
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTurning on hotspots: supracolloidal SERS probes made brilliant by an external activation mechanism†
Sophie
Jancke
 a,
Chen
Liu
bc,
Ruosong
Wang
a,
Swagato
Sarkar
a,
Quinn A.
Besford
a,
Tobias A. F.
König
adef,
Jürgen
Popp
bc,
Dana
Cialla-May
bc and
Christian
Rossner
*ade
a,
Chen
Liu
bc,
Ruosong
Wang
a,
Swagato
Sarkar
a,
Quinn A.
Besford
a,
Tobias A. F.
König
adef,
Jürgen
Popp
bc,
Dana
Cialla-May
bc and
Christian
Rossner
*ade
aLeibniz-Institut für Polymerforschung Dresden e.V., Institut für Physikalische Chemie und Physik der Polymere, D-01069 Dresden, Germany. E-mail: rossner@ipfdd.de
bLeibniz Institute of Photonic Technology, Member of Leibniz Health Technologies, Member of the Leibniz Centre for Photonics in Infection Research (LPI), Albert Einstein Straße 9, 07745 Jena, Germany
cInstitute of Physical Chemistry (IPC) and Abbe Center of Photonics (ACP), Friedrich Schiller University Jena, Member of the Leibniz Centre for Photonics in Infection Research (LPI), Helmholtzweg 4, 07743 Jena, Germany
dDresden Center for Intelligent Materials (DCIM), Technische Universität Dresden, D-01069 Dresden, Germany
eFaculty of Chemistry and Food Chemistry, Technische Universität Dresden, Bergstraße 66, 01069 Dresden, Germany
fCenter for Advancing Electronics Dresden (cfaed), Technische Universität Dresden, Helmholtzstraße 18, 01069 Dresden, Germany
First published on 2nd November 2023
Abstract
We achieved external activation of local hot-spot sites in supracolloidal assembly structures. The concept was demonstrated by boosting surface-enhanced Raman scattering (SERS) efficiency by one order of magnitude through a heating-induced process. Our approach involves assembling gold nanoparticles with distinct dimensions, i.e. 16 and 80 nm, into well-defined planet–satellite-type arrangement structures using thermoresponsive (poly(N-isopropylacrylamide)) star polymer linkers. Insights into the assembly process were obtained by calculations within the Derjaguin–Landau–Verwey–Overbeek (DLVO) theory framework. We observe one order of magnitude increase in SERS enhancement by a heating-induced volume-phase transition. This magnification aligns with simulations run using the finite-difference time-domain (FDTD) method. The implications of this adaptive supracolloidal concept are twofold: Firstly, our approach bypasses limitations of existing systems that are associated with the limited accessibility of electromagnetic hot-spot sites in strongly coupled, static assemblies of plasmonic nanoparticles, by providing the capability of dynamic hot-spot re-configuration. Second, these externally activated probes offer promising opportunities for the development of messenger materials and associated sensing strategies.
Introduction
The interaction between metal nanoparticles (NPs) and electromagnetic radiation of a suitable frequency leads to the excitation of localized surface plasmon resonance (LSPR) modes.1 Concomitantly with this excitation, an intense electromagnetic near-field emerges, which is significantly enhanced compared with the incident field intensity.2 This field enhancement can be amplified further through electrodynamic coupling interactions, that occur when two or more metal NPs are positioned in close proximity, giving rise to electromagnetic “hot-spots” in the inter-NP gaps.3 Clusters of metal NPs with short interparticle separations can therefore be used to drastically enhance light–matter interactions, for example in surface-enhanced vibrational spectroscopic methods, such as surface-enhanced Raman spectroscopy (SERS).4 Consequently, supracolloidal clusters of metal NPs hold great promise as field-enhanced biosensors for applications at the point of care.5 Substrate-based approaches6 that involve the patterning of surfaces with plasmonic NP clusters7 provide a versatile platform in this regard and paved the way to (bio-)sensor chips.8,9 For solution-based SERS, different strategies were developed, involving for example enhancement from cross-linked NP networks10 and NP chain assemblies.11 In this domain, achieving consistent SERS is essential. Part of the appeal of structurally well-defined so-called planet–satellite-type clusters is this capability of providing consistent SERS signals,12–14 making them proper candidates for quantitative SERS. Moreover, their architecture makes the concentration of many electromagnetic “hot-spots” in a small volume possible,15 giving rise to strong SERS enhancement (that scales with the number of attached satellites).16 Notably, comparably large SERS enhancement can be achieved using planet–satellite-type clusters,12,17 which allows the spectrum collection from single clusters,12 and the in situ chemical imaging of living cells.18,19 Although hot-spot-dominated SERS probes can typically fulfill the above-mentioned requirements of defined and consistent spectroscopic signals, ensuring the accessibility of the hot-spot sites for analyte molecules can be challenging. This accessibility of hot-spot sites in planet–satellite nanostructures depends on the synthetic route that is used for their fabrication, particularly on the choice of the particle-linking entities that connect the planet with the satellite NPs. These are typically chosen to produce small interparticle gap distances, leading to strong SERS enhancement. Among the different types of spacers, silica shell spacers provide a permanent barrier blocking the hot-spot sites. Therefore, the SERS reporter molecules must be placed within the hot-spots during planet–satellite-assembly.20,21 The use of folded and, therefore, highly compacted protein spacers15 can significantly impede analyte adsorption,17 greatly reducing the practicability of the produced SERS probes. Small molecule spacers typically result in dense surface grafted layers, which may also restrict analyte access toward the hot-spot. This is why in related studies22–24 they typically serve as SERS-active reporter molecules themselves. In contrast, synthetic macromolecular linkers25 represent planet–satellite linking entities that form swollen brush layers that are not dense in a good solvent environment.26 These solvent-swollen brush layers typically lead to relatively large planet–satellite separations resulting in decreased field enhancement in the hot-spots. However, it is possible to endow these macromolecular linkers with responsive features,27 such that conformational transitions in the linking brush layers lead to on-demand reduction of planet–satellite spacings upon application of suited external stimuli.28 RAFT (Reversible Addition–Fragmentation chain Transfer)29 polymers of N-isopropylacrylamide (NIPAM) with linear26,30,31 and star topology26,28,32,33 have recently been introduced as such responsive linkers in planet–satellite nanostructures.Based on this precedent, we developed a planet–satellite-type SERS platform with responsive polymer linkers that provide hot-spot accessibility under conditions in which the polymer linker is solvent-swollen, with the possibility to reduce the planet– satellite separation after successful analyte adsorption. Thus, we present a prototype platform for activating electromagnetic hot-spots in plasmonic nanoparticle clusters on-demand via an external activation mechanism. We submit that this platform can be adapted to other kinds of external stimuli by adjusting the chemical nature of the linking polymer,27 thereby offering potential for applications in e.g. process-control or biosensing.
Experimental section
Materials
Hydrogen tetrachloroaurate(III) trihydrate (≥99.9%, HAuCl4·3H2O), sodium citrate tribasic dihydrate (≥99%), pentaerythritol tetrakis[2-(dodecylthiocarbonothioylthio)-2-methylpropionate] (4-arm star RAFT agent), 2,2′-azobis(2-methylpropionitril) (AMPN), pure Tween 20 solution, and sodium chloride (≥99%) were purchased from Sigma-Aldrich and used as received. Toluene (≥99%), N,N-dimethylformamide (DMF, ≥99%), acetone (≥99%), and methanol (≥99%) were purchased from Acros Organics (Germany), and n-hexane (≥99%) was purchased from Merck KGaA (Germany). N-isopropylacrylamide (NIPAM) was recrystallized before use from toluene/n-hexane (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3), including hot filtration, cooling, and supernatant removal, after which the needle-like crystals were washed with hexane, dried under vacuum, and stored refrigerated in a sealed container. The other chemicals were used as received. Milli-Q water was used in all experiments. All glassware for preparation of AuNPs was cleaned with aqua regia and rinsed extensively with Milli-Q water before use.
:3), including hot filtration, cooling, and supernatant removal, after which the needle-like crystals were washed with hexane, dried under vacuum, and stored refrigerated in a sealed container. The other chemicals were used as received. Milli-Q water was used in all experiments. All glassware for preparation of AuNPs was cleaned with aqua regia and rinsed extensively with Milli-Q water before use.
Methods
:0.072:200:600 and purged with nitrogen for 10 min before being heated to 65 °C for 120 min. The polymer was purified by repeated precipitation. Size exclusion chromatography revealed a number–average molar mass Mn of 30 kg mol−1 (see Table S1 and Fig. S1, ESI†).

Here, ε is the dielectric constant, ε0 the vacuum permittivity, κ the inverse Debye length, ψi the surface potential of phase i, and p is a regulation parameter that quantifies the extent of the charge variation of the surface as they approach each other; p1 = p2 = 1 yields the classic boundary condition of constant charge. We assume the assumption of low potentials for 1:1 electrolyte, that the satellite-to-satellite interactions are negligible, and we neglect effects mediated by the intervening polymer. We convert W(L) to an interaction force, Fe, via the Derjaguin approximation, where F = 2πRW(L), and R is the effective radius given by R = R1R2/(R1 + R2), where Ri is the radius of particle i. The DLVO relation is obtained with a combination of the van der Waals attraction,
| F = FvdW + Fe |
Characterization
Results and discussion
Formation of planet–satellite-type clusters
For this work, 4-arm star-poly(NIPAM) was synthesized following a core-first R-group approach,40 where the star topology is already implemented in the commercial RAFT agent (Fig. 1) and the polymer average molar mass was confirmed with size exclusion chromatography (Table S1, and Fig. S1†). Gold nanospheres of about 80 nm diameter were synthesized by a modified Frens method.34 The nanospheres were functionalized with the RAFT 4-arm star-poly(NIPAM) following a “grafting-to” strategy41 that involved immersing overnight in methanol solution (see Experimental section). Subsequently, we attempted the binding of citrate-stabilized satellite particles (citrate@Au16) to the free sulfur-containing RAFT end groups.26,32 In pure MilliQ water, this step did not yield the targeted planet–satellite clusters, and we speculated that surface charges represent a major inhibiting factor. Han et al.30 successfully tethered satellite nanoparticles to poly(NIPAM)-stabilized planets by adding NaCl and the non-ionic surfactant Tween20.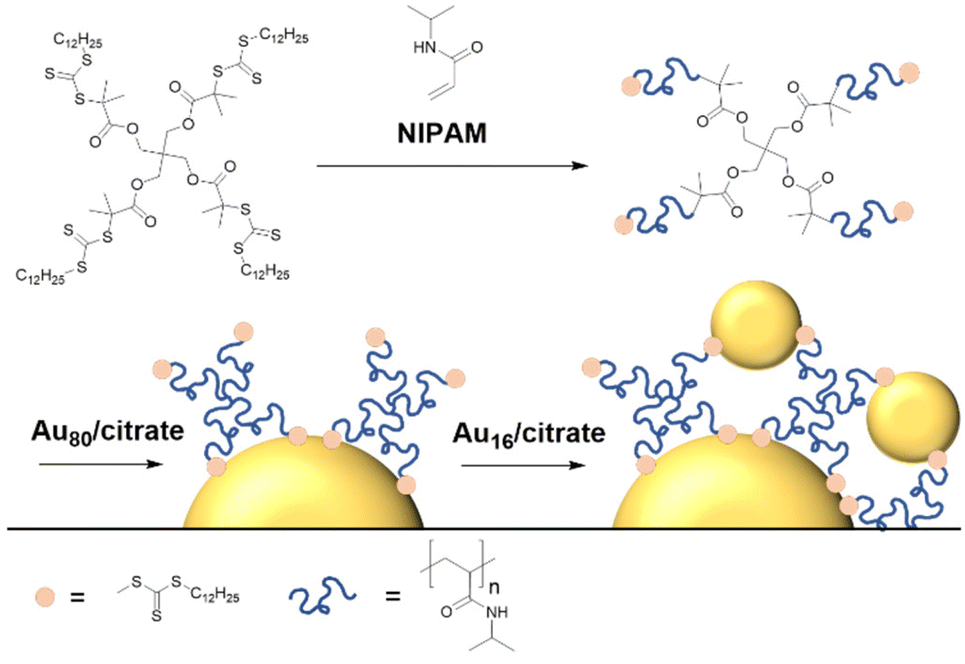 | ||
| Fig. 1 Synthesis scheme of 4-arm star RAFT poly(NIPAM), functionalization of gold 80 nm particles and tethering of 16 nm gold nanoparticles as satellites. | ||
To achieve an understanding of the involved colloidal interactions, we performed electrophoretic (zeta-potential) measurements for the involved colloidal building blocks in both MilliQ water (pH = 5.3) and a solution of NaCl (6.3 mM) (Table 1). These measurements use the movement of charged nanoparticles as a reaction to an external electrical field and provide an experimental approach to estimate the surface charges.42 Following the approach of Trefalt et al.,35 the electrostatic double layer free energy from differently charged particles was calculated (for details, see Methods section). This approach neglects structuring forces in water,43 and interfacial interactions that differ from bulk,44 as well as any effects from the polymer, but nonetheless, it was observed that in the solution with no added salt, repulsive forces dominate for all but very close distances (Fig. 2a, red) meaning that the particles will repel each other unless forced together, and that no strong Au-to-Au interactions contribute to the assembly. Therefore, particles are not inclined to approach each other toward shorter distances, a prerequisite for satellite tethering. Only with increased ionic strength the curve is pushed towards the negative regime, where attractive forces dominate at all length scales and allow the particles to get in sufficient proximity for the tethering of satellites (Fig. 2a, blue). Therefore, NaCl (6.3 mM) was added to the satellite solution prior to tethering to increase the ionic strength and allow for this assembly to occur. Additionally, the non-ionic surfactant Tween 20 was used to stabilize the satellite particles against electrolyte-induced aggregation, as reported by Han et al.30 The functionalized planets were then added to the satellite solution, and after immersion overnight and removal of unbound excess satellite particles (see Experimental section), the final planet–satellite clusters formed (Fig. 2b–d). While there is a dispersion with regard to the number of satellite particles attached, we find that on average 3 satellite particles are grabbed by the polymer layer surrounding the central “planet” particle (Fig. S3†).
 | ||
| Fig. 2 (a) Force–distance functions resulting from DLVO theory calculations with and without addition of 6.3 mM NaCl electrolyte solution. The upper and lower bounds in each curve correspond to variations in the Hamaker constant. (b–d) TEM images of final planet–satellite clusters with 80 nm Au cores, poly(NIPAM) linker and 16 nm Au satellites, taken at different magnifications. | ||
| Poly(NIPAM)@Au80 (planets) | Citrate@Au16 (satellites) | |
|---|---|---|
| Size [nm] | 80 ± 12.0 | 16 ± 2.3 |
| ζ no salt [mV] | −10.3 ± 0.5 | −32.9 ± 2.0 |
| ζ 6.3 mM [mV] | −1.4 ± 2.3 | −15.3 ± 1.3 |
Dynamic hot-spot re-configuration
When heated above the lower critical solution temperature (LCST), the poly(NIPAM) spacer undergoes a phase transition,45 resulting in a globular state with reduced spacing between planets and satellites.28,30 The distance change between the plasmonic nanoparticles results in a change of the extinction profile at visible frequencies.3,30 Therefore, extinction spectroscopy can be used to study the magnitude and reversibility of the distance change.30The optical extinction properties can be modulated reversibly over several heating/cooling cycles (Fig. 3a and c, Fig. S4a†). Upon heating, we find reduced extinction values at the original peak maximum and increased extinction toward higher wavelengths, apparent in a tailing of the spectra toward the red (Fig. 3a, and Fig. S4a†). The simulated extinction spectra, although showing more narrow resonances, show similar behavior (Fig. 3b, and Fig. S4b†). The simulated spectra furthermore reveal increased absorbance toward higher wavelengths (Fig. S4c†) and a decreased scattering contribution at higher temperature (Fig. S4d†). Because of the observed reversibility, we attribute the tailing in the experimentally observed extinction spectra (Fig. 3a, and Fig. S4a†) to the dispersity of the experimental system,3 rather than uncontrolled aggregation, which would be expected to be irreversible. The temperature-dependent size-switching behavior of the clusters was revealed using dynamic light scattering, which provided the hydrodynamic diameter of the entire cluster assembly (see Fig. S5† for the intensity-distributions). The overall cluster dimension in the expanded state is reasonable considering the distribution in the “planet” particle dimension (Fig. S2†) and the expected polymer shell dimension together with the bias of the DLS method toward larger sizes. Repeated switching of the particle dimension can be observed for several heating/cooling cycles, while the count rate of scattered photons shows no significant changes (Fig. 3d). Thereby, strong aggregation during the heating/cooling cycles can be ruled out, as this would (i) result in an increase in the scattering intensity and (ii) change of the Z-average to significantly larger dimension, as the dimensions determined by DLS are intensity-weighted and therefore highly sensitive to large aggregates.
 | ||
| Fig. 3 Reversible switching behaviour revealed: (a) Experimental extinction spectra below and above LCST; (b) FDTD simulation of extinction spectra at 25 °C and 40 °C; (c) change in absorption maximum from visible light extinction spectroscopy over multiple cycles; and (d) change in cluster diameter estimated from dynamic light scattering. (e) SERS signal of the clusters with 4-MBN as marker molecule at 25 °C and 40 °C. | ||
The DLS results reveal a reversible swelling/contraction of the hydrodynamic diameter of the overall nanostructures by ∼30 nm, i.e. a change in polymer layer thickness of ∼15 nm. To put this value into perspective with regard to particle spacings, we can rely on previously determined26 end-monomer distributions and satellite-particle distributions. It is established that the former decays to zero ∼5 nm farther from the planet particle than the latter distribution.26I.e., in the solvent-swollen state, the satellite particles can penetrate roughly 5 nm into the brush. Thus, the reversible diameter change relates to a change in the planet–satellite particle spacing of ∼10 nm. Furthermore, it is verified that switching occurs on time scales that are smaller than those of one light scattering measurement (few minutes), which is in line with the very fast (∼10 to 100 ns) switching dynamics46 observed for simpler gold–core–poly(NIPAM)–shell structures.
In the next step, we evaluated the SERS capabilities of the produced nanostructures. For the SERS experiments, 4-mercaptobenzonitrile (4-MBN) was chosen, structurally closely related to 4-mercaptobenzoic acid (4-MBA), known as a strongly adsorbing, universal SERS reporter molecule47 that is commonly used in colloidal systems.15 Incubation with 4-MBN was done at 25 °C so that the polymer spacer is in a swollen state and analyte access is facilitated. SERS spectra were then recorded at 25 °C and 40 °C using excitation at 785 nm. The pure clusters without 4-MBN were first recorded to observe Raman vibrations that might be present from adsorbed carbonaceous species. As no distinct peaks were observed, the respective spectra of the pure clusters at different temperatures were used for background correction. The data shown in Fig. 3e is the average of 120 separate spectra acquisitions respectively, and the shaded area is the standard deviation of the 120 spectra. The characteristic 4-MBN signals can be distinguished in both curves, especially the peaks at 1067 cm−1 and 1574 cm−1 that can be assigned to ring modes of the reporter molecule and the 2226 cm−1 band, which originates from the C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N-stretch mode.48–50 To evaluate the SERS signal intensity increase for the transition from the expanded to the contracted state of the clusters, four distinct peaks (Fig. 3e) were considered, and an intensity ratio was calculated respectively (for details, see ESI Fig. S6 and Table S2†). The intensity increase upon temperature change obtained is roughly one order of magnitude, on average around 30, and up to 67 for the 2220 cm−1 band from the CN-stretch mode (Table 2). Notably, thermally-induced noise does not seem to impair the signal amplification, as the spectrum shows distinct features that are significantly above the background, i.e. a sufficient signal-to-noise ratio.
N-stretch mode.48–50 To evaluate the SERS signal intensity increase for the transition from the expanded to the contracted state of the clusters, four distinct peaks (Fig. 3e) were considered, and an intensity ratio was calculated respectively (for details, see ESI Fig. S6 and Table S2†). The intensity increase upon temperature change obtained is roughly one order of magnitude, on average around 30, and up to 67 for the 2220 cm−1 band from the CN-stretch mode (Table 2). Notably, thermally-induced noise does not seem to impair the signal amplification, as the spectrum shows distinct features that are significantly above the background, i.e. a sufficient signal-to-noise ratio.
| Peak | Mode | Intensity ratio |
|---|---|---|
| 1 | ν(C–S) stretching/ν(C–C–C) bending mode | 7 |
| 2 | ν(C–H)in-plane ring bending mode | 16 |
| 3 | ν(C–C)in-plane ring stretching mode | 29 |
| 4 | ν(CN) stretching mode |
66 |
To study the hot-spot activation above the critical temperature and ensuing SERS enhancement, the electrical field around the clusters in the contracted and expanded states was simulated using the FDTD method. For a ‘core–shell–satellite’ model with an 80 nm AuNP core, 16 nm AuNP satellite and a poly(NIPAM) shell with a temperature-dependent variable shell thickness, the effect of the shell thickness towards field enhancements is evaluated for two different polarization configurations. The FDTD model corresponds to the experimental realization with 3 to 4 satellites per planet particle. The poly(NIPAM) layer of varying thickness can serve as a plasmonic hot-spot51,52 to enable local field enhancements. However, the polarization direction plays an essential role in this enhancement; electric fields parallel to the nanometric gap can allow confining of the plasmonic modes. Due to the random distribution of such ‘core–shell–satellite’ systems, experimental outcomes under the unpolarized excitation (785 nm SERS laser excitation) can have the incident polarization vector in all possible directions. To cover the orientation dependence, we systematically study the two principal cases where the electric field vector is either collinear or non-collinear with the core and satellites. For a simplified model, the core and the satellites are arranged in the same X–Y plane with the core at the origin (0, 0) and the four satellites at (+X, 0), (−X, 0), (+Y, 0), (−Y, 0) coordinates. Because of the spherically symmetric core, the X and Y take the same value, given by ‘rC + rS + t’, where rC and rS are the radius of the core and satellite AuNPs, and t is the poly(NIPAM) shell thickness. In the expanded state, the planet–satellite distance was estimated to be t25 °C = 14 nm based on the molar mass of the polymer and established distance-scaling behavior.26 Above the critical phase transition temperature, the polymer is dense, so the known density of poly(NIPAM) (dP = 1.1 g cm−3)53 was assumed and the shell thickness calculated following simple stoichiometry and geometry considerations (ESI†). With a grafting density of ∼0.1 nm−1 estimated based on previous results,26,54 a distance of t40 °C = 4 nm was calculated and this value used in the FDTD simulation. This variation in the shell thickness leads to two different configurations of plasmonic nanocavities with varying cavity length that has also been reflected in the experimental SERS intensity enhancement. The effect of varying the shell thickness for the situation where the electric field vector is kept parallel to the X axis, with the incident beam propagating along the ‘−Z’ direction is shown in Fig. 4(a, i and ii). To estimate the SERS intensity increase, we measure the electric field and the corresponding light-intensity enhancements  ;
;  is the incident electric field, and
is the incident electric field, and  is the resultant field after the plasmonic interaction. According to the color contour plots (Fig. 4(a, i and ii)), plasmonic hot-spots are created only between the core and the two satellites lying collinearly with the AuNP core and the electric field direction. Nodes with intensity minima can be found for the two other satellites, placed orthogonally with the X-axis. Such nodes are generally formed perpendicular to the electric field vector.55 For a quantitative analysis of the electromagnetic field enhancement, we define the REF as the ratio between the intensity for the two different thicknesses, here REF = It_40 °C/It_25 °C. Comparing the maximum intensity (Imax) from the two cases, we obtain a REF = 266/79 = 3.4. However, for SERS activity, the electric field associated with the core surface is of higher importance. Thus, using a magnified plot (given in the inset), we calculate REFsurf = 99.3/19.3 = 5.0. Since the SERS enhancement scales with the fourth power of the associated electric field,56,57 the SERS intensity ratio can be approximated as REF2surf ∼ 25.0. Further, to estimate the random orientation of the incident electric field, the non-collinear model is studied in Fig. 4(b, i and ii). For an incident polarization of 45°, the electric field along X and Y axis can now be estimated as E0x = E0cos45° = E0/√2 and E0y = E0sin45° = E0/√2. This leads to the simultaneous excitation of plasmonic hot-spot for all four satellites; the nodes can still be found perpendicular to the electric field vector orientation. The reduced electric field (by a factor of 1/√2) interacting with the core-satellite has resulted in reduced Imax (by a factor of ∼1/2), leading to an almost similar REF = 138/41 = 3.4, REFsurf = 49.7/9.9 = 5.0 and a corresponding SERS intensity ratio of again ∼25.0.
is the resultant field after the plasmonic interaction. According to the color contour plots (Fig. 4(a, i and ii)), plasmonic hot-spots are created only between the core and the two satellites lying collinearly with the AuNP core and the electric field direction. Nodes with intensity minima can be found for the two other satellites, placed orthogonally with the X-axis. Such nodes are generally formed perpendicular to the electric field vector.55 For a quantitative analysis of the electromagnetic field enhancement, we define the REF as the ratio between the intensity for the two different thicknesses, here REF = It_40 °C/It_25 °C. Comparing the maximum intensity (Imax) from the two cases, we obtain a REF = 266/79 = 3.4. However, for SERS activity, the electric field associated with the core surface is of higher importance. Thus, using a magnified plot (given in the inset), we calculate REFsurf = 99.3/19.3 = 5.0. Since the SERS enhancement scales with the fourth power of the associated electric field,56,57 the SERS intensity ratio can be approximated as REF2surf ∼ 25.0. Further, to estimate the random orientation of the incident electric field, the non-collinear model is studied in Fig. 4(b, i and ii). For an incident polarization of 45°, the electric field along X and Y axis can now be estimated as E0x = E0cos45° = E0/√2 and E0y = E0sin45° = E0/√2. This leads to the simultaneous excitation of plasmonic hot-spot for all four satellites; the nodes can still be found perpendicular to the electric field vector orientation. The reduced electric field (by a factor of 1/√2) interacting with the core-satellite has resulted in reduced Imax (by a factor of ∼1/2), leading to an almost similar REF = 138/41 = 3.4, REFsurf = 49.7/9.9 = 5.0 and a corresponding SERS intensity ratio of again ∼25.0.
 | ||
| Fig. 4 FDTD calculated intensity-enhancements at λSERS = 785 nm: (a, i and ii) Plasmonic core–shell-satellite models (AuNP-poly(NIPAM)@AuNP) with varied shell thickness (t) excited with electric field vector (red arrow) parallel to the X-axis. (b, i and ii) Similar plasmonic configurations for excitation with electric field vectors having components along both X- and Y-axis. The intensity maxima for the colour plots (in logarithmic scale) are inscribed along with the insets showing intensity values on the surface of the AuNP core. | ||
Thus, one can conclude that for different non-collinear interactions (even if there is an additional Z component due to the asymmetric orientation of satellites around the core), the REF2 – representing the SERS signal intensity increase upon heating above the LCST – can be expected to always be in the same range that is in reasonable agreement with the experimentally obtained value of ∼one order of magnitude.
Conclusions
We fabricated Au-poly(NIPAM)@Au planet–satellite clusters using an increase in the ionic strength of the solution and achieved a quantitative understanding of involved interactions in the framework of DLVO theory. The employed poly(NIPAM) linker and spacer allowed for reversible, temperature-mediated switching of the dimension of planet–satellite clusters based on the swelling state of the employed polymeric linker, associated with a modulation of their plasmonic signature. We demonstrate that this mechanism can be used to achieve an order of magnitude SERS signal intensity increase above the polymer's LCST, i.e. in the contracted non-swollen state. These experimental findings align well with calculations of electromagnetic field enhancement.In conclusion, we have established a concept for the “on-demand” activation of local electromagnetic hot-spot sites in functional hybrid nanostructures, as demonstrated by the capability to boost SERS capability through external activation. The developed approach features the advantage of facilitated analyte access in an initial solvent-swollen state, combined with high SERS signal intensity after switching to a contracted state. The described in situ hot-spot activation is mediated by a polymeric linker, the critical phase transition temperature of which could be fine-tuned based on the intended application via co-polymerization. Given the significant signal increase in response to critical temperature change, the investigated supracolloidal SERS markers also exhibit potential as highly sensitive messenger materials.58 These markers can report local overheating above a critical threshold temperature in complex environments, with the option to synthetically alter the LCST. Because of the underlying modular design, the hot-spot sites could as well be dynamically manipulated by macromolecular linkers that respond to different external parameters, like for example pH. This provides further prospects in biosensing, for example as nano-pH-sensor for tumor detection.59
Author contributions
Conceptualization: D.C.-M. and C.R.; methodology: S.J., C.L., S.S., Q.A.B. Formal analysis: S.J., C.L., S.S., Q.A.B. Investigation: S.J., C.L., R.W., S.S., Q.A.B. Data curation: S.J., C.L., R.W., S.S., Q.A.B. writing—original draft preparation: S.J., C.L., S.S., Q.A.B., C.R. writing—review and editing: S.J., C.L., R.W., S.S., Q.A.B., T.A.F.K., J.P, D.C.-M., C.R. visualization: S.J., S.S., Q.A.B., D.C.-M. supervision: T.A.F.K., J.P., D.C.-M., C.R.; funding acquisition: Q.A.B., T.A.F.K., D.C.-M., C.R. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) supported this work under grants 496201730 (Q. A. B.), 465289819 (D. C. M.), and 466182523 (C. R.). The Volkswagen Foundation financially supported T. A. F. K. through a Freigeist Fellowship. C. R. acknowledges receipt of a Liebig fellowship (Fonds der Chemischen Industrie).References
- M. A. Garcia, J. Phys. D: Appl. Phys., 2011, 44, 283001 CrossRef.
- K. L. Kelly, E. Coronado, L. L. Zhao and G. C. Schatz, J. Phys. Chem. B, 2003, 107, 668–677 CrossRef CAS.
- C. Rossner, T. A. F. König and A. Fery, Adv. Opt. Mater., 2021, 9, 2001869 CrossRef CAS.
- M. Blanco-Formoso, N. Pazos-Perez and R. A. Alvarez-Puebla, Nanoscale, 2020, 12, 14948–14956 RSC.
- V. Tran, B. Walkenfort, M. König, M. Salehi and S. Schlücker, Angew. Chem., Int. Ed., 2019, 58, 442–446 CrossRef CAS PubMed.
- H. Dang, S.-G. Park, Y. Wu, N. Choi, J.-Y. Yang, S. Lee, S.-W. Joo, L. Chen and J. Choo, Adv. Funct. Mater., 2021, 31, 2105703 CrossRef CAS.
- R. Wang, P. Zimmermann, D. Schletz, M. Hoffmann, P. Probst, A. Fery, J. Nagel and C. Rossner, Nano Sel., 2022, 3, 1502–1508 CrossRef CAS.
- F. L. Yap, P. Thoniyot, S. Krishnan and S. Krishnamoorthy, ACS Nano, 2012, 6, 2056–2070 CrossRef CAS PubMed.
- S.-G. Park, X. Xiao, J. Min, C. Mun, H. S. Jung, V. Giannini, R. Weissleder, S. A. Maier, H. Im and D.-H. Kim, Adv. Funct. Mater., 2019, 29, 1904257 CrossRef CAS.
- D. Caprara, F. Ripanti, A. Capocefalo, A. Sarra, F. Brasili, C. Petrillo, C. Fasolato and P. Postorino, Colloids Surf., A, 2020, 589, 124399 CrossRef CAS.
- R. W. Taylor, R. Esteban, S. Mahajan, J. Aizpurua and J. J. Baumberg, Optimizing SERS from Gold Nanoparticle Clusters, J. Phys. Chem. C, 2016, 120, 10512–10522 CrossRef CAS.
- N. Pazos-Perez, J. M. Fitzgerald, V. Giannini, L. Guerrini and R. A. Alvarez-Puebla, Nanoscale Adv., 2019, 1, 122–131 RSC.
- C. Kuttner, R. P. M. Höller, M. Quintanilla, M. J. Schnepf, M. Dulle, A. Fery and L. M. Liz-Marzán, Nanoscale, 2019, 11, 17655–17663 RSC.
- S.-Y. Chen and A. A. Lazarides, J. Phys. Chem. C, 2009, 113, 12167–12175 CrossRef CAS.
- R. P. M. Höller, M. Dulle, S. Thomä, M. Mayer, A. M. Steiner, S. Förster, A. Fery, C. Kuttner and M. Chanana, ACS Nano, 2016, 10, 5740–5750 CrossRef PubMed.
- P. Dey, S. Zhu, S. Zhu, K. J. Thurecht, P. M. Fredericks and I. Blakey, J. Mater. Chem. B, 2014, 2, 2827–2837 RSC.
- R. P. M. Höller, I. J. Jahn, D. Cialla-May, M. Chanana, J. Popp, A. Fery and C. Kuttner, ACS Appl. Mater. Interfaces, 2020, 12, 57302–57313 CrossRef PubMed.
- L. Xu, H. Kuang, C. Xu, W. Ma, L. Wang and N. A. Kotov, J. Am. Chem. Soc., 2012, 134, 1699–1709 CrossRef CAS PubMed.
- D. Radziuk, R. Schuetz, A. Masic and H. Moehwald, Phys. Chem. Chem. Phys., 2014, 16, 24621–24634 RSC.
- M. Schütz and S. Schlücker, Phys. Chem. Chem. Phys., 2015, 17, 24356–24360 RSC.
- M. Gellner, D. Steinigeweg, S. Ichilmann, M. Salehi, M. Schütz, K. Kömpe, M. Haase and S. Schlücker, Small, 2011, 7, 3445–3451 CrossRef CAS PubMed.
- N. Gandra, A. Abbas, L. Tian and S. Singamaneni, Nano Lett., 2012, 12, 2645–2651 CrossRef CAS PubMed.
- N. Gandra and S. Singamaneni, Chem. Commun., 2012, 48, 11540–11542 RSC.
- J. H. Yoon, J. Lim and S. Yoon, ACS Nano, 2012, 6, 7199–7208 CrossRef CAS PubMed.
- A. M. T. S. Juan, S. R. Chavva, D. Tu, M. Tircuit, G. Coté and S. Mabbott, Nanoscale Adv., 2021, 4, 258–267 RSC.
- C. Rossner, Q. Tang, O. Glatter, M. Müller and P. Vana, Langmuir, 2017, 33, 2017–2026 CrossRef CAS PubMed.
- G. Moad, Polym. Chem., 2016, 8, 177–219 RSC.
- C. Rossner, O. Glatter and P. Vana, Macromolecules, 2017, 50, 7344–7350 CrossRef CAS.
- J. Chiefari, Y. K. Chong, F. Ercole, J. Krstina, J. L. Jeffery, T. P. T. Le, R. T. A. Mayadunne, G. F. Meijs, C. L. Moad, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 1998, 31, 5559–5562 CrossRef CAS.
- F. Han, S. R. C. Vivekchand, A. H. Soeriyadi, Y. Zheng and J. J. Gooding, Nanoscale, 2018, 10, 4284–4290 RSC.
- F. Han, T. Armstrong, A. Andres-Arroyo, D. Bennett, A. Soeriyadi, A. A. Chamazketi, P. Bakthavathsalam, R. D. Tilley, J. J. Gooding and P. J. Reece, Nanoscale, 2020, 12, 1680–1687 RSC.
- C. Rossner and P. Vana, Angew. Chem., Int. Ed., 2014, 53, 12639–12642 CrossRef CAS PubMed.
- C. Rossner, V. Roddatis, S. Lopatin and P. Vana, Macromol. Rapid Commun., 2016, 37, 1742–1747 CrossRef CAS PubMed.
- N. G. Bastús, J. Comenge and V. Puntes, Langmuir, 2011, 27, 11098–11105 CrossRef PubMed.
- G. Trefalt, F. J. M. Ruiz-Cabello and M. Borkovec, J. Phys. Chem. B, 2014, 118, 6346–6355 CrossRef CAS PubMed.
- J. N. Israelachvili, Intermolecular and Surface Forces, Academic Press, Amsterdam, 3rd edn, 2011 Search PubMed.
- P. B. Johnson and R. W. Christy, Phys. Rev. B: Solid State, 1972, 6, 4370–4379 CrossRef CAS.
- Y. Brasse, M. B. Müller, M. Karg, C. Kuttner, T. A. F. König and A. Fery, ACS Appl. Mater. Interfaces, 2018, 10, 3133–3141 CrossRef CAS PubMed.
- L. Scarabelli, A. Sánchez-Iglesias, J. Pérez-Juste and L. M. Liz-Marzán, J. Phys. Chem. Lett., 2015, 6, 4270–4279 CrossRef CAS PubMed.
- R. T. A. Mayadunne, J. Jeffery, G. Moad and E. Rizzardo, Macromolecules, 2003, 36, 1505–1513 CrossRef CAS.
- B. Ebeling and P. Vana, Macromolecules, 2013, 46, 4862–4871 CrossRef CAS.
- R. M. Pashley and M. E. Karaman, in Applied Colloid and Surface Chemistry, John Wiley & Sons, Ltd, 2004, pp. 93–125 Search PubMed.
- Q. A. Besford, A. J. Christofferson, M. Liu and I. Yarovsky, J. Chem. Phys., 2017, 147, 194503 CrossRef PubMed.
- Q. A. Besford, A. J. Christofferson, J. Kalayan, J.-U. Sommer and R. H. Henchman, J. Phys. Chem. B, 2020, 124, 6369–6375 CrossRef CAS PubMed.
- G. Zhang and C. Wu, J. Am. Chem. Soc., 2001, 123, 1376–1380 CrossRef CAS.
- S. Murphy, S. Jaber, C. Ritchie, M. Karg and P. Mulvaney, Langmuir, 2016, 32, 12497–12503 CrossRef CAS PubMed.
- S. E. J. Bell, G. Charron, E. Cortés, J. Kneipp, M. Lamy de la Chapelle, J. Langer, M. Procházka, V. Tran and S. Schlücker, Angew. Chem., Int. Ed., 2019, 59, 5454–5462 CrossRef PubMed.
- S. Zhu, C. Fan, J. Wang, J. He and E. Liang, Appl. Phys. A, 2014, 117, 1075–1083 CrossRef CAS.
- S. Wang, L.-L. Tay and H. Liu, Analyst, 2016, 141, 1721–1733 RSC.
- M. F. Mrozek, S. A. Wasileski and M. J. Weaver, J. Am. Chem. Soc., 2001, 123, 12817–12825 CrossRef CAS PubMed.
- S. Sarkar, V. Gupta, T. Tsuda, J. Gour, A. Singh, O. Aftenieva, A. M. Steiner, M. Hoffmann, S. Kumar, A. Fery, J. Joseph and T. A. F. König, Adv. Funct. Mater., 2021, 31, 2011099 CrossRef CAS.
- V. Gupta, S. Sarkar, O. Aftenieva, T. Tsuda, L. Kumar, D. Schletz, J. Schultz, A. Kiriy, A. Fery, N. Vogel and T. A. F. König, Adv. Funct. Mater., 2021, 31, 2105054 CrossRef CAS.
- H. G. Schild, Prog. Polym. Sci., 1992, 17, 163–249 CrossRef CAS.
- Q. Tang, C. Rossner, P. Vana and M. Müller, Biomacromolecules, 2020, 21, 5008–5020 CrossRef CAS PubMed.
- D. Radziuk and H. Moehwald, Phys. Chem. Chem. Phys., 2015, 17, 21072–21093 RSC.
- Y. S. Yamamoto, Y. Ozaki and T. Itoh, J. Photochem. Photobiol., C, 2014, 21, 81–104 CrossRef CAS.
- S. Franzen, J. Phys. Chem. C, 2009, 113, 5912–5919 CrossRef CAS.
- Q. A. Besford, C. Rossner and A. Fery, Adv. Funct. Mater., 2023, 33, 2214915 CrossRef CAS.
- P. Mi, F. Wang, N. Nishiyama and H. Cabral, Macromol. Biosci., 2017, 17, 1600305 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3nr05121h |
| This journal is © The Royal Society of Chemistry 2023 |