
Stereoselective synthesis of the spirocyclic core of 13-desmethyl spirolide C using an aza-Claisen rearrangement and an exo-selective Diels–Alder cycloaddition†
Andrew D. W.
Earl
 a,
Freda F.
Li
*ab,
Chao
Ma
a,
Daniel P.
Furkert
ab and
Margaret A.
Brimble
*ab
a,
Freda F.
Li
*ab,
Chao
Ma
a,
Daniel P.
Furkert
ab and
Margaret A.
Brimble
*ab
aSchool of Chemical Sciences, The University of Auckland, 23 Symonds Street, Auckland 1010, New Zealand. E-mail: m.brimble@auckland.ac.nz; freda.li@auckland.ac.nz
bMaurice Wilkins Centre for Molecular Biodiscovery, The University of Auckland, 3A Symonds Street, Auckland 1010, New Zealand
First published on 5th January 2023
Abstract
13-Desmethyl spirolide C is a marine natural product of the cyclic imine class that demonstrates remarkable bioactivity against several biomarkers of Alzheimer's Disease, which renders its [7,6]-spirocyclic imine pharmacophore of significant synthetic interest. This work describes a facile and efficient synthesis of the [7,6]-spirocyclic core of 13-desmethyl spirolide C from inexpensive starting materials, featuring an aza-Claisen rearrangement to simultaneously set both stereocentres of the dimethyl moiety with complete atom economy, and a highly exo-selective Diels–Alder cycloaddition to construct the challenging contiguous tertiary and quaternary stereocentres of the spirocyclic core of 13-desmethyl spirolide C. A comprehensive study of the key Diels–Alder reaction was also performed to evaluate the stereoselectivity and reactivity of various functionalised dienes and protected lactam dienophiles, wherein the first successful exo-selective Diels–Alder cycloaddition to construct spirocyclic structures using a bromodiene and α-exo-methylene dienophiles is reported. This strategy not only establishes a more efficient stereoselective access to the spirocyclic core that can be used for the total synthesis of 13-desmethyl spirolide C, but also serves as a sound platform for convenient preparations of a range of spirocyclic analogues required for a comprehensive biological evaluation of this desirable pharmacophore.
Introduction
The cyclic imine class of marine natural products consists of a diverse array of polyketides which have garnered significant attention due to their fascinating range of bioactivity and complex molecular structures. Members of the cyclic imine class contain a characteristic [5,6]-, [6,6]-, or [7,6]-spirocyclic imine moiety (in blue) as illustrated by portimine A (1),1 gymnodimine A (2),2 pinnatoxin A (3),3 and 13-desmethyl spirolide C (4) (Fig. 1).4 In general, these marine phycotoxins act as potent subtype-selective inhibitors of nicotinic acetylcholine receptors.5,6 However, unique to 13-desmethyl spirolide C (4) is the additional capability to induce beneficial effects on several markers of Alzheimer's Disease, including both amyloid beta and hyperphosphorylated tau protein, as demonstrated in both in vitro and in vivo studies.7,8 Such intriguing bioactivity towards a currently unmet medical need9 renders this member of the cyclic imines, and in particular its spirocyclic imine pharmacophore,10 of great synthetic interest. Despite this, no total synthesis of 13-desmethyl spirolide C (4) has yet been reported, although total syntheses of related pinnatoxins have been achieved by the groups of Kishi,11,12 Hirama (formal),13 and Zakarian.6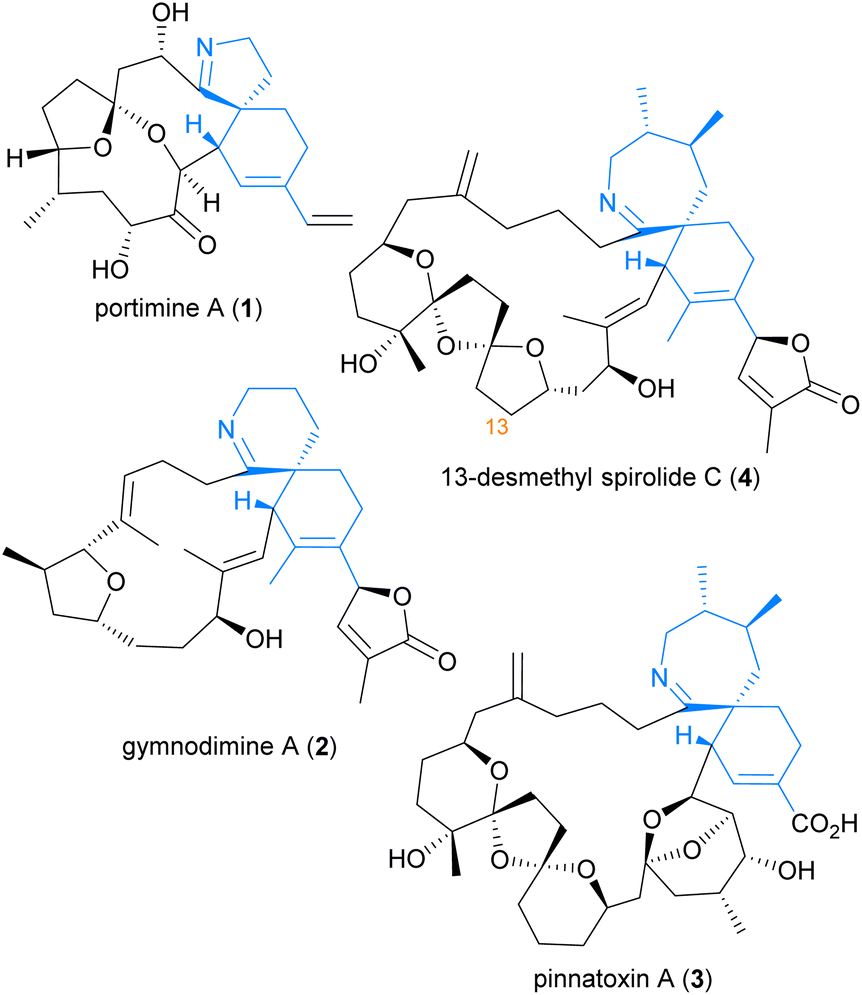 | ||
| Fig. 1 Representative members of the cyclic imine natural products. | ||
In addition to the total syntheses of the pinnatoxins, the [7,6]-spirocyclic imine pharmacophore has received particular attention due to its importance for the bioactivity of these marine phycotoxins. Synthesis of these spirocyclic imine fragments requires the construction of not only the challenging contiguous tertiary and quaternary stereocentres, but also the chiral vicinal dimethyl moiety. The groups of Evans,14 Ishihara,15 and Murata16,17 have recently reported synthetic studies towards the [7,6]-spirocyclic subunit of the spirolides and pinnatoxins, and approaches to the challenging chiral motifs have varied (Scheme 1, dimethyl moiety in blue). In 2011, Evans et al.14 utilised a low-yielding asymmetric oxidative homocoupling of imide 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 18 to construct the chiral dimethyl moiety of cyclic iminium dienophile 6, which then underwent an endo-selective Diels–Alder cycloaddition to afford the spirocyclic imine adduct 7, unfortunately bearing the undesired syn-stereochemistry. More recently, Ishihara et al.15 and Murata et al.16,17 each independently reported the synthesis lactam dienophile 8 from (S)-(−)-citronellic acid 9 or (S)-(−)-citronellol 10via common intermediate 11. The subsequent exo-selective Diels–Alder cycloaddition by Ishihara et al.15 afforded spirocyclic lactam adduct 12, whereas Murata et al.16,17 has employed Diels–Alder cycloadditions with both a silyl enol ether and silatrane diene to afford adduct 13. However, the previously reported syntheses of α-exo-methylene lactam 8 by Ishihara et al.15 and Murata et al.16 required linear construction of the dimethyl moiety from expensive chiral pool starting materials,‡ and the approach to the dimethyl moiety employed by Evans et al. only proceeded in low yield.14,18 We therefore aimed to develop a more economical and robust synthetic strategy to access the required lactam dienophile for synthesis of the spirocyclic fragment of 13-desmethyl spirolide C (4) using an exo-selective Diels–Alder reaction. We report herein a facile, inexpensive synthesis of α-exo-methylene lactam dienophile 8 which employs a key asymmetric aza-Claisen rearrangement19 to establish the required chiral dimethyl moiety in amide 15a. Dienophile 8 was then utilised in an exclusively exo-selective Diels–Alder cycloaddition to construct the spirocyclic core 16a of 13-desmethyl spirolide C (4).
18 to construct the chiral dimethyl moiety of cyclic iminium dienophile 6, which then underwent an endo-selective Diels–Alder cycloaddition to afford the spirocyclic imine adduct 7, unfortunately bearing the undesired syn-stereochemistry. More recently, Ishihara et al.15 and Murata et al.16,17 each independently reported the synthesis lactam dienophile 8 from (S)-(−)-citronellic acid 9 or (S)-(−)-citronellol 10via common intermediate 11. The subsequent exo-selective Diels–Alder cycloaddition by Ishihara et al.15 afforded spirocyclic lactam adduct 12, whereas Murata et al.16,17 has employed Diels–Alder cycloadditions with both a silyl enol ether and silatrane diene to afford adduct 13. However, the previously reported syntheses of α-exo-methylene lactam 8 by Ishihara et al.15 and Murata et al.16 required linear construction of the dimethyl moiety from expensive chiral pool starting materials,‡ and the approach to the dimethyl moiety employed by Evans et al. only proceeded in low yield.14,18 We therefore aimed to develop a more economical and robust synthetic strategy to access the required lactam dienophile for synthesis of the spirocyclic fragment of 13-desmethyl spirolide C (4) using an exo-selective Diels–Alder reaction. We report herein a facile, inexpensive synthesis of α-exo-methylene lactam dienophile 8 which employs a key asymmetric aza-Claisen rearrangement19 to establish the required chiral dimethyl moiety in amide 15a. Dienophile 8 was then utilised in an exclusively exo-selective Diels–Alder cycloaddition to construct the spirocyclic core 16a of 13-desmethyl spirolide C (4).
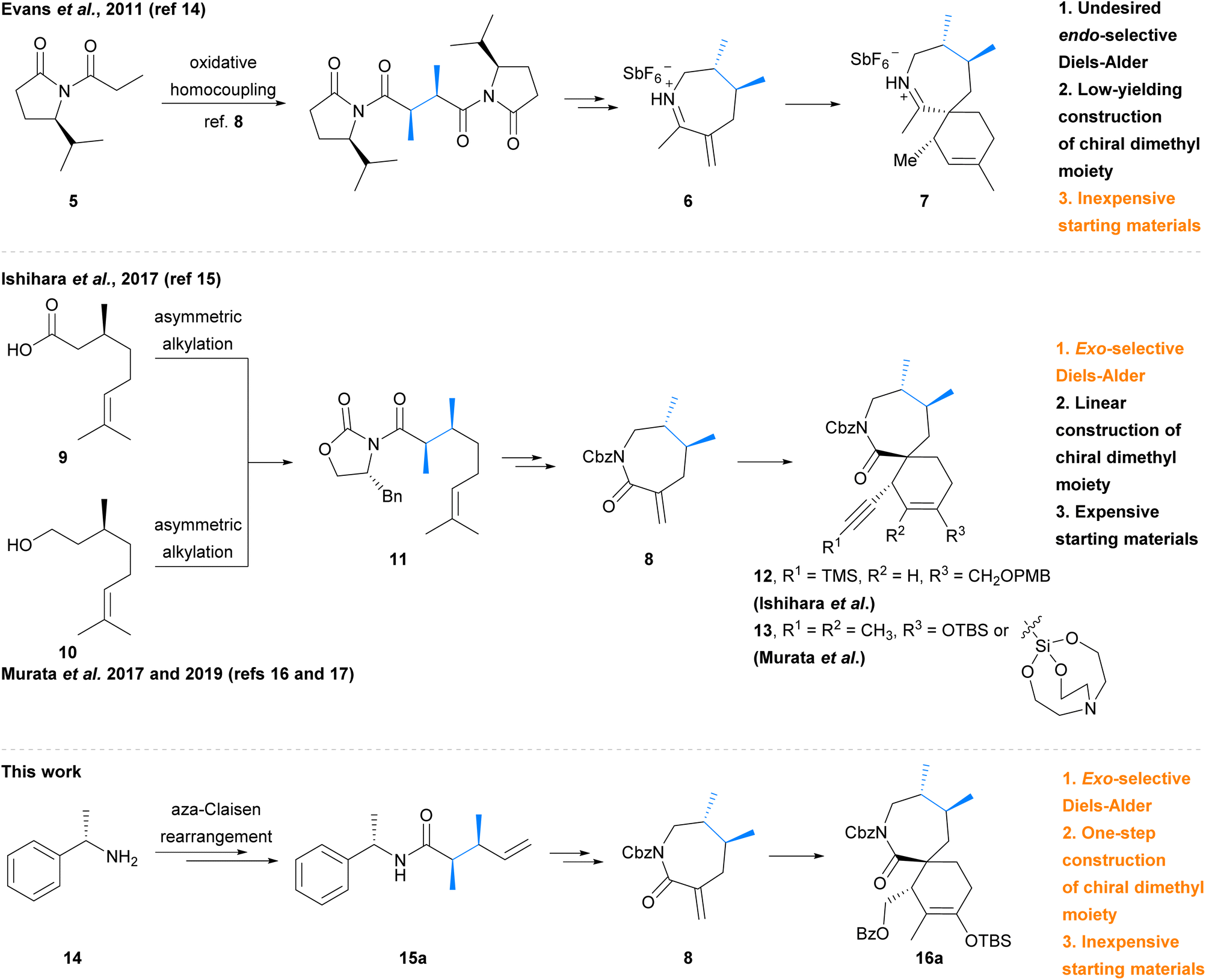 | ||
| Scheme 1 Previous and present syntheses of dimethyl [7,6]-spirocyclic systems. | ||
Results and discussion
Our research group has previously explored the use of in situ-generated α,β-unsaturated iminium dienophiles to construct [5,6]-, [6,6]- and [7,6]-spirocyclic systems.20–22 These studies revealed the strong preference of these dienophiles for the endo-transition state,23 which was also observed by Evans et al. in 2011.14 Investigating the use of alternative dienophiles such as α-exo-methylene lactams, which have been successfully employed in exo-selective Diels–Alder cycloadditions,15–17 was therefore of interest to us. We envisioned that the [7,6]-spirocyclic fragment 17 of 13-desmethyl spirolide C (4) could be accessed through an exo-selective Diels–Alder reaction of an α-exo-methylene lactam dienophile (18) with bromodiene 19 (Scheme 2). The vinyl bromide moiety of spirocyclic lactam 17, arising from use of bromodiene 19 in the Diels–Alder cycloaddition, would later enable a simplified and flexible approach to the desired pendant butenolide via transition-metal mediated coupling reactions. Chiral dienophile 18 would itself be synthesised from chiral amide 15a, which in turn was to be accessed from the aza-Claisen rearrangement of amide 20 previously reported by Tsunoda et al.19 wherein the dimethyl moiety could be efficiently established in a single transformation with complete atom economy. Employing the aza-Claisen rearrangement to construct the chiral vicinal dimethyl moiety would enable a novel and robust synthesis of the desired dienophile from inexpensive starting materials, overcoming the shortfalls of previously reported syntheses of similar [7,6]-spirocyclic fragments.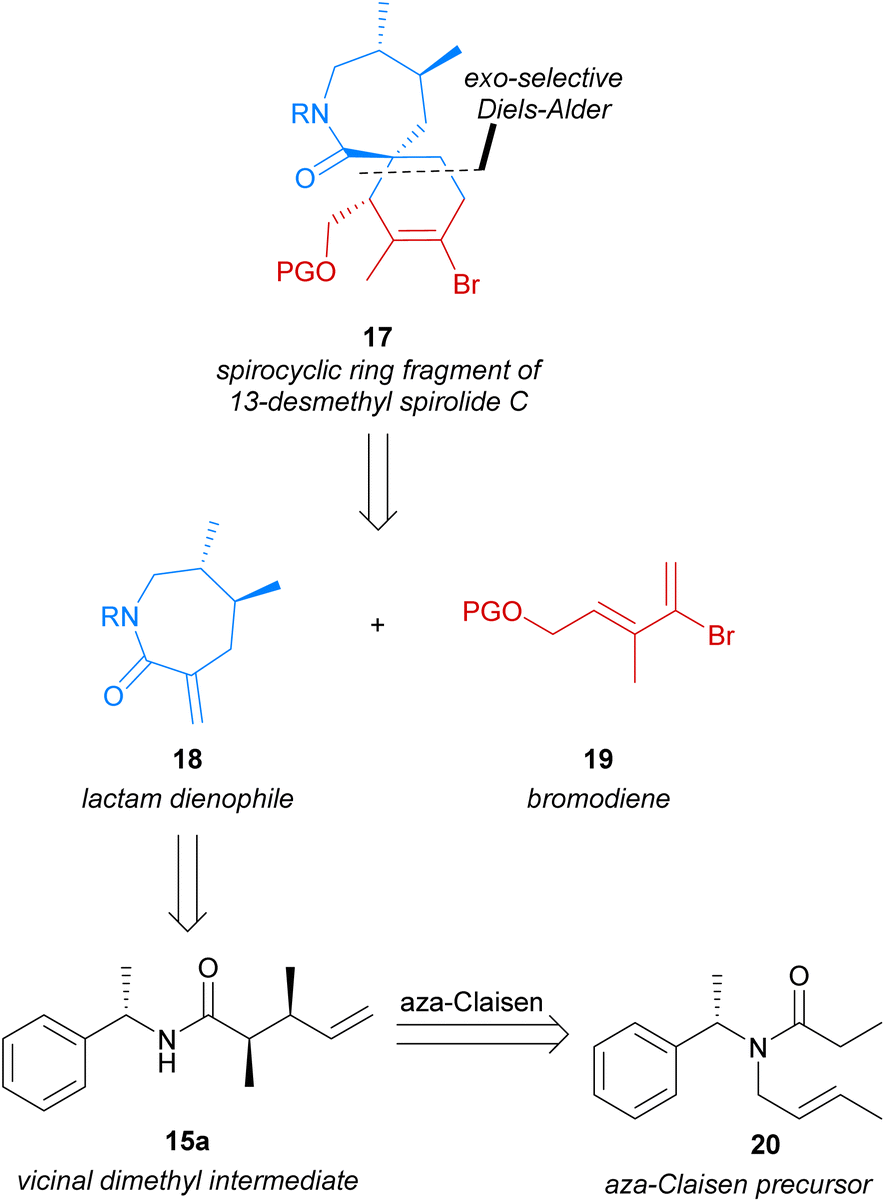 | ||
| Scheme 2 Retrosynthesis of [7,6]-spirocyclic fragment 17 of 13-desmethyl spirolide C (4). | ||
Use of the aza-Claisen rearrangement would also render our strategy particularly amenable to the future synthesis of structural and stereochemical analogues of this important lactam dienophile, as this transformation has been previously employed to access a diverse range of related amide products bearing variable stereochemistry and substitution at the chiral positions.28,29 These related amides could be similarly utilised to access analogues of dienophile 18, and therefore spirocyclic lactam 17.
Our synthesis began with preparation of aza-Claisen precursor amide 20, which was achieved in two steps from inexpensive commercially available (S)-(−)-α-methylbenzylamine (14)‡ using literature conditions24,25 (Scheme 3). Subsequent aza-Claisen rearrangement to synthesise known amide 15a initially proved low-yielding using previously reported conditions on larger, multigram scales.19,24,26 During reaction optimisation, it was found that use of a pressure vessel was imperative for the reaction to proceed, and an extended period at elevated temperature was necessary for complete consumption of the starting material. Employing commercially available LiHMDS solution in toluene19 was also found to improve the yield of this reaction compared to the previously reported26in situ formation of LiHMDS from HMDS and n-butyllithium in hexanes. Our optimised conditions proceeded smoothly on a multigram scale, affording the desired syn-dimethyl diastereomer 15a alongside a small portion of the undesired syn-dimethyl diastereomer 15b. The two diastereomers 15a and 15b were easily separable by flash chromatography, and formation of the anti-diastereomers was not observed under these conditions. While the aza-Claisen rearrangement had proceeded with excellent yield and selectivity, elaboration of similar α,β-dimethyl amides has been previously reported as a challenging endeavour26 and our initial attempts to reduce the amide moiety of amide 15a were also unfortunately problematic (Scheme 4). Although the resulting reduction of amide 15a to the corresponding amine proved sluggish, reduction was eventually achieved in excellent yield using lithium aluminium hydride in diethyl ether at reflux overnight. Protection of the resulting amine under standard conditions then afforded N-Boc amine 21 in good yield, albeit with incomplete consumption of the intermediate amine. Attempts to effect complete consumption of the intermediate amine through use of more forcing conditions resulted in reduced yield of the desired Boc-protected product (21). Oxidative cleavage of the alkene of N-Boc amine 21 to the corresponding aldehyde was immediately followed by Wittig reaction, without purification, to afford α,β-unsaturated ester 22 in good yield. Purification of α,β-unsaturated ester 22 proved absolutely essential, as a phosphorus-containing impurity of similar polarity poisoned the palladium catalyst of the ensuing hydrogenation and prevented efficient removal of the chiral auxiliary. The subsequent palladium-catalysed hydrogenation efficiently reduced the alkene moiety and cleaved the chiral auxiliary to afford N-Boc aminoester 23. Attempts to effect a direct cyclisation of N-Boc aminoester 23 were unsuccessful, necessitating a deprotection-reprotection sequence. Pleasingly, Boc deprotection proceeded smoothly under standard conditions in the presence of anisole, and the resulting free aminoester readily cyclised using triethylamine in a dilute mixture of 2:1 toluene/MeCN at reflux overnight to afford chiral lactam 24. Lactam 24 was then subjected to Cbz-protection and subsequent methylenation using previously reported procedures16,27 to afford the desired Cbz-protected chiral dienophile 8. We therefore finally achieved a robust preparation of key enantiopure dienophile 8 from the inexpensive commercially available amine 14.
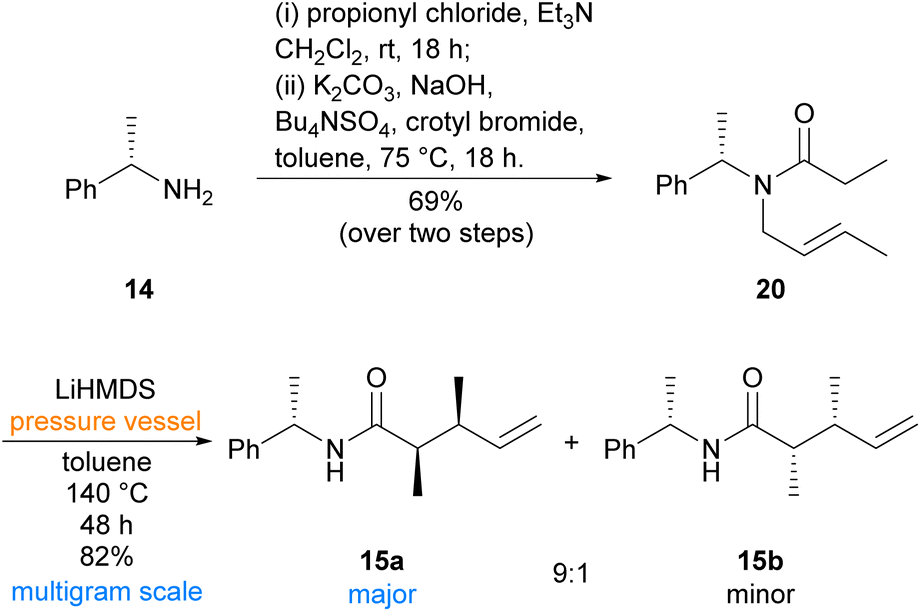 | ||
| Scheme 3 Construction of the syn-dimethyl moiety of amide 15a using an aza-Claisen rearrangement. | ||
 | ||
| Scheme 4 Synthesis of α-exo-methylene dienophile 8. | ||
Enantiopure dienophile 8 was then employed in a Diels–Alder cycloaddition in an attempt to construct a [7,6]-spirocyclic fragment. Recently, our group achieved a successful Diels–Alder/cross-coupling sequence from bromodiene 25 (Scheme 5) and acyclic dienophiles to synthesise a [5,6]-spirocyclic imine fragment of the portimines, albeit with unnatural endo-stereochemistry.30 However, the use of bromodienes with an appropriate dienophile to construct a spirocyclic fragment bearing the desired exo-stereochemistry has not been reported. It was envisioned that the desired exo-selective Diels–Alder cycloaddition could be achieved with the use of a cyclic exo-methylene dienophile (e.g., 8), which reportedly favours the exo-transition state in Diels–Alder cycloadditions31 particularly under thermal conditions.32 Unfortunately, following synthesis of bromodienes 25 or 26 using our previously reported methodology,30 attempts to effect the desired Diels–Alder cycloaddition using lactam 8 afforded only trace amounts of the respective cycloadducts, resulting instead in polymerisation of the bromodiene before cycloaddition occurred (Scheme 5). The simplified dienophiles 2733 and 28 were also employed to further probe this Diels–Alder cycloaddition. However no successful reaction was observed, and only trace formation of the respective cycloadducts was seen even with portionwise addition of excess amount of bromodiene over 6 hours. Interestingly, bromodiene 2930 bearing an extended sidechain demonstrated greater stability than the short-sidechain analogues (25 and 26), being stable for over 72 h under these conditions without noticeable polymerisation. Use of bromodiene 29 in a Diels–Alder cycloaddition with simplified dienophiles 27 and 28 afforded the desired exo-cycloadducts (±)-30 and (±)-31, albeit in poor yields of 18% and 11% respectively.
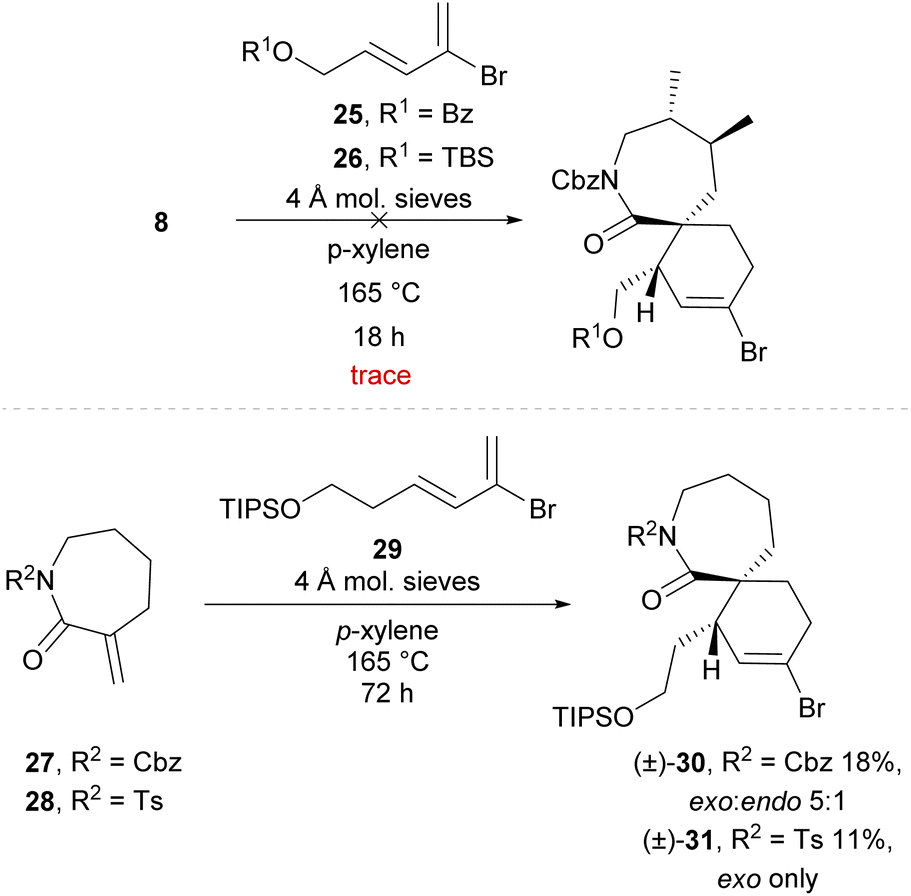 | ||
| Scheme 5 Diels–Alder cycloadditions of α-exo-methylene lactams with bromodienes. | ||
Alternatively, use of a more reactive diene bearing a similarly convenient synthetic handle, such as an analogous borate or boronic ester,34,35 was also considered. Unfortunately, use of commercially available furyl boronic esters and furyl trifluoroborates as electron-rich models of these diene scaffolds in Diels–Alder cycloadditions with simplified dienophiles 27 and 28 only returned unreacted starting materials (for details, see the ESI†).
The low yield of Diels–Alder cycloadditions using lactam dienophiles 27 and 28 with either bromodienes or boron-substituted furans suggested the generally low reactivity of these 7-membered lactam dienophiles towards cycloaddition, which would necessitate a more reactive diene counterpart. Our strategy was therefore revised to instead employ a more electron-rich silyl enol ether diene scaffold, which has previously been successfully utilised by our group in endo-selective Diels–Alder cycloadditions with α,β-unsaturated iminium dienophiles,21,22 and by Murata et al. in an exo-selective Diels–Alder reaction with lactam 8 using a chiral copper(II) bisoxazoline catalyst.16 For this approach, a Diels–Alder cycloaddition under Mg(OTf)2 catalysis was investigated. A recent investigation in 2019 by Murata et al.17 of the Diels–Alder cycloaddition of lactam dienophile 8 and a silatrane diene demonstrated that use of Mg(OTf)2 catalysis provides the best reaction outcomes compared to other metal triflate or copper(II) bisoxazoline catalysts; however Mg(OTf)2 catalysis was not explored with a silyl enol ether diene. We sought to adopt the use of these conditions employing a silyl enol ether diene scaffold, as the cycloadducts arising from use of these dienes (e.g.32–34, Scheme 6) themselves contain a useful silyl enol ether synthetic handle. This silyl enol ether synthetic handle can be readily elaborated to the corresponding enol triflate and utilised in subsequent coupling reactions.22 Silyl enol ether dienes 32–34 could be readily prepared via literature procedures,21,22 and the ensuing Diels–Alder cycloadditions of lactam dienophile 27 proved successful under Mg(OTf)2 catalysis (Scheme 6). Use of silyl enol ether diene 32 bearing a silyl-protected alcohol sidechain afforded the greatest yield (61%) of cycloadduct (±)-35 as a 3:1 mixture of diastereomers. The exo-stereochemistry of the crystalline major product (±)-35 was first determined through observation of NOE correlations between H-1′ and H-6, H-3 and H-8, and H-12 and H-8 (Scheme 6), and was later unambiguously confirmed by obtaining an X-ray crystal structure (Fig. 2). Similar analysis by NOE indicated that the exo-diastereomer was also the major cycloadduct formed in the Diels–Alder cycloadditions of lactam 27 with dienes 33 and 34. Compared to TBS-protected diene 32, use of PMB-protected diene 33 and benzoyl-protected diene 34 resulted in improved diastereoselectivity but reduced yield for their respective cycloadducts (±)-36 (34%, dr 10:1) and (±)-37 (39%, dr 5:1). On the other hand, cycloaddition of Ts-protected lactam dienophile 28 to dienes 32–34 under these conditions was found to be unsuccessful, returning only unreacted starting materials. Bidentate chelation of magnesium by the two carbonyl moieties of Cbz-protected lactam dienophiles has been previously suggested by Murata et al. to be an important contributor to dienophile activation,17 and the lack of reactivity displayed by the N-Ts lactam 28 under these conditions provides further evidence for its importance.
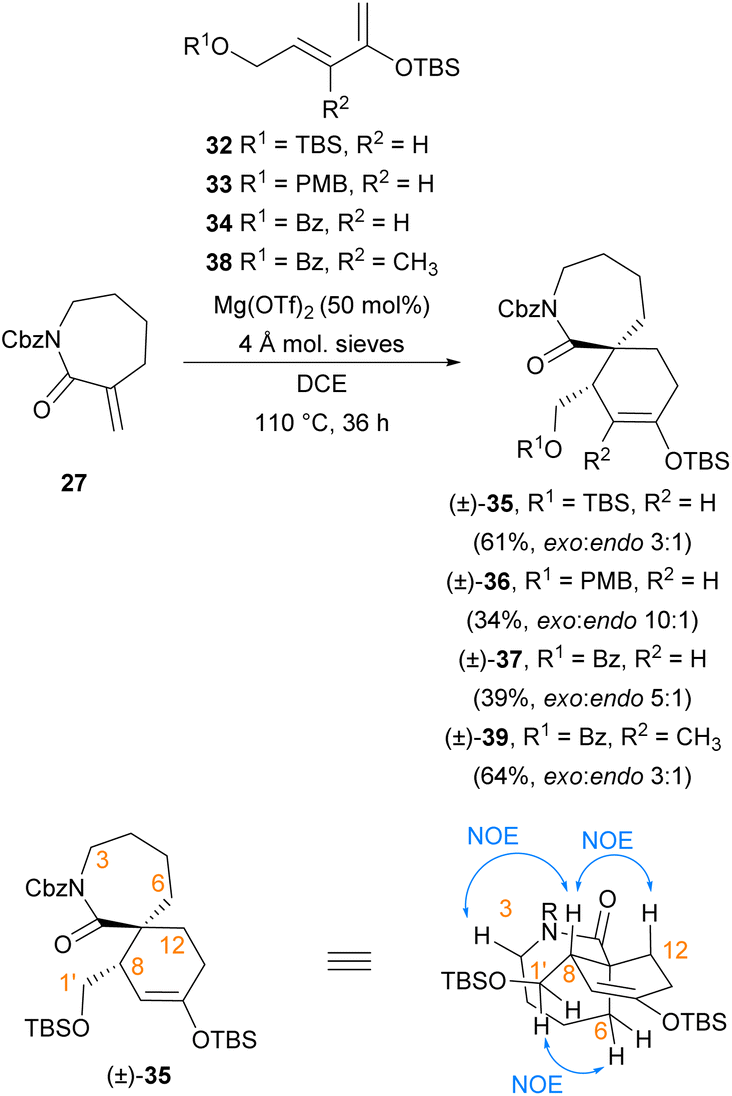 | ||
| Scheme 6 Diels–Alder cycloadditions of α-exo-methylene lactam 27 with silyl enol ether dienes. | ||
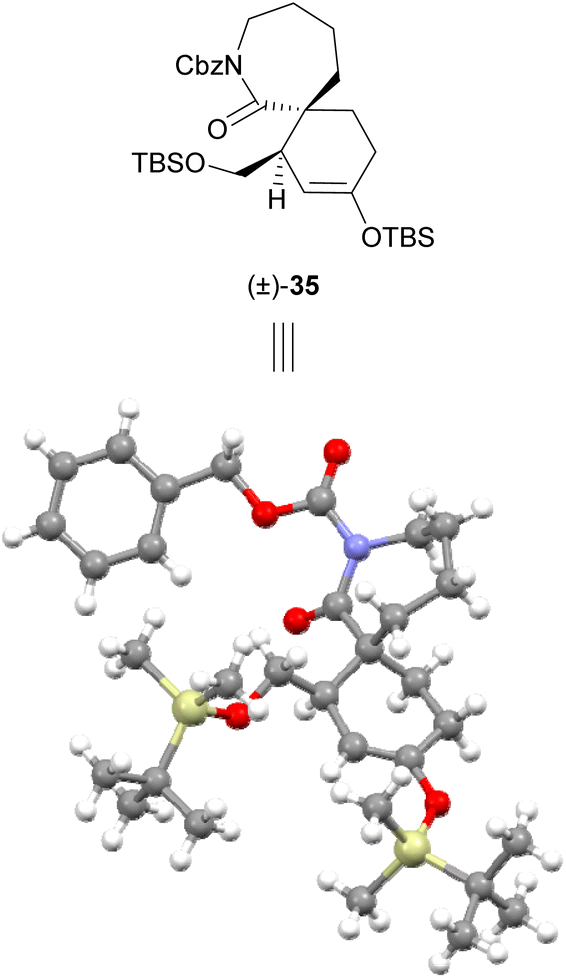 | ||
| Fig. 2 Crystal structure of exo-cycloadduct (±)-35.§ Enantiomer shown for clear comparison with X-ray crystal structure. | ||
Based on the relatively good balance of reactivity and diastereoselectivity observed in the Diels–Alder cycloaddition of diene 34 containing a benzoyl-protected sidechain, we elected to include this sidechain in more advanced diene 38 (Scheme 6), which bears the extra methyl substituent required for synthesis of the [7,6]-spirocyclic fragment of 13-desmethyl spirolide C (4). Inclusion of a benzoyl-protected alcohol sidechain would also allow for facile retention of alkene regiochemistry during elaboration of the spirocyclic unit. This sidechain has been previously demonstrated22 to enable regioselective deprotonation of the spirocyclic ketones arising from deprotection of the silyl enol ether moiety, which permits the selective formation of synthetic handles such as enol triflates with the desired alkene regiochemistry. Pleasingly, following synthesis of diene 38 using known methods,21 Diels–Alder cycloaddition between benzoyl-protected diene 38 and lactam 27 afforded the corresponding cycloadducts in an improved yield of 64%, although with reduced selectivity of 3:1 for the desired exo-adduct (±)-39 when compared to the analogous benzoyl-protected diene 34 (dr 5:1). This increased yield is likely due to a combination of stabilisation of the reactive s-cis diene conformation required for cycloaddition and increased electron density across the diene system arising from the extra methyl substituent of 38.
Following the successful synthesis of exo-adduct (±)-39 from simplified dienophile 27, our Diels–Alder cycloaddition reaction system was next applied to the synthesis of the spirocyclic core of 13-desmethyl spirolide C (4) using chiral lactam dienophile 8 (Scheme 7). In contrast to earlier cycloadditions (Scheme 6) with the simplified dienophile analogue 27, the Diels–Alder cycloaddition of lactam 8 bearing the chiral dimethyl moiety to diene 38 proceeded with complete exo-selectivity with no evidence of endo-cycloadduct formation observed. Exo-cycloadducts 16a and 16b were afforded as a 6:1 mixture of inseparable diastereomers in 47% yield, with the major exo-cycloadduct 16a bearing the desired (7R,8S)-stereochemistry of the natural product. The stereochemical identity of cycloadducts 16a and 16b as exo-cycloadducts was suggested through observation of similar NOE correlations to those seen in previously prepared exo-adducts (±)-35−(±)-37 and (±)-39, such as the important NOE correlation between H-8 and H-3 (Scheme 7). Major cycloadduct 16a was suggested to be the desired (7R,8S)-cycloadduct by observation of a key NOE correlation between H-5 and Hb-3, indicating that these were co-located on the same face of the lactam ring which was only expected in this diastereomer. This same key NOE correlation was not observed for minor cycloadduct 16b, supporting its assignment as the undesired (7S,8R)-cycloadduct. The facial selectivity of this reaction is likely to be dependent on the chiral dimethyl moiety, which renders one face of the lactam dienophile (8) more accessible to the approaching diene.17 When the facial selectivity imposed by the chiral dimethyl motif is combined with the inherent exo-preference of α-exo-methylene cyclic dienophiles,31 this results in a highly diastereoselective Diels–Alder cycloaddition between chiral lactam 8 and diene 38 which selectively affords the desired exo-cycloadduct 16a. The vinyl –OTBS group of cycloadduct 16a arising from use of diene 38 provides a convenient synthetic handle that can be elaborated to the required pendant butenolide of 13-desmethyl spirolide C (4) using methods previously reported by Romo et al. in the total synthesis of gymnodimine A (2).36 Additionally, as we have observed in previous work22 the inclusion of the benzoyl-protected sidechain assists with the regioselective deprotonation of spirocyclic ketones, such as that arising from silyl deprotection of cycloadduct 16a. Therefore, use of cycloadduct 16a will enable us to pursue coupling-based approaches towards the pendant butenolide that have previously met with limited success due to an inability to regioselectively deprotonate the corresponding spirocyclic ketone and install reactive functionalities such as enol triflates that are primed for subsequent coupling reactions.36
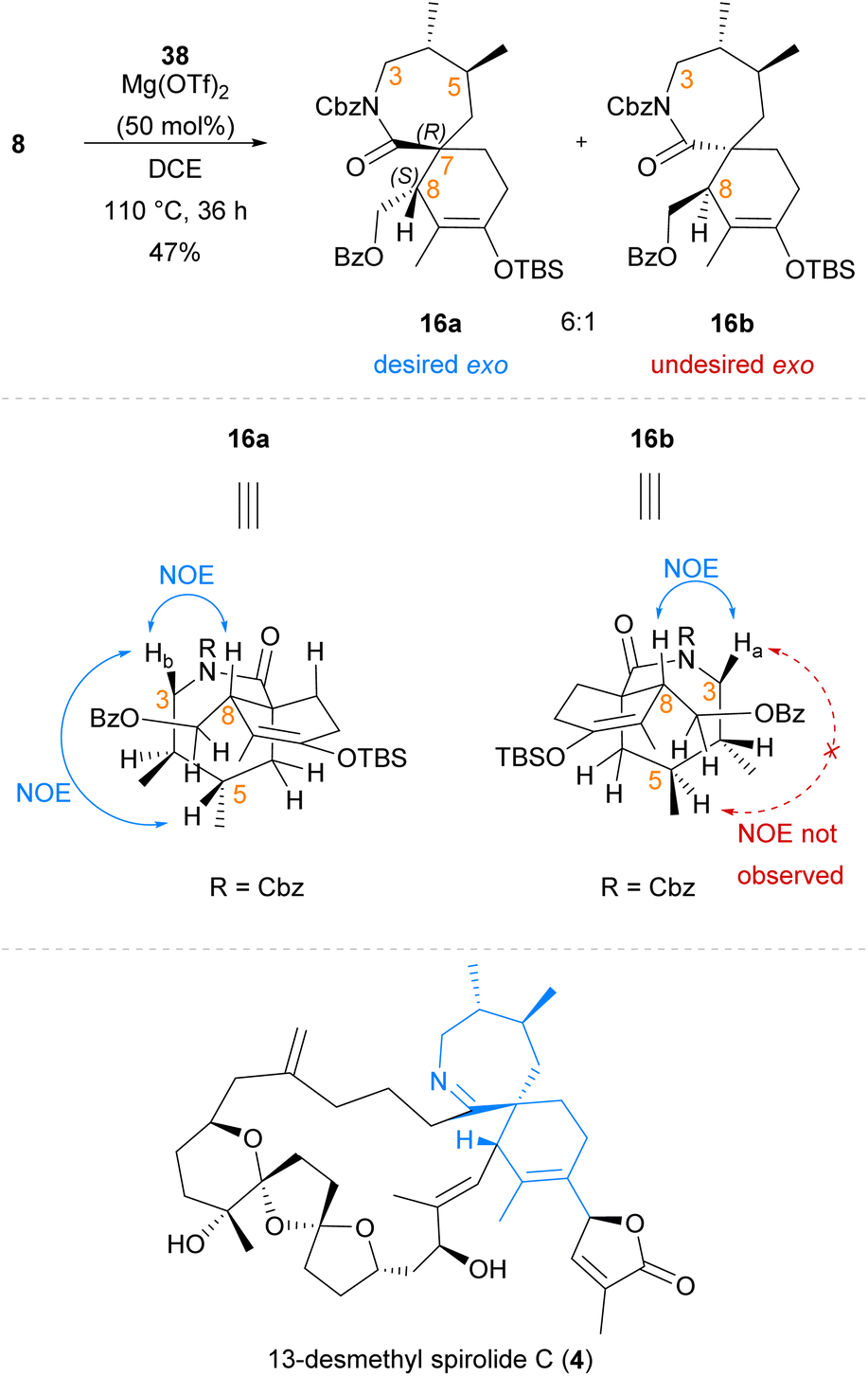 | ||
| Scheme 7 Synthesis of spirocyclic fragment 16 of 13-desmethyl spirolide C (4) via exo-selective Diels–Alder cycloaddition. | ||
Conclusions
In summary, an exclusively exo-selective Diels–Alder reaction of α-exo-methylene lactam 8 with diene 38 has been used to construct the spirocyclic fragment 16a bearing the desired (7R,8S)-stereochemistry of 13-desmethyl spirolide C (4). A novel approach to a chiral 7-membered α-exo-methylene lactam dienophile required for synthesis of the spirocyclic cores of the pinnatoxins, pteriatoxins, and spirolides was developed. Our approach begins from inexpensive starting materials and proceeds through a key aza-Claisen rearrangement to simultaneously install both stereocentres of the vicinal dimethyl moiety of the 7-membered ring with complete atom economy. The use of an aza-Claisen rearrangement to construct the chiral lactam dienophile (8) and a highly exo-selective Diels–Alder cycloaddition to set up the contiguous stereocentres of the spirocyclic core (16a) renders our overall strategy flexible and convergent. This approach is particularly suitable for convenient preparations of a range of spirocyclic analogues bearing diverse functionality and stereochemistry at the chiral positions, which will enable a comprehensive biological evaluation of this pharmacophore of 13-desmethyl spirolide C. A comprehensive study of the key Diels–Alder reaction was also undertaken to evaluate the stereoselectivity and reactivity of various functionalised dienes and protected lactam dienophiles. This included investigations into unprecedented exo-selective Diels–Alder cycloadditions between bromodienes and α-exo-methylene lactams. While use of less stable short-chain bromodienes in this reaction proved unsuccessful, the first synthesis of exo-cycloadducts was achieved with the use of a bromodiene bearing an extended sidechain, albeit in low yields. The resulting spirocyclic adducts possess a convenient vinyl bromide handle and can be readily elaborated to complex natural product intermediates with a full control of the alkene regiochemistry. Further investigations of the use of bromodienes in exo-selective Diels–Alder reactions are ongoing and will be reported in due course.Experimental
General information
All reactions were performed under an oxygen-free atmosphere of argon or nitrogen, dried by passage through a cylinder of anhydrous calcium chloride, in oven- or flame-dried glassware, unless otherwise stated. Solvents were dried using a solvent purifier (LC technology Solution Inc. SP-1 Standalone Solvent Purifier System). Solvents used for extraction and purification by flash chromatography were used as received unless otherwise noted. Commercially available starting materials and reagents were used as received unless otherwise noted. Reactions performed at low temperature were either cooled with an acetone/dry ice bath to reach −78 °C, or a water ice bath to reach 0 °C. The progress of reactions was monitored by analytical TLC using Merck 0.2 mm silica gel 60 F254 aluminium-backed plates with visualisation by ultraviolet irradiation (254 nm) followed by staining with potassium permanganate solution, or ethanolic solution of vanillin, or dinitrophenylhydrazine solution, or ninhydrin. Separation of mixtures was performed by flash chromatography using silica gel (60, 230–400 mesh) with the denoted solvent system. Yields refer to chromatographically and spectroscopically (1H NMR) homogenous materials, unless otherwise stated. Melting points were determined on a Kofler hot-stage apparatus. Optical rotations of chiral compounds were obtained on a PerkinElmer® 341 polarimeter, using sodium-D line (589 nm) at the indicated temperature in the indicated solvent. Concentrations are quoted in grams per 100 mL. NMR spectra were recorded at 21 °C, unless otherwise noted, on a Bruker® DRX400 spectrometer operating at 400 MHz for 1H nuclei and 100 MHz for 13C nuclei. All chemical shifts are recorded in parts per million from tetramethylsilane (δ 0.00) measured relative to the chloroform reference peaks at δ 7.26 and δ 77.16, or the DMSO reference peaks at δ 2.50 and δ 39.52, for 1H and 13C spectra, respectively. Coupling constants are reported in Hertz. 1H NMR spectra are reported as chemical shift in ppm, followed by relative integral, multiplicity (“s” singlet, “br s” broad singlet, “d” doublet, “dd” doublet of doublets, “ddd” doublet of doublet of doublets, “dt” doublet of triplets, “dtd” doublet of triplet of doublets, “dq” doublet of quartets, “t” triplet, “td” triplet of doublets, “q” quartet, “m” multiplet), and coupling constant where applicable. 13C NMR spectra are reported as chemical shift in ppm. High resolution mass spectrometry (HRMS) was performed using a VG-70SE spectrometer at a nominal accelerating voltage of 70 eV or on a Bruker micrOTOF-Q II mass spectrometer.:1) to afford the secondary amide (10.2 g, quant.) as a colourless, amorphous solid. Rf = 0.24 (pet. ether–EtOAc, 7:3); [α]21.0D −90.4 (c 0.95, CHCl3); (lit(37) [α]20D = −136 (c 1.0, CHCl3)); 1H NMR (400 MHz, CDCl3): δ 7.36–7.24 (m, 5H), 5.63 (br s, 1H), 5.14 (dq, J1 = J2 = 7.2 Hz, 1H), 2.21 (q, J = 7.6 Hz, 2H). 1.49 (d, J = 7.0 Hz, 3H) 1.16 (t, J = 7.5 Hz, 3H); 13C NMR (100 MHz, CDCl3): δ 172.7, 143.3, 128.7, 127.4, 126.2, 48.6, 29.8, 21.7, 9.8. The analytical data were in agreement with those reported in the literature.37
To a stirred solution of the secondary amide previously prepared (5.00 g, 28.2 mmol) in toluene (85 mL) was added K2CO3 (4.68 g, 33.9 mmol), NaOH (4.29 g, 107 mmol) and Bu4NHSO4 (1.25 g, 3.67 mmol). The resulting mixture was heated to 60 °C and a solution of crotyl bromide (85% w/w trans:cis, 7.42 g, 45.1 mmol) in toluene (10 mL) was then added dropwise. The mixture was then heated to 75 °C for 18 h before the mixture was filtered and the filtrate concentrated in vacuo. The crude product was purified by flash chromatography (pet. ether–EtOAc, 9:1) to afford tertiary amide 20 (4.53 g, 69%) as a yellow oil. Rf = 0.22 (pet. ether–EtOAc, 9:1); [α]21.0D −10.4 (c 1.25, CHCl3); (lit(26) [α]D −9.4 (c 1.06, CHCl3)); 1H NMR (400 MHz, CDCl3): δ 7.33–7.22 (m, 5H), 6.08 (q, J = 7.1 Hz, 1H), 5.46–5.39 (m, 1H), 5.18–5.14 (m, 1H), 3.76–3.38 (m, 2H), 2.35 (q, J = 7.3 Hz, 2H), 1.60 (d, J = 6.4 Hz, 3H), 1.49 (d, J = 7.1 Hz, 3H), 1.17 (t, J = 7.4 Hz, 3H); 13C NMR (100 MHz, CDCl3): δ 174.4, 141.3, 128.3, 128.1, 127.6, 127.4, 127.2, 50.9, 45.3, 27.0, 17.6, 16.8, 9.6. The analytical data were in agreement with those reported in the literature.26
:1 mixture of the crude diastereomers 15a and 15b. The crude product was purified by flash chromatography (pet. ether–EtOAc, 4:1) to afford the major diastereomer 15a (7.60 g, 76%) as an amorphous pale yellow solid. [α]21.0D −80.5 (c 0.79, MeOH) (lit(26) [α]16D −66.2 (c 1.00, MeOH)); 1H NMR (400 MHz, CDCl3): δ 7.36–7.24 (m, 5H), 5.82–5.73 (m, 1H), 5.61 (d, J = 6.5 Hz, 1H), 5.13 (dq, J1 = J2 = 7.3 Hz, 1H), 5.05–4.97 (m, 2H), 2.48–2.39 (m, 1H), 2.05 (dq, J = 7.1, 7.0 Hz, 1H), 1.47 (d, J = 6.9 Hz, 3H), 1.11 (d, J = 6.9 Hz, 3H), 1.02 (d, J = 6.9 Hz, 3H); 13C NMR (100 MHz, CDCl3): δ 174.4, 143.4, 141.9, 128.8, 127.5, 126.4, 114.5, 48.6, 47.0, 40.9, 21.8, 16.7, 14.7. The analytical data of 15a were in agreement with those reported in the literature.26 Minor diastereomer 15b was not isolated and its characterisation data was not obtained. The combined yield as 82% for 15a and 15b reported in Scheme 3 was calculated based on the isolated yield of 15a and the diasteromeric ratio observed in the 1H NMR spectrum of the crude product mixture.
:1, 1% Et3N) to afford the corresponding amine S12 (2.14 g, 93%) as a yellow oil. Rf = 0.66 (CH2Cl2–MeOH–NH3, 99:0.9:0.1); [α]20.0D −5.56 (c 1.8, CHCl3); νmax/cm−1: 3063, 3026, 2962, 2927, 2875, 1451, 1370, 1128, 909; 1H NMR (400 MHz, CDCl3): δ 7.32–7.30 (m, 4H), 7.25–7.20 (m, 1H), 5.74–5.65 (m, 1H), 4.94–4.89 (m, 2H), 3.71 (q, J = 6.6 Hz, 1H), 2.36 (ABX, ΔδAB = 0.35, JAB = 11.6 Hz, JAX = 8.2 Hz, JBX = 5.1 Hz, 2H), 2.18–2.10 (m, 1H), 1.60–1.53 (m, 1H), 1.33 (d, J = 6.6 Hz, 3H), 1.45–1.37 (m, 1H), 0.91 (d, J = 6.9 Hz, 3H), 0.85 (d, J = 6.8 Hz, 3H);13C NMR (100 MHz, CDCl3): δ 146.3, 143.6, 128.5, 126.9, 126.7, 113.3, 58.6, 52.2, 40.8, 38.2, 24.7, 15.7, 14.9. HRMS (ESI/Q-TOF) m/z: [M + H]+ calcd for C15H24N, 218.1903; found, 218.1911.
To a stirred solution of the intermediate amine S12 prepared above (1.17 g, 5.38 mmol) and triethylamine (1.1 mL, 8.1 mmol) in CH2Cl2 (36 mL) at 0 °C, di-tert-butyl dicarbonate (5.87 g, 26.9 mmol) was added portionwise over 10 min, followed by DMAP (0.723 g, 5.92 mmol). The reaction was then stirred at room temperature for 18 h. Following this, water (10 mL) was added, the layers were separated and the aqueous phase was extracted with CH2Cl2 (3 × 10 mL). The combined organic layers were dried over anhydrous Na2SO4, filtered, and concentrated in vacuo. The crude product was purified by flash chromatography (pet. ether–EtOAc, 9:1) to yield N-Boc amine 21 (1.30 g, 76%) as a colourless oil. Rf = 0.66 (pet. ether–EtOAc, 7:3); [α]20.0D −5.93 (c 1.50, CHCl3); νmax/cm−1: 2974, 2925, 2879, 1685, 1453, 1405, 1365, 1148, 1025, 762, 698; 1H NMR (400 MHz, D6-DMSO, 340 K): δ 7.35–7.22 (m, 5H), 5.68 (ddd, J = 16.9, 10.4, 7.2 Hz, 1H), 5.02 (q, J = 7.0 Hz, 1H), 4.95–4.88 (m, 2H), 3.08–3.02 (m, 1H), 2.95 (dd, J = 14.1, 9.0 Hz, 1H), 2.06–1.98 (m, 1H), 1.56–1.51 (m, 4H), 1.35 (br s, 9H), 0.83 (d, J = 6.9 Hz, 3H), 0.71 (d, J = 6.9 Hz, 3H); 13C NMR (100 MHz, D6-DMSO, 340 K): δ 155.0, 142.7, 142.1, 127.7, 126.4, 112.9, 78.3, 54.3, 48.5, 39.0, 36.1, 27.7, 17.4, 14.3, 12.9. HRMS (ESI/Q-TOF) m/z: [M + Na]+ calcd for C20H31NNaO2, 340.2247; found 340.2258.
:1 pentane:Et2O (100 mL). The resulting mixture was then filtered, and concentrated in vacuo. Purification by flash chromatography (hexanes–acetone, 4:1) afforded α,β-unsaturated ester 22 (2.71 g, 74%) as a colourless oil. Rf = 0.51 (pet. ether–EtOAc, 4:1); [α]20.5D −59.0 (c 1.00, CHCl3); νmax/cm−1: 2973, 2930, 1717, 1686, 1366, 1253, 1176, 1148, 1026, 986; 1H NMR (400 MHz, D6-DMSO, 340 K): δ 7.35–7.23 (m, 5H), 6.75 (dd, J = 15.7, 7.4 Hz, 1H), 5.72 (dd, J = 15.7, 1.3 Hz, 1H), 5.03 (q, J = 6.9 Hz, 1H), 4.12 (q, J = 7.1 Hz, 2H), 3.02 (ABX, ΔδAB = 0.09, JAB = 14.4, JAX = 6.0, JBX = 8.8 Hz, 2H), 2.26–2.17 (m, 1H), 1.65–1.59 (m, 1H), 1.53 (d, J = 7.1 Hz, 3H), 1.35 (s, 9H), 1.22 (t, J = 7.1 Hz, 3H), 0.87 (d, J = 6.8 Hz, 3H), 0.72 (d, J = 6.9 Hz, 3H); Significant signal broadening was observed in the 13C NMR spectrum of 22 due to the presence of rotamers, and the 13C signals are not reported. Attempts to obtain a clear 13C NMR at 340 K were not successful, as degradation of α,β-unsaturated ester 22 occurred during the experiment before resolution of rotamers; HRMS (ESI/Q-TOF) m/z: [M + Na]+ calcd for C23H35NNaO4, 412.2458; found 412.2446.
:1); [α]20.5D −90.4 (c 0.95, CHCl3); νmax/cm−1: 2963, 2927, 1706, 1259, 1172, 1092, 1014, 909, 795, 734; 1H NMR (400 MHz, CDCl3): δ 4.54 (s, 1H), 4.12 (q, J = 7.1 Hz, 2H), 3.10–2.94 (m, 2H), 2.37–2.22 (m, 2H), 1.70–1.60 (m, 2H), 1.55–1.46 (m, 2H), 1.43 (s, 9H), 1.25 (t, J = 7.1 Hz, 3H), 0.80 (d, J = 6.7 Hz, 6H); 13C NMR (100 MHz, CDCl3): δ 174.0, 156.2, 79.2, 60.4, 44.9, 37.4, 34.1, 32.7, 30.0, 28.6, 14.4, 14.1, 12.5; HRMS (ESI/Q-TOF) m/z: [M + Na]+ calcd for C15H29NNaO4, 310.1989; found, 310.1982.
:1) to afford lactam 24 (0.301 g, 64%) as a colourless crystalline solid. Rf = 0.15 (pet. ether–acetone, 1:1); m.p. 94–95 °C (lit. 96–97 °C); [α]21.2D −12.71 (c = 0.70, CHCl3); (lit(15) [α]24D −18.1 (c 0.54, CHCl3)); 1H NMR (400 MHz, CDCl3): δ 6.38 (br s, 1H), 3.06–3.03 (m, 2H), 2.51–2.37 (m, 2H), 1.84–1.79 (m, 1H), 1.45–1.28 (m, 3H), 0.98 (d, J = 6.0 Hz, 3H), 0.93 (d, J = 6.6 Hz, 3H); 13C NMR (100 MHz, CDCl3): δ 178.8, 48.3, 41.7, 40.4, 34.6, 30.8, 21.1, 18.4. The analytical data were in agreement with those reported in the literature.15
:3) to afford the corresponding N-Cbz lactam S13 (0.215 g, 74%) as a colourless oil. Rf = 0.47 (pet. ether–EtOAc, 1:1); [α]21.5D −69.0 (c = 0.88, CHCl3); (lit(16) [α]17D −70.0 (c 0.63, CHCl3)); 1H NMR (400 MHz, CDCl3): δ 7.44–7.42 (m, 2H), 7.39–7.31 (m, 3H), 5.28 (ABq, ΔδAB = 0.02 ppm, JAB = 12.6 Hz, 2H), 4.01–3.98 (m, 1H), 3.36 (dd, J = 15.3, 8.7 Hz, 1H), 2.69 (ABXY, ΔδAB = 0.09 ppm, JAB = 15.2, JAX = 11.3, JBY = 8.0, JAY = JBX = 2.0 Hz, 2H), 1.91–1.85 (m, 1H), 1.54–1.44 (m, 1H), 1.39–1.33 (m, 2H), 0.99 (d, J = 3.6 Hz, 3H), 0.98 (d, J = 3.2 Hz, 3H); 13C NMR (100 MHz, CDCl3): δ 175.5, 154.4, 135.7, 128.7, 128.3, 128.0, 68.6, 51.5, 41.3, 40.2, 38.1, 31.3, 20.9, 18.5. The analytical data were in agreement with those reported in the literature.16
To a stirred solution of the N-Cbz lactam S13 prepared above (0.250 g, 0.908 mmol) in THF (4 mL) was added LiHMDS (1.0 M in THF, 1.8 mL, 1.8 mmol) at −78 °C and the resulting mixture stirred for 1 h. 2,2,2-trifluoroethyl trifluoroacetate (1.10 mL, 8.1 mmol) was then added and the reaction mixture stirred at −78 °C for a further 40 min, before sat. aq. NH4Cl (12 mL) was added at −78 °C and the resulting mixture warmed to room temperature. The layers were separated and the aqueous layer was extracted with EtOAc (3 × 25 mL). The combined organic layers were dried over anhydrous Na2SO4, filtered, and concentrated in vacuo. The crude trifluoroacetate product obtained was redissolved in toluene (29 mL), to which was added K2CO3 (0.376 g, 2.72 mmol), 18-crown-6 (0.060 g, 0.227 mmol), and paraformaldehyde (1.09 g, 36.3 mmol). The resulting mixture was heated to 90 °C for 18 h, at which time the reaction mixture was allowed to cool to room temperature and sat. aq. NH4Cl (100 mL) was added. The layers were separated and the aqueous layer was extracted with EtOAc (3 × 165 mL). The organic layer was washed with brine (15 mL), dried over anhydrous Na2SO4, filtered, and concentrated in vacuo. Purification by flash chromatography (pet. ether–EtOAc 9:1) afforded α-exo-methylene lactam 8 (0.144 g, 55%) as a colourless oil. Rf = 0.63 (pet. ether–EtOAc, 1:1); [α]21.5D −94.5 (c = 0.96, CHCl3); (lit(15) [α]20D −92.8 (c 0.9, CHCl3)); 1H NMR (400 MHz, CDCl3): δ 7.44–7.29 (m, 5H), 5.85 (d, J = 1.3 Hz, 1H), 5.42 (d, J = 1.0 Hz, 1H), 5.29 (ABq, ΔδAB = 0.04 ppm, JAB = 12.6 Hz, 2H), 3.90 (dd, J = 15.0, 1.4 Hz, 1H), 3.28 (dd, J = 15.0, 8.4 Hz, 1H), 2.49 (dd, J = 13.7, 2.6 Hz, 1H), 2.14 (dd, J = 13.8, 9.4 Hz, 1H), 1.47–1.41 (m, 2H), 1.01 (d, J = 6.6 Hz, 3H), 0.97 (d, J = 6.5 Hz, 3H); 13C NMR (100 MHz, CDCl3): δ 172.3, 154.3, 145.3, 135.7, 128.7, 128.3, 127.9, 124.4, 68.5, 51.3, 41.9, 40.0, 39.8, 20.7, 17.9. The analytical data were in agreement with those reported in the literature.15
:1) to afford the corresponding N-Cbz lactam (3.62 g, 83%) as a pale yellow oil. 1H NMR (400 MHz, CDCl3): δ 7.44–7.29 (m, 5H), 5.28 (s, 2H), 3.85 (dd, J = 5.5, 4.0 Hz, 2H), 2.69 (dd, J = 6.8, 4.3 Hz, 2H), 1.80–1.73 (m, 6H); 13C NMR (100 MHz, CDCl3): δ 175.7, 154.5, 135.7, 128.7, 128.3, 128.0, 68.7, 46.5, 39.6, 29.3, 28.8, 23.6. The analytical data were in agreement with those reported in the literature.38 Methylenation of the intermediate protected lactam was performed at 500 mg scale via an analogous procedure to the synthesis of α-exo-methylene lactam 8. Purification by flash chromatography (pet. ether–EtOAc, 9:1) afforded α,β-unsaturated lactam 27 (0.687 g, 59%) as a colourless oil. Rf = 0.42 (pet. ether–EtOAc, 7:3); 1H NMR (400 MHz, CDCl3): δ 7.44–7.29 (m, 5H), 5.82 (s, 1H), 5.43 (d, J = 0.8 Hz, 1H), 5.29 (s, 2H), 3.75 (t, J = 5.4 Hz, 2H), 2.42 (t, J = 6.3 Hz, 2H), 1.80–1.72 (m, 4H); 13C NMR (100 MHz, CDCl3): δ 172.4, 154.2, 146.9, 135.8, 128.7, 128.3, 127.9, 124.0, 68.5, 46.5, 32.6, 29.2, 28.1. The analytical data were in agreement with those reported in the literature.33
:1) to afford the corresponding N-Ts lactam (9.65 g, 82%) as a colourless, amorphous solid. 1H NMR (400 MHz, CDCl3): δ 7.89–7.86 (m, 2H), 7.30 (d, J = 8.6 Hz, 2H), 4.02 (d, J = 5.0 Hz, 2H), 2.53 (t, J = 6.2 Hz, 2H), 2.42 (s, 3H), 1.84 (dt, J = 9.7, 5.3 Hz, 2H), 1.76–1.67 (m, 4H); 13C NMR (100 MHz, CDCl3): δ 174.9, 144.6, 136.8, 129.4, 128.7, 46.6, 38.9, 29.5, 29.4, 23.1, 21.8. The analytical data were in agreement with those reported in the literature.38 Methylenation of the intermediate protected lactam was performed at 500 mg scale via an analogous procedure to the synthesis of α-exo-methylene lactam 8. Purification by flash chromatography (pet. ether–EtOAc 9:1) afforded α-exo-methylene lactam 28 (0.534 g, 50%) as a colourless amorphous solid. Rf = 0.35 (pet. ether–EtOAc, 7:3); 1H NMR (500 MHz, CDCl3): δ 7.93 (d, J = 8.5 Hz, 2H), 7.32 (d, J = 8.3 Hz, 2H), 5.64 (s, 1H), 5.35 (d, J = 1 Hz, 1H), 3.85–3.83 (m, 2H), 2.43 (s, 3H), 2.39 (t, J = 5.8 Hz, 2H), 1.92–1.87 (m, 2H), 1.78–1.73 (m, 2H); 13C NMR (125 MHz, CDCl3): δ 171.5, 145.7, 144.8, 136.3, 129.5, 128.7, 123.7, 47.0, 31.8, 28.8, 28.0, 21.8. HRMS (ESI+) m/z: [M + Na]+ calculated for C14H17NNaO3S: 302.0821; found 302.0821.
:1) afforded a 5:1 mixture of inseparable diastereomers of cycloadduct (±)-30 (21 mg, 18%) as a colourless oil. Rf = 0.74 (pet. ether–EtOAc, 7:3); νmax/cm−1: 2942, 2866, 1711, 1466, 1380, 1267, 1103; 1H NMR (400 MHz, CDCl3): δ 7.38–7.28 (m, 5H), 6.19–6.17 (m, 0.8H), 6.14–6.12* (m, 0.2H), 5.24–5.15 (m, 2.4H), 3.89–3.81 (m, 1H), 3.68 (t, J = 6.2 Hz, 2.4H), 3.56–3.50 (m, 1H), 3.05–3.02 (m, 1H), 2.56–2.52* (m, 0.4H), 2.51–2.48 (m, 0.8H), 2.45–2.37 (m, 1H), 2.10–1.95 (m, 2H), 1.77–1.63 (m, 6H), 1.40–1.30 (m, 1H), 1.06–1.04 (m, 21H). 13C NMR (100 MHz, CDCl3): δ 182.8, 154.9, 136.1, 131.6, 128.7, 128.6, 128.2, 128.0, 127.9, 120.5, 68.2, 62.4, 50.7, 45.1, 43.5, 33.7, 32.2, 31.5, 27.5, 27.3, 23.2, 18.2, 12.1. HRMS (ESI/Q-TOF) m/z: [M + Na]+ calcd for C30H46BrNNaO4Si, 614.2272; found, 614.2257. *denotes minor diastereomer.
:1) afforded cycloadduct (±)-31 (12 mg, 11%) as a colourless oil. Rf = 0.73 (pet. ether–EtOAc, 7:3); νmax/cm−1: 2942, 2867, 1685, 1461, 1353, 1168, 1089, 973, 722, 681; 1H NMR (400 MHz, CDCl3): δ 7.86–7.83 (m, 2H), 7.28 (d, J = 8.2 Hz, 2H), 6.10 (d, J = 2.6 Hz, 1H), 4.24 (dd, J = 15.3, 5.4 Hz, 1H), 3.72–3.63 (m 3H), 3.02–2.98 (m, 1H), 2.41 (br s, 5H), 1.97–1.91 (m, 1H), 1.86–1.81 (m, 2H), 1.79–1.71 (m, 4H), 1.62–1.57 (m, 1H), 1.40 (dtd, J = 8.6, 6.4, 2.3 Hz, 1H), 1.31–1.27 (m, 1H), 1.07–1.04 (m, 21H); 13C NMR (100 MHz, CDCl3): δ 178.4, 144.3, 136.7, 131.3, 129.4, 128.6, 119.6, 62.4, 49.8, 45.1, 43.4, 33.5, 32.0, 31.4, 28.5, 26.5, 22.9, 21.8, 18.2, 12.1; HRMS (ESI/Q-TOF) m/z: [M + Na]+ calcd for C29H46BrNNaO4SSi, 634.1992; found, 634.1975.
General procedure 1 – preparation of Diels–Alder cycloadducts (±)-35–(±)-37, (±)-39, and 16
To a stirred mixture of dienophile (1 eq.), Mg(OTf)2 (50 mol%) and 4 Å mol. sieves (20 mg) in dry dichloroethane (0.3 M) was added a solution of diene (1.3 eq.) in dry dichloroethane (0.3 M) and the resulting mixture heated to 110 °C in a sealed pressure vessel for two days. The reaction mixture was then allowed to cool to room temperature, filtered, and concentrated in vacuo. The crude residue was purified by flash chromatography to afford the title compounds. In the syntheses of cycloadducts (±)-35–(±)-37, and (±)-39, the minor endo cycloadducts were inseparable from other components of the reaction mixture and were not isolated. The combined yields reported in Scheme 6 were calculated based on the isolated yield of the respective exo-adducts and the diasteromeric ratio observed in the 1H NMR spectrum of the crude product. The isolated yield of each exo-cycloadduct is indicated in the corresponding experimental procedure.:1) afforded exo-cycloadduct (±)-35 (52 mg, 46%) as a colourless crystalline solid. Rf = 0.80 (pet. ether–EtOAc, 7:3); m.p. = 69–70 °C; νmax/cm−1: 2952, 2929, 2857, 1709, 1672, 1251, 1168, 1100, 836, 776; 1H NMR (400 MHz, CDCl3): δ 7.39–7.29 (m, 5H), 5.22 (ABq, ΔδAB = 0.01 ppm, JAB = 12.6 Hz, 2H), 4.85 (d, J = 2.2 Hz, 1H), 3.97 (dd, J = 7.2, 7.2 Hz, 1H), 3.60 (dd, J = 9.4, 5.4 Hz, 1H), 3.48 (dd, J = 7.4, 6.3 Hz, 1H), 3.36–3.32 (m, 1H), 3.20–3.18 (m, 1H), 2.06–2.01 (m, 3H), 1.99–1.91 (m, 1H), 1.74–1.60 (m, 6H), 0.91 (s, 9H), 0.85 (s, 9H), 0.13 (s, 3H), 0.12 (s, 3H), −0.03 (s, 3H), −0.04 (s, 3H); 13C NMR (100 MHz, CDCl3): δ 182.9, 155.0, 149.3, 136.2, 128.6, 128.1, 127.7, 104.1, 68.1, 63.6, 49.8, 46.1, 44.4, 30.8, 27.1, 26.7, 26.0, 25.8, 25.2, 22.1, 18.3, 18.2, −4.3, −4.4, −5.3; HRMS (ESI/Q-TOF) m/z: [M + Na]+ calcd for C32H53NNaO5Si2, 610.3354; found, 610.3336. The minor endo-cycloadduct was inseparable from the enone arising from hydrolysis of diene 32 and the characterisation is not reported.
:1) afforded exo-cycloadduct (±)-36 (35 mg, 31%) as a colourless oil. Rf = 0.45 (pet. ether–EtOAc, 4:1); νmax/cm−1: 2954, 2932, 2856, 1709, 1250, 1170, 837; 1H NMR (400 MHz, CDCl3): δ 7.40–7.28 (m, 5H), 7.15–7.13 (m, 2H), 6.83–6.80 (m, 2H), 5.19 (ABq, ΔδAB = 0.07 ppm, JAB = 12.8 Hz, 2H), 4.71 (d, J = 2.0 Hz, 1H), 4.23 (ABq, ΔδAB = 0.10 ppm, JAB = 11.6 Hz, 2H), 3.98 (dd, J = 14.6, 6.8 Hz, 1H), 3.78 (s, 3H), 3.46–3.41 (m, 3H), 3.20–3.15 (m, 1H), 2.07–1.93 (m, 4H), 1.77–1.59 (m, 6H), 0.91 (s, 9H), 0.12 (s, 3H), 0.11 (s, 3H); 13C NMR (100 MHz, CDCl3): δ 183.1, 159.1, 154.9, 149.4, 136.2, 130.7, 129.3, 128.6, 128.1, 127.8, 113.8, 103.9, 72.4, 70.6, 68.1, 55.4, 49.5, 44.4, 43.6, 30.7, 27.3, 26.6, 25.8, 24.8, 22.0, 18.1, −4.2, −4.3; HRMS (ESI/Q-TOF) m/z: [M + Na]+ calcd for C34H47NNaO6Si, 616.3065; found, 616.3048. The minor endo-cycloadduct was afforded in a negligible amount and the characterisation is not reported.
:1) afforded exo-cycloadduct (±)-37 (36 mg, 31%) as a colourless oil. Rf = 0.63 (pet. ether–EtOAc, 7:3); νmax/cm−1: 2957, 2930, 2858, 1718, 1673, 1272, 1174, 839, 712; 1H NMR (400 MHz, CDCl3): δ 8.00–7.98 (m, 2H), 7.57–7.53 (m, 1H), 7.45–7.40 (m, 2H), 7.36–7.34 (m, 2H), 7.29–7.25 (m, 2H) 7.23–7.21 (m, 1H), 5.19 (ABq, ΔδAB = 0.07 ppm, JAB = 12.5 Hz, 2H), 4.81–4.80 (m, 1H), 4.21 (ABX, ΔδAB = 0.19 ppm, JAB = 10.4 Hz, JAX = 4.8 Hz, JBX = 8.8 Hz, 2H), 3.94–3.88 (m, 1H), 3.54–3.48 (m, 2H), 2.09–2.03 (m, 4H), 1.81–1.71 (m, 6H), 0.89 (s, 9H), 0.10 (s, 3H), 0.095 (s, 3H); 13C NMR (100 MHz, CDCl3): δ 182.6, 166.6, 154.8, 150.5, 135.9, 133.1, 130.2, 129.7, 128.6, 128.5, 128.2, 127.9, 103.1, 68.4, 65.8, 50.0, 44.7, 42.7, 30.4, 27.0, 26.6, 26.1, 25.8, 22.5, 18.1, −4.2, −4.4; HRMS (ESI/Q-TOF) m/z: [M + Na]+ calcd for C33H43NNaO6Si, 600.2752; found, 600.2736. The minor endo-cycloadduct was inseparable from the enone arising from hydrolysis of diene 34 and the characterisation is not reported.
:1) afforded cycloadduct (±)-39 (55 mg, 48%) as a colourless oil. Rf = 0.43 (pet. ether–EtOAc, 7:3); νmax/cm−1: 2956, 2928, 2856, 1719, 1272, 1177, 1111, 838, 712; 1H NMR (400 MHz, CDCl3) δ 8.02–7.99 (m, 2H), 7.59–7.54 (m, 1H), 7.46–7.42 (m, 2H), 7.36–7.28 (m, 4H), 7.25–7.22 (m, 1H), 5.16 (ABq, ΔδAB = 0.23 ppm, JAB = 12.4 Hz, 2H), 4.37 (ABX, ΔδAB = 0.19 ppm, JAB = 11.6 Hz, JAX = 5.6, JBX = 4.8 Hz, 2H), 3.98 (ddd, J = 14.2, 7.3, 2.0 Hz, 1H), 3.56 (ddd, J = 14.2, 10.2, 1.6 Hz, 1H), 3.03 (t, J = 5.0 Hz, 1H), 2.34–2.26 (m, 1H), 2.22–2.16 (m, 1H), 2.08–2.02 (m, 1H), 1.89 (dt, J = 13.5, 7.4 Hz, 2H), 1.83–1.76 (m, 3H), 1.65 (t, J = 1.7 Hz, 3H), 1.60–1.56 (m, 2H), 0.89–0.83 (m, 9H), 0.05 (s, 3H), 0.03 (s, 3H); 13C NMR (100 MHz, CDCl3): δ 181.2, 166.6, 155.1, 145.8, 136.3, 133.2, 130.2, 129.7, 128.6, 128.5, 127.9, 127.6, 110.8, 68.0, 65.2, 51.9, 45.6, 43.2, 32.9, 29.6, 27.4, 27.0, 25.9, 24.4, 18.3, 16.2, −3.6, −3.7; HRMS (ESI/Q-TOF) m/z: [M + Na]+ calcd for C34H45NNaO6Si, 614.2908; found, 614.2892. The minor endo-cycloadduct was inseparable from the enone arising from hydrolysis of diene 38 and the characterisation is not reported.
:1) afforded a 6:1 inseparable mixture of cycloadduct 16a and cycloadduct 16b as a colourless oil (41 mg, 47%). Rf = 0.43 (pet. ether–EtOAc, 4:1); [α]26.0D 33.2 (c 0.85, CHCl3); νmax/cm−1: 2958, 2925, 2854, 1720, 1460, 1383, 1273, 1192, 1177, 1110; 1H NMR (400 MHz, CDCl3): δ 8.02–7.96 (m, 2H), 7.57–7.52 (m, 1H), 7.42 (t, J = 7.7 Hz, 2H), 7.38–7.29 (m, 5H), 5.20* (ABq, ΔδAB = 0.03 ppm, JAB = 12.6 Hz, 0.3H), 5.18 (ABq, ΔδAB = 0.20 ppm, JAB = 12.7 Hz, 1.7H), 4.39 (ABX, ΔδAB = 0.11 ppm, JAB = 11.8 Hz, JAX = 6.1 Hz, JBX = 4.7 Hz, 2H), 4.01 (dd, J = 14.9, 2.3 Hz, 0.9H), 3.90* (dd, J = 15.1, 2.8 Hz, 0.1H) 3.57 (dd, J = 14.9, 7.1 Hz, 1H), 3.46–3.43* (m, 0.1H), 3.29* (dd, J = 15.2, 7.0 Hz, 0.1H), 3.20–3.18 (m, 0.9H), 2.32–2.25 (m, 0.9H), 2.13–2.05 (m, 2H), 1.91–1.77 (m, 2H), 1.66 (s, 3H), 1.60–1.57 (m, 1H), 1.40–1.37 (m, 1H), 1.34–1.29 (m, 1H), 0.99* (d, J = 6.5 Hz, 0.5H), 0.96 (d, J = 6.8 Hz, 2.5H), 0.94* (s, 1.3H), 0.89 (s, 7.7H), 0.85 (d, J = 6.5 Hz, 3H), 0.11* (s, 0.5H), 0.07 (s, 3H), 0.05 (s, 2.5H); 13C NMR (100 MHz, CDCl3): δ 178.8, 166.7, 155.6, 145.5, 136.1, 133.2, 130.2, 129.8, 128.6, 128.5, 128.1, 127.9, 110.6, 68.4, 65.4, 51.5, 50.8, 43.4, 42.2, 39.5, 36.3, 31.5, 27.5, 25.9, 21.3, 18.7, 18.3, 15.9, −3.6, −3.7; Distinct 13C signals arising from minor diastereomer 16b may not be observed due to the low amount present in the inseparable mixture. HRMS (ESI/Q-TOF) m/z: [M + Na]+ calcd for C36H49NNaO6Si, 642.3221; found, 642.3209. *denotes minor diastereomer 16b.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge funding support from The University of Auckland through the award of a Doctoral Scholarship to ADWE. The contributions of Tania Groutso for collection of crystallographic data and Liam Hunt for proofreading are acknowledged.References
- A. I. Selwood, A. L. Wilkins, R. Munday, F. Shi, L. L. Rhodes and P. T. Holland, Tetrahedron Lett., 2013, 54, 4705–4707 CrossRef CAS.
- T. Seki, M. Satake, L. Mackenzie, H. F. Kaspar and T. Yasumoto, Tetrahedron Lett., 1995, 36, 7093–7096 CrossRef.
- A. I. Selwood, C. O. Miles, A. L. Wilkins, R. van Ginkel, R. Munday, F. Rise and P. McNabb, J. Agric. Food Chem., 2010, 58, 6532–6542 CrossRef CAS PubMed.
- T. Hu, I. W. Burton, A. D. Cembella, J. M. Curtis, M. A. Quilliam, J. A. Walter and J. L. C. Wright, J. Nat. Prod., 2001, 64, 308–312 CrossRef CAS PubMed.
- T. A. Hauser, C. D. Hepler, D. C. Kombo, V. P. Grinevich, M. N. Kiser, D. N. Hooker, J. Zhang, D. Mountfort, A. Selwood, S. R. Akireddy, S. R. Letchworth and D. Yohannes, Neuropharmacology, 2012, 62, 2239–2250 CrossRef CAS PubMed.
- R. Araoz, D. Servent, J. Molgó, B. I. Iorga, C. Fruchart-Gaillard, E. Benoit, Z. Gu, C. Stivala and A. Zakarian, J. Am. Chem. Soc., 2011, 133, 10499–10511 CrossRef CAS PubMed.
- E. Alonso, C. Vale, M. R. Vieytes, F. M. Laferla, L. Giménez-Llort and L. M. Botana, Neurochem. Int., 2011, 59, 1056–1065 CrossRef CAS PubMed.
- E. Alonso, P. Otero, C. Vale, A. Alfonso, A. Antelo, L. Gimenez-Llort, L. Chabaud, C. Guillou and L. M. Botana, Curr. Alzheimer Res., 2013, 10, 279–289 CrossRef CAS PubMed.
- T. E. Golde, Mol. Neurodegener., 2022, 17, 18 CrossRef CAS PubMed.
- Y. Bourne, Z. Radic, R. Araoz, T. T. Talley, E. Benoit, D. Servent, P. Taylor, J. Molgo and P. Marchot, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 6076–6081 CrossRef CAS PubMed.
- J. A. McCauley, K. Nagasawa, P. A. Lander, S. G. Mischke, M. A. Semones and Y. Kishi, J. Am. Chem. Soc., 1998, 120, 7647–7648 CrossRef CAS.
- F. Matsuura, J. Hao, R. Reents and Y. Kishi, Org. Lett., 2006, 8, 3327–3330 CrossRef CAS PubMed.
- S. Sakamoto, H. Sakazaki, K. Hagiwara, K. Kamada, K. Ishii, T. Noda, M. Inoue and M. Hirama, Angew. Chem., Int. Ed., 2004, 43, 6505–6510 CrossRef CAS PubMed.
- D. Marcoux, P. Bindschädler, A. W. H. Speed, A. Chiu, J. E. Pero, G. A. Borg and D. A. Evans, Org. Lett., 2011, 13, 3758–3761 CrossRef CAS PubMed.
- J. Ishihara, S. Tojo, T. Makino, H. Sekiya, A. Tanabe, M. Shiraishi, A. Murai and S. Hatakeyama, Heterocycles, 2017, 95, 422 CrossRef CAS PubMed.
- H. Tsuchikawa, K. Minamino, S. Hayashi and M. Murata, Asian J. Org. Chem., 2017, 6, 1322–1327 CrossRef CAS.
- K. Minamino, M. Murata and H. Tsuchikawa, Org. Lett., 2019, 21, 8970–8975 CrossRef CAS PubMed.
- C. D. Lu and A. Zakarian, Org. Synth., 2008, 85, 158 CrossRef CAS.
- T. Tsunoda, M. Sakai, O. Sasaki, Y. Sako, Y. Hondo and S. Itô, Tetrahedron Lett., 1992, 33, 1651–1654 CrossRef CAS.
- P. O'Connor, K. Körber and M. Brimble, Synlett, 2008, 1036–1038 Search PubMed.
- Z. Wang, N. Krogsgaard-Larsen, B. Daniels, D. P. Furkert and M. A. Brimble, J. Org. Chem., 2016, 81, 10366–10375 CrossRef CAS PubMed.
- J. L. Freeman, M. A. Brimble and D. P. Furkert, Org. Biomol. Chem., 2019, 17, 2705–2714 RSC.
- J. L. Freeman, F. F. Li, D. P. Furkert and M. A. Brimble, Synlett, 2020, 657–671 CAS.
- T. Tsunoda, T. Nishii, M. Yoshizuka, C. Yamasaki, T. Suzuki and S. Itô, Tetrahedron Lett., 2000, 41, 7667–7671 CrossRef CAS.
- T. Nishii, F. Miyamae, M. Yoshizuka, H. Kaku, M. Horikawa, M. Inai and T. Tsunoda, Tetrahedron: Asymmetry, 2012, 23, 739–741 CrossRef CAS.
- C. E. Rye and D. Barker, J. Org. Chem., 2011, 76, 6636–6648 CrossRef CAS PubMed.
- G. Broggini, L. Garanti, G. Molteni, T. Pilati, A. Ponti and G. Zecchi, Tetrahedron: Asymmetry, 1999, 10, 2203–2212 CrossRef CAS.
- N. Dittrich, L. I. Pilkington, E. Leung and D. Barker, Tetrahedron, 2017, 73, 1881–1894 CrossRef CAS.
- M. V. Riofski, J. P. John, M. M. Zheng, J. Kirshner and D. A. Colby, J. Org. Chem., 2011, 76, 3676–3683 CrossRef CAS PubMed.
- H. Choi, H. J. Shirley, H. R. M. Aitken, T. Schulte, T. Söhnel, P. A. Hume, M. A. Brimble and D. P. Furkert, Org. Lett., 2020, 22, 1022–1027 CrossRef CAS PubMed.
- W. R. Roush and B. B. Brown, J. Org. Chem., 1992, 57, 3380–3387 CrossRef CAS.
- J. A. Berson, Z. Hamlet and W. A. Mueller, J. Am. Chem. Soc., 1962, 84, 297–304 CrossRef CAS.
- J. Ishihara, M. Horie, Y. Shimada, S. Tojo and A. Murai, Synlett, 2002, 0403–0406 CrossRef CAS.
- L. Wang and M. E. Welker, J. Org. Chem., 2012, 77, 8280–8286 CrossRef CAS PubMed.
- S. De, C. Day and M. E. Welker, Tetrahedron, 2007, 63, 10939–10948 CrossRef CAS.
- K. Kong, Z. Moussa, C. Lee and D. Romo, J. Am. Chem. Soc., 2011, 133, 19844–19856 CrossRef CAS PubMed.
- R. S. L. Chapman, J. D. Tibbetts and S. D. Bull, Tetrahedron, 2018, 74, 5330–5339 CrossRef CAS.
- J. Guo, J. D. Harling, P. G. Steel and T. M. Woods, Org. Biomol. Chem., 2008, 6, 4053 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details and spectra of novel compounds. CCDC 2205771. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2ob01992b |
| ‡ US60.7 per g (10) vs. US$4.1 per g (14), Merck Sigma Aldrich (New Zealand), 2022. |
| § For preparation of dienes 26, 32–34, and 38; details on attempted Diels–Alder cycloadditions using lactams 27 or 28 and boron-substituted furans; 1H and 13C NMR spectra of compounds 8, 15a, 16a, 16b, 21–24, 26, 28, and 30–39, key NOESY correlations of cycloadducts (±)-30, (±)-31, (±)-35–(±)-37, (±)-39, 16a and 16b; and details regarding the X-ray crystal structure of (±)-35. |
| This journal is © The Royal Society of Chemistry 2023 |