
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis and ring-opening metathesis polymerisation of o-alkoxy benzothiadiazole paracyclophane-1,9-dienes†
Yurachat
Janpatompong
,
Venukrishnan
Komanduri
,
Raja U.
Khan
and
Michael L.
Turner
 *
*
Organic Materials Innovation Centre (OMIC), Department of Chemistry, The University of Manchester, Oxford Road, Manchester, M13 9PL, UK. E-mail: michael.turner@manchester.ac.uk
First published on 21st March 2023
Abstract
ortho-Diethylhexyloxyphenylene benzothiadiazole paracyclophane-1,9-diene as a mixture of diastereomers was synthesized by a sequential benzyne-induced Stevens rearrangement, oxidation and pyrolysis of a dithia[3.3]paracyclophane. Reaction of these highly strained cyclophanedienes with the second generation Grubbs catalyst showed that they can be ring opened to alternating cis,trans-phenylenevinylene polymers. In situ NMR experiments showed that one isomer 8a polymerised to 90% conversion, whereas the other 8b gave only 9% conversion due to steric hindrance on both faces of the alkene bridges of this isomer. The resulting polymers can be readily isomerized in dilute solution using visible light to the all-trans isomer and the optical and electrochemical properties of these polymers were examined by theory and experiment.
Introduction
[2.2]Paracyclophanes contain distorted π-electron systems,1–5 in which the close proximity of the coplanar benzene rings leads to unique electronic properties that have been utilised in asymmetric catalysis for planar chiral derivatives, chemical sensors and liquid crystals.6–12 [2.2]Paracyclophanedienes, were first synthesised by Cram and Dewhirst in the 1960s13 and studies have shown that introducing a double bond into the bridges increases the π-interactions between the coplanar aromatic rings and the angles between the bridgehead carbons.1,14,15 The aromatic rings in many cyclophanes are not planar and are distorted into boat, chair and twisted conformations, consistent with the highly strained nature of these molecules.1,16–18 The release of this ring strain by metathesis of one of the bridging alkenes in the [2.2]paracyclophanedienes leads to these molecules being effective monomers for ring opening metathesis polymerisation (ROMP) to give well-defined poly(p-phenylenevinylenes) (PPVs).19–25 Paracyclophanedienes with benzothiadiazole (BT) and 2,5-dialkoxy phenylene rings have been reported by Elacqua and Turner et al.23,26 These asymmetric [2.2]paracyclophane-1,9-dienes with electron rich and electron poor building blocks enable the preparation of donor–acceptor PPVs by ROMP.23,26,27 The alternation of the donor and acceptor moieties within the polymer backbone reduces the polymer band gap and strongly red shifts the optical properties of the material.28–33To date most [2.2]paracyclophane-1,9-dienes reported are substituted at the 2,5-positions (para) of the phenylene rings. Substitution at the 2,3-positions (ortho) is little reported.24,25,34 but this substitution pattern is known to lead to PPVs that show significantly blue-shifted optical properties over the comparable 2,5-isomers. o-Dialkoxyphenylene benzothiazole paracyclophane-1,9-dienes are unprecedented and in this contribution we report the preparation of these highly strained compounds and examine the stereospecific ring opening of the alkene bridges by alkene metathesis (Scheme 1).
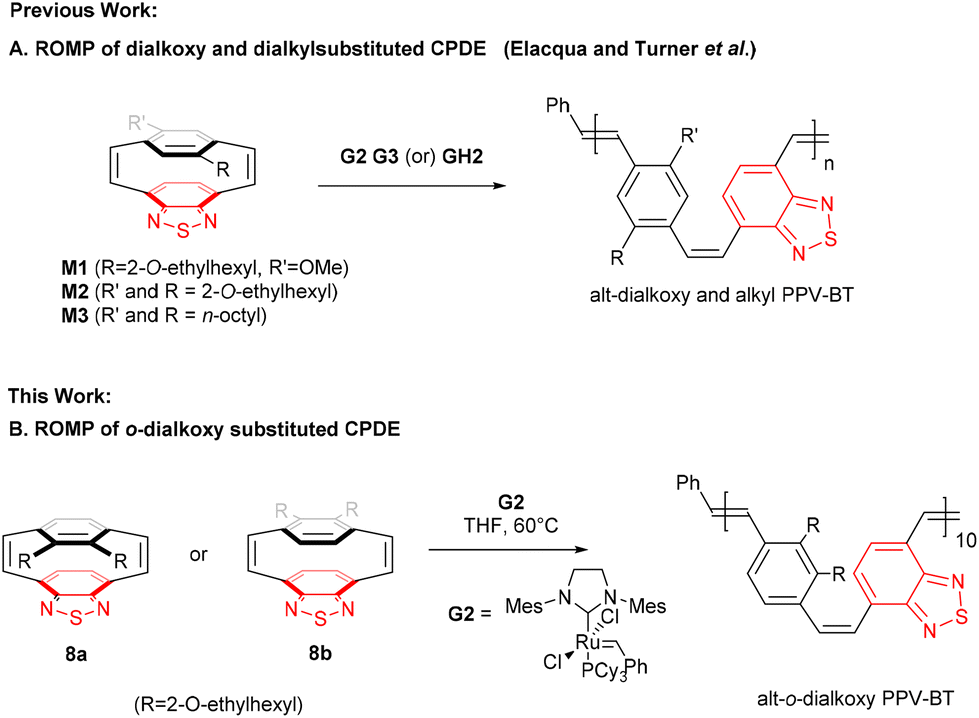 | ||
| Scheme 1 (A) ROMP approach to cis/trans D–A dialkoxy and dialkyl D–A PPVBT copolymers.23,26 (B) ROMP approach to o-alkoxy D–A PPVBT copolymer. | ||
Results and discussion
The synthesis of [3.3]dithiaparacyclophane 4 was achieved by slow addition of a deoxygenated toluene solution of 235 and 3 to a large volume of deoxygenated ethanol containing KOH (Scheme 2). The slow addition reduced the formation of linear and larger cyclic oligomers and favoured the desired intramolecular cyclisation between 2 and 3. | ||
| Scheme 2 Synthesis of monomers 8a and 8b. | ||
The 1H NMR spectrum of dithiacyclophane 4 is shown in Fig. 1. Several distinct regions are apparent corresponding to the structural units of the molecule and it is clear that only one isomer of 4 is isolated in high yield (75%) by column chromatography. Two singlets, integrating to two hydrogens, at 7.10 ppm (a) and 5.67 ppm (b) were assigned to the four hydrogens of the disubstituted aromatic rings. The doublets at 4.54 and 3.43 ppm integrating to two hydrogens can be assigned to the four thioether hydrogens connected to benzothiadiazole. The other four thioether hydrogens are observed as doublets at 4.04 and 3.94 ppm. The complex multiplets for the OCH2 hydrogens of the ethylhexyl side chains are observed at 3.98 and 3.80 ppm as shown in the inset of Fig. 1. Based on literature precedence36 and theoretical calculations (see ESI†), the isomer is assigned as shown in Fig. 1 and Fig. S1.†
 | ||
| Fig. 1 1H NMR spectrum of dithia[3.3]paracyclophane 4 in CDCl3. | ||
A benzyne-induced Stevens rearrangement was performed by the dropwise addition of tetrabutylammonium fluoride trihydrate to a solution of 2-(trimethysilyl)phenyl trifluoromethanesulfonate 5 and 4 in THF at room temperature over a period of 5 hours. After evaporation of the solvent the crude product was purified by flash column chromatography, initially eluting with petroleum ether to remove biphenylene (by-product from the dimerisation of benzyne), followed by elution with 30% DCM/petroleum ether to obtain the mixture of bis-phenyl sulfides 6 as a highly viscous yellow oil in 65% yield. The benzyne-induced Stevens rearrangement is not regioselective and migration of the phenyl sulfide group can occur to either α-carbons (either adjacent to a substituted or unsubstituted ring). Additionally each of these α-carbons is a stereogenic centre and when coupled with the planar chirality this results in a large number of possible isomers of 6, therefore, the 1H NMR spectrum of compound 6 is very complex with a large number of overlapping signals (see ESI, Fig. S8†). High resolution mass spectrometry (HRMS) gave a molecular ion of 739.2341 m/z for 6. This mixture of compounds 6 was then oxidized by addition of hydrogen peroxide (2.2 eq.) in acetic acid and toluene, at room temperature for 20 hours. This gave a complex mixture of compounds 7 as each sulfoxide group is also an additional stereogenic centre (see ESI, Fig. S9†). Thermal elimination of the phenyl sulfoxide groups from compound 7 to generate cis-vinylene bonds was achieved by heating to 150 °C in deoxygenated, anhydrous o-xylene, under argon for 20 hours. After solvent removal, the crude product was purified by flash column chromatography and an inseparable mixture of the syn- and anti-cyclophanedienes, 8a and 8b, in a ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 (by integration of the aromatic protons), was isolated as a reddish oil in a yield of 33%. The 1H NMR spectrum of the cyclophanedienes 8a and 8b is shown in Fig. 2. The hydrogens on the benzothiadiazole rings for the two isomers are observed as singlets at 6.60 ppm for 8a and 6.92 ppm for 8b in a ratio of 1:3. The hydrogens of the phenylene rings appear at 6.13 ppm for 8a and 5.13 for 8b, the upfield shift of the hydrogens associated with 8b is consistent with these hydrogens lying under the thiadiazole ring. The integration of these hydrogens, confirmed the 1:3 ratio of the two isomers of 8. The vinylene hydrogens were observed as two different doublets; at 7.16 ppm and 6.90 ppm with J = 10 Hz in 8a and 7.09 ppm and 6.85 ppm with J = 10 Hz in 8b. The protons of OCH2 group bonded directly to the aromatic ring are diastereotopic and in cyclophanediene 8a these OCH2 appear as two multiplets between 3.23–3.27 and 3.65–3.71 ppm. In 8b these OCH2 also appear as two multiplets between 3.57–3.64 ppm and 3.78–3.86 ppm. The temperature dependence of the mixture of isomers was studied by 1H NMR spectroscopy (Fig. S15–17†). This showed that the aromatic hydrogens on the phenylene rings (Ha′) and the cis-vinylene bonds (Hc′) for 8b appeared as multiplets at temperatures below 293 K. These signals coalesced as the temperature was raised to above 300 K resulting in a sharp singlet and a doublet at the average chemical shift of the multiplets observed at lower temperatures. When the sample was cooled, the original spectrum at room temperature was recovered, consistent with a conformational interconversion. There was no interconversion between isomers 8a and 8b observed even at the highest temperatures (325 K).
:3 (by integration of the aromatic protons), was isolated as a reddish oil in a yield of 33%. The 1H NMR spectrum of the cyclophanedienes 8a and 8b is shown in Fig. 2. The hydrogens on the benzothiadiazole rings for the two isomers are observed as singlets at 6.60 ppm for 8a and 6.92 ppm for 8b in a ratio of 1:3. The hydrogens of the phenylene rings appear at 6.13 ppm for 8a and 5.13 for 8b, the upfield shift of the hydrogens associated with 8b is consistent with these hydrogens lying under the thiadiazole ring. The integration of these hydrogens, confirmed the 1:3 ratio of the two isomers of 8. The vinylene hydrogens were observed as two different doublets; at 7.16 ppm and 6.90 ppm with J = 10 Hz in 8a and 7.09 ppm and 6.85 ppm with J = 10 Hz in 8b. The protons of OCH2 group bonded directly to the aromatic ring are diastereotopic and in cyclophanediene 8a these OCH2 appear as two multiplets between 3.23–3.27 and 3.65–3.71 ppm. In 8b these OCH2 also appear as two multiplets between 3.57–3.64 ppm and 3.78–3.86 ppm. The temperature dependence of the mixture of isomers was studied by 1H NMR spectroscopy (Fig. S15–17†). This showed that the aromatic hydrogens on the phenylene rings (Ha′) and the cis-vinylene bonds (Hc′) for 8b appeared as multiplets at temperatures below 293 K. These signals coalesced as the temperature was raised to above 300 K resulting in a sharp singlet and a doublet at the average chemical shift of the multiplets observed at lower temperatures. When the sample was cooled, the original spectrum at room temperature was recovered, consistent with a conformational interconversion. There was no interconversion between isomers 8a and 8b observed even at the highest temperatures (325 K).
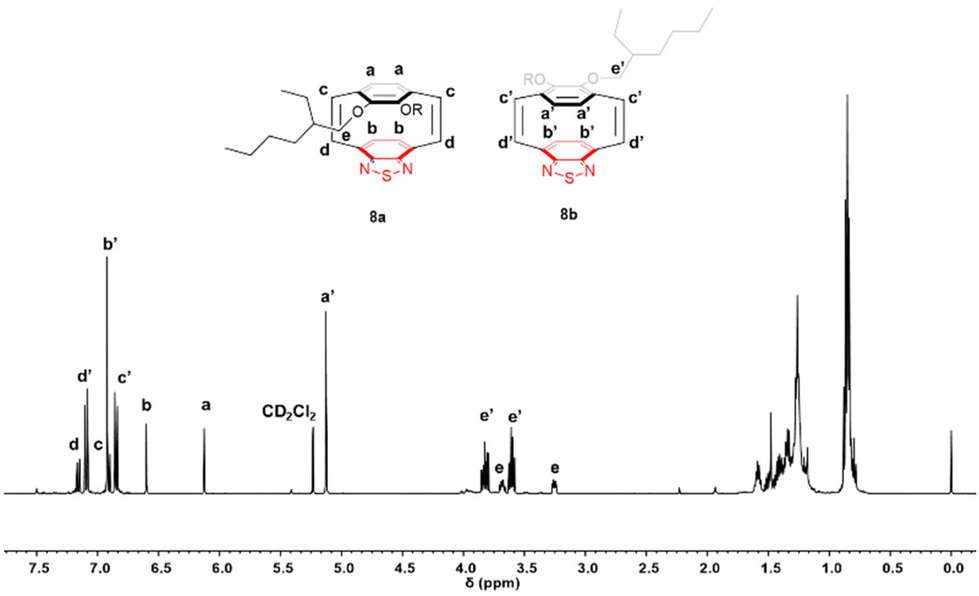 | ||
| Fig. 2 1H NMR spectrum of 8a and 8b in CD2Cl2. | ||
It was not possible to crystallise either 8a or 8b and determine the solid state structure. Hence the ground state geometry of 8a and 8b was calculated using density-functional theory calculations (B3LYP/6-311G(d,p)). Both aromatic systems display pseudo-boat conformations. The intramolecular distance between the two carbons of the phenylene group bonded to the vinylene bond is predicted to be 1.5 Å for both 8a and 8b. The vinylene bond length in both 8a and 8b are calculated to be around 1.35 Å which is in agreement with that of a standard cis-vinylene bond 1.32 Å.37 Each isomer shows a highly strained structure with estimated ring strains of 58 kcal mol−1 (8a) and 55 kcal mol−1 (8b). The calculated π–π distances for 8a and 8b are very short at 3.06 and 3.04 Å respectively due to the electrostatic attraction between the electron rich and electron poor aromatic rings (Fig. 3).
 | ||
| Fig. 3 DFT (Density-Functional Theory) optimized geometry of isomers of 8: (a and b) Wireframe views of 8a and 8b. (c and d) Space-filling views of 8a and 8b. | ||
Initial ROMP studies of monomers 8a and 8b with G2 initiator was conducted using an in situ1H NMR experiment (Fig. 4). Polymerization of monomer 8a and 8b was performed in THF-d8 at 60 °C ([8a]/[G2] = 10) and ([8b]/[G2] = 30); after 5 min a signal at δ 16.32 ppm corresponding to the presence of the O-chelated carbene intermediate and a signal at δ 17.31 ppm corresponding to the N-chelated carbene intermediate were observed but significant quantities of unreacted G2 remained. After 2 h the initiator was completely consumed and after 24 h the reaction was quenched by adding an excess of ethyl vinyl ether. The crude product was purified by precipitation (MeOH/Celite) followed by elution with chloroform and the desired polymer, 9, was obtained (Mn(calc.) = 5.2 kDa, Mn(obs.) = 5.8 kDa, Đ = 1.56). This polymer was derived by the polymerisation of isomer 8a and 8b. Specifically, ROMP of isomer 8a led to 90% conversion, whereas 8b afforded only 9% conversion (see Fig. S18†). We attribute the higher relative reactivity of 8a to the presence of an open face of the vinylene bridge (see red arrow) at which the catalyst is able to form the desired metallacyclobutane intermediate by a ring opening methathesis reaction (see Fig. 3c). A similar observation has been seen before in the polymerisation of the tetraalkoxy 2,5-substituted paracyclophanediene derivatives.17,36 By comparison in compound 8b, the optimised structure (Fig. 3d) shows steric crowding at both faces of the vinylene bridges by the o-alkoxy and benzothiazole groups.
 | ||
| Fig. 4 ROMP of monomer 8a and 8b with G2 catalyst; in situ1H NMR experiment – carbene region. | ||
The MALDI-TOF mass spectrum of the 2,3-dialkoxy PPV 9 (Fig. 5) showed a main series of peaks ( ) separated by an interval of 519 mass units, corresponding to the molecular weight of the monomer. This is consistent with the desired polymer terminated with phenyl and vinyl end groups as expected (total mass 104). A further minor series of peaks (
) separated by an interval of 519 mass units, corresponding to the molecular weight of the monomer. This is consistent with the desired polymer terminated with phenyl and vinyl end groups as expected (total mass 104). A further minor series of peaks ( ) was consistent with the formation of cyclic polymers via intramolecular secondary metathesis as the active chain end of a propagating polymer can react at the cis vinylenes on the same chain, creating a lower molecular weight polymer and a cyclic oligomer. A similar observation has been seen before in the polymerisation of 2,5-dialkoxy PPV polymers.36
) was consistent with the formation of cyclic polymers via intramolecular secondary metathesis as the active chain end of a propagating polymer can react at the cis vinylenes on the same chain, creating a lower molecular weight polymer and a cyclic oligomer. A similar observation has been seen before in the polymerisation of 2,5-dialkoxy PPV polymers.36
 | ||
Fig. 5 MALDI-TOF-MS of 9, ( ) peaks corresponding to polymers with phenyl and vinyl end groups, and ( ) peaks corresponding to polymers with phenyl and vinyl end groups, and ( ) peaks corresponding to cyclic oligomers. ) peaks corresponding to cyclic oligomers. | ||
The 1H NMR spectrum of 9 (Fig. 6) was recorded in CDCl3. The peak at 3.83 ppm can be assigned to the hydrogens of the methylene groups attached to the oxygen for the cis and trans vinylene links. Signals for the cis vinylene groups appear between 6.67 to 7.01 ppm, and those for the trans vinylene and other aromatic hydrogens appear further downfield after 7.01 ppm. The copolymer 9 was initially isolated with alternating cis/trans vinylene stereochemistry. Isomerization to the all trans form was achieved by vigorously stirring dilute solutions (1 mg mL−1 in dichloromethane) of 9 under exposure to visible light in the absence of oxygen. The cis vinylene and phenyl signals can be seen to disappear after isomerisation. In SEC analysis lower retention times were observed for the trans isomer relative to their cis/trans form due to larger hydrodynamic volume of the trans isomer (Fig. S23†).
 | ||
| Fig. 6 1H NMR spectra (CDCl3) of 9 taken before (bottom) and after (top) photoisomerization. | ||
The absorption and emission spectra of the D–A polymers were recorded in chloroform solution (Fig. 7). The cis/trans polymer exhibits a λmax of 479 nm, with the PL λmax at 605 nm, the trans polymer shows a λmax of 530 nm, with the PLmax at 621 nm (see ESI, Fig. S24 and Table S1†). The spectra were red shifted when compared to the analogous 2,3-dialkoxy PPV prepared by the ROMP of a 2,3-dialkoxyparacyclophanediene (absλmax = 384 nm and emλmax = 490 nm).25 They were also red-shifted over the 2,3-dialkoxy PPV polymers reported by Holmes et al., these polymers have two alkoxy groups in the 2,3 position of every phenylene ring, and they exhibited a λmax of 454 nm and a PLmax of 519 nm.34 Incorporation of the BT unit in 9 effectively red-shifts the absorption and emission maximum over the spectra of the reported 2,3-dialkoxy PPV polymers. By comparison the absorption and PL spectra of 9 and trans-9 are blue shifted relative to the optical properties of the 2,5-dialkoxy PPBTV polymers and MEH–PPBTV analogues.23,26 The blue shift of the absorption maxima can be attributed to the steric interactions of the ortho-alkoxy substituents. To investigate this further the ground state structures of oligomers were calculated by DFT using the B3LYP/6-311G(d,p) basis set. The calculations show that oligomers based on 9 and trans-9 are stabilized by intramolecular van der Waals and the electrostatic interactions between heteroatoms.38 The strong steric repulsions between adjacent sites in the phenylene ring lead to rotation of the substituents around the C(sp2)–O bonds and consequently hinder effective orbital overlap of the lone pair electrons of the alkoxy-oxygen with the aromatic rings. This causes a more twisted conformation of the polymer backbone (Fig. S2†), which directly affect the λmax.39
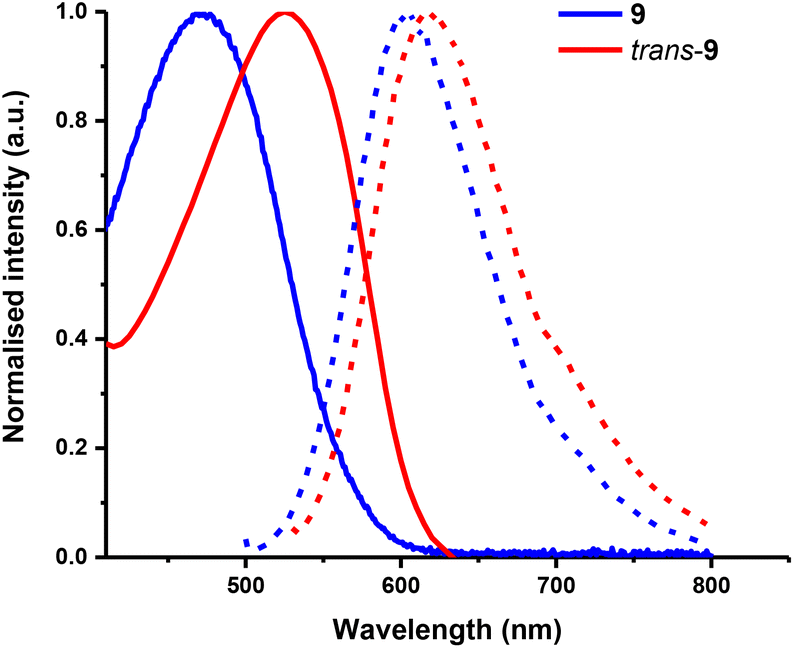 | ||
| Fig. 7 Absorption and emission profiles of copolymers 9 (Ex = 470 nm) and trans-9 (Ex = 520 nm) in CHCl3 solution. | ||
The electrochemical properties of 9 were investigated using cyclic voltammetry (CV) in an acetonitrile solution of tetrabutylammonium hexafluorophosphate (0.1 M) as electrolyte by drop casting a thin film of 9 on the platinum working electrode. Irreversible oxidation and reductions were observed for cis/trans9 and all-trans9 (Fig. S25–27†), against a Ag/AgNO3 reference electrode. The energy levels of the HOMO and the LUMO for the polymers were estimated from the onset of the oxidation and reduction peaks, respectively. The electrochemical bandgap of polymer 9 and trans-9 was observed to be 1.93 eV and 1.87 eV respectively, with the extended conjugation of the trans-9 leading to the smaller band gap, as expected.20,40
The nature of the frontier molecular orbitals were examined by DFT calculations using B3LYP/6-311G(d,p) and Fig. 8 shows a plot of the electron density contours of the calculated HOMO and LUMO orbitals. The ethylhexyl side chains of all the structures were replaced with propyl groups to reduce the computational costs without influencing the energy of HOMO and LUMO. For both cis/trans9, and trans-9 the HOMO is delocalized over both the dialkoxyphenylene and the benzothiazole unit while in the case of the LUMO orbital, electrons are predominately localised on the BT acceptor moiety. The energy gaps of the all trans structures are lower than alternative cis/trans conformation in gas phase due to extended conjugation. The calculated HOMO/LUMO energy levels are higher than those obtained from cyclic voltammetry,41 nonetheless the trends are consistent with the observed experimental data with a reduction around 0.3 eV expected on isomerisation from cis/trans to trans isomers.
 | ||
| Fig. 8 Calculated HOMO/LUMO energy levels of of cis/trans9 and all trans9. | ||
Conclusions
o-Dialkoxy benzothiadiazole [2.2]paracyclophane-1,9-dienes, 8, can be synthesised from a [3.3]dithiaparacyclophane (4). These highly strained molecules are isolated as a mixture of syn and anti isomers, 8a and 8b, that cannot be interconverted but can be ring opened by ruthenium metathesis catalyst (G2) to give regularly alternating cis/trans-PPV-BT copolymers, 9. However, 8a reacted substantially faster than 8b. The initially formed cis/trans polymer can be photoisomerised to the corresponding all trans isomer. The optical properties were studied in solution and HOMO–LUMO energy levels of the polymers determined by cyclic voltammetry and the orbital contributions investigated by DFT calculation. The all trans polymer 9 showed a red shifted absorption maximum with a smaller optical and electrochemical band gaps owing to the enhanced molecular orbital overlap.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank the Development and Promotion of Science and Technology Talents Project (DPST), Thailand for funding to Y. J. The authors would like to thank the European Commission for the award of a Marie-Curie International Fellowship (CyclAr) to V. K. and the EPSRC for funding of the NMR spectrometers under grant EP/K039547/1 and Carlo Bawn for technical advice.References
- H. Hopf, Angew. Chem., Int. Ed., 2008, 47, 9808–9812 CrossRef CAS PubMed.
- Y. Morisaki, K. Inoshita, S. Shibata and Y. Chujo, Polym. J., 2015, 47, 278–281 CrossRef CAS.
- I. Majerz and T. Dziembowska, Mol. Divers., 2020, 24, 11–20 CrossRef CAS PubMed.
- M. Hasegawa, K. Kobayakawa, H. Matsuzawa, T. Nishinaga, T. Hirose, K. Sako and Y. Mazaki, Chem. – Eur. J., 2017, 23, 3267–3271 CrossRef CAS PubMed.
- S. Park, J. H. Heo, J. H. Yun, T. S. Jung, K. Kwak, M. J. Ko, C. H. Cheon, J. Y. Kim, S. H. Im and H. J. Son, Chem. Sci., 2016, 7, 5517–5522 RSC.
- Z. Hassan, E. Spuling, D. M. Knoll, J. Lahann and S. Bräse, Chem. Soc. Rev., 2018, 47, 6947–6963 RSC.
- Z. Hassan, E. Spuling, D. M. Knoll and S. Bräse, Angew. Chem., Int. Ed., 2020, 59, 2156–2170 CrossRef CAS PubMed.
- N. Khan, N. S. Sheikh, A. F. Khan, R. Ludwig, T. Mahmood, W. Rehman, Y. S. S. Al-Faiyz and K. Ayub, J. Mol. Model., 2015, 21, 148 CrossRef PubMed.
- U. H. F. Bunz, D. Mäker and M. Porz, Macromol. Rapid Commun., 2012, 33, 886–910 CrossRef CAS PubMed.
- S. Felder, S. Wu, J. Brom, L. Micouin and E. Benedetti, Chirality, 2021, 33, 506–527 CrossRef CAS PubMed.
- M. Gon, R. Sawada, Y. Morisaki and Y. Chujo, Macromolecules, 2017, 50, 1790–1802 CrossRef CAS.
- K. Mutoh, Y. Nakagawa, A. Sakamoto, Y. Kobayashi and J. Abe, J. Am. Chem. Soc., 2015, 137, 5674–5677 CrossRef CAS PubMed.
- K. C. Dewhirst and D. J. Cram, J. Am. Chem. Soc., 1958, 80, 3115–3125 CrossRef CAS.
- D. J. Cram, R. C. Helgeson, D. Lock and L. A. Singer, J. Am. Chem. Soc., 1966, 88, 1324–1325 CrossRef CAS.
- P. K. Gantzel and K. N. Trueblood, Acta Crystallogr., 1965, 18, 958–968 CrossRef CAS.
- A. de Meijere, S. I. Kozhushkov, K. Rauch, H. Schill, S. P. Verevkin, M. Kümmerlin, H.-D. Beckhaus, C. Rüchardt and D. S. Yufit, J. Am. Chem. Soc., 2003, 125, 15110–15113 CrossRef CAS PubMed.
- F. Menk, M. Mondeshki, D. Dudenko, S. Shin, D. Schollmeyer, O. Ceyhun, T.-L. Choi and R. Zentel, Macromolecules, 2015, 48, 7435–7445 CrossRef CAS.
- B. J. Lidster, D. R. Kumar, A. M. Spring, C.-Y. Yu, M. Helliwell, J. Raftery and M. L. Turner, Org. Biomol. Chem., 2016, 14, 6079–6087 RSC.
- B. J. Lidster, D. R. Kumar, A. M. Spring, C.-Y. Yu and M. L. Turner, Polym. Chem., 2016, 7, 5544–5551 RSC.
- C.-Y. Yu, M. Horie, A. M. Spring, K. Tremel and M. L. Turner, Macromolecules, 2010, 43, 222–232 CrossRef CAS.
- D. R. Kumar, B. J. Lidster, R. W. Adams and M. L. Turner, Polym. Chem., 2017, 8, 3186–3194 RSC.
- V. Komanduri, D. R. Kumar, D. J. Tate, R. Marcial-Hernandez, B. J. Lidster and M. L. Turner, Polym. Chem., 2019, 10, 3497–3502 RSC.
- V. Komanduri, D. J. Tate, R. Marcial-Hernandez, D. R. Kumar and M. L. Turner, Macromolecules, 2019, 52, 7137–7144 CrossRef CAS.
- A. Mann and M. Weck, ACS Macro Lett., 2022, 11, 1055–1059 CrossRef CAS PubMed.
- Y. Janpatompong, A. M. Spring, V. Komanduri, R. U. Khan and M. L. Turner, Macromolecules, 2022, 55, 10854–10864 CrossRef CAS PubMed.
- E. Elacqua and M. Gregor, Angew. Chem., Int. Ed., 2019, 58, 9527–9532 CrossRef CAS PubMed.
- V. Komanduri, Y. Janpatompong, R. Marcial-Hernandez, D. J. Tate and M. L. Turner, Polym. Chem., 2021, 12, 6731–6736 RSC.
- T. Marszalek, M. Li and W. Pisula, Chem. Commun., 2016, 52, 10938–10947 RSC.
- M. Kim, S. U. Ryu, S. A. Park, K. Choi, T. Kim, D. Chung and T. Park, Adv. Funct. Mater., 2020, 30, 1904545 CrossRef CAS.
- K. Xu, H. Sun, T.-P. Ruoko, G. Wang, R. Kroon, N. B. Kolhe, Y. Puttisong, X. Liu, D. Fazzi, K. Shibata, C.-Y. Yang, N. Sun, G. Persson, A. B. Yankovich, E. Olsson, H. Yoshida, W. M. Chen, M. Fahlman, M. Kemerink, S. A. Jenekhe, C. Müller, M. Berggren and S. Fabiano, Nat. Mater., 2020, 19, 738–744 CrossRef CAS PubMed.
- J. Miao, H. Li, T. Wang, Y. Han, J. Liu and L. Wang, J. Mater. Chem. A, 2020, 8, 20998–21006 RSC.
- J. Rao, L. Yang, X. Li, L. Zhao, S. Wang, H. Tian, J. Ding and L. Wang, Angew. Chem., Int. Ed., 2021, 60, 9635–9641 CrossRef CAS PubMed.
- K. Klaue, W. Han, P. Liesfeld, F. Berger, Y. Garmshausen and S. Hecht, J. Am. Chem. Soc., 2020, 142, 11857–11864 CrossRef CAS PubMed.
- B. S. Chuah, F. Cacialli, D. A. dos Santos, N. Feeder, J. E. Davies, S. C. Moratti, A. B. Holmes, R. H. Friend and J. L. Brédas, Synth. Met., 1999, 102, 935–936 CrossRef CAS.
- Z. A. Page, Y. Liu, E. Puodziukynaite, T. P. Russell and T. Emrick, Macromolecules, 2016, 49, 2526–2532 CrossRef CAS.
- C.-Y. Yu, M. Helliwell, J. Raftery and M. L. Turner, Chem. – Eur. J., 2011, 17, 6991–6997 CrossRef CAS PubMed.
- F. H. Allen, O. Kennard, D. G. Watson, L. Brammer, A. G. Orpen and R. Taylor, J. Chem. Soc., Perkin Trans. 2, 1987, S1–S19 RSC.
- A. S. Özen, C. Atilgan and G. Sonmez, J. Phys. Chem. C, 2007, 111, 16362–16371 CrossRef.
- P. C. Man, J. A. Crayston, M. Halim and I. D. W. Samuel, Synth. Met., 1999, 102, 1081–1082 CrossRef.
- F. Wang, F. He, Z. Xie, M. Li, M. Hanif, X. Gu, B. Yang, H. Zhang, P. Lu and Y. Ma, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 5242–5250 CrossRef CAS.
- H.-W. Tsai, K.-L. Hsueh, M.-H. Chen and C.-W. Hong, Crystals, 2021, 11, 1292 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ob01989b |
| This journal is © The Royal Society of Chemistry 2023 |