, Org. Biomol. Chem., 2017, 15, 2797–2803 RSC.
- C. N. Marrs and N. H. Evans, Org. Biomol. Chem., 2015, 13, 11021–11025 RSC.
- Lewis has subsequently reported yields of up 51% for analogous [2]catenane formation by using bis-amines with longer glycol chains: J. E. M. Lewis, Org. Biomol. Chem., 2019, 17, 2442–2447 RSC.
- M. J. Langton, L. C. Duckworth and P. D. Beer, Chem. Commun., 2013, 49, 8608–8610 RSC.
- In our hands, we obtained 1 in higher yield than previously reported. See ESI p. S3 for details.†.
- C. Deng, R. Fang, Y. Guan, J. Jiang, C. Lin and L. Wang, Chem. Commun., 2012, 48, 7973–7975 RSC.
- (a) H. M. Branderhorst, R. Ruijtenbeek, R. M. J. Liskamp and R. J. Pieter, ChemBioChem, 2008, 9, 1836–1844 CrossRef CAS PubMed.
- K. Hayashi, Y. Miyaoka, Y. Ohishi, T. Uchinda, M. Iwamura, K. Nozaki and M. Inouye, Chem. – Eur. J., 2018, 24, 14613–14616 CrossRef CAS PubMed.
- The corresponding author had previously prepared (but not reported) [2]catenane 15, preparing precursor 11 by a different route. See ESI pp. S48–S67 for details.†.
- To date it has not been possible to isolate pure samples of the macrocyclic product arising from cyclization of 11–13.
- By analogous reactions (in the absence of macrocycle 14), non-interlocked axles 22 and 24 were also prepared (in 80% and 79% yield respectively) to aid 1H NMR spectroscopic interpretation (see ESI pp. S3–S5†).
- (a) N. H. Evans, Chem. – Eur. J., 2018, 24, 3101–3112 CrossRef CAS PubMed; (b) N. Pairault and J. Niemeyer, Synlett, 2018, 689–698 CAS; (c) E. M. G. Jamieson, F. Modicom and S. M. Goldup, Chem. Soc. Rev., 2018, 47, 5266–5311 RSC; (d) K. Nakazono and T. Takata, Symmetry, 2020, 12, 144 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Further experimental procedures; copies of spectral characterization data. See DOI: https://doi.org/10.1039/d2ob02019j |
| This journal is © The Royal Society of Chemistry 2023 |