
Common xanthene fluorescent dyes are visible-light activatable CO-releasing molecules†
Marek
Martínek
ab,
Lucie
Ludvíková
a,
Mária
Šranková
c,
Rafael
Navrátil
d,
Lucie
Muchová
 c,
Jiří
Huzlík
e,
Libor
Vítek
cf,
Petr
Klán
ab and
Peter
Šebej
*a
c,
Jiří
Huzlík
e,
Libor
Vítek
cf,
Petr
Klán
ab and
Peter
Šebej
*a
aRECETOX, Faculty of Science, Masaryk University, Kamenice 735/5, D29, 625 00 Brno-Bohunice, Czech Republic. E-mail: sebej@recetox.muni.cz
bDepartment of Chemistry, Faculty of Science, Masaryk University, Kamenice 735/5, A08, 625 00 Brno-Bohunice, Czech Republic
cInstitute of Medical Biochemistry and Laboratory Diagnostics, General University Hospital in Prague and 1st Faculty of Medicine, Charles University, Kateřinská 32, 121 08 Praha 2, Czech Republic
dDepartment of Organic Chemistry, Faculty of Science, Charles University, Hlavova 2030/8, 128 43, Prague, Czech Republic
eTransport Research Centre (CDV), Líšeňská 33a, 636 00 Brno-Líšeň, Czech Republic
f4th Department of Internal Medicine, General University Hospital in Prague and 1st Faculty of Medicine, Charles University, Kateřinská 32, 121 08 Praha 2, Czech Republic
First published on 3rd November 2022
Abstract
Fluorescein, eosin Y, and rose bengal are dyes used in clinical medicine and considered (photo-)chemically stable. Upon extensive irradiation with visible light in aqueous solutions, we found that these compounds release carbon monoxide (CO) – a bioactive gasotransmitter – in 40–100% yields along with the production of low-mass secondary photoproducts, such as phthalic and formic acids, in a multistep degradation process. Such photochemistry should be considered in applications of these dyes, and they could also be utilized as visible-light activatable CO-releasing molecules (photoCORMs) with biological implications.
Introduction
One of the main prerequisites for the applications of organic dyes, such as fluorescent tags,1,2 laser dyes,3 or pH indicators,4,5 is their thermal and photochemical stability. Fluorescein (1), eosin Y (2), and rose bengal (3, Fig. 1) are frequently used xanthene dyes, but their photoreactivities are mostly neglected.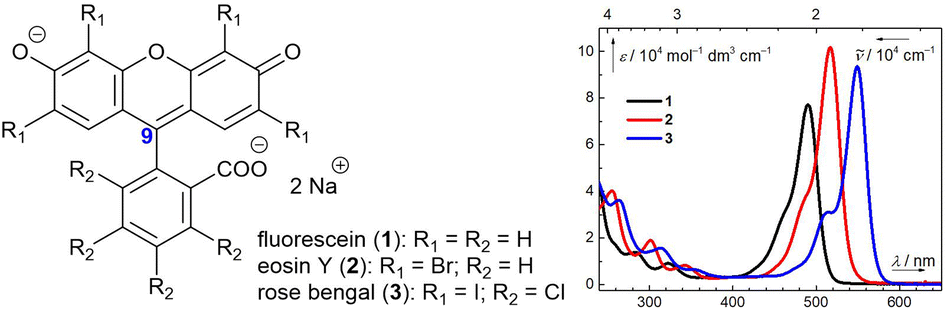 | ||
| Fig. 1 Dyes 1–3 and their absorption spectra in an aqueous solution at pH = 7.4. | ||
Early studies of the chemical behavior of excited xanthenes, such as eosin6,7 and 1,8 focused on initial reaction steps.9 Lindquist detected several different early intermediates in his thorough study,8 and Imamura et al. showed that the photodegradation of 1 is irreversible.10 However, to the best of our knowledge, stable products formed from 1–3 upon irradiation have never been identified.
CO is an essential gasotransmitter11 interacting with metalloproteins,12 bound to hemoglobin in mammals (∼80%),13 whereas the remaining part present in plasma has, under physiological concentrations, various regulatory and protective functions, such as protection from hypoxia,14 vasodilatory,15 bactericidal16 and anti-inflammatory effects,17,18 promotion of wound healing,19 or reducing tumor growth.20–22 CO has also emerged as a promising therapeutic agent for acute kidney injury, a previously unmet medical need.23 However, at higher blood concentrations,14 it is a deadly poison inhibiting the binding of oxygen to hemoglobin, thus causing tissue hypoxia24,25 and dysregulation of redox homeostasis.14
Inhalation of CO is the simplest but difficult to control delivery mode to mammalian organisms.26 Current efforts to develop more reliable CO-delivery methods focus on small molecules releasing CO upon activation (CO-releasing molecules; CORMs) allowing precise temporal and spatial control.27–30 The first known CORMs were metal-carbonyl complexes undergoing thermodynamic hydrolysis,31,32 usually leading to quick increase of CO concentration after dissolution, but not allowing spatial control over its release. Other strategies are based on use of an external trigger such as enzymatic activity, protonation, ligand exchange or electromagnetic heating which brings the possibility of temporal and partially spatial control over the CO release.29,30
Visible light can also trigger CO liberation, e.g., from carbonyl metal complexes33,34 or metal-free molecules.27,35–38 Rational design of such light absorbing xanthene35,39 or BODIPY photoCORMs56 allows fine-tuning of their optical and chemical properties.
The potential therapeutic use of CORMs involves the same legal and economic considerations and tests as for the other drugs. The clinical trials involve toxicity testing of drug candidates, their metabolites, and impurities. The potential phototoxicity and photo-induced adverse effects of drugs are often unreported, misdiagnosed,40,41 or underexplored.42 Their mechanisms include, in particular, photosensitization43 or degradation to a toxic product.44
Because diagnostic dyes are used in conjunction with irradiation, we re-examined photochemical properties of 1 (approved by the European Medicines Agency, U.S. Food and Drug Administration, and other regulatory bodies for use in human medicine, such as for diagnostic purposes in ophthalmology,45,46 urology47 or neurosurgery),482 (a common photocatalyst,49,50 and stain for histological fixatives),51 and 3 (an ophthalmological agent52 and a triplet sensitizer).53 We studied their photochemistry with optical spectroscopy, NMR, chromatographic, and MS analysis. We show that 1–3 produce CO and other low-mass photoproducts upon extensive irradiation. The related biological aspects of these transformations are discussed in a recently published article.54
Results and discussion
We irradiated 1–3 (c ∼ 1.4 × 10−5 mol dm−3) in PBS or methanol with LEDs (λirr = 400–700 nm; Fig. S83;† 21 °C) until no starting material was observed. The pseudo first-order degradation rate constants were obtained from a single-exponential fit of the dyes’ decay monitored at its absorption maximum. Comparison of the observed rate constants (corrected to the amount of absorbed light; Table S2†), calculated from a decrease of the absorption signals of the starting material (no other major absorption peaks appeared in the spectrum during irradiation), allowed us to calculate the degradation quantum yields (Table 1). We found that both 1 and 3 are consumed with similar quantum yields, which are about (3–5)-fold higher than that of 2. CO-release cross-sections of 1–3 (Table 1) are comparable to that observed for cyanine-flavonol hybrids,55 isoelectronic xanthene-9-carboxylic acid,35 or meso-BODIPY-carboxylates,56 as the only reported small-molecule non-metal organic photoCORMs absorbing above 500 nm.27| Dye | Φ decomp /10−4 | εΦ | [CO]/eq. |
|---|---|---|---|
| a The aliquots for CO quantification were taken from the headspace after no starting material was observed (UV/vis absorption) and analyzed by GC. Samples were aerated unless stated otherwise. Each experiment was run in triplicate or more replications, and the averaged values are shown. b Absolute quantum yields of degradation of 1-3 were determined in aerated solutions using chemical actinometer (ESI†). c εΦ/mol−1 dm3 cm−1, CO-release cross-section (the product of the molar absorption coefficient at λmax(abs) (ESI†) and the corresponding quantum yield of dye degradation). d Irradiation of diluted (c ∼ 1.5 × 10−5 mol dm−3) solutions; quantification by GC-RGA. In parentheses: Irradiation of more concentrated (c ∼ 1–3 × 10−4 mol dm−3) solutions. e Aerated. f Argon-bubbled solutions; analyzed by GC-MS. g See ESI† for more details on the statistical analysis. | |||
| 1 | 6.7 | 53 | 0.41 ± 0.11d (0.33 ± 0.14e; 0.11 ± 0.04f)g |
| 2 | 1.3 | 13 | 1.04 ± 0.16d (0.52 ± 0.07e) p < 0.001 |
| 3 | 4.0 | 38 | 0.81 ± 0.16d (0.43 ± 0.06e) p < 0.001 |
Solutions of 1–3 (c ∼ 1–3 × 10−5 mol dm−3, V = 0.5 L) in an aq triethylammonium acetate buffer (cbuf = 0.1 mol dm−3, pH = 7.0) in a custom-made glass reactor (Fig. S81†) were irradiated with LEDs (Fig. S83†) for 48 h. The headspace was transferred into an IR gas cuvette, and the IR spectra were recorded (Fig. S28–S30†). A well-resolved absorption band with a fine structure was found in the region of 2220–2060 cm−1. The spectral features of this band match those of the authentic sample (10 ppm CO in dry N2(g)) measured independently under the same conditions (Fig. S27†). The remaining signals were attributed to gases of ambient air (CO2, water vapor, etc.) or buffer constituents’ vapors. No other gaseous products were found in significant amounts. However, this method is restricted only to qualitative analysis due to large measurement uncertainty.
To quantify CO, solutions of 1–3 (c ∼ 1.5 × 10−5 mol dm−3, PBS) were irradiated by white LEDs (Fig. S84†) in headspace vials for 16 h (until no starting material was observed). The amount of CO released into the vial headspace was measured by GC with a reducing gas analyzer (RGA). The molar amounts n(CO) were calculated from the calibration curves and compared to the starting molar amounts of 1–3 (Table 1).
We found the chemical yields of released CO from 1 to be independent of the dye concentration in the range from 1 × 10−5 to 1 × 10−4 mol dm−3. In contrast, they decreased by a factor of ∼2 for higher concentrated solutions of 2–3 (Table 1; ESI†), as also reported for 3 elsewhere.57 We hypothesized that it is connected to competitive bimolecular photoprocesses which are more efficient than CO liberation. Dyes with high intersystem crossing (isc) quantum efficiency, e.g., 3 (Φisc(PBS) = 0.98),58 readily transfer the excitation energy in the solution and produce 1O2, which might participate in decomposition.57
Oxygen was reported to play only a minor role in the photochemical transformations of dyes with inefficient isc, such as 1 (Φisc ∼ 0.03–0.06).59,60 Upon prolonged irradiation of 1 in an O2-free environment, there are contradicting observations of (1) no irreversible changes,10 and (2) very inefficient photodecomposition in the presence of other oxidizing agents (as well as in presence of oxygen).8 We observed, that the yields of released CO dropped to about 10% in deoxygenated samples of 1 (Ar-purged), compared to ∼40% in aerated solutions (Table 1), supporting the hypothesis that 1 is indeed reacting in absence of oxygen, but with lower efficiency.
To further test the reactivity of 1–3 with singlet oxygen, thermostated methanol solutions (T = 40 °C) with a naphthalene-based 1O2-source61 (4, c = 5.3 × 10−3 mol dm−3; ∼100 eq.)62 were monitored for 20 h in the dark (ESI†). We observed only an insignificant decrease in the concentrations of 1–3 (<7%; evaluated at λabs(max); Fig. S6†) and concluded that 1O2 plays only a minor role in the primary reaction of photolysis of 1–3. The dyes (which are also O2 sensitizers; Table S1†) in identical aerated aq and methanol solutions completely degraded upon irradiation during the same time period.591O2 was reported to accelerate degradation of 3,57 but its role as an oxidant is presumably important only in the later degradation steps.
We attempted to find out which carbon atom of 1 is primarily responsible for the CO formation. A solution of 13C2-1 (containing ∼50% of 13C in the carboxylic group and ∼50% of 13C in the C-9 atom, Scheme 1; see ESI† for synthesis) was irradiated under the same conditions as in the previous experiments. The total yield of 13CO reached ∼5% (Fig. S85†). We thus conclude that decarbonylation of the carboxylate group or oxidation of the C-9 atom are not major contributors of produced CO.
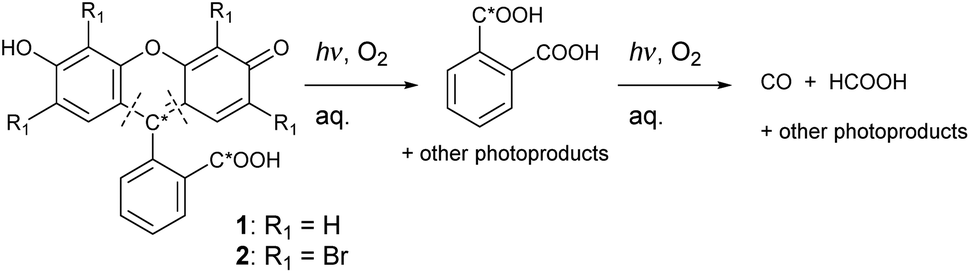 | ||
| Scheme 1 Photochemistry of 1, 13C2-1 and 2 in PBS (products identified by NMR and EI-MS). Note: * denotes 13C label (only one position in 1 is labeled in a particular molecule). Dashed lines show disconnections leading to a multistep oxidative release of phthalic acid. | ||
We analyzed photolyzed mixtures to identify photoproducts by NMR. Solutions of 1, 13C2-1, 2, or 3 (c ∼ 6 × 10−3 mol dm−3) in D2O-based PBS were irradiated with LEDs (Fig. S83†) until no starting material was observed. Upon irradiation of 1, 13C2-1 or 2, we observed a simple set of signals in both aromatic and aliphatic regions of 1H NMR, with a distinct multiplet at δ = 7.5–7.7 ppm and a singlet at δ ∼ 8.4 ppm (Fig. S39, S41, and S43†). However, 13C{1H} NMR spectrum of the reaction mixture from 13C2-1 revealed only one major signal at δ 169.2 ppm, which we assigned to phthalic acid (Fig. S34†), indicating that concentrations of other compounds bearing the 13C atom are very low. Further NMR (incl. 2D) and MS analyses (Fig. S38†) allowed us to identify the formation of phthalic acid in ∼95% yield at ∼30% conversion of 2 (Fig. 2). The singlet at δ ∼ 8.4 ppm was confirmed to be formic acid using an authentic sample. A rather complicated set of signals with a broad cluster at δ = 7.5–8.0 ppm (Fig. S32, S33, S40, and S41†) was observed, but no other photoproducts were identified. A similar product distribution was found upon exhaustive irradiation of 1 and 13C2-1 in CD3OD (Fig. S36 and S37†). Upon irradiation of 3 in PBS, the corresponding signal at δ ∼8.4 ppm assigned to formic acid was found (Fig. S46†), assuming that analogous tetrachlorophthalic acid can be produced from 3.
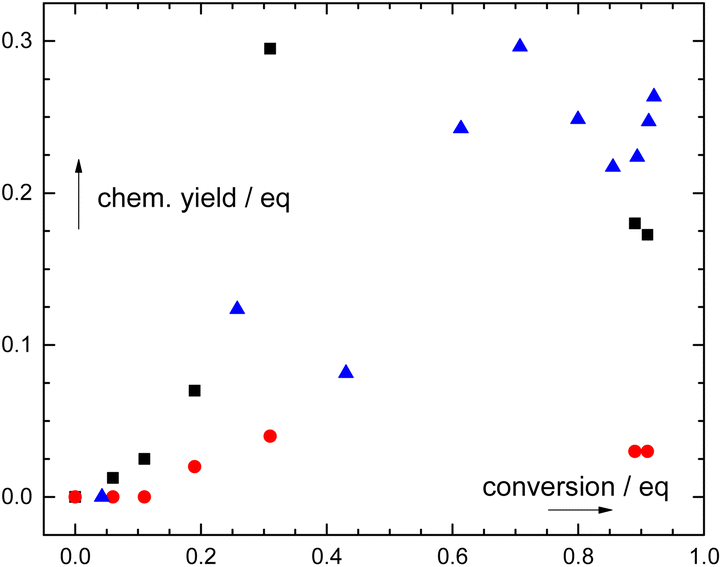 | ||
| Fig. 2 Dependence of the chemical yields of phthalic acid (black squares), formic acid (red circles), and CO (blue triangles) on the photodegradation of 2. Concentrations of photoproducts were determined independently – by quantitative 1H NMR for phthalic and formic acids, and by GC-MS for CO (ESI†). | ||
The photoproduct distribution was not constant during the photolysis of 1–3 (1H NMR; Fig. S39, S43, and S45†). Phthalic acid emerged at early stages of irradiation in high yields (up to ∼95% at ∼30% conversion of 2; Fig. 2) and slowly disappeared upon prolonged irradiation. Additionally, formic acid appeared only at higher conversions. Both CO and formic acid must thus originate from secondary photochemical processes (Scheme 1) because no elementary step leads to their formation. However, this kinetic analysis is not a prove of a sequence of the steps and does not bring any information on the particular carbon atom or fragment transformation as a source of CO or formic acid.
LC-MS analyses of irradiated solutions of 1 and 13C2-1 (Fig. S59–S80; ESI†) identified phthalic acid (Fig. S60 and S65†) as a photoproduct in accordance with our NMR data. Several other relatively intense signals appeared, and we attempted to structurally assign them to specific intermediates. We considered several different modes of the bonds’ disconnections in 1 and suggested potential structures derived from the xanthene core (e.g., Fig. S8†), such as various diphenylethers, carboxylic acids, and (poly)phenols or quinones. None of the observed MS signals corresponded with the exact proposed structures (ESI†) but the products must be closely related. Anticipated diphenylethers and phenols are likely to undergo efficient secondary photodegradation. For example, phenol can be readily oxidized by 1O2 to quinones.63 Such subsequent reactions hamper their identification.
When 13C2-1 was photolyzed to give labeled phthalic acid but not CO or HCOOH to a more significant extent (<5% 13C in both CO and HCOOH; Fig. S85†), the products must preferentially originate from other structural parts of 1 than the C9 atom and the carboxylate group. Still, the major degradation pathway involves opening the central 2H-pyran ring. The secondary reactions are most likely oxidative, involving either singlet oxygen sensitized by a triplet-excited dye or ground-state oxygen reacting with a triplet-excited or radical species. Nevertheless, <5% yield of 13C in CO suggests that at least one of the labeled carbons still partially contributes to its formation.
We also evaluated the photochemistry of 1 in the gas phase using photodissociation and mass spectroscopies. From all acid–base forms, only a cation (not present in aqueous media under physiological pH; Scheme S2†) was detectable. The photodissociation spectra of 1+ feature a low-intensity maximum at λobs(abs) = 420.8 nm corresponding to the 0–0 transition (Fig. S9†). A He-tagging spectrum suggested that the nature of the excited state nature is most likely complex (ESI†).
The MS analysis further showed several fragmentations of excited 1+ such as a loss of 46 mass units (Fig. S10†), which could correspond to the dissociation of formic acid from the carboxylate (ESI†). However, we were unable to draw more detailed conclusions or establish structures of other putative products. The experimental setup hindered our attempts to examine the anions 1-H− and 12−.
Upon heating to 500 °C, we identified the release of only CO2 and H2O as major products but no CO (Fig. S47–S58†), ruling out thermal decomposition of 1–3 as a possible dark pathway.
Experimental
The detailed experimental and analytical procedures are in the ESI.†Conclusion
In conclusion, we demonstrate that extensive irradiation of fluorescein, eosin Y, and rose bengal with green and white light leads to the release of CO in amounts that can affect physiological processes in cells.54 The release of CO was evidenced and quantified by independent optical spectroscopy, chromatography, and MS techniques. Also, 13C labeling was utilized to identify the origin of CO, suggesting that at least two individual reaction pathways are responsible. We found phthalic and formic acids as other photoproducts, but cannot draw a plausible detailed mechanism of these transformations now. We conclude that the main reaction pathway leading to the release of CO and other products is a complex multistep reaction involving multiple excitations and several oxidative degradation steps. Our data suggest that the photodegradation of these dyes may be of biological importance and should be considered in medicinal optical applications.Author contributions
M. M., L. L., M. Š., R. N., L. M., J. H. and P. Š. performed experiments and analysed the data. P. Š. conceived the idea, designed the research and wrote the manuscript with inputs from all other co-authors. M. M., L. L., L. M., and L. V. designed particular experiments. All authors revised the final draft.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by Czech Science Foundation (GJ20-30004Y), grant GAUK 314621, Ministry of Transport (program of long-term conceptual development of research institutions), RECETOX RI (No. LM2018121) and CETOCOEN EXCELLENCE project (No. CZ.02.1.01/0.0/0.0/17_043/0009632) financed by the MEYS, and the EU's Horizon 2020 Research and Innovation Programme (No. 857560). This publication reflects only the author's view, and the European Commission is not responsible for any use that may be made of the information it contains.References
- L. D. Lavis, Biochemistry, 2017, 56, 5165–5170 CrossRef CAS
.
- S. B. Wagh, V. A. Maslivets, J. J. La Clair and A. Kornienko, ChemBioChem, 2021, 22, 3109–3139 CrossRef CAS
- M. Yamashita and H. Kashiwagi, IEEE J. Quantum Electron., 1976, 12, 90–95 Search PubMed
- J. Han and K. Burgess, Chem. Rev., 2010, 110, 2709–2728 CrossRef CAS
-
J. R. Lakowicz, Principles of Fluorescence Spectroscopy, Springer US, 3rd edn, 2006 Search PubMed
- L. I. Grossweiner and E. F. Zwicker, J. Chem. Phys., 1959, 31, 1141–1142 CrossRef CAS
- K. Uchida, S. Kato and M. Koizumi, Nature, 1959, 184, 1620–1621 CrossRef CAS
- L. Lindqvist, Ark. Kemi, 1960, 16, 79–137 CAS
- S. Boudin, J. Chem. Phys., 1930, 27, 285–290 CAS
- M. Imamura and M. Koizumi, Bull. Chem. Soc. Jpn., 1955, 28, 117–124 CrossRef CAS
-
A. A. Untereiner, L. Wu and R. Wang, in Gasotransmitters: Physiology and Pathophysiology, Springer, Berlin, Heidelberg, 2012, pp. 37–70 Search PubMed
- N. Chim, P. M. Johnson and C. W. Goulding, J. Inorg. Biochem., 2014, 133, 118–126 CrossRef CAS
- L. Wu and R. Wang, Pharmacol. Rev., 2005, 57, 585–630 CrossRef CAS
- R. Motterlini and L. E. Otterbein, Nat. Rev. Drug Discovery, 2010, 9, 728 CrossRef CAS PubMed
- S. M. Lowson, Br. Med. Bull., 2004, 70, 119–131 CrossRef CAS
- D. Nguyen, T.-K. Nguyen, S. A. Rice and C. Boyer, Biomacromolecules, 2015, 16, 2776–2786 CrossRef CAS PubMed
- R. Motterlini, B. Haas and R. Foresti, Med. Gas Res., 2012, 2, 28 CrossRef CAS PubMed
- L. E. Otterbein, F. H. Bach, J. Alam, M. Soares, H. T. Lu, M. Wysk, R. J. Davis, R. A. Flavell and A. M. K. Choi, Nat. Med., 2000, 6, 422–428 CrossRef CAS
- A. Halilovic, K. A. Patil, L. Bellner, G. Marrazzo, K. Castellano, G. Cullaro, M. W. Dunn and M. L. Schwartzman, J. Cell. Physiol., 2011, 226, 1732–1740 CrossRef CAS PubMed
- J. A. Campbell, J. Pathol. Bacteriol., 1932, 35, 379–394 CrossRef CAS
- L. Vítek, H. Gbelcová, L. Muchová, K. Váňová, J. Zelenka, R. Koníčková, J. Suk, M. Zadinova, Z. Knejzlík, S. Ahmad, T. Fujisawa, A. Ahmed and T. Ruml, Dig. Liver Dis., 2014, 46, 369–375 CrossRef PubMed
- M. Allanson and V. E. Reeve, Cancer Immunol. Immunother., 2007, 56, 1807–1815 CrossRef CAS PubMed
- X. Yang, M. de Caestecker, L. E. Otterbein and B. Wang, Med. Res. Rev., 2019, 40, 1147–1177 CrossRef PubMed
- S. Kourembanas, Antioxid. Redox Signal., 2002, 4, 291–299 CrossRef CAS PubMed
- I. Blumenthal, J. R. Soc. Med., 2001, 94, 270–272 CrossRef CAS PubMed
- F. Gullotta, A. di Masi and P. Ascenzi, IUBMB Life, 2012, 64, 378–386 CrossRef CAS
- P. Šebej and T. Slanina, Photochem. Photobiol. Sci., 2018, 17, 692–710 CrossRef
- W. Adach and B. Olas, Future Med. Chem., 2018, 11, 61–73 CrossRef
- C. C. Romão, W. A. Blättler, J. D. Seixas and G. J. L. Bernardes, Chem. Soc. Rev., 2012, 41, 3571–3583 RSC
- S. H. Heinemann, T. Hoshi, M. Westerhausen and A. Schiller, Chem. Commun., 2014, 50, 3644–3660 RSC
- R. Motterlini, J. E. Clark, R. Foresti, P. Sarathchandra, B. E. Mann and C. J. Green, Circ. Res., 2002, 90, e17–e24 CrossRef CAS PubMed
- S. García-Gallego and G. J. L. Bernardes, Angew. Chem., Int. Ed., 2014, 53, 9712–9721 CrossRef
- M. Wrighton, Chem. Rev., 1974, 74, 401–430 CrossRef CAS
- M. A. Wright and J. A. Wright, Dalton Trans., 2016, 45, 6801–6811 RSC
- L. A. P. Antony, T. Slanina, P. Šebej, T. Šolomek and P. Klán, Org. Lett., 2013, 15, 4552–4555 CrossRef CAS PubMed
-
R. S. Givens, in Organic Photochemistry, ed. A. Padwa, Marcel Dekker, 1981, pp. 227–346 Search PubMed
- R. G. W. Norrish and C. H. Bamford, Nature, 1937, 140, 195–196 CrossRef CAS
- M. Martínek, J. Váňa, P. Šebej, R. Navrátil, T. Slanina, L. Ludvíková, J. Roithová and P. Klán, ChemPlusChem, 2020, 85, 2230–2242 CrossRef PubMed
- L. Xing, B. Wang, J. Li, X. Guo, X. Lu, X. Chen, H. Sun, Z. Sun, X. Luo, S. Qi, X. Qian and Y. Yang, J. Am. Chem. Soc., 2022, 144, 2114–2119 CrossRef CAS
- A. F. Monteiro, M. Rato and C. Martins, Clin. Dermatol., 2016, 34, 571–581 CrossRef
- S. Goetze, C. Hiernickel and P. Elsner, Skin Pharmacol. Physiol., 2017, 30, 76–80 CrossRef CAS PubMed
- W. B. Kim, A. J. Shelley, K. Novice, J. Joo, H. W. Lim and S. J. Glassman, J. Am. Acad. Dermatol., 2018, 79, 1069–1075 CrossRef CAS
- G. A. Hofmann and B. Weber, JDDG, J. Dtsch. Dermatol. Ges., 2021, 19, 19–29 Search PubMed
- G. Cosa, M. Lukeman and J. C. Scaiano, Acc. Chem. Res., 2009, 42, 599–607 CrossRef CAS PubMed
- R. Brancato, F. Bandello and R. Lattanzio, Surv. Ophthalmol., 1997, 42, 41–70 CrossRef CAS
-
WHO, World Health Organization model list of essential medicines: 21st list 2019, 21st edn, 2019 Search PubMed
- G. A. Sonn, S.-N. E. Jones, T. V. Tarin, C. B. Du, K. E. Mach, K. C. Jensen and J. C. Liao, J. Urol., 2009, 182, 1299–1305 CrossRef PubMed
- L. M. Wang, M. A. Banu, P. Canoll and J. N. Bruce, Front. Oncol., 2021, 11, 666734 CrossRef PubMed
- M. Majek, F. Filace and A. J. von Wangelin, Beilstein J. Org. Chem., 2014, 10, 981–989 CrossRef PubMed
- V. Srivastava and P. P. Singh, RSC Adv., 2017, 7, 31377–31392 RSC
- E. K. W. Schulte and D. H. Wittekind, Stain Technol., 1989, 64, 255–256 CrossRef CAS PubMed
- R. P. G. Feenstra and S. C. G. Tseng, Arch. Ophthalmol., 1992, 110, 984–993 CrossRef CAS PubMed
- F. Wilkinson, W. P. Helman and A. B. Ross, J. Phys. Chem. Ref. Data, 1993, 22, 113–262 CrossRef CAS
- M. Šranková, A. Dvořák, M. Martínek, P. Šebej, P. Klán, L. Vítek and L. Muchová, Int. J. Mol. Sci., 2022, 23, 1504 CrossRef
- L. Štacková, M. Russo, L. Muchová, V. Orel, L. Vítek, P. Štacko and P. Klán, Chem. – Eur. J., 2020, 26, 13184–13190 CrossRef
- E. Palao, T. Slanina, L. Muchová, T. Šolomek, L. Vítek and P. Klán, J. Am. Chem. Soc., 2016, 138, 126–133 CrossRef CAS PubMed
- L. Ludvíková, P. Friš, D. Heger, P. Šebej, J. Wirz and P. Klán, Phys. Chem. Chem. Phys., 2016, 18, 16266–16273 RSC
- P. Murasecco-Suardi, E. Gassmann, A. M. Braun and E. Oliveros, Helv. Chim. Acta, 1987, 70, 1760–1773 CrossRef CAS
- K. Gollnick and G. O. Schenck, Pure Appl. Chem., 1964, 9, 507–526 CrossRef CAS
- Y. Usui, Chem. Lett., 1973, 2, 743–744 CrossRef
- N. J. Turro and M. F. Chow, J. Am. Chem. Soc., 1981, 103, 7218–7224 CrossRef CAS
- R. Bernstein and C. S. Foote, J. Phys. Chem. A, 1999, 103, 7244–7247 CrossRef CAS
- J. Al-Nu'airat, B. Z. Dlugogorski, X. Gao, N. Zeinali, J. Skut, P. R. Westmoreland, I. Oluwoye and M. Altarawneh, Phys. Chem. Chem. Phys., 2019, 21, 171–183 RSC
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ob01823c |
| This journal is © The Royal Society of Chemistry 2023 |