Persistent azulene α-carbocations: synthesis from aldehydes, spectroscopic and crystallographic properties†
Received
16th September 2022
, Accepted 21st December 2022
First published on 5th January 2023
Abstract
The non-benzenoid aromatic system azulene is sufficiently nucleophilic at C1 that it can react with a protonated aldehyde to form an α-azulenyl alcohol. This in turn may be protonated and undergo loss of water to give an azulene α-carbocation. We report the isolation of such azulenyl cations as salts with non-coordinating anions. The salts have been characterised by NMR, UV/Vis absorption and (in certain cases) X-ray crystallography. Reduction of representative salts to afford azulenyl(aryl) methylenes has been demonstrated.
Introduction
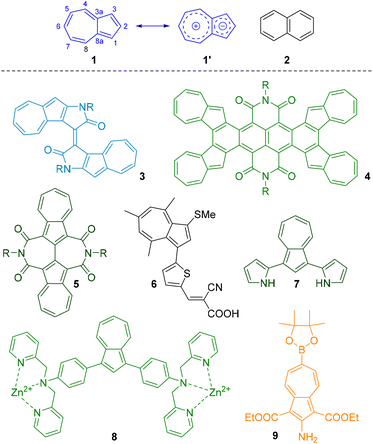
Azulene 1 is a bicyclic non-benzenoid aromatic hydrocarbon with a 10-electron π-system, which is isomeric with naphthalene 2. Its properties differ from those of benzenoid 2 in several significant ways.1 It is unusually polar for a molecule devoid of heteroatoms, possessing a dipole moment of 1.08 D,2 which arises from the contribution to the resonance hybrid of structure 1′, in which the two rings are both individual 6-electron aromatic systems. It also exhibits the anomalous fluorescence property of emitting as a result of the S0 ← S2 transition, in violation of Kasha's rule.3 Strikingly, whereas 2 is colourless, 1 is a deep blue colour, as a result of its HOMO–LUMO gap being in the visible region. Ring substituents that perturb the azulene chromophore give rise to derivatives of many different colours. The relationship between the electronic nature of a substituent, its point of attachment and resultant shift in the absorption maximum has been studied.4 Many new dyes based on azulene have been reported, and selected examples are shown in Fig. 1, such as azulenoisoindigo (3),5 azulene-fused tetracene diimide 4,6 and biazulene diimides 5.7 Azulene dyes such as 6 have been exploited in dye-sensitized solar cells.8 The changes in optical properties (both absorbance and fluorescence) that occur as a result of changes to the substituents make azulene derivatives ideal for use as chemosensors and chemodosimeters. For example, bis(pyrrolyl)azulene 7 is a fluorescent and colorimetric sensor for fluoride,9 AzuFluor® 435-DPA-Zn (8) is a fluorescent sensor for ADP10 and AzuFluor® 483-Bpin (9) is a fluorescent dosimeter for peroxynitrite.11
 |
| | Fig. 1 Structures of azulene, naphthalene and azulene-based dyes. | |
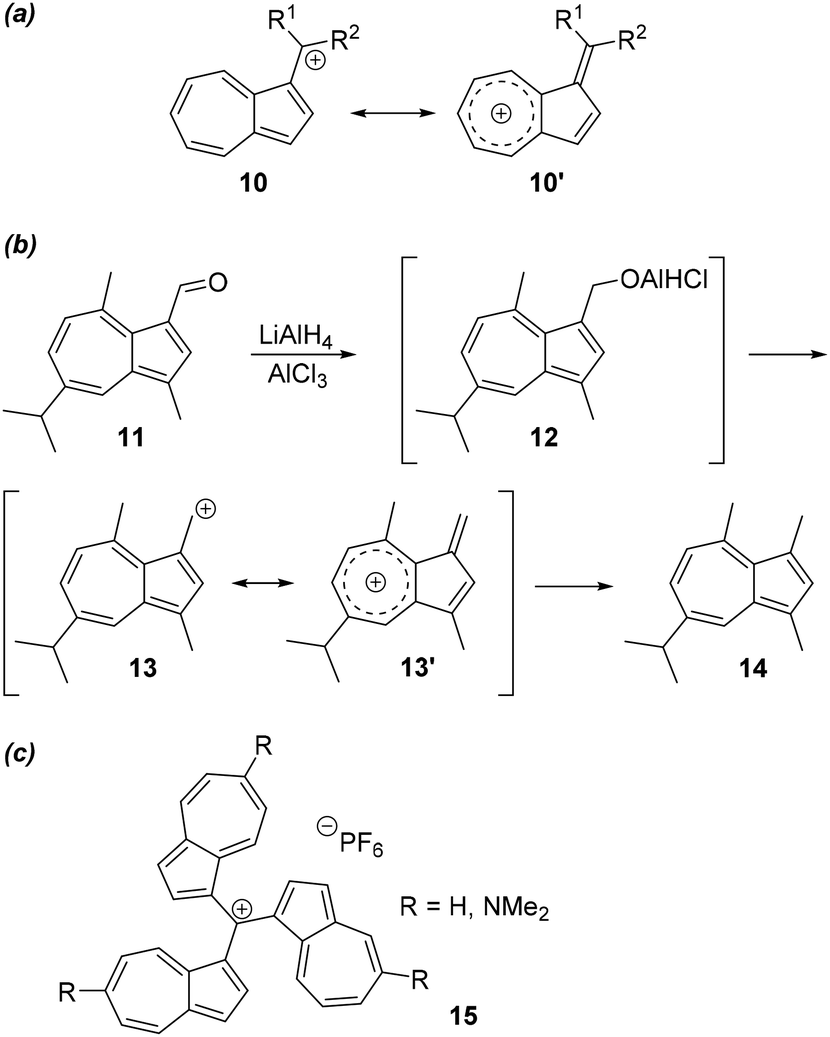
In any azulene 10 with a carbon substituent at the 1- or 3-position, a positive charge on that α-aryl carbon will be stabilised due to the existence of a resonance structure comprising a tropylium ring, 10′ (Scheme 1a). The stabilisation of such cations explains various aspects of azulene reactivity that are distinct from benzenoid aromatic systems.12 For example, 3-formylguaiazulene (11) may be reduced twice (Scheme 1b), first to alkoxy intermediate 12, and then to 3-methylguaiazulene (14).13 This second reduction is enabled by the stability of cationic intermediate 13 (Scheme 1b). The intermediacy of cations such as 10 also enables reactions such as hydration of 1-azulenylalkynes,14 formation of azuleno[2,1-b]thiophenes,15 formation of calix[n]azulenes,16 pyrrole-mediated azulene decarbonylation,17 and protection of thiols with azulene-based protecting groups.18 If cations of type 10 possess R1 and R2 substituents which are sufficiently cation-stabilising, then they may be isolable as stable salts with non-nucleophilic anions. A notable example are the tris(azulen-1-yl)cations 15, which display exceedingly high pKR+ values as a result of the stability conferred by the three cation-stabilising azulene substituents (Scheme 1C). For example, 15 (R = H)19 has pKR+ = 11.3, whereas the effect of additional electron donating groups is clearly shown for 15 (R = NMe2),20 with pKR+ = 24.3.
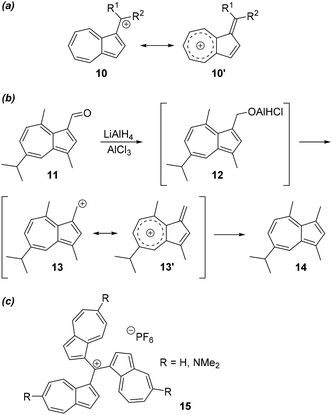 |
| | Scheme 1 Stability of α-aryl cations at the azulene 1- or 3-position. | |
Cations of type 10 can be stable enough to be isolated even when R1 = H, as long as R2 ≠ H. Such cations can be conveniently prepared by reaction of an azulene with the requisite aldehyde in the presence of an appropriate Brønsted acid (i.e. one with a non-coordinating anion, such as HBF4, HPF6 or HClO4) as shown in Scheme 2. Protonation of the aldehyde 16 activates it towards an SEAr reaction with an azulene 17 to afford alcohol 19. This in turns undergoes a second protonation, followed by loss of water to give persistent cations 20 (Scheme 2). The outcome of the reaction is dependent on stoichiometry, since if an excess of the azulene is used, a second SEAr reaction can occur to give bis(azulen-1-yl)methines of type 21. Products of type 20 have been studied by Reid21 and by Stepanow,22 who have demonstrated the applicability of this process to various aromatic and aliphatic aldehydes. Subsequently, Takekuma also disclosed extensive studies on this class of compounds.23 Applications of salts of type 20 have been described in the patent literature, such as in optical data storage,24 in thermal-transfer printing,25 in photocrosslinkable resins,26 in cancer phototherapy27 and in retinal tissue staining.28 These salts have also been employed for the synthesis of various heterocycles29 and in chemical sensing of various anions.30 The wide substrate scope of the reaction shown in Scheme 2 led us to prepare and study novel cations of type 20 derived from aldehydes that have not previously been used in this process. The results of these investigations are reported here.
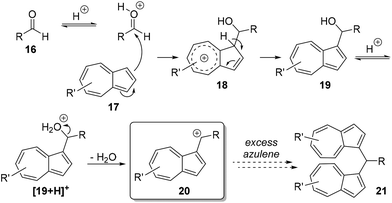 |
| | Scheme 2 Synthesis of azulene α-aryl cations from aryl aldehydes. | |
Results and discussion
We chose guaiazulene 22 as the substrate for cation formation, since not only is it appreciably cheaper than 1 (as a result of the availability of 22 from natural sources31), but the presence of the C1-methyl substituent precludes oligomerisation reactions.32 Application of previously reported reaction conditions (HPF6, MeOH, room temperature, 2 h) gave the expected products with a wide range of both aryl and heteroaryl aldehydes, including those bearing both electron-donating and electron-withdrawing substituents (Scheme 3). In the first instance we employed p-dimethylaminobenzaldehyde to prepare salt 24. Other salt forms of 24 have been reported previously (with BF4−, ClO4−, or I− counteranions).21b,23f,r Salt 24 was comprehensively characterised by NMR spectroscopy (see ESI†), which allowed for assignment of each proton and carbon resonance in the structure (Fig. 2).
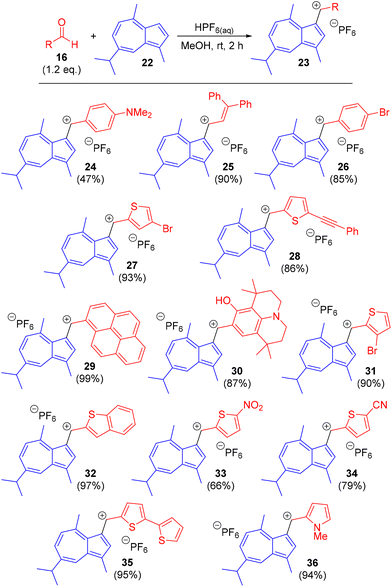 |
| | Scheme 3 Synthesis of arylazulenyl cations 24–36. | |
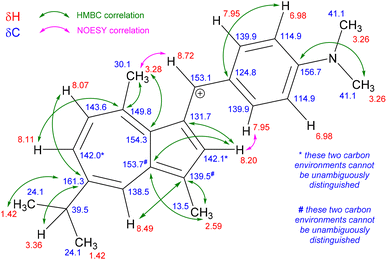 |
| | Fig. 2 Structure of 24, with selected HMBC and NOESY interactions indicated. | |
We next applied the reaction conditions to a selection of (hetero)aromatic aldehydes to synthesise novel salts 25–36. These were isolated as air-stable crystalline solids in good yields (79–99%), with the exception of 33, for which the yield was moderate (66%). This lower yield may be attributable to the presence of the highly electron-withdrawing nitro substituent. There are few reports of α-aryl cations in conjugation with nitro groups, but such salts can be isolable when other electron-donating groups are also present.33 We next sought to characterise the novel salts crystallographically and were able to obtain X-ray structures for 25 (Fig. 3), 26 (Fig. 4), 30 (Fig. 5) and 32 (Fig. 6).
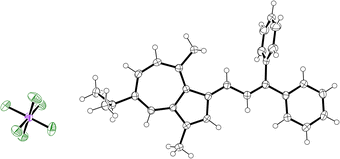 |
| | Fig. 3 ORTEP representation of the X-ray structure of 25. Ellipsoids are shown at 50% probability. Hydrogens are shown as spheres of arbitrary radius. CCDC 2129313.† | |
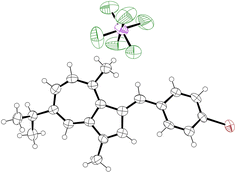 |
| | Fig. 4 ORTEP representation of the X-ray structure of 26. Ellipsoids are shown at 50% probability. Hydrogens are shown as spheres of arbitrary radius. Only one of 3 independent molecules in the unit cell is shown for clarity. CCDC 2129312.† | |
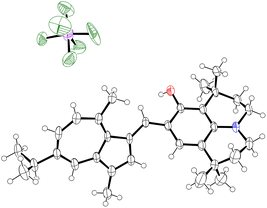 |
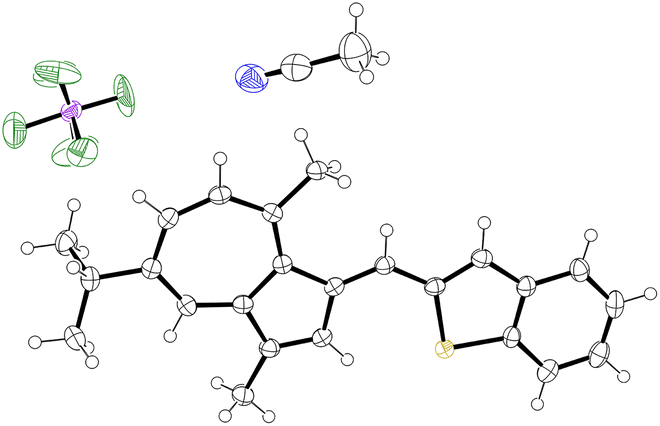
| | Fig. 5 ORTEP representation of the X-ray structure of 30. Ellipsoids are shown at 50% probability. Hydrogens are shown as spheres of arbitrary radius. Only one of 2 independent molecules in the unit cell is shown. Disordered water molecules and disorder in PF6− anion are omitted for clarity. CCDC 2129314.† | |
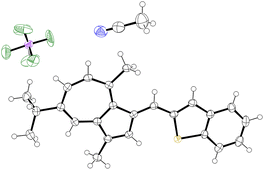 |
| | Fig. 6 ORTEP representation of the X-ray structure of 32·MeCN. Ellipsoids are shown at 50% probability. Hydrogens are shown as spheres of arbitrary radius. Disorder in PF6− anion omitted for clarity. CCDC 2129315.† | |
The structure of 25 reveals that in the solid state the π-system is not planar. Rather, the (Z)-phenyl substituent is oriented almost perpendicular to the propenyl chain; the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–Cipso–Cortho dihedral angle is 85.1(3)°. This is in keeping with the trend observed for all 1,1-diphenylethenes for which X-ray crystallographic data have been reported, i.e. there are no examples in which this motif has the two phenyl rings oriented coplanar. Such an orientation would of course lead to the occlusion of the two ortho-hydrogen atoms, but it is nevertheless surprising that the deviation from planarity in 25 is as significant as it is.
C–Cipso–Cortho dihedral angle is 85.1(3)°. This is in keeping with the trend observed for all 1,1-diphenylethenes for which X-ray crystallographic data have been reported, i.e. there are no examples in which this motif has the two phenyl rings oriented coplanar. Such an orientation would of course lead to the occlusion of the two ortho-hydrogen atoms, but it is nevertheless surprising that the deviation from planarity in 25 is as significant as it is.
As per 25, the structures of 26 and 30 also adopt conformations in the solid state where the π-system exhibits a degree of deviation from planarity (Table 1); the CAz–C+–CAr(α)–CAr(β) dihedral angle being between 28(1)° and 30(2)° for 26 and between 22.1(6)° and 24.9(6)° for 30.‡ In both cases, this deviation from planarity can again be explained through occlusion of hydrogen atoms, in this case the phenyl ortho-hydrogen and the hydrogen at the azulene 2-position. In contrast, the π-system is close to fully planar for 32, the CAz–C+–CAr(α)–S dihedral angle being 3.5(4)°. We suggest such a planar conformation is now more favourable as the sulfur atom lacks a substituent to occlude the azulene 2-position hydrogen.
Table 1 Selected parameters for X-ray structures of 25, 26, 30 and 32; (X = C, S)
| Salt |
CAz–C+ bond length‡/Å |
C+–Cα bond length‡/Å |
CAz–C+–CAr(α)–XAr(β) dihedral angle‡ |
|
25
|
1.372(3) |
1.430(3) |
n/a |
|
26
|
1.36(1)–1.39(1) |
1.45(2) |
28(1)°–30(2)° |
|
30
|
1.385(6)–1.391(6) |
1.421(4)–1.426(5) |
22.1(6)°–24.9(6)° |
|
32
|
1.373(3) |
1.428(3) |
3.5(4)° |
We next characterised novel salts 24–36 by UV-vis absorption spectroscopy (Fig. 7 and Table 2). The diverse array of colours exhibited by 24–36 is reflected by the variation in absorption maxima that were observed. Notably, the two salts exhibiting the highest extinction coefficients in the visible region were 24 and 30, which are the two salts bearing the most electron-donating substituents, i.e. amino groups. The difference in absorption maxima may be ascribable to the fact that 30 bears an additional conjugated electron-donating substituent, i.e. a hydroxyl group.
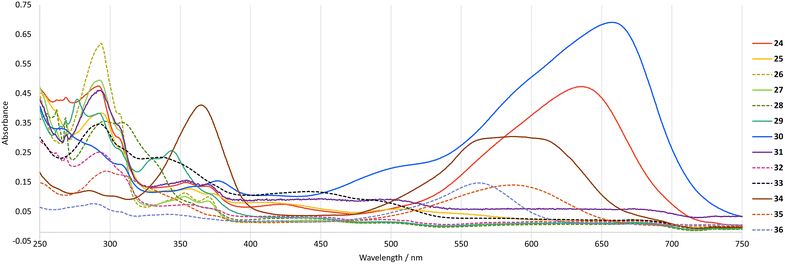 |
| | Fig. 7 UV/vis absorption spectra for salts 24–36 (15 μM) in MeCN. | |
Table 2 Absorption maxima and extinction coefficients for salts 24–36 in MeCN
| Compound |
λ
max/nm (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ε) ε) |
|
24
|
640 (4.50), 421 (3.70), 354 (4.00) |
|
25
|
506 (3.59), 409 (3.74), 357 (3.93) |
|
26
|
669 (3.02), 447 (3.16), 370 (3.82) |
|
27
|
676 (2.89), 445 (3.11), 370 (3.76) |
|
28
|
670 (2.79), 439 (3.28) |
|
29
|
669 (2.94), 490 (3.01) |
|
30
|
657 (4.66), 377 (4.01) |
|
31
|
656 (3.59), 499 (3.79), 355 (4.02) |
|
32
|
674 (3.10), 492 (3.19), 353 (3.69) |
|
33
|
672 (3.17), 441 (3.90) |
|
34
|
586 (4.31), 365 (4.44) |
|
35
|
587 (3.97), 298 (4.10) |
|
36
|
563 (3.99), 289 (3.71) |
We investigated the reduction of salts of type 23 with a hydridic reducing agent. Thus, two representative example salts (27 and 29) were treated with sodium borohydride to afford the corresponding methylene compounds 37 and 38 (Scheme 4). In both cases the desired reduction occurred rapidly at room temperature, with the product precipitating from solution in sufficient purity that after filtration, no further purification was necessary. Both 37 and 38 were blue in the solid state, as a consequence of the azulene motif no longer being conjugated to the other aromatic system. We were able to obtain an X-ray structure for 38 (Fig. 8).
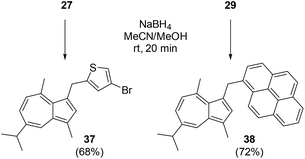 |
| | Scheme 4 Reduction of salts 27 and 29. | |
 |
| | Fig. 8 ORTEP representation of the X-ray structure of 38. Ellipsoids are shown at 50% probability. Hydrogens are shown as spheres of arbitrary radius. CCDC 2129310.† | |
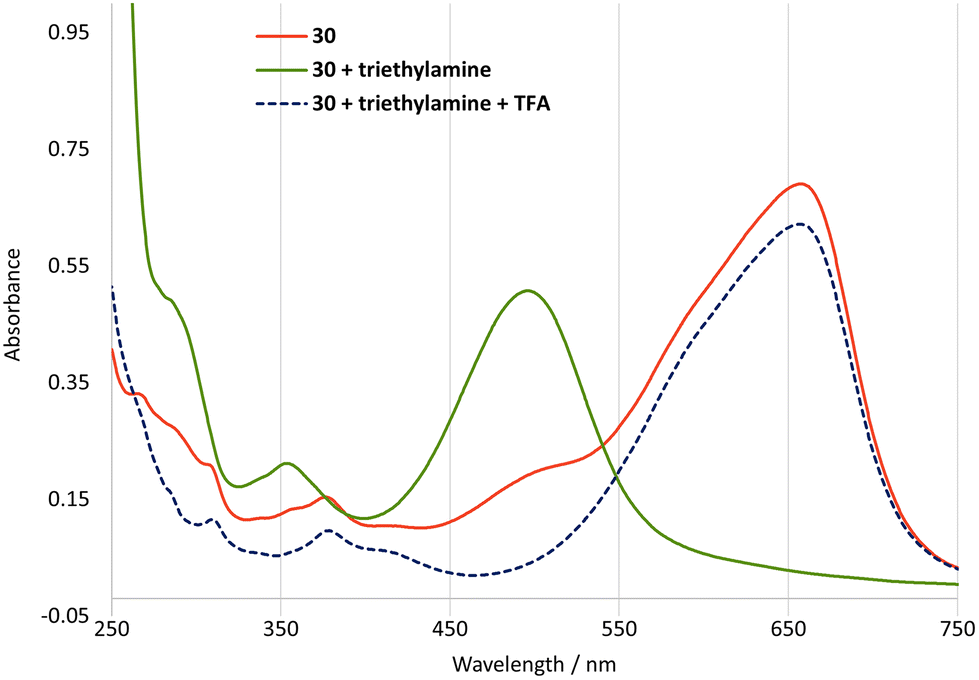
The protons on the C4-methyl group of guaiazulene are acidic,34 and in cations 24–36 they would be expected to be even more acidic due to the presence of the conjugated cation. However, deprotonation at the C4-methyl group of 24–36 would give an azulenoquinone dimethide system 39, which would be expected to be unstable with respect to self-reaction35 or other decomposition pathways (Scheme 5a). Of the salts 24–36, julolidine derivative 30 is unique in possessing an alternative site of deprotonation, namely the hydroxyl group. In this case, deprotonation of 30 would lead to neutral ortho-quinone methide 40, in which aromaticity of the azulene motif is retained (Scheme 5b). In the event treatment of a solution of 30 in CH2Cl2 with triethylamine led from a rapid colour change from blue to red, which we ascribe to formation of 40. The red colour of the solution remained unchanged upon prolonged standing under air, and the halochromic behaviour was shown to be reversible, with addition of trifluoroacetic acid leading to regeneration of the blue colour of 30 (Fig. 9). This process could be repeated multiple times. Azulenes directly conjugated to para-quinone methides have been reported,36 but to our knowledge 40 constitutes the first example of an azulene directly conjugated to an ortho-quinone methide.
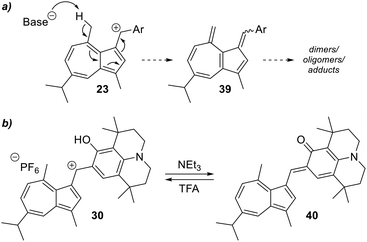 |
| | Scheme 5 (a) Deprotonation at C4-methyl group leading to 39, anticipated to be unstable. (b) Deprotonation of 30 at the hydroxyl group, leading to o-quinone methide 40. | |
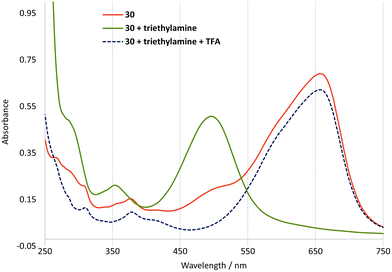 |
| | Fig. 9 UV/vis absorption spectra for salts 30 (15 μM) upon treatment with base and subsequent acid in MeCN. | |
Conclusions
We have reported the synthesis and characterisation of highly conjugated azulene-substituted cations, which are readily isolated as hexafluorophosphate salts. They are stable in the solid state, with no decomposition observed after being stored on the bench >1 year (with no protection from air/light). The cations persist in both protic (MeOH) and aprotic (MeCN, CH2Cl2) solvents. The synthesis of these salts requires only one step and employs a cheap, naturally-occurring azulene starting material. Furthermore, salts with high extinction coefficients and different absorption maxima in the visible region (and hence different colours) are accessible by varying the aldehyde starting material. As such salts of the type described here may find applications as low-cost dyestuffs. Salts such as 30, with an appropriate acidic proton, can display halochromic behaviour as a result of deprotonation to form neutral quinoidal systems, such as 40. Further studies on salts of this type are ongoing in our laboratory and the results will be reported in due course.
Experimental
Materials and methods
See the ESI† for general details of the materials and methods used.
Synthetic procedures
General procedure A for synthesis of compounds 24–36.
To a solution of guaiazulene 22 (100 mg, 0.50 mmol, 1 eq.) in MeOH (2 mL) was added a solution of aldehyde (0.61 mmol, 1.2 eq.) in MeOH (4 mL) containing HPF6 (55% aqueous solution, 0.12 mL, 2.7 eq.). The reaction mixture was stirred at room temperature under air until the starting material was consumed, as shown by TLC (EtOAc/petrol 1:4). The reaction mixture was diluted with diethyl ether (10 mL), then cooled to 0 °C for the specified time. Precipitated material was isolated by filtration, dried under vacuum and then purified as specified.
[4-(Dimethylamino)phenyl]-3-guaiazulenylmethylium hexafluorophosphate 24.
General procedure A was used with p-diaminobenzaldehyde (90 mg); reaction time 1.5 h; cooling time 10 min. Recrystallisation from MeCN/diethyl ether gave 24 as green crystals (113 mg, 47% yield). M.pt 181 °C; 1H-NMR (500 MHz, CD3CN) δ = 8.72 (s, 1H), 8.49 (d, J = 2.1 Hz, 1H), 8.20 (s, 1H), 8.11 (dd, J = 11.1, 2.1 Hz, 1H), 8.07 (d, J = 11.1 Hz, 1H), 7.95 (d, J = 9.3 Hz, 2H), 6.98 (d, J = 9.3 Hz, 2H), 3.36 (hept, J = 6.9, 1H), 3.28 (s, 3H, 14), 3.26 (s, 6H), 2.59 (d, J = 1.0 Hz, 3H), 1.42 (d, J = 6.8 Hz, 6H) ppm; 13C-NMR (126 MHz, CD3CN) δ = 161.3, 156.7, 154.3, 153.7, 153.1, 149.8, 143.6, 142.0, 142.1, 139.8, 139.4, 138.5, 124.8, 114.9, 41.0, 39.4, 30.0, 24.0, 13.4 ppm. NMR data in agreement with those previously reported for the tetrafluoroborate salt.23f
(2,2-Diphenylethenyl)-3-guaiazulenylmethylium hexafluorophosphate 25.
General procedure A was used with β-phenylcinnamaldehyde (126 mg); reaction time 3 h; cooling time 10 min. Recrystallisation from MeCN/diethyl ether gave 25 as red crystals (244 mg, 90% yield). M.pt 199 °C; 1H-NMR (500 MHz, CD3CN) δ = 8.56 (s, 1H), 8.34–8.26 (m, 4H), 7.92 (d, J = 12.0 Hz, 1H), 7.65–7.47 (m, 8H), 7.43–7.39 (m, 2H), 3.45 (hept, J = 6.9 Hz, 1H), 2.86 (s, 3H), 2.56 (s, 3H), 1.43 (d, J = 6.8 Hz, 6H) ppm; 13C-NMR (126 MHz, CD3CN) δ = 169.4, 163.5, 160.3, 156.6, 152.4, 149.4, 148.1, 144.4, 143.9, 141.3, 140.1, 139.8, 139.5, 139.0, 132.3, 131.9, 131.0, 130.6, 129.9, 129.7, 126.6, 40.1, 28.8, 23.8, 13.9 ppm. IR νmax 2967, 1553, 1397, 1339, 1295, 1255, 1210, 1109, 1046, 985, 825, 780, 705, 596, 555 cm−1. HRMS (ESI+): calc. for C30H29 [M]+ 389.2264, found 389.2264.
(4-Bromophenyl)-3-guaiazulenylmethylium hexafluorophosphate 26.
General procedure A was used with 4-bromobenzaldehyde (112 mg); reaction time 4 h; cooling time 10 min. 26 was obtained as orange crystals (219 mg, 85% yield); no further purification was necessary. M.pt 173 °C; 1H-NMR (500 MHz, CD3CN) δ = 8.69 (s, 1H), 8.59 (d, J = 2.2 Hz, 1H), 8.56 (d, J = 11.3 Hz, 1H), 8.45 (dd, J = 11.3, 2.2 Hz, 1H), 7.93 (s, 1H, 7), 7.80–7.77 (m, 2H), 7.72–7.69 (m, 2H), 3.51 (hept, J = 6.9 Hz, 1H), 3.36 (s, 3H), 2.51 (s, 3H), 1.46 (d, J = 6.9 Hz, 6H) ppm; 13C-NMR (126 MHz, CD3CN) δ = 172.6, 162.0, 158.4, 153.8, 151.3, 148.8, 146.8, 145.4, 141.0, 140.1, 135.6, 135.1, 133.5, 127.3, 40.4, 29.7, 23.8, 13.9 ppm. One aromatic carbon was not observed. IR νmax 2974, 1573, 1552, 1454, 1403, 1344, 1313, 1195, 1071, 1047, 1005, 983, 826, 743, 662, 635, 555, 502 cm−1. HRMS (ESI+): calc. for C22H2279Br [M]+ 365.0900, found 365.0901.
(4-Bromothiophen-2-yl)-3-guaiazulenylmethylium hexafluorophosphate 27.
General procedure A was used with 4-bromo-2-thiophenecarboxaldehyde (116 mg); reaction time 4 h; cooling time 10 min. 27 was obtained as orange crystals (246 mg, 93% yield); no further purification was necessary. M.pt 178 °C; 1H-NMR (500 MHz, CD3CN) δ = 8.63 (s, 1H), 8.57 (d, J = 2.2 Hz, 1H), 8.49 (d, J = 11.2 Hz), 8.40 (dd, J = 11.2, 2.2 Hz, 1H), 8.16 (br s, 1H), 8.01–8.00 (m, 1H), 7.85 (br s, 1H), 3.49 (hept, J = 6.8 Hz, 1H), 3.32 (s, 3H), 2.56 (s, 3H), 1.45 (d, J = 6.8 Hz, 6H) ppm; 13C-NMR (126 MHz, CD3CN) δ = 171.4, 160.8, 157.7, 154.0, 150.7, 146.7, 145.2, 142.2, 141.3, 140.2, 139.1, 137.7, 136.1, 113.6, 40.2, 29.7, 23.8, 14.0 ppm. One aromatic carbon was not observed. IR νmax 3115, 2963, 1568, 1398, 1344, 1294, 1168, 1118, 1045, 825, 742, 585, 555 cm−1. HRMS (ESI+): calc. for C20H2081BrS [M]+ 373.0444, found 373.0445.
3-Guaiazulenyl-(5-(phenylethynyl)thiophen-2-yl)methylium hexafluorophosphate 28.
General procedure A was used with 5-(2-phenyleth-1-ynyl)thiophene-2-carbaldehyde (116 mg); reaction time 4 h; cooling time 10 min. Recrystallisation from MeCN/diethyl ether gave 28 as dark green lustrous crystals in two batches (148 mg and 87 mg; 86% total yield). M.pt 198 °C; 1H-NMR (500 MHz, CD3CN) δ = 8.74 (s, 1H), 8.57 (d, J = 2.2 Hz, 1H), 8.43 (d, J = 11.2 Hz, 1H), 8.36 (dd, J = 11.2, 2.2 Hz, 1H), 8.21 (br s, 1H), 7.91 (d, J = 4.2 Hz, 1H), 7.61–7.59 (m, 2H), 7.50 (d, J = 4.2 Hz, 1H), 7.50–7.45 (m, 3H), 3.47 (hept, J = 6.9 Hz, 1H), 3.34 (s, 3H), 2.58 (s, 3H), 1.45 (d, J = 6.9 Hz, 6H) ppm; 13C-NMR (126 MHz, CD3CN) δ = 170.1, 160.0, 157.3, 153.7, 149.8, 146.1, 144.8, 143.4, 141.6, 140.2, 140.1, 139.2, 136.7, 136.4, 135.2, 132.6, 131.1, 130.0, 122.4, 101.8, 83.1, 40.1, 29.8, 23.8, 14.0 ppm. IR νmax 2965, 2198, 1980, 1601, 1565, 1400, 1377, 1343, 1175, 1101, 1042, 837, 822, 806, 747, 688, 665, 551, 517 cm−1. HRMS (ESI+): calc. for C28H25S [M]+ 393.1672, found 393.1672.
3-Guaiazulenyl-(1-pyrenyl)methylium hexafluorophosphate 29.
General procedure A was used with pyrene-1-carbaldehyde (139 mg); reaction time 4 h; cooling time 10 min. Recrystallisation from MeCN/diethyl ether gave 29 as burgundy crystals (279 mg, 99% yield). M.pt 187 °C; 1H-NMR (500 MHz, CD3CN) δ = 9.65 (s, 1H), 8.55–8.49 (m, 3H), 8.44–8.37 (m, 5H), 8.33–8.30 (m, 2H), 8.24 (d, J = 8.9 Hz, 1H), 8.16 (app t, J = 7.7 Hz, 1H), 7.89 (s, 1H), 3.51 (hept, J = 6.9 Hz, 1H), 3.48 (s, 3H), 2.48 (s, 3H), 1.49 (d, J = 6.9 Hz, 6H) ppm; 13C-NMR (126 MHz, CD3CN) δ = 171.7, 161.2, 157.9, 150.5, 148.3, 146.2, 145.0, 142.6, 141.5, 139.8, 135.2, 132.7, 132.2, 131.7, 131.5, 131.0, 130.7, 128.40, 128.36, 128.20, 128.17, 126.5, 125.4, 124.6, 40.3, 29.9, 23.8, 13.9 ppm. IR νmax 2972, 1560, 1532, 1431, 1387, 1340, 1318, 1258, 1235, 1208, 1113, 1043, 829, 718, 681, 658, 629, 598, 556 cm−1. HRMS (ESI+): calc. for C32H27 [M]+ 411.2108, found 411.2112.
3-Guaiazulenyl-(8-hydroxy-1,1,7,7-tetramethyl-2,3,6,7-tetrahydro-1H,5H-pyrido[3,2,1-ij]quinolin-9-yl)methylium hexafluorophosphate 30.
General procedure A was used with 8-hydroxy-1,1,7,7-tetramethyljulolidine-9-carboxaldehyde (165 mg); reaction time 4 h; cooling time 10 min. 30 was obtained as lustrous green-gold crystals (262 mg, 87% yield); no further purification was necessary. M.pt 170 °C; 1H-NMR (500 MHz, CD3CN) δ = 8.82 (s, 1H), 8.40 (d, J = 2.2 Hz, 1H), 7.91 (s, 1H), 7.88 (dd, J = 10.9, 2.2 Hz, 1H), 7.72 (d, J = 10.9 Hz, 1H), 7.64 (br s, 1H), 7.61 (s, 1H), 3.65–3.62 (m, 2H), 3.55–3.53 (m, 2H), 3.27 (hept, J = 6.9 Hz, 1H), 3.17 (s, 3H), 2.61 (s, 3H), 1.84–1.79 (m, 4H), 1.46 (s, 6H), 1.40 (d, J = 6.9 Hz, 6H), 1.29 (s, 6H) ppm; 13C-NMR (126 MHz, CD3CN) δ = 160.9, 155.3, 154.3, 151.8, 148.6, 145.3, 145.0, 142.3, 140.3, 138.1, 137.5, 134.5, 132.8, 131.5, 127.7, 119.8, 118.0, 50.0, 49.1, 39.1, 38.6, 35.0, 32.9, 32.7, 29.7, 28.8, 27.9, 24.3, 13.3 ppm. IR νmax 3574, 2957, 1611, 1528, 1500, 1462, 1312, 1257, 1089, 1046, 904, 824, 777, 648, 592, 554 cm−1. HRMS (ESI+): calc. for C32H40NO [M]+ 454.3105, found 454.3107.
(3-Bromothiophen-2-yl)-3-guaiazulenylmethylium hexafluorophosphate 31.
General procedure A was used with 3-bromothiophene-2-carboxaldehyde (116 mg); reaction time 4 h; cooling time 10 min. 31 was obtained as burgundy crystals (235 mg, 90% yield); no further purification was necessary. M.pt 169 °C; 1H-NMR (500 MHz, CD3CN) δ = 8.83 (s, 1H), 8.59 (d, J = 2.2 Hz, 1H), 8.51 (d, J = 11.2 Hz, 1H,), 8.41 (dd, J = 11.2, 2.2 Hz, 1H), 8.23 (s, 1H), 8.16 (d, J = 5.4 Hz, 1H), 7.41 (d, J = 5.4 Hz, 1H), 3.49 (hept, J = 6.9 Hz, 1H), 3.39 (s, 3H), 2.57 (s, 3H), 1.46 (d, J = 6.9 Hz, 6H) ppm. 13C-NMR (126 MHz, CD3CN) δ = 171.1, 160.5, 157.3, 154.2, 150.6, 146.9, 145.1, 140.3, 139.7, 138.6, 138.3, 137.1, 135.1, 133.6, 127.3, 40.2, 29.8, 23.8, 14.1 ppm. IR νmax 3121, 2967, 1599, 1563, 1447, 1398, 1377, 1296, 1211, 1158, 1103, 1042, 979, 819, 738, 659, 608, 554 cm−1. HRMS (ESI+): calc. for C20H2081BrS [M]+ 373.0444, found 373.0443.
(Benzothiophen-2-yl)-3-guaiazulenylmethylium hexafluorophosphate 32.
General procedure A was used with benzothiophene-2-carboxaldehyde (98 mg); reaction time 4 h; cooling time 10 min. 32 was obtained as red crystals (238 mg, 97% yield); no further purification was necessary. M.pt 192 °C; 1H-NMR (500 MHz, CD3CN) δ = 8.80 (s, 1H), 8.52 (d, J = 2.2 Hz, 1H), 8.43 (d, J = 11.2 Hz, 1H), 8.36 (dd, J = 11.2, 2.2 Hz, 1H), 8.23 (s, 1H), 8.19 (s, 1H), 7.92 (d, J = 7.4 Hz, 1H), 7.91 (d, J = 8.2 Hz, 1H), 7.47–7.44 (m, 1H), 7.37 (app t, J = 7.6 Hz, 1H), 3.48 (hept, J = 6.9 Hz, 1H), 3.33 (s, 3H), 2.57 (s, 3H), 1.47 (d, J = 6.9 Hz, 6H) ppm. 13C-NMR (126 MHz, CD3CN) δ = 170.8, 160.4, 157.6, 153.6, 150.3, 146.2, 145.6, 145.0, 141.5, 141.2, 140.2, 140.0, 139.6, 139.3, 137.8, 130.1, 127.3, 127.1, 123.7, 40.2, 29.8, 23.8, 14.1 ppm. IR νmax 2972, 1605, 1568, 1494, 1308, 1342, 1299, 1173, 1160, 1120, 1042, 825, 765, 658, 556 cm−1. HRMS (ESI+): calc. for C24H23S [M]+ 343.1515, found 343.1517.
3-Guaiazulenyl-(5-nitrothiophen-2-yl)methylium hexafluorophosphate 33.
General procedure A was used with 5-nitrothiophene-2-carboxaldehyde (95 mg); reaction time 4 h; cooling time 10 min. 33 was obtained as burgundy crystals (162 mg, 66% yield); no further purification was necessary. M.pt 189 °C; 1H-NMR (500 MHz, CD3CN) δ = 8.59 (s, 1H), 8.57 (d, J = 11.2 Hz, 1H), 8.46 (dd, J = 11.2, 2.2 Hz, 1H), 8.15–8.11 (m, 1H), 8.07 (d, J = 4.4 Hz, 1H), 7.80 (dd, J = 4.4, 0.9 Hz, 1H), 3.51 (hept, J = 6.9 Hz, 1H), 3.34 (s, 3H), 2.57 (s, 3H), 1.46 (d, J = 6.8 Hz, 6H) ppm. 13C-NMR (126 MHz, CD3CN) δ = 173.7, 162.1, 158.6, 154.3, 152.1, 149.0, 145.9, 144.8, 140.9, 140.5, 138.7, 138.4, 137.9, 130.6, 40.5, 29.6, 23.7, 14.1 ppm. One aromatic carbon was not observed. IR νmax 3105, 2980, 1605, 1574, 1522, 1494, 1455, 1419, 1404, 1334, 1319, 1250, 1200, 1109, 1048, 985, 814, 730, 653, 523, 555 cm−1. HRMS (ESI+): calc. for C20H20NO2S [M]+ 338.1210, found 338.1210.
3-Guaiazulenyl-(5-cyanothiophen-2-yl)methylium hexafluorophosphate 34.
General procedure A was used with 5-cyanothiophene-2-carboxaldehyde (83 mg); reaction time 4 h; cooling time 10 min. 34 was obtained as orange-red crystals (185 mg, 79% yield); no further purification was necessary. M.pt 194 °C; 1H-NMR (500 MHz, CD3CN) δ = 8.66 (s, 1H), 8.59 (d, J = 2.2 Hz, 1H), 8.55 (d, J = 11.2 Hz, 1H), 8.44 (dd, J = 11.2, 2.2 Hz, 1H), 8.11 (s, 1H), 7.89 (d, J = 4.1 Hz, 1H), 7.86 (d, J = 4.1 Hz, 1H), 3.50 (hept, J = 6.6 Hz, 1H), 3.33 (s, 3H), 2.56 (s, 3H), 1.46 (d, J = 6.9 Hz, 6H) ppm. 13C-NMR (126 MHz, CD3CN) δ = 173.1, 161.8, 158.4, 154.3, 151.7, 148.4, 145.7, 145.6, 140.4, 140.1, 140.0, 139.4, 138.6, 137.7, 114.2, 40.4, 29.7, 23.7, 14.1 ppm. One aromatic carbon was not observed. IR νmax 3640, 3570, 3084, 2962, 2286, 2165, 1626, 1548, 1517, 1417, 1362, 1330, 1197, 1022, 811, 750, 669, 558 cm−1. HRMS (ESI+): calc. for C21H20NS [M]+ 318.1311, found 318.1312.
3-Guaiazulenyl-((2,2′-bithiophen)-5-yl)methylium hexafluorophosphate 35.
General procedure A was used with 5-cyanothiophene-2-carboxaldehyde (116 mg); reaction time 3 h; cooling time 10 min. 35 was obtained as dark green powder (247 mg, 95% yield); no further purification was necessary. M.pt 194–196 °C; 1H-NMR (500 MHz, CD3CN) δ = 8.80 (1H, s), 8.56 (1H, d, J = 2.2 Hz), 8.37 (1H, d, J = 11.2 Hz), 8.32 (1H, dd, J = 11.2, 2.2 Hz), 8.31 (1H, br s), 7.93 (1H, d, J = 4.2 Hz), 7.65–7.62 (2H, m), 7.52 (1H, d, J = 4.1 Hz), 7.21 (1H, dd, J = 5.0, 3.7 Hz), 3.45 (1H, hept, J = 6.9 Hz), 3.34 (3H, s), 2.60 (3H, s), 1.45 (6H, d, J = 6.9 Hz) ppm. 13C-NMR (126 MHz, CD3CN) δ = 168.1, 158.5, 156.4, 152.7, 152.6, 148.6, 146.0, 144.5, 144.2, 141.1, 139.6, 139.3, 139.0, 136.3, 134.7, 130.7, 130.2, 128.7, 126.8, 39.9, 29.8, 23.9, 14.0 ppm. IR νmax 3129, 2967, 1376, 1497, 1223, 1040, 734, 611 cm−1. HRMS (ESI+): calc. for C24H23S2 [M]+ 375.1236, found 375.1241.
3-Guaiazulenyl-(N-methylpyrrol-2-yl)methylium hexafluorophosphate 36.
General procedure A was used with N-methylpyrrole-2-carboxaldehyde (65 mg); reaction time 3 h; cooling time 10 min. 36 was obtained as dark purple crystals (218 mg, 94% yield); no further purification was necessary. M.pt 168–171 °C; 1H-NMR (500 MHz, CD3CN) δ = 8.60 (1H, s), 8.53 (1H, s), 8.27 (1H, br s), 8.19 (2H, br s), 7.61 (1H, d, J = 4.5 Hz), 7.55 (1H, br s), 6.65 (1H, ddd, J = 4.5, 2.2, 0.9 Hz), 3.92 (3H, s), 3.40 (1H, hept, J = 6.9 Hz), 3.33 (3H, s), 2.59 (3H, s), 1.43 (6H, d, J = 6.9 Hz) ppm. 13C-NMR (126 MHz, CD3CN) δ = 163.6, 154.5, 151.2, 145.4, 142.8, 141.4, 140.8, 139.9, 139.1, 136.5, 136.4, 133.9, 130.8, 127.1, 117.1, 39.6, 35.6, 29.8, 24.0, 13.6 ppm. IR νmax 3129, 2967, 1583, 1377, 1225, 1041, 828, 651 cm−1. HRMS (ESI+): calc. for C21H24N [M]+ 290.1903, found 290.1896.
General procedure B for synthesis of compounds 37–38.
To a solution of the relevant guaiazulenyl carbocation (0.008 mmol, 1 eq.) in MeCN (2.0 mL) was added a solution of NaBH4 (7 mg, 0.17 mmol, 2.1 eq.) in EtOH (2 mL). The mixture was stirred at room temperature under air until the starting material was consumed, as shown by TLC (EtOAc/petrol 1:4), approximately 20 min. Precipitation of solid was observed, which was isolated by filtration, then dried under vacuum to afford the desired product; no further purification was necessary.
4-Bromo-2-((5-isopropyl-3,8-dimethylazulen-1-yl)methyl)thiophene 37.
General procedure B was used with cation 27. Product 37 was obtained as a navy blue solid (21 mg, 68% yield). M.pt 104 °C; 1H-NMR (500 MHz, CDCl3) δ = 8.12 (d, J = 2.1 Hz, 1H), 7.47 (s, 1H), 7.30 (dd, J = 10.8, 2.1 Hz, 1H, 6), 7.01 (d, J = 1.4 Hz, 1H), 6.86 (d, J = 10.7 Hz, 1H), 6.51 (d, J = 1.3 Hz, 1H), 4.67 (s, 2H), 3.04 (hept, J = 6.9 Hz, 1H), 2.87 (s, 3H), 2.62 (s, 3H), 1.35 (d, J = 6.9 Hz, 6H) ppm. 13C-NMR (126 MHz, CDCl3) δ = 149.2, 145.3, 140.7, 139.7, 138.1, 135.1, 133.9, 132.9, 126.95, 126.93, 124.5, 123.8, 121.0, 109.3, 37.9, 31.9, 26.7, 24.7, 13.0 ppm. IR νmax 3084, 2962, 1626, 1548, 1517, 1417, 1362, 1330, 1197, 1022, 811, 750, 667, 558 cm−1. HRMS (ESI+): calc. for C20H2181BrS [M + H]+ 375.0600, found 375.0593.
1-((5-Iso-propyl-3,8-dimethylazulen-1-yl)methyl)pyrene 38.
General procedure B was used with cation 29. Product 38 was obtained as a light blue solid (27 mg, 72% yield). M.pt 149 °C; 1H-NMR (500 MHz, CDCl3) δ = 8.36 (d, J = 9.2 Hz, 1H), 8.18 (d, J = 7.6 Hz, 2H), 8.12 (d, J = 2.2 Hz, 1H), 8.11 (d, J = 9.2 Hz, 1H), 8.04 (d, J = 7.7 Hz, 1H), 8.03 (s, 2H), 8.00 (app t, J = 7.6 Hz, 1H), 7.55 (d, J = 8.0 Hz, 1H), 7.30 (dd, J = 10.8, 2.2 Hz, 1H), 7.19 (s, 1H), 6.85 (d, J = 10.7 Hz, 1H), 5.36 (s, 2H), 3.05 (hept, J = 7.0 Hz, 1H), 2.92 (s, 3H), 2.54 (s, 3H), 1.54 (s, 2H), 1.37 (d, J = 6.8 Hz, 6H); ppm. 13C-NMR (126 MHz, CDCl3) δ = 145.8, 141.0, 139.1, 137.9, 136.9, 135.1, 133.7, 133.5, 131.6, 131.1, 129.9, 128.7, 127.75, 127.74, 127.6, 126.8, 126.5, 126.0, 125.8, 125.14, 125.13, 125.10, 125.08, 124.96, 124.5, 123.7, 37.9, 35.4, 26.9, 24.8, 13.0 ppm. IR νmax 3035, 2952, 2859, 1588, 1547, 1517, 1444, 1312, 1244, 1181, 1098, 1055, 1003, 774, 843, 753, 706, 690, 644 cm−1. HRMS (ESI+): calc. for C32H28 [M + H]+ 413.2264, found 413.2250.
Author contributions
SEL conceived the project and acquired funding; CH, JLH and LCM carried out synthesis and spectroscopic characterisation. GKK acquired and interpreted X-ray crystallographic data. SEL wrote the manuscript with input from all authors.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We thank EPSRC for IAA funding (EP/R51164X/1) and for DTG PhD studentships to L. C. M. and J. L. H. The authors acknowledge the Material and Chemical Characterisation Facility (MC2) at University of Bath (https://doi.org/10.15125/mx6j-3r54).
References
- R. S. H. Liu, J. Chem. Educ., 2002, 79, 183 CrossRef CAS.
- A. G. Anderson, Jr. and B. M. Steckler, J. Am. Chem. Soc., 1959, 81, 4941 CrossRef.
-
(a) M. Beer and H. C. Longuet-Higgins, J. Chem. Phys., 1955, 23, 1390 CrossRef CAS;
(b) B. D. Wagner, D. Tittelbach-Helmrich and R. P. Steer, J. Phys. Chem., 1992, 96, 7904 CrossRef CAS;
(c) K. Veys and D. Escudero, J. Phys. Chem. A, 2020, 124, 7228 CrossRef CAS.
- R. S. H. Liu, R. S. Muthyala, X.-S. Wang and A. E. Asato, Org. Lett., 2000, 2, 269–271 CrossRef CAS.
-
(a) B. Hou, J. Li, X. Yang, J. Zhang, H. Xin, C. Ge and X. Gao, Chin. Chem. Lett., 2022, 33, 2147 CrossRef CAS;
(b)
X. Gao, B. Hou, J. Li and C. Ge, Chin. Pat, CN113402442, 2021 Search PubMed.
- T. Koide, M. Takesue, T. Murafuji, K. Satomi, Y. Suzuki, J. Kawamata, K. Terai, M. Suzuki, H. Yamada, Y. Shiota, K. Yoshizawa and F. Tani, ChemPlusChem, 2017, 82, 1010 CrossRef CAS PubMed.
- H. Xin, C. Ge, X. Yang, H. Gao, X. Yang and X. Gao, Chem. Sci., 2016, 7, 6701 RSC.
- P. Cowper, A. Pockett, G. Kociok-Köhn, P. J. Cameron and S. E. Lewis, Tetrahedron, 2018, 74, 2775 CrossRef CAS.
- H. Salman, Y. Abraham, S. Tal, S. Meltzman, M. Kapon, N. Tessler, S. Speiser and Y. Eichen, Eur. J. Org. Chem., 2005, 2207 CrossRef CAS.
- C. M. López-Alled, S. J. Park, D. J. Lee, L. C. Murfin, G. Kociok-Köhn, J. L. Hann, J. Wenk, T. D. James, H. M. Kim and S. E. Lewis, Chem. Commun., 2021, 57, 10608 RSC.
- L. C. Murfin, M. Weber, S. J. Park, W. T. Kim, C. M. López-Alled, C. L. McMullin, F. Pradaux-Caggiano, C. L. Lyall, G. Kociok-Köhn, J. Wenk, S. D. Bull, J. Yoon, H. M. Kim, T. D. James and S. E. Lewis, J. Am. Chem. Soc., 2019, 141, 19389 CrossRef CAS.
- For reviews, see:
(a) A. C. Razus, Symmetry, 2021, 13, 526 CrossRef CAS;
(b) S. Ito and N. Morita, Eur. J. Org. Chem., 2009, 4567 CrossRef CAS.
-
(a) K. Satoh, I. Ogura and M. Yamaguchi, Nippon Kagaku Kaishi, 1982, 1982, 1206, DOI:10.1246/nikkashi.1982.1199;
(b) P. Uebelhart and H.-J. Hansen, Helv. Chim. Acta, 1992, 75, 2493 CrossRef.
- T. Shoji, M. Tanaka, T. Araki, S. Takagaki, R. Sekiguchi and S. Ito, RSC Adv., 2016, 6, 78303 RSC.
- T. Shoji, K. Miura, A. Ohta, R. Sekiguchi, S. Ito, Y. Endo, T. Nagahata, S. Mori and T. Okujima, Org. Chem. Front., 2019, 6, 2801 RSC.
-
(a) D. A. Colby and T. D. Lash, J. Org. Chem., 2002, 67, 1031 CrossRef CAS;
(b) Y. Jin, K. Akagawa, T. Mutai, I. Yoshikawa and K. Kudo, Tetrahedron, 2021, 84, 132146 CrossRef.
- S. Ito, N. Morita and T. Asao, Bull. Chem. Soc. Jpn., 1999, 72, 2543 CrossRef CAS.
- Y. Jin, K. Akagawa and K. Kudo, Tetrahedron, 2021, 84, 131998 CrossRef CAS.
- S. Ito, N. Morita and T. Asao, Bull. Chem. Soc. Jpn., 1995, 68, 1409 CrossRef CAS.
- S. Ito, S. Kikuchi, N. Morita and T. Asao, J. Org. Chem., 1999, 64, 5815 CrossRef CAS.
-
(a) D. H. Reid, W. H. Stafford, W. L. Stafford, G. McLennan and A. Voigt, J. Chem. Soc., 1958, 1110 RSC;
(b) E. C. Kirby and D. H. Reid, J. Chem. Soc., 1960, 494 RSC;
(c) E. C. Kirby and D. H. Reid, J. Chem. Soc., 1961, 1724 RSC;
(d) E. C. Kirby and D. H. Reid, J. Chem. Soc., 1961, 3579 RSC;
(e) M. Fraser and D. H. Reid, J. Chem. Soc., 1963, 1421 RSC.
-
(a) F. N. Stepanow and N. A. Aldanowa, Russ. J. Gen. Chem., 1959, 29, 344 Search PubMed;
(b) F. N. Stepanow and N. A. Aldanowa, Angew. Chem., 1959, 71, 125 CrossRef.
-
(a) S.-i. Takekuma, H. Takekuma, Y. Matsubara, A. Hirai, H. Yamamoto and T. Nozoe, Nippon Kagaku Kaishi, 1996, 1996, 419, DOI:10.1246/nikkashi.1996.419;
(b) S.-i. Takekuma, H. Takekuma, Y. Hanaoka and H. Yamamoto, Nippon Kagaku Kaishi, 1996, 1996, 659, DOI:10.1246/nikkashi.1996.659;
(c) S.-i. Takekuma, H. Takekuma, Y. Hatanaka, J. Kawaguchi and H. Yamamoto, Nippon Kagaku Kaishi, 1998, 1998, 275, DOI:10.1246/nikkashi.1998.275;
(d) S.-i. Takekuma, M. Sasaki, H. Takekuma and H. Yamamoto, Chem. Lett., 1999, 28, 999 CrossRef;
(e) S.-i. Takekuma, M. Sasaki, H. Takekuma and H. Yamamoto, Nippon Kagaku Kaishi, 2000, 2000, 107, DOI:10.1246/nikkashi.2000.107;
(f) M. Sasaki, M. Nakamura, T. Uriu, H. Takekuma, T. Minematsu, M. Yoshihara and S.-i. Takekuma, Tetrahedron, 2003, 59, 505 CrossRef CAS;
(g) M. Nakamura, M. Sasaki, H. Takekuma, T. Minematsu and S.-i. Takekuma, Bull. Chem. Soc. Jpn., 2003, 76, 2051 CrossRef CAS;
(h) M. Sasaki, M. Nakamura, G. Hannita, H. Takekuma, T. Minematsu, M. Yoshihara and S.-i. Takekuma, Tetrahedron Lett., 2003, 44, 275 CrossRef CAS;
(i) S.-i. Takekuma, K. Sasaki, M. Nakatsuji, M. Sasaki, T. Minematsu and H. Takekuma, Bull. Chem. Soc. Jpn., 2004, 77, 379 CrossRef CAS;
(j) M. Nakatsuji, Y. Hata, T. Fujihara, K. Yamamoto, M. Sasaki, H. Takekuma, M. Yoshihara, T. Minematsu and S.-i. Takekuma, Tetrahedron, 2004, 60, 5983 CrossRef CAS;
(k) S.-i. Takekuma, Y. Hata, T. Nishimoto, E. Nomura, M. Sasaki, T. Minematsu and H. Takekuma, Tetrahedron, 2005, 61, 6892 CrossRef CAS;
(l) S.-i. Takekuma, K. Takahashi, A. Sakaguchi, Y. Shibata, M. Sasaki, T. Minematsu and H. Takekuma, Tetrahedron, 2005, 61, 10349 CrossRef CAS;
(m) S.-i. Takekuma, K. Sonoda, C. Fukuhara and T. Minematsu, Tetrahedron, 2007, 63, 2472 CrossRef CAS;
(n) S.-i. Takekuma, K. Tone, M. Sasaki, T. Minematsu and H. Takekuma, Tetrahedron, 2007, 63, 2490 CrossRef CAS;
(o) S.-i. Takekuma, K. Mizutani, K. Inoue, M. Nakamura, M. Sasaki, T. Minematsu, K. Sugimoto and H. Takekuma, Tetrahedron, 2007, 63, 3882 CrossRef CAS;
(p) S.-i. Takekuma, M. Tamura, T. Minematsu and H. Takekuma, Tetrahedron, 2007, 63, 12058 CrossRef CAS;
(q) S.-i. Takekuma, K. Sonoda, T. Minematsu and H. Takekuma, Tetrahedron, 2008, 64, 3802 CrossRef CAS;
(r) S.-i. Takekuma, N. Ijibata, T. Minematsu and H. Takekuma, Bull. Chem. Soc. Jpn., 2009, 82, 585 CrossRef CAS;
(s) S.-i. Takekuma, K. Fukuda, Y. Kawase, T. Minematsu and H. Takekuma, Bull. Chem. Soc. Jpn., 2009, 82, 879 CrossRef CAS;
(t) S.-i. Takekuma, K. Fukuda, T. Minematsu and H. Takekuma, Bull. Chem. Soc. Jpn., 2009, 82, 1398 CrossRef CAS;
(u) S.-i. Takekuma, M. Kaibara, T. Minematsu and H. Takekuma, Tetrahedron, 2011, 67, 4780 CrossRef CAS;
(v) S.-i. Takekuma, I. Miyamoto, A. Hamasaki and T. Minematsu, Tetrahedron, 2011, 67, 9719 CrossRef CAS;
(w) S.-i. Takekuma, M. Yamamoto, A. Nakagawa, T. Iwata, T. Minematsu and H. Takekuma, Tetrahedron, 2012, 68, 8318 CrossRef CAS;
(x) S.-i. Takekuma, N. Kobayashi, T. Minematsu and H. Takekuma, Bull. Chem. Soc. Jpn., 2013, 86, 968 CrossRef CAS;
(y) S.-i. Takekuma, A. Seki and T. Minematsu, Bull. Chem. Soc. Jpn., 2014, 87, 511 CrossRef CAS.
-
(a)
Y. Ogawa, Jpn. Pat, JP08187940, 1996 Search PubMed;
(b)
T. Santo and C. Hioki, Jpn. Pat, JP01071791, 1989 Search PubMed;
(c)
K. Takano, Jpn. Pat, JP63252791, 1988 Search PubMed.
-
S. Kawakami and Y. Kojima, Jpn. Pat, JP04153086, 1992 Search PubMed.
-
Y. Oguchi and K. Katagiri, Jpn. Pat, JP61262737, 1986 Search PubMed.
-
T. Okamoto, N. Yamamoto and M. Kawaguchi, Eur. Pat, EP659407, 1995 Search PubMed.
-
K. Watanabe, T. Shintou, T. Nomoto, T. Miyazaki, T. Tanaka, Y. Nishimura, Y. Shimada and N. Nishimura, US Pat, US20110236310, 2011 Search PubMed.
- S. Yang, X.-S. Xiong and D.-L. Wang, Heterocycles, 2021, 102, 2179 CrossRef CAS.
-
S. H. Kim, Y. A. Son, J. S. Bae and D. H. Lee, Korea Pat, KR2014078467, 2014 Search PubMed.
- See: D. Chen, S. Yu, L. van Ofwegen, P. Proksch and W. Lin, J. Agric. Food Chem., 2012, 60, 112 CrossRef CAS PubMed and references therein.
- S. Takekuma, Z. Zhao, Y. Matsubara, D. Makihara, H. Yamamoto and T. Nozoe, Nippon Kagaku Kaishi, 1994, 1994, 399, DOI:10.1246/nikkashi.1994.399.
-
(a) S. Ito, R. Sekiguchi, A. Mizushima, K. Kudo, J. Kawakami and T. Shoji, ARKIVOC, 2018, 2018(2), 145, DOI:10.24820/ark.5550190.p010.163;
(b) H. Noguchi, T. Hirose, S. Yokoyama and K. Matsuda, CrystEngComm, 2016, 18, 7377 RSC;
(c) S. Nakatsuji, H. Nakazumi, K. Nakashima and S. Akiyama, Nippon Kagaku Kaishi, 1992, 1992, 1279, DOI:10.1246/nikkashi.1992.1279;
(d) S. Nakatsuji, N. Okamoto, K. Nakashima and S. Akiyama, Chem. Lett., 1986, 15, 329 CrossRef;
(e) J. R. Gandler, J. Am. Chem. Soc., 1985, 107, 8218 CrossRef CAS.
- G. E. Williams, G. Kociok-Köhn, T. D. James and S. E. Lewis, Org. Biomol. Chem., 2021, 19, 2502 RSC.
-
(a) R. Luhowy and P. M. Keehn, Tetrahedron Lett., 1976, 17, 1043 CrossRef;
(b) N. Kato, Y. Fukazawa and S. Itô, Tetrahedron Lett., 1976, 17, 2045 CrossRef;
(c) R. Luhowy and P. M. Keehn, J. Am. Chem. Soc., 1977, 99, 3797 CrossRef CAS;
(d) N. Kato, H. Matsunaga, S. Oeda, Y. Fukazawa and S. Itô, Tetrahedron Lett., 1979, 20, 2419 CrossRef;
(e) T. Koenig, K. Rudolf, R. Chadwick, H. Geiselmann, T. Patapoff and C. E. Klopfenstein, J. Am. Chem. Soc., 1986, 108, 5024 CrossRef CAS.
-
(a) G. Laus, H. Schottenberger, K. Wurst, J. Schütz, K.-H. Ongania, U. E. I. Horvath and A. Schwärzler, Org. Biomol. Chem., 2003, 1, 1409 RSC;
(b) S. Ito, S. Kikuchi, H. Kobayashi, N. Morita and T. Asao, J. Org. Chem., 1997, 62, 2423 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: NMR spectra, X-ray crystallographic data. CCDC 2129310–2129315. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2ob01695h |
| ‡ Ranges are given for these dihedral angles since for 26 and 30 the unit cells comprise multiple independent molecules. |
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  a,
Jodie L.
Hann
a,
Jodie L.
Hann