
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of mono-nitroxides and of bis-nitroxides with varying electronic through-bond communication†
Angeliki
Giannoulis
 *ab,
Katrin
Ackermann
b,
Alexey
Bogdanov
a,
David B.
Cordes
b,
Catherine
Higgins
b,
Joshua
Ward
b,
Alexandra M. Z.
Slawin
b,
James E.
Taylor
*bc and
Bela E.
Bode
*b
*ab,
Katrin
Ackermann
b,
Alexey
Bogdanov
a,
David B.
Cordes
b,
Catherine
Higgins
b,
Joshua
Ward
b,
Alexandra M. Z.
Slawin
b,
James E.
Taylor
*bc and
Bela E.
Bode
*b
aDepartment of Chemical and Biological Physics, Weizmann Institute of Science, Rehovot, 76100, Israel. E-mail: angeliki.giannoulis@weizmann.ac.il
bEaStCHEM School of Chemistry, Biomedical Sciences Research Complex and Centre of Magnetic Resonance, University of St Andrews, North Haugh, St Andrews, KY16 9ST, UK. E-mail: beb2@st-andrews.ac.uk
cDepartment of Chemistry, University of Bath, Claverton Down, Bath, BA2 7AY, UK. E-mail: j.e.taylor@bath.ac.uk
First published on 8th December 2022
Abstract
Nitroxides are a unique class of persistent radicals finding a wide range of applications, from spin probes to polarizing agents, and recently bis-nitroxides have been used as proof-of-concept molecules for quantum information processing. Here we present the syntheses of pyrroline-based nitroxide (NO) radicals and give a comparision of two possible synthetic routes to form two key intermediates, namely 2,2,5,5-tetramethylpyrroline-1-oxyl-3-acetylene (TPA) and 1-oxyl-2,2,5,5-tetramethylpyrroline-3-carboxylic acid (TPC). TPC and TPA were then used as precursors for the synthesis of three model compounds featuring two distant NO groups with a variable degree of conjugation and thus electronic communication between them. Using relatively facile synthetic routes, we produced a number of mono- and bis-nitroxides with the structures of multiple compounds unambiguously characterized by X-ray crystallography, while Continuous Wave Electron Paramagnetic Resonance (CW-EPR) allowed us to quantify the electronic communication in the bis-nitroxides. Our study expands the repertoire of mono- and bis-nitroxides with possibilities of exploiting them for studying quantum coherence effects and as polarizing agents.
Introduction
Electronic communication is a common effect in nature; for example, electron transfer in proteins can lead to charge separation and subsequent recombination.1 In general, high π-conjugation favours efficient electronic communication and possibly enables coherent quantum pathways between the electrons. Towards understanding quantum coherence (QC) phenomena, proof-of-concept paramagnetic molecules have been designed and electronic communication has been studied via electron paramagnetic resonance (EPR) spectroscopy which allows direct measurement of the exchange energy between two singly occupied molecular orbitals.2,3 In recent years pulse dipolar EPR spectroscopy (PDS) has also emerged to analyse such molecules for quantum information processing.2,4,5 For example, three molecular assemblies with similar macromolecular structure and flexibility differing only in the degree of the electronic communication via π-conjugation were used as proof-of-principle compounds for demonstrating constructive QC where a four-fold increase of the exchange coupling (J) was found using two conjugated linkers rather than a single one, a value in accordance with the theoretical increase in conductance.4 Such experiments have been sparked by new methodologies for creating and analysing different magnitudes of weak exchange interactions and their distribution.6–8 Additionally, the exchange interaction is of interest in dynamic nuclear polarization (DNP) experiments, where the nuclear magnetic resonance (NMR) spectroscopy sensitivity is enhanced by applying microwave irradiation to samples containing electron spins. Therefore, bis-nitroxides with a small but non-negligible J along with orthogonal geometry of the paramagnetic moieties determined by design are under development for DNP applications.9Here we were particularly interested in the effect of ester versus acetylene linkages in breaking conjugation on a 2 nm scale. While double ester-linked model systems were shown to suppress the exchange coupling present in double ethyne linked analogues,3 the introduction of a single ester does not appear to fully diminish the exchange coupling but to considerably lessen it.10,11 Thus, we set out to revisit the synthesis of the ethyne- and carboxylic acid-functionalised spin-labels 2,2,5,5-tetramethyl-pyrrolin-1-oxyl-3-acetylene (TPA) and 1-oxyl-2,2,5,5-tetramethylpyrroline-3-carboxylic acid (TPC) and their use in producing three model systems with different levels of conjugation between the radical bearing pyrroline moieties. Besides the different degree of π-conjugation between the nitroxides (NOs), the bis-nitroxides feature very similar geometries and two electron spins per molecule. The rod-like geometry is afforded by the biphenyl core and the conjugation degree is tuned by attaching different combinations of ester or ethyne groups at the central biphenyl bridge (Fig. 1a). As J is dependent on orbital overlap it will decay exponentially with the distance between the unpaired electrons. However, exchange can be mediated through the σ- or π-bond network (or both) and this makes predictions inherently difficult and extended conjugation has been shown to remain measurable over several nm.12 5-Ring nitroxides (e.g., pyrrolidine-based) have been shown to retain larger conjugation and J than identical linkers with 6-ring-based nitroxides and were selected in this case. Predictions based on the linking groups are equally challenging as σ- or π-interactions will be attenuated differently by e.g., insertion of a CH2 group.13 Computational predictions are hampered by the very small magnitude of long-range exchange couplings that lie within the numeric noise of common ab initio and DFT calculations. Nevertheless, the π-conjugation between biphenyl bridged pyrroline-nitroxides is expected to be significantly reduced when replacing ethyne linkages by esters. Thus, the π-conjugation and therefore J is expected to follow the trend: 1 < 2 < 3.
 | ||
| Fig. 1 (a) Bis-nitroxide targets and (b) key intermediates. | ||
A key synthetic intermediate in the synthesis of linear bis-nitroxides 1–3 is TPA 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 14–18 (Fig. 1b), which can be introduced via palladium-catalysed Sonogashira cross-coupling to form spin-labelled substrates suitable for EPR studies.16,19–25 The alkyne within TPA 4 has previously been installed through elimination of the corresponding vinyl chloride using either lithium diisopropylamide (LDA)18 or potassium tert-butoxide.16 Hideg and co-workers prepared TPA 4via 1,2-bromination of the corresponding vinyl nitroxide, followed by double elimination using potassium hydroxide in ethanol.15 Alternatively, TPA 4 can be prepared directly from aldehyde 5 (Fig. 1b) using the Ohira–Bestmann reagent.17 Nitroxide 5 can itself be obtained from either TPC 6 or its amide derivatives.26–29
14–18 (Fig. 1b), which can be introduced via palladium-catalysed Sonogashira cross-coupling to form spin-labelled substrates suitable for EPR studies.16,19–25 The alkyne within TPA 4 has previously been installed through elimination of the corresponding vinyl chloride using either lithium diisopropylamide (LDA)18 or potassium tert-butoxide.16 Hideg and co-workers prepared TPA 4via 1,2-bromination of the corresponding vinyl nitroxide, followed by double elimination using potassium hydroxide in ethanol.15 Alternatively, TPA 4 can be prepared directly from aldehyde 5 (Fig. 1b) using the Ohira–Bestmann reagent.17 Nitroxide 5 can itself be obtained from either TPC 6 or its amide derivatives.26–29
Herein, we report the syntheses and characterisation of rigid bis-nitroxides 1–3. Two synthetic routes for the preparation of key intermediates TPA 4 and TPC 6 are investigated and compared. The key intermediates are then used in Sonogashira cross-coupling reactions and Steglich esterifications, respectively, for the efficient preparation of the bis-nitroxide targets. Most of the key intermediates and the linear bis-nitroxide products were characterised by X-ray crystallographic analysis.
Results and discussion
Synthesis of TPA 4 and aldehyde 5
First, nitroxide 5 was prepared from commercially available 2,2,6,6-tetramethyl-4-piperidone 7 following known literature procedures (Scheme 1a).14,16,30,31 Bromination followed by Favorskii rearrangement with aqueous ammonia could be performed on a large scale (77 mmol, 12 g) to give amide 8 in good yield over the two steps. Oxidation with hydrogen peroxide in the presence of catalytic sodium tungstate (3 mol%) and EDTA gave nitroxide 9 after purification by recrystallisation in 73% yield. Aqueous hydrolysis of 9 gave TPC 6, which is a synthetically useful precursor for the introduction of nitroxide spin labels via amidation or esterification.32 Reduction of TPC 6 with Red-Al®16 gave alcohol 10 with subsequent Swern oxidation16 giving the desired aldehyde 5 in good yield over the two steps. The structures of nitroxides 5, 6, a mixture of 8 and 9, and 10 were unambiguously confirmed by single crystal X-ray analysis.33 Although this route gave access to the desired aldehyde 5, an alternative and more succinct pathway was sought to improve the overall yield (Scheme 1b). Bromination of 7 followed by Favorskii rearrangement in the presence of N,O-dimethylhydroxylamine hydrochloride gave Weinreb amide 11 in 65% yield over the two steps,30,34 which was oxidised as previously to give nitroxide 12 in 75% yield. Reduction of 12 with DIBAL-H proceeded smoothly30 on a multi-gram scale to give aldehyde 5 (17 mmol, 2.8 g) in an excellent 95% yield. Overall, while the first route (Scheme 1a) gives access to 5via synthetically versatile TPC 6, aldehyde 5 is itself most conveniently prepared on a gram-scale in four steps from commercial 7 using the route depicted in Scheme 1b.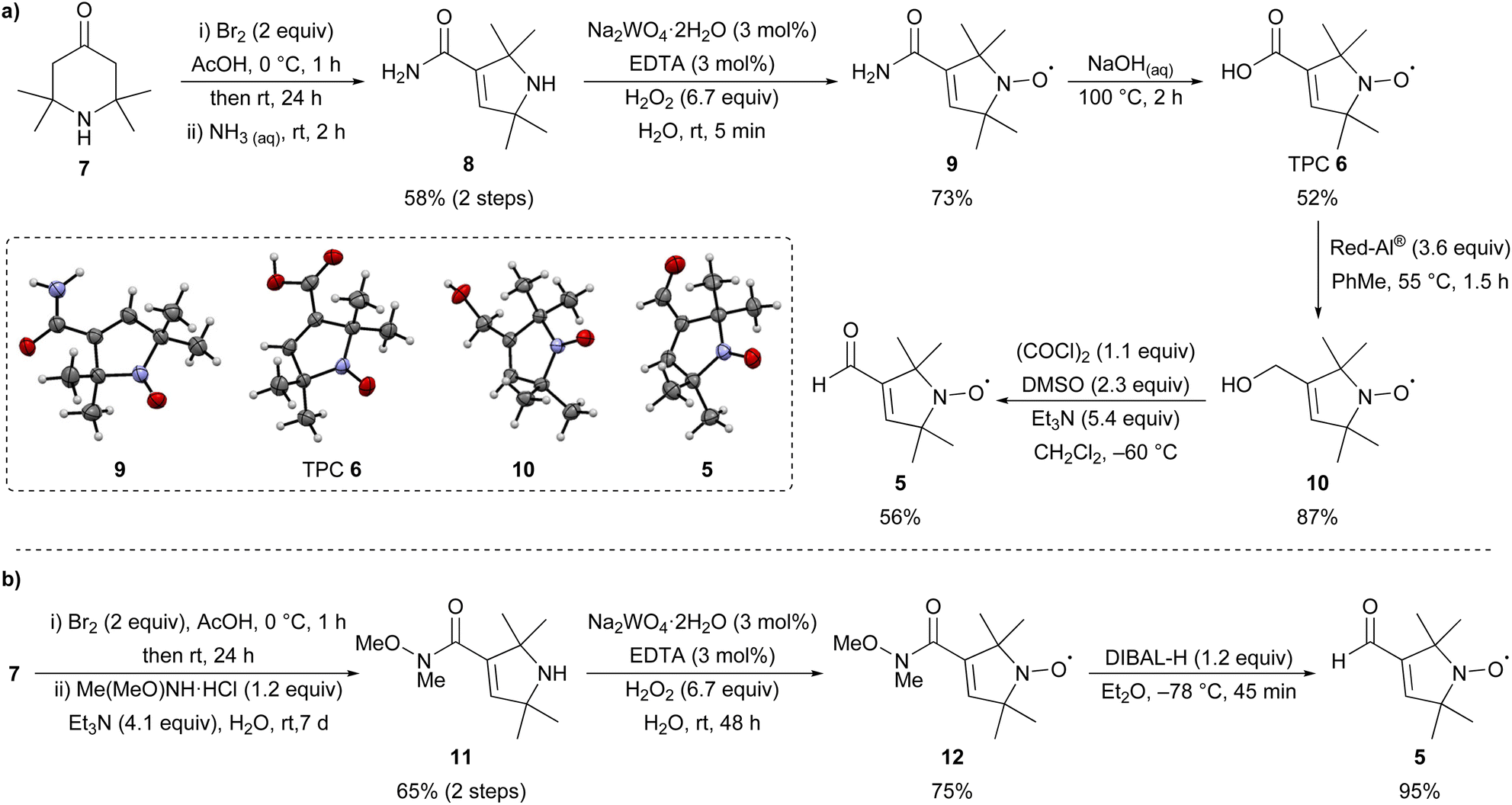 | ||
| Scheme 1 Two routes for the synthesis of nitroxide 5 (a) via TPC 6 and (b) via Weinreb amide 11. | ||
Next, the synthesis of TPA 4 from aldehyde 5 was investigated using a literature procedure.14,16 Reaction of 5 with an excess of the ylide derived from chlorotriphenylphosphonium chloride 13 gave chloro-alkene 14, previously reported as a 1:1 mixture of E/Z stereoisomers,18 in an excellent 98% yield (Scheme 2a). Elimination from nitroxide 14 using KOt-Bu in THF gave access to TPA 4 after purification by column chromatography. Although this route gave access to the desired spin-label 4, the yield of the elimination step was variable upon multiple repeats. Moreover, intermediate nitroxide 14 was obtained as viscous yellow oil, which proved difficult to handle. Therefore, a new route to TPA 4 from aldehyde 5 was explored. Treating 5 under standard Corey–Fuchs conditions35 using an excess of carbon tetrabromide and triphenylphosphine gave dibromide 15 in 66% yield (Scheme 2b). Pleasingly, nitroxide 15 is a crystalline solid, which was unambiguously characterised by single-crystal X-ray analysis,36 and is significantly easier to handle compared with 14. Treating 15 with two equivalents of n-butyl lithium resulted in elimination to form TPA 4 in 10% yield. While the yield of the elimination was low, this new route proceeds via a more favourable intermediate and provides access to sufficient quantities of TPA 4 for further reactions.
 | ||
| Scheme 2 Synthesis of TPA 4 (a) via Literature procedure (ref. 14, 16) and (b) via Corey-Fuchs conditions (ref. 35). | ||
Synthesis of linear bis-nitroxides 1–3
Having compared synthetic routes towards the synthesis of the key nitroxide spin labels 4 and 5, we turned to the preparation of linear bis-nitroxides 1–3 with differing degrees of conjugation between the two radicals. Diester 1 was prepared as described previously in 82% yield through double Steglich esterification of biphenol 16 with TPC 6 using N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDCI·HCl) as the coupling reagent (Scheme 3a).32 Synthesis of the linear nitroxide 2 is for the first time described here using a two-step procedure starting from 4′-iodo-[1,1′-biphenyl]-4-ol 17 (Scheme 3b). Esterification of phenol 17 with TPC 6 using EDCI·HCl proceeded smoothly, forming intermediate 18 in 80% yield.37 Sonogashira cross-coupling of intermediate 18 with TPA 4 gave the desired bis-nitroxide 2 in a reasonable 28% yield after purification by column chromatography, with the structure confirmed by single crystal X-ray analysis.38 Diethyne 3 was synthesized similarly as described previously,3i.e., by reacting 4,4′-diiodo-1,1′-biphenyl 19 under similar Sonogashira conditions as for 2, using two equivalents of TPA 4 (Scheme 3c). Purification of 3 from the undesired homocoupled product of TPA 4 was found to be difficult, nonetheless separation was achieved in 35% yield, which is better than the one reported previously (7% yield).3 Additionally, the homocoupled product 20 was isolated for the first time here in 21% yield, with the identity of both products confirmed by X-ray analysis.39 | ||
| Scheme 3 Synthesis of bis-nitroxide (a) 1, (b) 2, and (c) 3 and 20. | ||
Continuous-wave EPR (CW-EPR) of linear bis-nitroxides 1–3, 20
Next, we set out to characterize the electronic communication in the bis-nitroxides 1–3, 20 using continuous-wave EPR (CW-EPR) aiming at resolving and characterizing different spectral features as a result of different exchange coupling. Bis-nitroxide 1 was used as a reference were no exchange coupling should be present as shown previously on 13 and on a CuII–NO10 pair, where the CuII and NO spins where separated by a linker featuring an ester group. Compounds 2, 3 are expected to have (very) small spin–spin communication mediated though the ethyne bond(s). Finally, short bis-ethyne 20 should serve as reference for the two spins showing stronger exchange interaction. The experimental CW-EPR spectra of 1–3, 20 and results of their numerical simulations are presented in Fig. 2. Simulations were performed using software package Orthos40 taking into account Brownian rotational diffusion, anisotropic Zeeman, hyperfine and spin–spin dipolar interactions, as well as the isotropic exchange interaction (where applicable) of the two nitroxide fragments.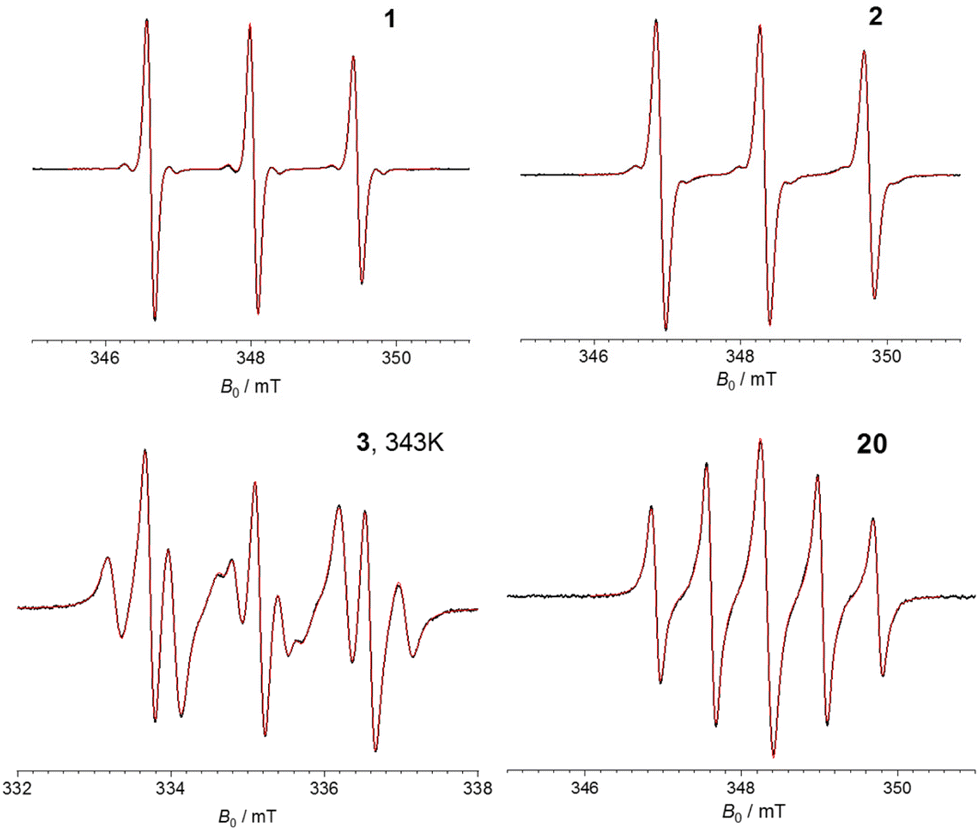 | ||
| Fig. 2 CW-EPR spectra of bis-nitroxides 1–3 and 20 recorded at X-band frequencies (9.7 GHz) at room temperature (298 K), apart from 3 which was measured at 343 K. The black and red lines indicate the experimental and simulated spectra, respectively. | ||
The CW-EPR spectra of 1–2 exhibit three peaks, characteristic of the NO EPR signal at X-band arising from the hyperfine interaction of the electron spin with the 14N nucleus (AN) with a hyperfine coupling of 39.8 MHz (1.42 mT). Additionally, the 13C coupling of 16.8 MHz (0.60 mT) was well resolved for 1 and to a lesser extent for 2. In addition to the diminished 13C couplings, the spectrum of 2 is slightly broader compared to that of 1 (by 0.5 MHz, 0.002 mT). These effects could be an indication of a very small J in 2 of <1 MHz. However, we cannot exclude that the broadening might originate from a non-infinitely diluted sample or due to the presence of a small amount of oxygen in the sample. In the simulation of 2J was not taken into account and the broadening was of Lorentzian shape further suggesting it does not originate from J. The CW-EPR spectrum of 3 features a more complicated line splitting where each of the three 14N lines is further split, suggesting J is comparable to AN. Indeed, our simulations suggest a J of +21.9 MHz (+0.78 mT) and AN = 40.1 MHz (1.43 mT), with the features reproduced in three different temperatures (298, 323 and 343 K). The exchange coupling of +21.7 MHz is in agreement with previously published data on 341 where J was found from X-band CW-EPR at 338 K to be 21.3 ± 0.8 MHz. Here, the positive J value indicates the singlet state to be the ground state. The CW-EPR spectrum of 20 shows five lines with a spacing of 19.6 MHz (0.7 mT), i.e., half of the AN, and the peaks appear with ratio 1:1.5:1.8:1.5:1, characteristic of the case where J ≫ AN. This is not surprising considering the presence of the two acetylene groups and the short NO–NO distance (RNO–NO) of 1.22 nm (from the middle of each NO group) found from X-ray. Simulations of the experimental spectrum suggest a J of at least 1100 MHz (40 mT). It should be noted that for 3 and 20 the dipolar coupling was simulated in the point dipolar approximation, with the distance between the NO fragments and their relative orientations taken directly from the solved X-ray structures of the biradicals (Scheme 3c).
The J value of 3 is comparable to the one found previously for compound 21 which features a shorter RNO–NO and the linker contained two C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bonds (Fig. 3 and Table 1). A comparison between 3 and 20 makes evident that the biphenyl core reduces J by 1100 MHz. Compound 22 is structurally similar to 3 apart from the NO ring and, interestingly, it was found to have a J of 12.5 MHz; this suggests the presence of the six-membered NO ring reduced J by almost 40% compared to the corresponding five-membered ring. Compound 20 features a larger exchange interaction compared to 23 and 24 having a shorter RNO–NO and one or two amide groups, respectively, in the linker between the NO-labelled rings. This suggests that the amide group reduces inter-spin communication compared to the alkyne bond.
N bonds (Fig. 3 and Table 1). A comparison between 3 and 20 makes evident that the biphenyl core reduces J by 1100 MHz. Compound 22 is structurally similar to 3 apart from the NO ring and, interestingly, it was found to have a J of 12.5 MHz; this suggests the presence of the six-membered NO ring reduced J by almost 40% compared to the corresponding five-membered ring. Compound 20 features a larger exchange interaction compared to 23 and 24 having a shorter RNO–NO and one or two amide groups, respectively, in the linker between the NO-labelled rings. This suggests that the amide group reduces inter-spin communication compared to the alkyne bond.
 | ||
| Fig. 3 Structure of small bis-nitroxides exhibiting exchange interaction between the paramagnetic nitroxide groups. | ||
| Compound | J/mT (MHz) |
R
NO–NOa/nm |
|---|---|---|
| a Refers to the middle of each N–O bond. b Presented here. c Based on geometrical model. d Based on DFT calculation. e Based on X-ray structure. f Based on simulations of rigid limit CW-EPR spectrum. | ||
| 2 | 0–0.04 (0–1)b | 2.04b |
| 3 | 0.77 (21.7)b and ref. 41, 0.43 (12)3 | 2.11b,e, 2.08 (trans), 2.07 (cis)41,c 2.13,c |
| 20 | 40 (≥1121)b | 1.22b,e |
| 21 | 0.7 (19.6)43 | 1.1043,d,f |
| 22 | 0.44 (12.5)41,44 | 2.2341,44,d, 2.3341,44,d |
| 23 | 30 (>850)45 | 0.8845,f |
| 24 | 21 (>600)45 | 0.9245,f |
| 25 | 128 (≥3600)46 | 0.8246,f 0.8546,d |
| 26 | 54 (≥1500)46 | 1.1046,e,f |
Compounds 25 and 26 having the six-membered NO ring were found to have a J of 3600 and 1500 MHz, respectively. Accounting for the ×0.6 difference between 3 and 22 as discussed above, this means that the bis-nitroxides with the five-membered ring NO groups would be expected to afford J values of 5760 and 2400 MHz, therefore allowing a better fine-tuning of J. Accurate determination of J is dependent not only on the interspin distance, where J decays with the distance, but also on the linkage between the spins, the relative orientation of the NO moieties, as well as the nature of the NO-rings. For example it is well established that the ethyne moiety enhances J, compared to ethylene or CH2 groups due to the increased π orbital overlap of the former and 5-ring nitroxides exhibit larger J than 6-ring. The discussion of bis-nitroxides having J is not exhaustive; however, it becomes evident that the ‘pool’ of bis-nitroxides and their characterization using CW-EPR techniques42 allow us synthesize bis-nitroxides with desirable J for use in quantum information processing and DNP experiments.9
Conclusions
In this work we expanded the repertoire of mono- and bis-nitroxide compounds with the NO on the pyrroline ring, we unambiguously characterized the compounds with X-ray crystallography and quantified the J-coupling in the bis-nitroxides with CW-EPR techniques. Linear rigid bis-nitroxides 1–3 were readily prepared from the key synthetic precursors TPA 4 and TPC 6 using Sonogashira cross-coupling and Steglich esterification to introduce the nitroxide functionality, respectively. Nitroxide containing aldehyde 5, a key precursor to TPA 4, is most conveniently prepared from reduction of the corresponding Weinreb amide 12. A new method of preparing TPA 4 from aldehyde 5 using a Corey–Fuchs strategy proceeds via crystalline vinyl dibromide intermediate 15, which is easier to handle than the alternative vinyl chloride 14. CW-EPR showed diminishing of the exchange coupling in 2 compared to 3, i.e. by substituting one triple bond with one ester bond. This suggests one triple bond does not suffice inter-spin communication when the inter-spin distance is in the 2 nm range, even in the case of a fully conjugated system. We also found that removal of the biphenyl core reduces J by ∼1 GHz as evidenced form the CW-EPR spectra and respective simulations of 3 and 20. Such observations are important in the design and synthesis of molecules with desired J values, typically sought in quantum information processing technologies, as well as in DNP experiments and our findings provide new options for fine-tuning J interactions in bis-nitroxide systems.Experimental
General information
Reactions involving moisture- and air-sensitive reagents were carried out in flame-dried glassware under an inert atmosphere (N2 or Ar) using standard vacuum line techniques. Anhydrous solvents (CH2Cl2, Et2O, PhMe, THF) were obtained after passing through an alumina column (MBraun SPS-800). Other anhydrous solvents (DMF, DMSO, MeOH) were purchased and used as received. Anhydrous Et3N was obtained by distillation from CaH2. All other solvents and commercial reagents were used as received without further purification.Room temperature (rt) refers to 20–25 °C. Temperatures of 0 °C and −78 °C were obtained using ice/water and CO2(s)/acetone baths, respectively. Reflux conditions were obtained using DrySyn® blocks equipped with a contact thermometer.
Flash column chromatography purification was performed using silica gel 60 (Merck or Crawford Scientific), alumina gel (Sigma Aldrich) activated with 4% H2O or Biotage® IsoleraTM 4, using Biotage® Snap Ultra or Biotage® KP Sil columns (CV = column volume) under the solvent system stated. Analytical thin layer chromatography was performed on pre-coated polystyrene silica TLC sheets (POLYGRAM® SIL G/UV254) or aluminium TLC plates (Merck or Fluorochem). Plates were visualised under UV light (254 nm) or by staining with 1% aq. KMnO4 or 6% aq. vanillin followed by heating. Petrol is defined as petroleum ether 40–60 °C.
Melting points (mp) were recorded on an Electrothermal 9100 melting point apparatus and are uncorrected; dec refers to decomposition.
Infrared spectra (νmax) were recorded on a Shimadzu IRAffinity-1 Fourier transform IR spectrophotometer using either thin film or solid using Pike MIRacle ATR accessory. Analysis was carried out using Shimadzu IRsolution v1.50 and only characteristic peaks are reported. The following abbreviations are used: s, strong; sh, sharp; w, weak; m, medium; br, broad.
1H NMR spectra were acquired on either a Bruker Avance 300 (δH 300 MHz), a Bruker Avance II 400 (δH 400 MHz, δC 100 MHz), or a Bruker Ultrashield 500 (δH 500 MHz, δC 126 MHz) spectrometer at ambient temperature in the deuterated solvent stated. The data were analysed using Bruker TopSpin 4.1.1. Chemical shifts, δ, are quoted in parts per million (ppm) and are referenced to the residual solvent peak and abbreviation s denotes singlet.
CW-EPR spectra were recorded on deoxygenated samples at room temperature, unless otherwise stated, on a Bruker EMXplus EPR spectrometer with an ELEXSYS super-high-sensitivity probehead (Bruker ER4122SHQE) and nitrogen evaporator variable temperature system (Bruker ER4131VT) operating at ∼9.8 GHz (X-band). All samples were prepared in deuterated PhMe at 500 μM concentration for mono-nitroxides, at 400 μM for 1 and 100 μM for 2, 3 and 20. Samples were contained in 4 mm OD quartz tubes sealed with rubber septa and deoxygenated by saturation with nitrogen or argon gas. For mono-nitroxides the experimental parameters were: modulation amplitude 0.05 mT for 4, 6, 9, 10, 12 and 0.4 mT for 5, 14, 15, time constant and conversion time 20.48 ms for all. The number of scans varied among different samples and DPPH (2,2-diphenyl-1-picrylhydrazyl, g = 2.0036) was used to estimate the g-values of the mono-radicals. For bis-nitroxides the experimental parameters were: modulation amplitude 0.05 mT for 1, 0.06 mT for 2, 0.01 mT for 3 and 0.08 mT for 20, time constant and conversion time, respectively, 40.96 and 40.96 ms for 1, 2 and 20 and 20.48 and 4.00 ms for 3, number of scans: 1 for 1, 5 for 2 and 20 and 10 for 3.
The simulations of CW EPR spectra of the mono-nitroxides were run with Easyspin47 using the ‘garlic’ function assuming fast molecular tumbling and isotropic g- and hyperfine-tensors, with electron spin S = ½ and considering hyperfine couplings from 14N, 2 inequivalent 1H (denoted as 1H(1), 1H(2)) and 13C. The number of 14N, 1H(1), 1H(2) and 13C nuclei coupled to the electron spin are for all molecules 1, 12, 1, 4, respectively. Additionally, a Voigtian spectral linewidth was added. All parameters were optimized using ‘esfit’ function. For molecules 4, 6, 9, 10, 12 the optimized parameters were obtained from the spectra shown in Fig. S1.† For molecules 5, 14, 15 that are presented with 0.4 mT modulation amplitude (Fig. S1†) an additional spectrum with 0.05 mT modulation amplitude was obtained (data not shown). For these molecules all optimized parameters were obtained from these additional EPR spectra while the 13C hyperfine couplings were optimized from the 0.4 mT modulation amplitude EPR spectra. The parameters of the simulated spectra are given in ESI, Table S1.† The simulations of 1, 2, 3 and 20 were performed using software package Orthos.40 The theoretical spectra were calculated by solving Stochastic Liouville Equation using the Brownian rotational diffusion as relaxation model. 1 and 2 were simulated as mono-radicals with an isotropic diffusion coefficient (Riso) of 2.1 × 109 s−1 and 2.2 × 109 s−1, respectively and a Voigtian spectral lineshape with Gaussian contribution of 0.1 mT for both and Lorentzian contribution of 0.009 mT and 0.046 mT for 1 and 2, respectively. 3 and 20 were simulated as biradicals. For 20 isotropic rotational diffusion with Riso = 4.7 × 109 s−1 was sufficient to describe the spectra within the experimental uncertainty. For 3 the rotation anisotropy was taken into account by using axial rotation diffusion tensor with R∥ > 5 × 1010 s−1 and R⊥ = 2 × 109 s−1. All experimental and simulated spectra are shown after baseline correction and normalization.
Mass spectrometry (m/z) data were acquired by atmospheric pressure chemical ionisation (APCI) and nanospray ionisation (NSI) at the EPSRC UK National Mass Spectrometry Facility at Swansea University.
Elemental analysis was carried out at the London Metropolitan University where the solid samples were weighed using a Mettler Toledo high-precision scale and analysed using ThermoFlash 2000. The analysis determined the carbon, hydrogen and nitrogen percentage (%CHN). All values are quoted in mass percentage (%).
X-ray diffraction data for all compounds were collected at 173 K using either a Rigaku FR-X Ultrahigh Brilliance Microfocus RA generator/confocal optics with XtaLAB P200 diffractometer [Mo Kα radiation (λ = 0.71073 Å)], or a Rigaku MM-007HF High Brilliance RA generator/confocal optics with XtaLAB P100 diffractometer [Cu Kα radiation (λ = 1.54187 Å)]. Intensity data were collected using ω steps or both ω and φ steps accumulating area detector images spanning at least a hemisphere of reciprocal space. Data were collected and processed (including correction for Lorentz, polarization and absorption) using CrystalClear.48 Structures were solved by charge-flipping (Superflip49), direct (SIR-201150), dual-space (SHELXT51) or Patterson methods (PATTY52) and refined by full-matrix least-squares against F2 (SHELXL-2018/353). Non-hydrogen atoms were refined anisotropically, and hydrogen atoms were refined using a riding model. All calculations were performed using the Olex254 interface. CCDC 2212061–2212068 contains the supplementary crystallographic data for this paper.†
The research data supporting this publication can be accessed at https://doi.org/10.17630/75719a96-4ec5-4e45-a838-7f34642d4151.55
Compound data
Intermediate dibromide (22.8 g) was suspended in 35% aq. NH3 (70 mL) and stirred at rt for 2 h. The solution was filtered, and the filtrate was saturated with KOH pellets until pH ∼ 14 was obtained. The resulting suspension was filtered and concentrated under reduced pressure to give the crude product, which was purified by recrystallisation from PhMe to give 8 (7.5 g, 44.7 mmol, 58% yield (over two steps)) as brown crystals. mp 177–178 {Lit.56 179–180 °C}; vmax (cm−1) 1359s, 1598s, 1650s, 1662s, 2962br, 3178–3346br; 1H NMR (400 MHz, CDCl3) δH: 1.29 (6 H, s, CH3), 1.45 (6 H, s, CH3), 6.17 (1 H, s, CCH); 13C NMR (100 MHz, CDCl3) δC: 30.2 (2 × CH3), 30.3 (2 × CH3), 63.5 (CNH), 66.9 (CNH), 68.9 (CCHC), 142.3 (CCO), 166.0 (CO).
A solution of N,O-dimethylhydroxylamine hydrochloride (9.9 g, 102 mmol, 1.2 equiv.) in H2O (58 mL) was cooled to 0 °C before NEt3 (49 mL, 352 mmol, 4.1 equiv.) was added. The dibromide intermediate (33.5 g, 85 mmol, 1.0 equiv.) was then added in portions over the course of 7 h at rt. The reaction was stirred at rt for 7 days. The pH was adjusted to between 9.0 and 9.5 using 40% w/v aq. NaOH and the solution extracted with EtOAc (3 × 60 mL). The combined organic phases were dried over Na2SO4 and concentrated under reduced pressure. Product 11 (13.0 g, 62 mmol, 65% over two steps) was pure by TLC analysis and was used without further purification as a light orange oil. Upon multiple repeats of the reaction the crude product was not as pure, but could be purified by flash column chromatography on silica gel (CH2Cl2/MeOH 98:2 to 90:10). vmax (cm−1) 750sh, 972m, 1178m, 1352s, 1610m, 1654m, 2962w; 1H NMR (500 MHz, CDCl3) δH: 1.24 (6 H, s, 2 × CH3), 1.34 (6 H, s, 2 × CH3), 3.16 (3 H, s, NCH3), 3.56 (3 H, s, OCH3), 6.04 (1 H, s, CCHvinC); m/z (NSI+) 213 ([M + H]+, 100%); HRMS (NSI+) C11H21N2O2 ([M + H]+) found 213.1594, calculated 213.1598 (−1.7 ppm).
From 10. A solution of oxalyl chloride (0.25 mL, 2.6 mmol, 1.1 equiv.) in CH2Cl2 (3 mL) was cooled to −60 °C under an atmosphere of N2 before a solution of anhydrous DMSO (0.4 mL, 5.6 mmol) in CH2Cl2 (3 mL) was added dropwise over the course of 7 min. The reaction mixture was stirred for 3 min before a solution of 10 (0.41 g, 2.4 mmol, 1.0) in CH2Cl2 was added. The reaction was stirred at −60 °C for 15 min before NEt3 (2 mL, 14 mmol, 5.4 equiv.) was added and the solution was stirred for 5 min. The solution was allowed to warm to rt before being quenched with H2O (25 mL). The phases were separated and the aqueous extracted with CH2Cl2. The combined organic phases were washed with brine (20 mL), 1% w/w sulfuric acid (9 mL), H2O (9 mL), and 5% w/v NaHCO3 (9 mL), dried over MgSO4 and concentrated under reduced pressure. The crude product was purified by sublimation at 50 °C to give 5 (0.23 g, 1.34 mmol, 56%) as yellow crystals. mp 76–78 °C (dec) {Lit.30 77–79 °C}; vmax (cm−1) 765sh, 869sh, 952w, 1116w, 1161w, 1278m, 1350m, 1622w, 1680s; 2978w; m/z (APCI+) 168 ([M]+, 100%); HRMS (APCI+) C9H14NO2 ([M]+) found 168.1016, calculated 168.1019 (−1.8 ppm).
From 12. A solution of 12 (3.9 g, 17.4 mmol, 1.0 equiv.) in Et2O (22 mL) was cooled to −78 °C under an atmosphere of N2 before a solution of DIBAL-H (18.0 mL, 21.6 mmol, 1.2 m in PhMe, 1.2 equiv.) was added dropwise over the course of 30 min. After stirring for 15 min, the reaction was quenched by slowly pouring the mixture into a solution of aq. 2 M HCl (22.0 mL) at 0 °C. The mixture was allowed to warm to rt and the phases separated. The aqueous was extracted with EtOAc (3 × 30 mL), and the combined organics were dried over MgSO4 and concentrated under reduced pressure to give 5 (2.8 g, 16.6 mmol, 95%) as yellow crystals. The product was used for the next step without further purification. Data as above.
:MeOH 10:0 to 9:1) to give 14 (1.63 g, 8.1 mmol, 98%) as an orange oil. vmax (cm−1) 626s, 694m, 740m, 844s, 941s, 1160s, 1224, 1284w, 1357s, 1465m, 1610w, 2929w, 2974s; m/z (APCI+) 201 ([M + H]+, 100%); HRMS (APCI+) C10H16NOCl ([M + H]+) found 201.0911, calculated 201.0915 (−2.0 ppm).
From 14. A solution of 14 (0.15 g, 0.7 mmol, 1.0 equiv.) in THF (6 mL) was subjected to three freeze–pump–thaw cycles. KOt-Bu (0.5 g, 4.1 mmol, 5.8 equiv.) was added at rt before the reaction was heated to 55 °C for 2 h. The solution was allowed to cool to rt before being quenched with aqueous NH4Cl (0.3 mL, 1 M) and dried under reduced pressure. The dried product was re-dissolved in Et2O (8 mL) and washed with H2O (3 × 4 mL) and brine (4 mL), and the organic phase dried over MgSO4 and concentrated under reduced pressure. The crude product was purified by sublimation (51 °C) to give TPA 4 (0.03 g, 0.17 mmol, 26%) as yellow solid. mp 122–123 °C {Lit.15 122–123 °C}; vmax (cm−1) 752s, 879s, 1087w, 1159s, 1274m, 1359s, 1438m, 1456m, 2092w, 2933w 2980s, 3194s; m/z (APCI+) 164 ([M]+, 100%); HRMS (APCI+) C10H14NO ([M]+) found 164.1066, calculated 164.1070 (−2.4 ppm).
From 15. A solution of 15 (0.47 g, 1.5 mmol, 1.0 equiv.) in THF (20 mL) was cooled to −78 °C under an atmosphere of N2 before n-BuLi (1.5 mL, 2.9 mmol, 1.9 m in hexanes, 1.9 equiv.) was added dropwise over the course of 5 min. The reaction was stirred at −78 °C for 45 min before being allowed to warm to rt and stirred for a further 3 h, followed by quenching with H2O (20 mL). The phases were separated. The aqueous was extracted with Et2O (3 × 20 mL), and the combined organics dried over Na2SO4 and concentrated under reduced pressure. The crude product was purified by flash column chromatography on silica gel (CH2Cl2) to give TPA 4 (0.023 g, 0.14 mmol, 10%). Data as above.
:40) to give 2 (0.01 g, 0.021 mmol, 28%) as yellow solid. Crystals suitable for X-ray analysis were obtained after recrystallisation from CHCl3. mp 230–232 °C (dec); vmax (cm−1) 759m, 800s, 840m, 875m, 1000sh, 1149s, 1246w, 1286m, 1384m, 1446w, 1490n, 1732s, 2978w; m/z (APCI+) 499 ([M + H]+, 100%); HRMS (APCI+) C31H35N2O4 ([M + H]+) found 499.2585, calculated 499.2591 (−1.3 ppm); C31H34N2O4 calculated C, 74.67; H, 6.87; N, 5.62%; found C, 74.67; H, 6.95; N, 5.51%.
:0 to 85:15 12 CV, 85:15 7 CV, 75:25 3 CV, 65:35 5 CV, 50:50 5CV)] to give 3 (0.02 g, 0.04 mmol, 35%) as beige crystalline solid. Crystals suitable for X-ray analysis were obtained after recrystallisation from hexane/EtOAc (1:1). mp 237–238 °C (dec); vmax (cm−1) 759w, 820s, 881w, 1002w, 1163m, 1247m, 1357m, 1462w, 2850w, 2922m, 2976w, 3051w; m/z (APCI+) 479 ([M + H]+, 100%); HRMS (APCI+) C32H35N2O2 ([M + H]+) found 479.2689, calculated 479.2693 (−0.8 ppm); C32H34N2O2 calculated C, 80.3; H, 7.16; N, 5.85%; found C, 80.42; H, 7.02; N, 5.87%.
A second fraction containing 3,3′-(buta-1,3-diyne-1,4-diyl)bis(2,2,5,5-tetramethyl-2,5-dihydro-1H-pyrrol-1-oxyl) 20 (0.08 g, 0.024 mmol, 21%) was also obtained as a yellow solid. Crystals suitable for X-ray analysis were obtained after recrystallisation from petrol/EtOAc (60:40). mp 225–227 °C (dec); vmax (cm−1) 885w, 997w, 1093s, 1155m, 1438s, 1479m, 2320br; m/z (NSI+) 327 ([M + H]+, 100%); HRMS (APCI+) C20H27N2O2 ([M + H]+) found 327.2074, calculated 327.2072 (+0.6 ppm).
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
AG acknowledges the UK-MRBT-CDT funded by EPSRC (EP/J500045/1), BEB acknowledges the Wellcome Trust Institutional Strategic Support fund (204821/Z/16/Z), JET acknowledges the Leverhulme Trust (Early Career Fellowship; ECF-2014-005). We also thank the EPSRC UK National Mass Spectrometry Facility at Swansea University, Mr Steven Boyer for elemental analyses at London Metropolitan University and Dr Thomas Lebl and Mrs Melanja Smith at the NMR facility at the University of St-Andrews.References
- I. A. Balabin and J. N. Onuchic, Science, 2000, 290, 114–117 CrossRef CAS.
- E. Zaytseva, I. Timofeev, O. Krumkacheva, D. Parkhomenko, D. Mazhukin, K. Sato, H. Matsuoka, T. Takui and E. Bagryanskaya, Appl. Magn. Reson., 2019, 50, 967–976 CrossRef CAS.
- A. Weber, O. Schiemann, B. Bode and T. F. Prisner, J. Magn. Reson., 2002, 157, 277–285 CrossRef CAS PubMed.
- S. Richert, J. Cremers, I. Kuprov, M. D. Peeks, H. L. Anderson and C. R. Timmel, Nat. Commun., 2017, 8, 14842 CrossRef CAS.
- M. E. Boulon, A. Fernandez, E. M. Pineda, N. F. Chilton, G. Timco, A. J. Fielding and R. E. P. Winpenny, Angew. Chem., Int. Ed., 2017, 56, 3876–3879 CrossRef CAS PubMed.
- K. Keller, I. Ritsch, H. Hintz, M. Hulsmann, M. Qi, F. D. Breitgoff, D. Klose, Y. Polyhach, M. Yulikov, A. Godt and G. Jeschke, Phys. Chem. Chem. Phys., 2020, 22, 21707–21730 RSC.
- I. Ritsch, H. Hintz, G. Jeschke, A. Godt and M. Yulikov, Phys. Chem. Chem. Phys., 2019, 21, 9810–9830 RSC.
- S. J. Lockyer, S. Nawaz, A. Brookfield, A. J. Fielding, I. J. Vitorica-Yrezabal, G. A. Timco, N. A. Burton, A. M. Bowen, R. E. P. Winpenny and E. J. L. McInnes, J. Am. Chem. Soc., 2020, 142, 15941–15949 CrossRef CAS PubMed.
- C. Ysacco, E. Rizzato, M. A. Virolleaud, H. Karoui, A. Rockenbauer, F. Le Moigne, D. Siri, O. Ouari, R. G. Griffin and P. Tordo, Phys. Chem. Chem. Phys., 2010, 12, 5841–5845 RSC.
- B. E. Bode, J. Plackmeyer, M. Bolte, T. F. Prisner and O. Schiemann, J. Organomet. Chem., 2009, 694, 1172–1179 CrossRef CAS.
- B. E. Bode, J. Plackmeyer, T. F. Prisner and O. Schiemann, J. Phys. Chem. A, 2008, 112, 5064–5073 CrossRef CAS.
- P. Wautelet, J. Le Moigne, V. Videva and P. Turek, J. Org. Chem., 2003, 68, 8025–8036 CrossRef CAS.
- L. J. Berliner and J. Reuben, Biological Magnetic Resonance Spin Labeling Theory and Applications, Plenum Press, New York, 1989 Search PubMed.
- M. Azarkh, V. Singh, O. Okle, I. T. Seemann, D. R. Dietrich, J. S. Hartig and M. Drescher, Nat. Protoc., 2013, 8, 131–147 CrossRef CAS PubMed.
- T. Kalai, M. Balog, J. Jeko and K. Hideg, Synthesis, 1999, 973–980 CrossRef CAS.
- O. Schiemann, N. Piton, J. Plackmeyer, B. E. Bode, T. F. Prisner and J. W. Engels, Nat. Protoc., 2007, 2, 904–923 CrossRef CAS PubMed.
- G. Ur, T. Kalai, M. Balog, B. Bognar, G. Gulyas-Fekete and K. Hideg, Synth. Commun., 2015, 45, 2122–2129 CrossRef CAS.
- A. Spaltenstein, B. H. Robinson and P. B. Hopkins, Biochemistry, 1989, 28, 9484–9495 CrossRef CAS PubMed.
- J. Fritscher, M. Beyer and O. Schiemann, Chem. Phys. Lett., 2002, 364, 393–401 CrossRef CAS.
- E. A. Jaumann, S. Steinwand, S. Klenik, J. Plackmeyer, J. W. Bats, J. Wachtveitl and T. F. Prisner, Phys. Chem. Chem. Phys., 2017, 19, 17263–17269 RSC.
- S. Obeid, M. Yulikov, G. Jeschke and A. Marx, Angew. Chem., Int. Ed., 2008, 47, 6782–6785 CrossRef CAS PubMed.
- N. Piton, O. Schiemann, Y. Mu, G. Stock, T. Prisner and J. W. Engels, Nucleosides, Nucleotides Nucleic Acids, 2005, 24, 771–775 CrossRef CAS.
- T. Strube, O. Schiemann, F. MacMillan, T. Prisner and J. W. Engels, Nucleosides, Nucleotides Nucleic Acids, 2001, 20, 1271–1274 CrossRef CAS.
- T. Kalai, M. Balog, J. Jeko, W. L. Hubbell and K. Hideg, Synthesis, 2002, 2365–2372 CAS.
- T. Kalai, J. Schindler, M. Balog, E. Fogassy and K. Hideg, Tetrahedron, 2008, 64, 1094–1100 CrossRef CAS.
- S. Sato, M. Tsunoda, M. Suzuki, M. Kutsuna, K. Takido-uchi, M. Shindo, H. Mizuguchi, H. Obara and H. Ohya, Spectrochim. Acta, Part A, 2009, 71, 2030–2039 CrossRef.
- K. Zhang, B. B. Noble, A. C. Mater, M. J. Monteiro, M. L. Coote and Z. Jia, Phys. Chem. Chem. Phys., 2018, 20, 2606–2614 RSC.
- R. W. Murray and M. Singh, Tetrahedron Lett., 1988, 29, 4677–4680 CrossRef CAS.
- K. Oyaizu, T. Kawamoto, T. Suga and H. Nishide, Macromolecules, 2010, 43, 10382–10389 CrossRef CAS.
- S. W. Stork and M. W. Makinen, Synthesis, 1999, 1309–1312 CrossRef CAS.
- E. G. Rosantsev, Free nitroxyl radicals, Plenum Press, New York, 1970 Search PubMed.
- S. Valera, J. E. Taylor, D. S. Daniels, D. M. Dawson, K. S. Athukorala Arachchige, S. E. Ashbrook, A. M. Slawin and B. E. Bode, J. Org. Chem., 2014, 79, 8313–8323 CrossRef CAS PubMed.
- X-ray crystallographic data for 5, 6, 8–9, and 10 has been deposited with the Cambridge Crystallographic Data Centre and can be accessed via CCDC numbers 2212063, 2212064, 2212065, and 2212066, respectively.†.
- T. Kumamoto, K. Suzuki, S. K. Kim, K. Hoshino, M. Takahashi, H. Sato, H. Iwata, K. Ueno, M. Fukuzumi and T. Ishikawa, Helv. Chim. Acta, 2010, 93, 2109–2114 CrossRef CAS.
- E. J. Corey and P. L. Fuchs, Tetrahedron Lett., 1972, 3769–3772 CrossRef CAS.
- X-ray crystallographic data for 15 has been deposited with the Cambridge Crystallographic Data Centre and can be accessed via CCDC number 2212067.†.
- K. Ackermann, A. Giannoulis, D. B. Cordes, A. M. Z. Slawin and B. E. Bode, Chem. Commun., 2015, 51, 5257–5260 RSC.
- X-ray crystallographic data for 2 has been deposited with the Cambridge Crystallographic Data Centre and can be accessed via CCDC number 2212061.†.
- X-ray crystallographic data for 3 and 20 has been deposited with the Cambridge Crystallographic Data Centre and can be accessed via CCDC numbers 2212062 and 2212068, respectively.†.
- A. V. Bogdanov and A. K. Vorobiev, Phys. Chem. Chem. Phys., 2016, 18, 31144–31153 RSC.
- A. I. Kokorin, O. I. Gromov, T. Kalai and K. Hideg, Appl. Magn. Reson., 2016, 47, 1283–1293 CrossRef CAS.
- M. Abe, Chem. Rev., 2013, 113, 7011–7088 CrossRef CAS PubMed.
- A. V. Bogdanov, B. Y. Mladenova Kattnig, A. K. Vorobiev, G. Grampp and A. I. Kokorin, J. Phys. Chem. B, 2020, 124, 11007–11014 CrossRef CAS.
- A. I. Kokorin, O. I. Gromov, A. E. Putnikov, P. V. Dorovatovskii, Y. V. Zubavichus and V. N. Khrustalev, Russ. J. Phys. Chem. B, 2021, 15, 212–218 CrossRef CAS.
- J. P. Y. Kao, W. Moore, L. B. Woodcock, N. D. A. Dirda, E. A. Legenzov, S. S. Eaton and G. R. Eaton, Appl. Magn. Reson., 2022, 53, 221–232 CrossRef CAS.
- O. I. Gromov, E. N. Golubeva, V. N. Khrustalev, T. Kalai, K. Hideg and A. I. Kokorin, Appl. Magn. Reson., 2014, 45, 981–992 CrossRef CAS.
- S. Stoll and A. Schweiger, J. Magn. Reson., 2006, 178, 42–55 CrossRef CAS.
- CrystalClear-SM Expert v2.1, Rigaku Americas, The Woodlands, Texas, USA, and Rigaku Corporation, Tokyo, Japan, 2015 Search PubMed.
- L. Palatinus and G. Chapuis, J. Appl. Crystallogr., 2007, 40, 786–790 CrossRef CAS.
- M. C. Burla, R. Caliandro, M. Camalli, B. Carrozzini, G. L. Cascarano, C. Giacovazzo, M. Mallamo, A. Mazzone, G. Polidori and R. Spagna, J. Appl. Crystallogr., 2012, 45, 357–361 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef.
- P. T. Beurskens, G. Beurskens, R. de Gelder, S. Garcia-Granda, R. O. Gould, R. Israel and J. M. M. Smits, DIRDIF-99, Crystallography Laboratory, University of Nijmegen, The Netherlands, 1999 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- A. Giannoulis, K. Ackermann, A. Bogdanov, D. B. Cordes, C. Higgins, J. Ward, A. M. Z. Slawin, J. E. Taylor and B. E. Bode, Synthesis of mono-nitroxides and of bis-nitroxides with varying electronic through-bond communication (dataset), Dataset, University of St Andrews Research Portal, 2022. DOI:10.17630/75719a96-4ec5-4e45-a838-7f34642d4151.
- C. Sandris and G. Ourisson, Bull. Soc. Chim. Fr., 1958, 3, 338–344 Search PubMed.
- H. O. Hankovszky, K. Hideg, L. Lex, G. Kulcsar and H. A. Halasz, Can. J. Chem., 1982, 60, 1432–1438 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Additional CW-EPR data, characterization data (NMR, mass spectrometry and IR spectra). CCDC 2212061–2212068. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2ob01863b |
| This journal is © The Royal Society of Chemistry 2023 |