
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLigand induced chirality in In2S3 nanoparticles†
Lorenzo
Branzi
 *a,
Oriane
Lavet
b and
Yurii K.
Gun'ko
*a
*a,
Oriane
Lavet
b and
Yurii K.
Gun'ko
*a
aSchool of Chemistry, CRANN and AMBER Research Centres, Trinity College Dublin, College Green, Dublin 2, Ireland. E-mail: branzil@tcd.ie; igounko@tcd.ie
bChemistry Department, University of Clermont Auvergne, Antenne du Puy en Velay, 43009 Le Puy en Velay Cedex, France
First published on 1st November 2023
Abstract
Chiral inorganic nanostructures have attracted a lot of attention over the last few years. Here we report the first observation of chirality in indium sulfide nanoparticles, which have been produced by a co-precipitation reaction in the presence of cysteine as a chiral agent. The process resulted in the production of spherical nanoparticles with an average diameter of around 3.6 nm. Circular dichroism spectroscopy of the nanoparticles showed an intense chiroptical signal corresponding to the indium sulfide excitonic transition, confirming the successful transfer of chirality to the In2S3 inorganic matrix. Nuclear magnetic resonance analysis of a colloidal solution of the nanoparticles demonstrated critical evidence of chemisorption of the chiral ligand on the surface of the nanoparticles and revealed a characteristic fast chemical exchange between the ligand chemisorbed on the surface of the nanoparticles and the free ligand in solution. Finally, the effect of the chiral ligand's structure on the transfer of chirality was investigated, with consideration of other amino acid ligands, and the critical role of the thiolate group in the optimisation of the chiral transfer was observed. This research is expected to stimulate further development and applications of new chiral semiconductor nanomaterials.
Introduction
Chiral inorganic nanostructures have recently attracted a great deal of attention due to their promising potential applications1–6 and extensive investigation of the introduction of chirality in several inorganic systems.7,8 Due to the intrinsic complexity of these systems, the origin of chirality in inorganic nanostructures can be attributed to multiple sources. In general, it is possible to distinguish between the chirality originating from: (i) structure, solids that crystallise in non-centrosymmetric space groups can present a chiral arrangement of atoms;9–11 (ii) morphology, opportune control of the nanoparticle growth has been used to promote the formation of nanoparticles having characteristic chiral shapes;12,13 (iii) interface, chirality originating at the nanoparticle–ligand interface is known as ligand-induced chirality; in this case, a chiral ligand is employed to passivate the nanoparticle's surface and, under suitable conditions, the ligand transfers chirality to the nanoparticle via electronic coupling or structural distortion14–16 or (iv) assembly, achiral nanoparticles that are arranged in a chiral pattern can produce collective chirality.8,17–19 Apart from these main categories, multiple sources of chirality can exist simultaneously in more complex systems. Regardless of the origin of chirality, chiral ligands are commonly employed to induce symmetry breaking during synthesis to both introduce chirality in the final system and selectively promote the formation of an enantiomeric excess in intrinsically chiral materials.10 However, in the case of ligand-induced chirality systems, the interaction between the chiral ligand and the nanoparticle's surface is critical to guaranteeing the particle's chirality and related chiroptical properties. For this reason, recent activity in this field has been mainly focused on the investigation of the different factors that play a critical role in the transfer of chirality.20 In particular, in the case of semiconductor nanoparticles, some authors have focused their attention predominantly on inorganic nanoparticles, considering the nanoparticle size,21,22 shape23–25 and structure14,26 (core–shell systems characterised by different band alignment types and shell thicknesses). Instead, others have been more focused on the effect of the ligand, involving the ligand structure,27–29 binding modes and chemisorption equilibrium.28,30,31Indium sulfide is a III–VI type wide band gap semiconductor material, largely investigated due to its high environmental stability combined with its potential use as a toxic-metal free semiconductor for a broad range of applications in optoelectronics, including photocatalysis,32 photovoltaics33 and photodetectors.34 During the last decade, several reports dealing with the control of the particle's morphology have been published. Small nanoparticles or quantum dots have been prepared by co-precipitation reactions in water or observed as intermediate species in the growth of more complex nanostructures.32,35–37 There are several articles on morphological control to obtain 1D and 2D nanostructures. The formation of 1D nanorods was observed by the thermal decomposition of di-tert-butyl disulfide in oleylamine at 180 °C in the presence of indium acetylacetonate.38 Nanoribbons with a giant aspect ratio have been produced by the reaction of sulfur-oleylamine solution and indium chloride at 215 °C; in this case, the authors observed the critical role of water as a growth-directing agent to induce the formation of highly anisotropic nanocrystals.39 Under similar reaction conditions, apart from a rigorously anhydrous environment, the formation of 2D ultrathin nanoplates is promoted.39,40 Moreover, the formation of nanosheets, nanocoils and self-assembled structures has also been investigated.41–43 Despite the large amount of work dealing with the control of the morphology of indium sulfide nanoparticles and the significant potential of the materials for applications in photonics, the chiroptical activity in indium sulfide nanoparticles has never been reported.
In this article, we report the first observation of chiroptically active indium sulfide nanoparticles (NPs). The chirality was introduced via a co-precipitation reaction in the presence of cysteine as a chiral ligand. The process resulted in the formation of a colloidal solution composed of spherical NPs with a diameter of around 3.6 nm. The chirality of the NPs was verified by circular dichroism spectroscopy which showed high chiroptical activity in the excitonic transition region, confirming the successful transfer of chirality to the inorganic In2S3 matrix. G-factors of −5.11 × 10−4 and 5.66 × 10−4 were calculated for the NPs produced in the presence of L and D-cysteine, respectively. The observed values are in line with the expected order of magnitude for chiral inorganic nanosystems characterised by ligand-induced chirality. Further details on the interaction between the inorganic NPs and the chiral ligand were collected using nuclear magnetic resonance (NMR) spectroscopy in a colloidal solution. In particular, the 1H-NMR spectrum showed characteristic evidence of ligand chemisorption on the surface of the NPs which is characterised by a fast chemical exchange. Nuclear Overhauser effect spectroscopy was employed to further highlight the contribution of the overall NMR signal of the population of the chiral ligand chemisorbed on the surface of the NPs. Finally, the effect of the ligand's structure on the chiroptical activity was investigated by a ligand displacement study with other amino acids and a comparison of the effect of the ligand substitution on the circular dichroism. This study also provides evidence of the fundamental role of sulfur donor groups in the transfer of chirality in indium sulfide NPs. These observations can be applied to the induction and optimisation of chirality in other novel excitonic nanomaterials of great interest for a broad range of potential applications.
Results and discussion
Indium sulfide nanoparticles (NPs) were produced by a co-precipitation process in water using cysteine as a chiral ligand. A schematic of the synthesis method is presented in Fig. 1. In the first step, the precipitation of indium sulfide was triggered by the rapid addition of sodium sulfide to a diluted indium acetate solution in the presence of cysteine. L or D-cysteine were used as chiral inductors and according to the ligand stereochemistry, chiral hydrophilic L-In2S3 or D-In2S3 NPs were produced. Apart from its role as a chiral inductor, cysteine plays a critical role in both preventing the precipitation of hydroxide species and controlling the growth and colloidal stability of the NPs in an aqueous solution. In the second synthetic step, the NPs were thermally annealed under mild conditions (80 °C) under an argon atmosphere, and this step improved the crystallinity of the NPs. The process brought about the formation of yellow colloidal solutions with high colloidal stability under alkaline conditions (Fig. S1†). | ||
| Fig. 1 Scheme of the synthetic process for the production of L and D-In2S3 NPs. | ||
The TEM micrographs of L-In2S3 and D-In2S3 are presented in Fig. 2ai and aii, respectively; both samples are composed of spherical particles. The size distribution analysis (Fig. 2b) shows similar sizes for the particles prepared in the presence of L and D-cysteine, showing an average size of 3.6 ± 0.7 nm and 3.7 ± 0.7 nm for L-In2S3 and D-In2S3 respectively.
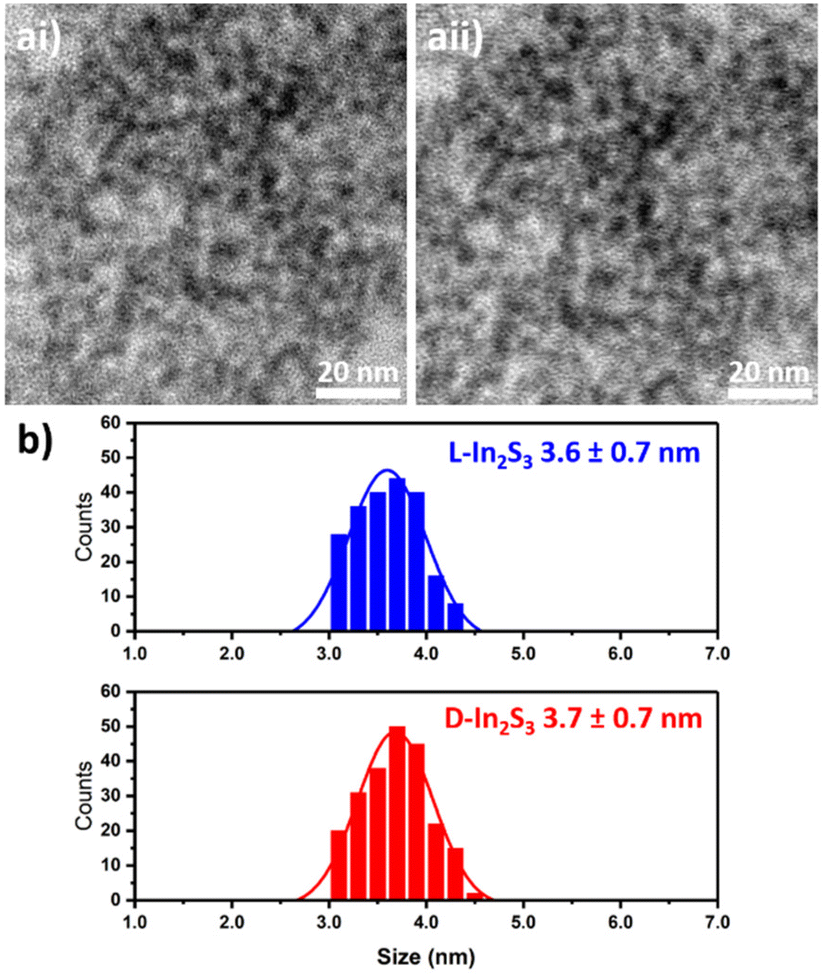 | ||
| Fig. 2 Representative TEM micrographs of L-In2S3 (ai) and D-In2S3 (aii) NPs. (b) Results of the size distribution analysis. | ||
The crystallographic structure of the NPs was investigated by X-ray diffraction (XRD). The indium sesquisulfide system is characterised by complex polymorphism with multiple crystallographic phases stable at room temperature. Among them, cubic α (Fd![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m) and tetragonal β (I41/amd) are the most commonly observed structures. Both phases can be described as spinel structures; however, in the case of the α phase, In3+ atoms occupy all the octahedral sites (Oh) and 2/3 of the tetrahedral (Td) sites with statistically distributed vacancies (□) occupying the remaining Td sites, thus the compound is described as [In2]Oh[In2/3□1/3]TdS4.44 Instead, according to the observation reported by Rooymans45 and Steigmann et al.,46 the β phase is characterised by a vacancy ordering along the 41 screw axis and the structure can be described as adopting a quadratic super cell composed of three spinel blocks stacked along the c axis and the formula [In6]Oh[In2□]TdS12. Due to the similar XRD patterns of these two structures, the distinction between the α cubic and the β tetragonal phases is extremely challenging without high-quality X-ray data and especially limited in the case of nanocrystals that are affected by intrinsic sources of line broadening.39 Other crystallographic phases are known, such as the rhombohedral ε (Rc) or the hexagonal γ (Pm1) phase, the latter is a structure stable at high temperatures, obtained by heating the beta phase above 750 °C.47,48 The diffraction patterns of L and D-In2S3 NPs are presented in Fig. 3 along with the reference patterns related to the α, β and γ phases. The observed pattern is characterised by broad peaks that are likely related to the small nanocrystal size. The main features include a broad peak centred at around 31° and a second peak at 47°, this pattern is characteristic of small spherical NPs and it is related to the tetragonal β-In2S3 phase or to the cubic α-In2S3 phase. As mentioned above the distinction between the two structures is limited to the disorder of the vacancy along the c axis and in the case of nanosystems, the distinction between these two structures is especially challenging.32,39 The first diffraction can be related to the overlap of the (2 1 3) and (2 2 0) diffraction planes located at 27.39° and 33.18° respectively. Instead, the peak observed at higher angles is related to the (2 2 12) plane at 47.64°. Instead, in the case of the α phase, the first broad peak is related to the (3 1 1) and (4 0 0) diffraction planes located at 27.30° and 33.08° and the third peak is related to the diffraction originated by the (4 4 0) plane located at 47.48°. The high resolution transmission electron microscopy (HR-TEM) micrographs of L and D-In2S3 NPs (Fig. S2†) show electron diffraction fringes with a spacing of 0.32 nm that can be related to the (2 1 3) plane of β-In2S3 or the (3 1 1) plane of α-In2S3. Similar observations have also been reported by other authors for indium sulfide nanoparticles produced using different methods and with different morphologies.36,40,42 The effect of the crystal structure on the chiroptical activity has been investigated on CdSe nanoplatelets by Gao and coworkers.49 They observed large differences in the number and sign of the CD transitions between wurtzite and zinc blende nanoplatelets, along with a significant effect on the intensity of the CD transitions. The authors investigated the system using a nondegenerate coupled-oscillator model and related the effects on the chiroptical activity to the different conformations of cysteine on the surface of the nanoplatelets. In the case of indium sulfide polymorphism, the tetragonal β-In2S3 and the cubic α-In2S3 phases are characterised by a similar atomic disposition, with the difference being limited to the ordering of the vacancy on the tetrahedral site (Fig. S3†). For this reason, we do not expect a significant variation in the chiroptical activity between these two different phases. However, further investigation of the trigonal γ-In2S3 phase, which is characterised by a significantly different structure, may lead to the observation of interesting variations in the chiroptical activity.
m) and tetragonal β (I41/amd) are the most commonly observed structures. Both phases can be described as spinel structures; however, in the case of the α phase, In3+ atoms occupy all the octahedral sites (Oh) and 2/3 of the tetrahedral (Td) sites with statistically distributed vacancies (□) occupying the remaining Td sites, thus the compound is described as [In2]Oh[In2/3□1/3]TdS4.44 Instead, according to the observation reported by Rooymans45 and Steigmann et al.,46 the β phase is characterised by a vacancy ordering along the 41 screw axis and the structure can be described as adopting a quadratic super cell composed of three spinel blocks stacked along the c axis and the formula [In6]Oh[In2□]TdS12. Due to the similar XRD patterns of these two structures, the distinction between the α cubic and the β tetragonal phases is extremely challenging without high-quality X-ray data and especially limited in the case of nanocrystals that are affected by intrinsic sources of line broadening.39 Other crystallographic phases are known, such as the rhombohedral ε (Rc) or the hexagonal γ (Pm1) phase, the latter is a structure stable at high temperatures, obtained by heating the beta phase above 750 °C.47,48 The diffraction patterns of L and D-In2S3 NPs are presented in Fig. 3 along with the reference patterns related to the α, β and γ phases. The observed pattern is characterised by broad peaks that are likely related to the small nanocrystal size. The main features include a broad peak centred at around 31° and a second peak at 47°, this pattern is characteristic of small spherical NPs and it is related to the tetragonal β-In2S3 phase or to the cubic α-In2S3 phase. As mentioned above the distinction between the two structures is limited to the disorder of the vacancy along the c axis and in the case of nanosystems, the distinction between these two structures is especially challenging.32,39 The first diffraction can be related to the overlap of the (2 1 3) and (2 2 0) diffraction planes located at 27.39° and 33.18° respectively. Instead, the peak observed at higher angles is related to the (2 2 12) plane at 47.64°. Instead, in the case of the α phase, the first broad peak is related to the (3 1 1) and (4 0 0) diffraction planes located at 27.30° and 33.08° and the third peak is related to the diffraction originated by the (4 4 0) plane located at 47.48°. The high resolution transmission electron microscopy (HR-TEM) micrographs of L and D-In2S3 NPs (Fig. S2†) show electron diffraction fringes with a spacing of 0.32 nm that can be related to the (2 1 3) plane of β-In2S3 or the (3 1 1) plane of α-In2S3. Similar observations have also been reported by other authors for indium sulfide nanoparticles produced using different methods and with different morphologies.36,40,42 The effect of the crystal structure on the chiroptical activity has been investigated on CdSe nanoplatelets by Gao and coworkers.49 They observed large differences in the number and sign of the CD transitions between wurtzite and zinc blende nanoplatelets, along with a significant effect on the intensity of the CD transitions. The authors investigated the system using a nondegenerate coupled-oscillator model and related the effects on the chiroptical activity to the different conformations of cysteine on the surface of the nanoplatelets. In the case of indium sulfide polymorphism, the tetragonal β-In2S3 and the cubic α-In2S3 phases are characterised by a similar atomic disposition, with the difference being limited to the ordering of the vacancy on the tetrahedral site (Fig. S3†). For this reason, we do not expect a significant variation in the chiroptical activity between these two different phases. However, further investigation of the trigonal γ-In2S3 phase, which is characterised by a significantly different structure, may lead to the observation of interesting variations in the chiroptical activity.
 | ||
| Fig. 3 XRD patterns of L-In2S3 (blue) and D-In2S3 (red) NPs. The reference patterns: tetragonal β (I41/amd) ICSD 133317 (blue), cubic α (Fdm) ICSD 133338 (green) and γ (P3m1) ICSD 244280 (pink). | ||
The chemical composition of L-In2S3 NPs was analysed by energy dispersive X-ray spectroscopy (EDS) on a powder sample produced after extensive cleaning of the nanoparticles to reduce the content of cysteine adsorbed on the particle surface. The spectra shown in Fig. S4† confirm an indium-to-sulfur ratio In![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :S of 1:1.5. The presence of a cysteine ligand on the NP surface was confirmed by Fourier-transform infrared (FTIR) spectroscopy (Fig. S5†). The broad bands between 3500 and 3000 cm−1 are related to the νN–H of the amine group and νO–H due to water contamination. The narrow intense band located at 1587 cm−1 is related to the νCO− asymmetric stretch of the carboxylate group and the symmetric stretch is observed at 1390 cm−1.50,51 The narrow bands at a lower energy i.e., 1290 cm−1 are related to the amine deformation modes and the band between 1120 and 1050 cm−1 can be related to νC–N.50,51
:S of 1:1.5. The presence of a cysteine ligand on the NP surface was confirmed by Fourier-transform infrared (FTIR) spectroscopy (Fig. S5†). The broad bands between 3500 and 3000 cm−1 are related to the νN–H of the amine group and νO–H due to water contamination. The narrow intense band located at 1587 cm−1 is related to the νCO− asymmetric stretch of the carboxylate group and the symmetric stretch is observed at 1390 cm−1.50,51 The narrow bands at a lower energy i.e., 1290 cm−1 are related to the amine deformation modes and the band between 1120 and 1050 cm−1 can be related to νC–N.50,51
The UV-Vis spectra of the colloidal solution of NPs (Fig. 4a) show a broad band centred at around 340 nm and a tail that extends in the visible range; similar spectral features were observed for In2S3 hexagonal nanoplates40 and spherical nanoparticles.35 The analysis of (αhν)2vs. energy plot (Fig. S6†) reveals a band gap of 3.60 eV, considering a direct allowed band gap; similar observations have been reported for In2S3 NPs characterised by a similar size.35,36 The value observed for L-In2S3 NPs is larger than the reported data for the bulk β-In2S3 of 2.0–2.2 eV.33,36,39 The blue-shift is attributed to quantum confinement effects considering that the observed NP size is smaller than the excitonic Bohr radius of In2S3which has been estimated to be 33.8 nm.37
 | ||
| Fig. 4 UV-Vis absorption (a) and CD (b) spectra of L-In2S3 (blue) and D-In2S3 colloidal solutions of NPs. | ||
The NP's chirality was investigated by circular dichroism (CD) spectroscopy (Fig. 4b). Both the enantiomers show an intense CD signal in the nanoparticles’ excitonic transition region. The CD signal of L-In2S3 NPs is dominated by a negative peak located at 370 nm and a second negative signal located at around 260 nm, the opposite signal was recorded for D-In2S3 NPs prepared using D-cysteine as a chiral inductor. The CD activity in the nanoparticles’ excitonic transition region confirms the successful transfer of chirality; the chiral inductor L or D cysteine is characterised by CD active transition in the UV range centred at 211 nm (Fig. S7†).52 The g-factor values of −5.11 × 10−4 and 5.66 × 10−4 are calculated for L-In2S3 and D-In2S3 NPs, respectively, considering the transition located at 390 nm. These values are in line with the expected magnitude of the chirality in ligand-induced chirality systems with anisotropic morphology53 and centrosymmetric crystallographic structures.9,54 Interestingly, the observed g-factor values are higher than those of other semiconductor nanoparticles characterised by similar size, shape and origin of chirality, such as cadmium sulfide,28 cadmium selenides21,26,28,31 and silver indium sulfide27 which typically present a g-factor around one order of magnitude lower than L/D-In2S3 NPs. However, assessing the origin of chirality in small NPs is challenging and usually multiple sources of chirality are involved simultaneously.8 Due to the high surface-to-volume ratio observed in the sub 10 nm size regime, a critical role is ascribed to interface chemistry and the effect of the chiral ligand chemisorbed on the particle surface; this is the typical case of the ligand-induced chirality mechanism.20 The NP can be described as a hybrid organic–inorganic system composed of an achiral inorganic core surrounded by an organic chiral shell. This description was employed by Elliott et al.15 to model the origin of chiroptical activity in CdS nanocrystals stabilised by D-penicillamine. In this case, the chiral ligand chemisorbed on the NP's surface can affect the NP's electronic state or introduce some structural distortions on the surface.14,15 The presence of stable chiral defects that could allow a chiral memory effect was investigated via a ligand substitution process as previously done by other authors.55 For this purpose, L and D-In2S3 NPs were transferred from water to cyclohexane using 1-dodecanethiol as an achiral ligand, washed several times and suspended in cyclohexane (see the Ligand exchange and chiral memory section in the ESI† for further details), and then the absence of cysteine in the NPs after ligand exchange was verified via FTIR (Fig. S9a†). The CD analysis after phase transfer (Fig. S9b†) reveals a certain degree of chiroptical activity in the excitonic transition region, even when the NPs are stabilised by achiral 1-dodecanethiol. This may be related to chiral defects that are introduced by cysteine during the synthesis and that are preserved after the ligand exchange process.55,56 However, a reduction of the g-factor is observed after the phase transfer; the g-factor of L-In2S3 NPs decreases from −5.11 × 10−4 to −1.2 × 10−4 which indicates that a significant contribution to the NPs’ chirality is directly related to the ligand binding. However, further investigations are required to quantify the remaining content of cysteine after the phase transfer and a detailed study on the chiral memory of In2S3 NPs is beyond the scope of this report. Finally, to further confirm the origin of chirality, linear dichroism (LD) was recorded. The LD spectra (Fig. S10†) do not show chiroptical activity in the investigated range, proving that the observed CD signal is due to the chirality of indium sulfide NPs without contributions from other sources of anisotropy.
The interaction between the chiral ligand and the surface of NPs was investigated by nuclear magnetic resonance spectroscopy (NMR) in a colloidal solution. The 1H-NMR spectra (Fig. 5a) show a single set of frequencies that can be related to the ligand, similar to what is observed for other colloidal systems.31 Cysteine shows a characteristic rapid chemical exchange between the ligand chemisorbed on the particle surface and the free ligand in solution. Under these conditions, it is not possible to collect individual data from the different ligand forms and the observed signal is an average weighting of the different ligand populations.57–59 The spectra of L-In2S3 NPs in the presence of a ligand concentration of 8.0 mM show a remarkable shift of Hα from 3.60 to 3.81 ppm, and a less pronounced shift can be observed for Hβ from 2.79 ppm to 2.92 ppm merging to form a band at 2.95 ppm. A similar downfield shift of the proton frequencies was observed for In2S3 nanoparticles stabilised by 1-mercaptoglycerol, as reported by Nagesha and coworkers.35 This effect on the chemical shift is related to the interaction with the surface of the NPs; in particular, and the observed deshielding is produced by the electronic density donation from the ligand toward the In3+ cations exposed on the surface of the NPs. In contrast to previous observations on 1-mercaptoglycerol, which reported the strongest shift for the methylenic protons in the vicinal position of the thiolate group,35 in the case of cysteine, the strongest shift is observed for Hα (0.21 ppm or 85 Hz). The Hβ, which are in the proximity of the thiolate group, experience a minor shift (around 0.09 ppm or 36 Hz). This can be explained by the interaction with the particle surface mediated by the combination of amine and carboxylate groups, which promotes multidentate binding and strongly affects the dynamic and local magnetic environments of the Hα. Similar observations were reported in the investigation of the chemisorption of cysteine on silver indium sulfide nanoparticles.31 Moreover, the large broadening and the lack of resolution of the fine structure are related to the contribution of the ligand chemisorbed on the surface of the NPs.57 This effect is associated with multiple sources of line broadening, and among these, the main role is related to the reduction of the spin–spin relaxation time (T2) due to the increase of the rotational correlation time of the ligand molecules that are chemisorbed on the surface of the NPs.35,60 Moreover, the distribution of chemical shifts due to the interaction of the ligand with different sites on the surface of the NP as well as the different packing of the ligand molecules on the particle surface are other sources of line broadening.61–63 Apart from the signals arising from cysteine, small signals related to the minor content of the cysteine disulfide dimer (marked in Fig. 5a with a star) can be seen in the spectra. This contamination is normally present in commercial cysteine and its amount in solution can be largely affected by the catalytic activity of nanoparticles or metal ions toward cysteine oxidation.64,65 The correlated spectroscopy (COSY) analysis (Fig. S11a†) further confirmed the attribution of the spin system, and the HαHβ cross peak was clearly observed, as expected due to the strong scalar coupling between these protons. The analysis of the peaks of lower intensity also allowed the spin system related to the cysteine disulfide dimer to be resolved (Fig. S11b†). Finally, nuclear Overhauser effect spectroscopy (NOESY) was employed to gain further information on the ligand chemisorbed on the particle surface. NOESY spectroscopy was successfully used to identify the contribution of the chemisorbed ligand in the presence of rapid chemical exchange in different colloidal nanosystems.31,66 In particular, this technique is highly sensitive to variations in the tumbling dynamics of small organic molecules which are strongly affected by their interaction with the nanoparticle surface.59 The interaction between small organic molecules, like the common ligands, and the nanoparticle's surface largely affects the dynamics of the ligand in solution promoting the rapid build-up of intense negative peaks.57–59 The NOESY spectra reported in Fig. 5b show an intense negative HαHβ cross peak, indicating that the protons are characterised by a slow tumbling dynamics which confirms the interaction between the chiral ligand and the nanoparticle surface in solution. By contrast, small molecules such as free cysteine are characterised by fast tumbling dynamics and the spectrum (Fig. 5c) shows only the antiphase fine structure characteristic of zero-quantum coherence artefacts that are commonly observed for spins involved in strong scalar coupling.59
 | ||
| Fig. 5 (a) 1H-NMR analysis of L-In2S3 NPs in the presence of 8.0 mM L-cysteine concentration and the reference of L-cysteine; the signals related to the cysteine disulfide dimer are marked with a star. (b) NOESY spectra of L-In2S3 NPs in the presence of 8.0 mM L-cysteine and (c) only 8.0 mM L-cysteine. All the spectra were recorded at 25 °C in D2O and the pH was adjusted to 9.0. | ||
The effect of the chiral ligand structure on the transfer of chirality was investigated by a ligand displacement study (Fig. 6a), using a high concentration of a second chiral ligand to promote the substitution of the cysteine chemisorbed on the surface of the NPs. Before the addition of the second chiral ligand, chiral In2S3 NPs were washed several times to remove the excess cysteine ligand producing partially unpassivated NPs. The most strongly bonded ligand remains attached to the surface of the NPs as can be observed by FTIR analysis (Fig. S12†). L and D-In2S3 NPs after ligand removal show a slightly modified CD signal compared to the NPs in the presence of an excess ligand. In particular, a new negative CD peak appears at around 330 nm as well as a small reduction of the g-factor to −4.8 × 10−4 (Fig. 7). The variation of the CD signal of the NPs with the chiral ligand concentration can be related to the variation of the conformation of the chiral ligand chemisorbed on the surface of the NPs.30,31 Studies on cadmium selenide and silver indium sulfide nanoparticles show a significant dependence of the CD signal on the ligand concentration, affecting the CD signal intensity and shape.30,31 The competition for co-ordination to the particle's surface was investigated by dispersing the L-In2S3 NPs, after ligand removal, in the presence of a high concentration (48 mM) of a second chiral ligand. Under these conditions, the substitution of cysteine with the second chiral ligand is promoted. This post-synthetic approach allows investigating the effect of the chiral inductor's structure and in this case the amino acid lateral chain on the CD signal, minimising other effects such as variations in the particle size or morphology that are commonly observed when changing the chiral inductor during the synthetic step. For this purpose, chiral ligands with similar structural components were chosen, such as lysine (Lys), glutamic acid (Glu), methionine (Met) and penicillamine (Pen). All the investigated ligands were characterised by the same amino and carboxylate backbone structure and stereochemistry with the only differences localised on the lateral chain (Fig. 6b).67 The UV-Vis spectra (Fig. S13†) of the NPs after ageing in the presence of the second chiral ligand show the same absorption spectra in the excitonic transition region, confirming that the process does not affect the size of the NPs. The CD signals of the NPs in the presence of the different ligands are shown in Fig. 7. All the spectra are characterised by two negative peaks located at around 385 and 330 nm and a positive one located at 295 nm. For all the samples, a variation of the CD signal with respect to the nanoparticles after cleaning processes was observed, indicating that the presence of the new chiral inductor affects the interface of the NPs and therefore the particle chirality. In the presence of L-Lys and L-Glu the particle chirality is reduced and the CD signals at 390 nm for L-Lys and L-Glu are −1.81 and −1.40 mdeg respectively. Small variations in the CD activity observed in the presence of L-Lys and L-Glu suggest a low affinity of these amino acids for the particle surface; however, in both cases the presence of the second chiral ligand causes a reduction of the chirality of the NPs. In the case of L-Met the CD signal was more structured and the transition at 390 nm reached −2.12 and the highest chiroptical activity was observed in the case of L-Pen with −2.30 mdeg at 390 nm and a significant increase of the intensity for the other two transitions as well. This evidenced the critical role played by thiol and thioether in the transfer of chirality. Similar observations were made using D-amino acids (D-Lys, D-GLu, D-Met and D-Pen) for the substitution on D-In2S3 NPs. The NPs exposed to a high concentration of anionic and cationic amino acids, such as L-Lys and L-Glu, show a reduction in particle chirality despite the fact that both can potentially coordinate to the particle surface as X-type ligands and the coordination can be promoted by electrostatic interactions. This evidence supports the hypothesis that the large effect observed in the NMR analysis for the Hα chemical shift is mainly associated with the synergistic role of both amine and carboxylate groups that promotes multidentate binding in the amino acid backbone with respect to the intrinsic affinity of the individual group for the surface of the NPs as observed in the case of the lateral chain of L-Lys and L-Glu. Instead, a small increase in the chirality of the NPs was observed in the case of L-Met whose chemisorption is mediated by the coordination of a thioether group as an L-type ligand involving the donation of electronic density from a lone pair in the sulfur atom. The highest performance was observed in the case of L-Pen via thiolate coordination, where a g-factor of −6.26 × 10−4 was reached at 390 nm. This evidence supports the critical role of sulphur atoms as a donor group and in particular, the thiolate group for the coordination to indium sulfide NPs to improve the efficiency of the transfer of chirality. In particular, a strong interaction via the thiolate group can promote tridentate coordination which is considered the most effective binding mode to promote the transfer of chirality.30 Finally, to compare the results of the post-synthetic ligand displacement with the direct synthesis approach, we produced chiral In2S3 NPs using L and D-Pen via a direct synthesis. The CD spectra shown in Fig. S14† confirm the successful chiral induction in Pen stabilised In2S3. Interestingly, L-Pen stabilised In2S3 shows a positive CD signal between 250 and 410 nm with a maximum located at around 270 nm and a shoulder at 340 nm, in contrast to L-cysteine stabilised NPs that show a negative CD signal. This can be related to the conformational variation of the chiral ligand on the surface of the NPs, similar to the inversion of chirality observed in CdS and CdSe stabilised by cysteine or homocysteine and N-acetyl cysteine.28 This would explain the partial degree of inversion for the signal observed via post-synthetic ligand displacement (Fig. 7) which shows a positive signal at 290 nm in the presence of penicillamine. Regarding the other ligands (Lys, Glu and Met), their application in the direct synthesis of chiral In2S3 NPs using a similar protocol was not successful and significant variations to the synthetic procedure are required to prevent the precipitation of indium hydroxide when using these ligands.
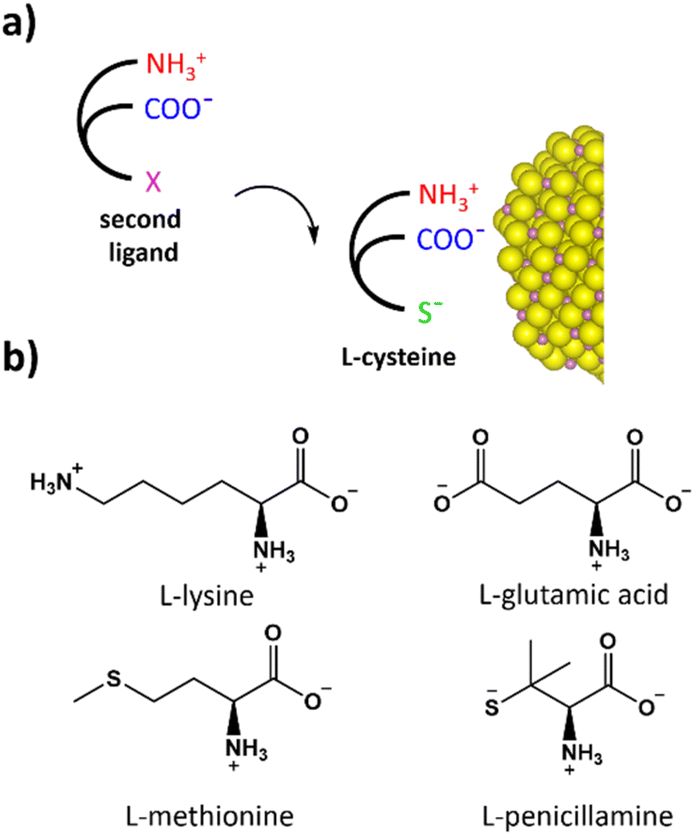 | ||
| Fig. 6 (a) Schematic representation of ligand chemisorption competition. (b) Structure of the L enantiomers of the chiral ligand was investigated considering the prevalent ionisation form at pH = 9.0 according to ref. 55. | ||
 | ||
| Fig. 7 CD spectra of L (solid line) and D (dashed line) In2S3 NPs post ligand removal treatment and in the presence of a second L (solid line) or D (dashed line) chiral ligand: lysine (Lys), glutamic acid (Glu), methionine (Met) and penicillamine (Pen). | ||
Conclusions
The first observation of chirality in chiral indium sulfide NPs has been reported. The chiral nanoparticles were produced via direct synthesis in the presence of cysteine as a chiral inductor. TEM analysis reveals that the NPs produced by this method present a spherical shape and an average size of around 3.6–3.7 nm. The analysis of the NPs’ chiroptical activity shows remarkable particle chirality in the correspondence of the exciton region, confirming the successful transfer of chirality in the indium sulfide matrix. The inversion of the CD signal is observed by changing the stereochemistry of the chiral inductor, and g-factors of −5.11 × 10−4 and 5.66 × 10−4 were observed for L-In2S3 and D-In2S3 NPs respectively. The chiroptical activity, morphology and crystallographic structure of this nanosystem suggest a ligand-induced chirality and further details of the interaction between the chiral inductor and nanoparticle surface were collected by NMR spectroscopy in a colloidal solution. The analysis highlights a characteristic rapid chemical exchange between the ligand chemisorbed on the particle surface and the free ligand in solution. Finally, the effect of the chiral inductor structure was investigated by a ligand displacement study on partially unpassivated nanoparticles after exposure to a relatively high concentration of a second chiral ligand. The analysis evidences the role of different functional groups present in the lateral chain of amino acid-type ligands, and the critical role of thiolate ligands (such as cysteine and penicillamine) was observed to promote interaction with the particle surface. We believe that this study can promote the design of novel chiral inorganic nanostructures based on ligand-induced chirality and the application of these novel nanomaterials in several fields of great technological interest.Materials and methods
Reagents
Indium acetate 99.99%, sodium sulfide anhydrous, L-cysteine 98.5%, sodium hydroxide, hydrochloric acid 37%, D2O 99.9%, sodium deuteroxide 40% solution in D2O 99.5 atom %, L-penicillamine 99%, L-lysine monohydrochloride 99.5%, L-glutamic acid 99% and L-methionine 99.5% were purchased from Sigma Aldrich (Merck). D-Cysteine 97% was purchased from Fluorochem. D-Penicillamine 99%, D-lysine 99%, D-methionine and D-glutamic acid 99% were purchased from Acros Organics (Thermo Fisher Chemicals). All the chemicals were used without further purification.Synthesis of chiral In2S3 NPs
In the general synthesis of L-In2S3 NPs, 0.01 M indium acetate solution (24.0 ml) was diluted with 90.0 ml of Milli-Q water. 1.0 M sodium hydroxide (0.6 ml) and 0.16 M L-cysteine (6.0 ml) were added and the reaction mixture was stirred at room temperature for 15 min. Then, 1.0 M sodium sulfide solution (0.4 ml) was rapidly added and the reaction mixture was stirred at room temperature for one hour. The nanoparticles were precipitated by reducing the pH to around 6 with the addition of a few drops of 1.0 M hydrochloric acid, and collected by centrifugation at 9000 rpm for 5.0 min. The nanoparticles were cleaned three times with 10 ml of Milli-Q water. Then, the nanoparticles were dispersed in 10.0 ml of Milli-Q water in the presence of 48 mM L-cysteine, and the final pH was adjusted to 9.0 with the addition of a few microliters of 1.0 M sodium hydroxide. Mild thermal annealing improves the crystallinity of the nanoparticles, and for this treatment, the colloidal nanoparticle solution was transferred into a three-necked round bottom flask and placed under argon, purging the atmosphere four times. The reaction mixture was heated to 80 °C for one hour under argon. After the treatment, the reaction mixture was cooled to room temperature in a water bath. The nanoparticles were precipitated by reducing the pH to around 6 with the addition of a few drops of 1.0 M hydrochloric acid, and collected by centrifugation at 9000 rpm for 5.0 min. The nanoparticles were cleaned three times with 10 ml of Milli-Q water. Then, the nanoparticles were dispersed in 10.0 ml of Milli-Q water in the presence of 48 mM L-cysteine. The final colloidal solution was stored in a fridge for further use. D-In2S3 NPs were produced following the above procedure, with D-cysteine in the place of L-cysteine. Penicillamine-stabilised In2S3 NPs were produced using L or D-pencillamine instead of L or D-cysteine.Ligand displacement study
L-In2S3 NPs (10.0 ml) were precipitated by the slow addition of 1.0 M hydrochloric acid to a final pH of 6. The nanoparticles were collected by centrifugation at 9000 rpm for 5.0 min. The pellet was washed six times using 10.0 ml of Milli-Q water. After these cleanings, the nanoparticles were dispersed in 10.0 ml of Milli-Q water in the presence of the new ligand (L-lysine, L-glutamic acid, L-methionine or L-penicillamine) at a concentration of 48 mM and left to equilibrate overnight before optical characterization.Characterisation
TEM and HR-TEM micrographs were collected using a Jeol 2100 TEM working with a beam voltage of 200 kV. Energy dispersive X-ray spectroscopy (EDS) was used for the chemical composition analysis of the nanoparticles. The samples for the analysis were prepared by the deposition of a thick layer of powder on carbon tape and analysed using a Zeiss sigma VP field emission scanning electron microscope using a Bruker Quantarax 200 detector working with an ETH voltage of 10 kV. Fourier-transform infrared (FTIR) spectra were collected using a PerkinElmer Spectrum 100 FT-IR spectrometer equipped with an attenuated total reflectance accessory. The circular and linear dichroism spectra of colloidal solutions of the nanoparticles were recorded using a Jasco J815 spectropolarimeter working with a scan speed of 50 nm min−1, a data pitch of 1.0 nm, a DIT of 2.0 s and a bandwidth of 2.0 nm. UV-Vis spectra were collected using an Agilent Cary 60 UV-Vis spectrophotometer. A 3.0 ml quartz cuvette with an optical path length of 1.0 cm was used for both CD and UV-Vis spectroscopy. The g-factor value was calculated according to:28where AL and AR are the absorptions of left and right circular polarized light, respectively, and A is the absorbance of unpolarized light. Nuclear magnetic resonance investigations were performed using a Bruker Avance III 400 MHz spectrometer (proton frequency of 400.23 MHz) equipped with a 5.0 mm BBOF probe.

Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge the Science Foundation Ireland, SFI (project: SFI-20/FFP-A/8904) and the SFI Bioeconomy Research Centre, Biorbic (project: SFI 16/RC/3889) for financial support. The authors would also like to thank the Advanced Microscopy Laboratory (AML), Trinity College, Dublin for the microscopy services.References
- J. Liu, L. Yang, P. Qin, S. Zhang, K. K. L. Yung and Z. Huang, Adv. Mater., 2021, 33, 1–12 Search PubMed.
- F. Wang, X. Yue, Q. Ding, H. Lin, C. Xu and S. Li, Nanoscale, 2023, 15, 2541–2552 RSC.
- S. Jiang and N. A. Kotov, Adv. Mater., 2022, 2108431 Search PubMed.
- C. Hao, X. Wu, M. Sun, H. Zhang, A. Yuan, L. Xu, C. Xu and H. Kuang, J. Am. Chem. Soc., 2019, 141, 19373–19378 CrossRef CAS PubMed.
- T. Yasukawa, A. Suzuki, H. Miyamura, K. Nishino and S. Kobayashi, J. Am. Chem. Soc., 2015, 137, 6616–6623 CrossRef CAS PubMed.
- H. Al-Bustami, S. Khaldi, O. Shoseyov, S. Yochelis, K. Killi, I. Berg, E. Gross, Y. Paltiel and R. Yerushalmi, Nano Lett., 2022, 22, 5022–5028 CrossRef CAS PubMed.
- N. A. Kotov, L. M. Liz-Marzán and P. S. Weiss, ACS Nano, 2021, 15, 12457–12460 CrossRef CAS.
- W. Ma, L. Xu, A. F. De Moura, X. Wu, H. Kuang, C. Xu and N. A. Kotov, Chem. Rev., 2017, 117, 8041–8093 CrossRef CAS PubMed.
- J. Kuno, K. Miyake, S. Katao, T. Kawai and T. Nakashima, Chem. Mater., 2020, 32, 8412–8419 CrossRef CAS.
- U. Hananel, A. Ben-Moshe, H. Diamant and G. Markovich, Proc. Natl. Acad. Sci. U. S. A., 2019, 166, 11159–11164 CrossRef PubMed.
- A. Ben-Moshe, S. G. Wolf, M. B. Sadan, L. Houben, Z. Fan, A. O. Govorov and G. Markovich, Nat. Commun., 2014, 5, 4302 CrossRef CAS PubMed.
- Y. Y. Lee, N. H. Cho, S. W. Im, H. E. Lee, H. Y. Ahn and K. T. Nam, ChemNanoMat, 2020, 6, 362–367 CrossRef CAS.
- H. E. Lee, H. Y. Ahn, J. Mun, Y. Y. Lee, M. Kim, N. H. Cho, K. Chang, W. S. Kim, J. Rho and K. T. Nam, Nature, 2018, 556, 360–364 CrossRef CAS PubMed.
- A. Ben-Moshe, A. Teitelboim, D. Oron and G. Markovich, Nano Lett., 2016, 16, 7467–7473 CrossRef CAS PubMed.
- S. D. Elliott, M. P. Moloney and Y. K. Gun'ko, Nano Lett., 2008, 8, 2452–2457 CrossRef CAS PubMed.
- M. Sujith, E. K. Vishnu, S. Sappati, M. S. Oliyantakath Hassan, V. Vijayan and K. G. Thomas, J. Am. Chem. Soc., 2022, 144, 5074–5086 CrossRef CAS PubMed.
- B. Auguié, J. L. Alonso-Gómez, A. Guerrero-Martínez and L. M. Liz-Marzán, J. Phys. Chem. Lett., 2011, 2, 846–851 CrossRef PubMed.
- K. W. Smith, H. Zhao, H. Zhang, A. Sánchez-Iglesias, M. Grzelczak, Y. Wang, W. S. Chang, P. Nordlander, L. M. Liz-Marzán and S. Link, ACS Nano, 2016, 10, 6180–6188 CrossRef CAS PubMed.
- Á. Coogan, L. Hughes, F. Purcell-Milton, S. Cardiff, V. Nicolosi and Y. K. Gun'Ko, J. Phys. Chem. C, 2022, 126, 18980–18987 CrossRef.
- V. Kuznetsova, Y. Gromova, M. Martinez-Carmona, F. Purcell-Milton, E. Ushakova, S. Cherevkov, V. Maslov and Y. K. Gun'Ko, Nanophotonics, 2020, 10, 797–824 CrossRef.
- U. Tohgha, K. K. Deol, A. G. Porter, S. G. Bartko, J. K. Choi, B. M. Leonard, K. Varga, J. Kubelka, G. Muller and M. Balaz, ACS Nano, 2013, 7, 11094–11102 CrossRef CAS PubMed.
- A. Ben Moshe, D. Szwarcman and G. Markovich, ACS Nano, 2011, 5, 9034–9043 CrossRef CAS PubMed.
- X. Gao, X. Zhang, K. Deng, B. Han, L. Zhao, M. Wu, L. Shi, J. Lv and Z. Tang, J. Am. Chem. Soc., 2017, 139, 8734–8739 CrossRef CAS PubMed.
- J. E. Govan, E. Jan, A. Querejeta, N. A. Kotov and Y. K. Gun'ko, Chem. Commun., 2010, 46, 6072–6074 RSC.
- J. Hao, Y. Li, J. Miao, R. Liu, J. Li, H. Liu, Q. Wang, H. Liu, M. H. Delville, T. He, K. Wang, X. Zhu and J. Cheng, ACS Nano, 2020, 14, 10346–10358 CrossRef CAS PubMed.
- F. Purcell-Milton, A. K. Visheratina, V. A. Kuznetsova, A. Ryan, A. O. Orlova and Y. K. Gun'Ko, ACS Nano, 2017, 11, 9207–9214 CrossRef CAS PubMed.
- K. Varga, S. Tannir, B. E. Haynie, B. M. Leonard, S. V. Dzyuba, J. Kubelka and M. Balaz, ACS Nano, 2017, 11, 9846–9853 CrossRef CAS PubMed.
- J. K. Choi, B. E. Haynie, U. Tohgha, L. Pap, K. W. Elliott, B. M. Leonard, S. V. Dzyuba, K. Varga, J. Kubelka and M. Balaz, ACS Nano, 2016, 10, 3809–3815 CrossRef CAS PubMed.
- L. Branzi, A. Kavanagh, M. Back, A. Speghini, Y. K. Gun'ko and A. Benedetti, Chem. Commun., 2023, 6024–6027 RSC.
- V. A. Kuznetsova, E. Mates-Torres, N. Prochukhan, M. Marcastel, F. Purcell-Milton, J. O'Brien, A. K. Visheratina, M. Martinez-Carmona, Y. Gromova, M. Garcia-Melchor and Y. K. Gun'Ko, ACS Nano, 2019, 13, 13560–13572 CrossRef CAS PubMed.
- L. Branzi, F. Purcell-Milton, C. Cressoni, M. Back, E. Cattaruzza, A. Speghini, Y. K. Gun'ko and A. Benedetti, Nanoscale, 2022, 14, 12174–12182 RSC.
- Y. Tian, L. Wang, H. Tang and W. Zhou, J. Mater. Chem. A, 2015, 3, 11294–11301 RSC.
- K. Hara, K. Sayama and H. Arakawa, Sol. Energy Mater. Sol. Cells, 2000, 62, 441–447 CrossRef CAS.
- J. Tang, G. Konstantatos, S. Hinds, S. Myrskog, A. G. Pattantyus-Abraham, J. Clifford and E. H. Sargent, ACS Nano, 2009, 3, 331–338 CrossRef CAS PubMed.
- D. K. Nagesha, X. Liang, A. A. Mamedov, G. Gainer, M. A. Eastman, M. Giersig, J. J. Song, T. Ni and N. A. Kotov, J. Phys. Chem. B, 2001, 105, 7490–7498 CrossRef CAS.
- R. Li, L. Tang, Q. Zhao, T. H. Ly, K. S. Teng, Y. Li, Y. Hu, C. Shu and S. P. Lau, Nanoscale Res. Lett., 2019, 14, 161 CrossRef PubMed.
- W. Chen, J.-O. Bovin, A. G. Joly, S. Wang, F. Su and G. Li, J. Phys. Chem. B, 2004, 108, 11927–11934 CrossRef CAS.
- M. A. Franzman and R. L. Brutchey, Chem. Mater., 2009, 21, 1790–1792 CrossRef CAS.
- L. Guillemeney, L. Lermusiaux, P. Davidson, A. Hubley, S. Pierini, D. Pierucci, G. Patriarche, B. Canut, E. Lhuillier, B. Mahler and B. Abécassis, Chem. Mater., 2022, 34, 9270–9281 CrossRef CAS.
- K. H. Park, K. Jang and S. U. Son, Angew. Chem., Int. Ed., 2006, 45, 4608–4612 CrossRef CAS PubMed.
- R. Wu, Y. Xu, R. Xu, Y. Huang and B. Zhang, J. Mater. Chem. A, 2015, 3, 1930–1934 RSC.
- X. Wang, P. P. Wang, Y. Yang and J. Zhuang, J. Am. Chem. Soc., 2013, 135, 6834–6837 CrossRef PubMed.
- B. Ni, H. Liu, P. P. Wang, J. He and X. Wang, Nat. Commun., 2015, 6, 1–8 Search PubMed.
- N. Barreau, Sol. Energy, 2009, 83, 363–371 CrossRef CAS.
- C. J. M. Rooymans, J. Inorg. Nucl. Chem., 1959, 11, 78–79 CrossRef CAS.
- G. A. Steigmann, H. H. Sutherland and J. Goodyear, Acta Crystallogr., 1965, 19, 967–971 CrossRef CAS.
- K. J. Range and M. Zabel, Z. Naturforsch., B: Anorg. Chem., Org. Chem., 1978, 33, 463–464 CrossRef.
- K. D. Kundra and S. Z. Ali, Phys. Status Solidi, 1976, 36, 517 CrossRef CAS.
- X. Gao, X. Zhang, L. Zhao, P. Huang, B. Han, J. Lv, X. Qiu, S. H. Wei and Z. Tang, Nano Lett., 2018, 18, 6665–6671 CrossRef CAS PubMed.
- G. Socrates, Infrared and Raman Characteristic Group Frequencies, John Wiley & Sons, Chichester, 3rd edn, 2001 Search PubMed.
- A. Pawlukojć, J. Leciejewicz, A. J. Ramirez-Cuesta and J. Nowicka-Scheibe, Spectrochim. Acta, Part A, 2005, 61, 2474–2481 CrossRef PubMed.
- N. Amdursky and M. M. Stevens, ChemPhysChem, 2015, 16, 2768–2774 CrossRef CAS PubMed.
- X. Gao, X. Zhang, K. Deng, B. Han, L. Zhao, M. Wu, L. Shi, J. Lv and Z. Tang, J. Am. Chem. Soc., 2017, 139, 8734–8739 CrossRef CAS PubMed.
- A. Ben-Moshe, A. O. Govorov and G. Markovich, Angew. Chem., Int. Ed., 2013, 52, 1275–1279 CrossRef CAS PubMed.
- T. Nakashima, Y. Kobayashi and T. Kawai, J. Am. Chem. Soc., 2009, 131, 10342–10343 CrossRef CAS PubMed.
- Y. Zhou, M. Yang, K. Sun, Z. Tang and N. A. Kotov, J. Am. Chem. Soc., 2010, 132, 6006–6013 CrossRef CAS PubMed.
- Z. Hens and J. C. Martins, Chem. Mater., 2013, 25, 1211–1221 CrossRef CAS.
- A. Hassinen, I. Moreels, C. De Mello Donegá, J. C. Martins and Z. Hens, J. Phys. Chem. Lett., 2010, 1, 2577–2581 CrossRef CAS.
- B. Fritzinger, I. Moreels, P. Lommens, R. Koole, Z. Hens and J. C. Martins, J. Am. Chem. Soc., 2009, 131, 3024–3032 CrossRef CAS PubMed.
- R. Grisorio, D. Quarta, A. Fiore, L. Carbone, G. P. Suranna and C. Giansante, Nanoscale Adv., 2019, 1, 3639–3646 RSC.
- X. Ji, D. Copenhaver, C. Sichmeller and X. Peng, J. Am. Chem. Soc., 2008, 130, 5726–5735 CrossRef CAS PubMed.
- M. Malicki, J. M. Hales, M. Rumi, S. Barlow, L. McClary, S. R. Marder and J. W. Perry, Phys. Chem. Chem. Phys., 2010, 12, 6267–6277 RSC.
- M. J. Hostetler, J. E. Wingate, C.-J. Zhong, J. E. Harris, R. W. Vachet, M. R. Clark, J. David Londono, S. J. Green, J. J. Stokes, G. D. Wignall, G. L. Glish, M. D. Porter, N. D. Evans and R. W. Murray, Langmuir, 1997, 14, 17–30 CrossRef.
- T. Ruks, C. Beuck, T. Schaller, F. Niemeyer, M. Zähres, K. Loza, M. Heggen, U. Hagemann, C. Mayer, P. Bayer and M. Epple, Langmuir, 2019, 35, 767–778 CrossRef CAS PubMed.
- X. Chen, N. C. Berner, C. Backes, G. S. Duesberg and A. R. McDonald, Angew. Chem., Int. Ed., 2016, 55, 5803–5808 CrossRef CAS PubMed.
- B. Fritzinger, I. Moreels, P. Lommens, R. Koole, Z. Hens and J. C. Martins, J. Am. Chem. Soc., 2009, 131, 3024–3032 CrossRef CAS PubMed.
- W. M. Haynes, D. R. Lide and T. J. Bruno, CRC Handbook of Chemistry and Physics, 2014 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3nr04320g |
| This journal is © The Royal Society of Chemistry 2023 |