
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Small extracellular vesicles administered directly in the brain promote neuroprotection and decreased microglia reactivity in a stroke mouse model†
Miguel M.
Lino
a,
Tiago
Rondão
 a,
Arnab
Banerjee
a,
Inês
Aires
a,
Magda
Rodrigues
a,
Tiago
Reis
a,
António
Santinha
a,
Dominique
Fernandes
a,
Débora
Serrenho
ab,
Tomás
Sobrino
c,
João
Sargento-Freitas
d,
Frederico C.
Pereira
def,
Ana Luísa
Carvalho
ag and
Lino
Ferreira
*ad
a,
Arnab
Banerjee
a,
Inês
Aires
a,
Magda
Rodrigues
a,
Tiago
Reis
a,
António
Santinha
a,
Dominique
Fernandes
a,
Débora
Serrenho
ab,
Tomás
Sobrino
c,
João
Sargento-Freitas
d,
Frederico C.
Pereira
def,
Ana Luísa
Carvalho
ag and
Lino
Ferreira
*ad
aCNC – Centre for Neuroscience and Cell Biology, CIBB – Centre for Innovative Biomedicine and Biotechnology, Coimbra, Portugal. E-mail: lino.ferreira@uc.pt
bPhD Programme in Experimental Biology and Biomedicine, Institute for Interdisciplinary Research (IIIUC), University of Coimbra, Portugal
cNeuroAging Laboratory (NEURAL), Clinical Neurosciences Research Laboratory (LINC), Health Research Institute of Santiago de Compostela (IDIS), Santiago de Compostela, Spain
dFaculty of Medicine, University of Coimbra, Coimbra, Portugal
eiCBR, Coimbra Institute for Clinical and Biomedical Research, CIBB – Centre for Innovative Biomedicine and Biotechnology, Coimbra, Portugal
fInstitute of Pharmacology and Experimental Therapeutics, Faculty of Medicine, University of Coimbra, Portugal
gDepartment of Life Sciences, University of Coimbra, Coimbra, Portugal
First published on 7th November 2023
Abstract
Herein, we investigate the bioactivity of small extracellular vesicles (sEVs), focusing on their local effect in the brain. sEVs from mononuclear cells (MNCs) showed superior effects in vitro to sEVs from mesenchymal stem cells (MSCs) and were able to promote neuroprotection and decrease microglia reactivity in a stroke mouse model.
Stroke is the second leading cause of both disability and death worldwide.1 Current standard-of-care procedures for the treatment of ischemic stroke are intravenous injection of recombinant tissue plasminogen activator (rtPA) to dissolve blood clots or thrombectomy, an intervention that includes the administration of a catheter through the artery to remove the clot by aspiration or fragmentation.2,3 Unfortunately, in many cases, patients who survive a stroke event have limited functional recovery due to a limited remodelling and restorative process in the lesion area. Neuroprotective strategies targeting the cascade of cellular and molecular events that leads to ischemic damage, and strategies to promote post-ischemic regeneration, have been pursued in the last few years, although with little or no efficacy in clinical trials.4 In this regard, most of the pharmacological interventions (e.g. free-radical trapping agents; magnesium; NA-1) target a single molecular target.4 Since post-ischemic brain damage involves multiple molecular events, one of the factors that might be contributing to the failure of neuroprotection strategies is related to the use of therapeutics that have a single target instead of drugs targeting multiple pathways.5
In the past 20 years, therapeutic interventions based on cell therapies have been pursued, including neural stem cells,6 bone marrow MNCs,7 and MSCs.8 Mostly in preclinical models, these therapies have shown positive effects by reducing the infarct size, promoting functional recovery and modulating the immune response.
Recently, the evidence that part of the effects observed in cell therapies are due to paracrine effects has led to increased interest in sEVs as a therapeutic strategy for ischemic stroke.9 sEVs are lipidic vesicles released by cells with sizes between 50 and 200 nm, carrying a cocktail of bioactive molecules (microRNAs, mRNAs, proteins, and lipids) that can target multiple pathways at the same time. Functional benefits of sEVs secreted by MSCs10–14 and neural stem cells (NSCs)15–17 have been observed after systemic administration in mice,10,15 rats11,12,14,18 and pigs,16 with cerebral ischemia induced by the occlusion of the middle cerebral artery (MCAO). sEVs have been reported to reduce the infarct volume, improve angiogenesis and neovascularization,10,11 reduce astrocyte activation12,17 and modulate peripheral immune responses.17,19
Although recent evidence shows that sEVs have the ability to cross the blood–brain barrier,20 brain accumulation of sEVs administered systemically is relatively low (in some cases, below 1% of the initial dose injected21). This suggests that most of the effects that have been observed in ischemic stroke models might be systemic and not local effects. Indeed, sEVs from different sources have a therapeutic effect by altering the systemic immune response.17 In line with this, it has recently been demonstrated that MSC-derived sEVs have an immunomodulatory activity, reducing leukocyte infiltration and its deleterious effect in the brain.19 These reports suggest that the peripheral immunomodulatory effect could contribute indirectly to brain regeneration. Nevertheless, an exacerbation of the stroke-induced immunosuppression might be detrimental.22 In addition, it is unclear whether a systemic effect would be enough to provide long-term neuroprotection and induce brain regeneration. The therapeutic effect of sEVs administered locally in the brain, by intracerebral administration, remains elusive. Although the direct administration of sEVs by stereotaxic injection into the brain parenchyma has been reported,14 the interaction of sEVs with different brain cells and the consequent effects have not been investigated yet.
Herein, we investigate in vitro and in vivo the therapeutic activity of sEVs, focusing on their local effect in the brain parenchyma, specifically their role in neuroprotection and the local immune response. We started by evaluating the prosurvival effect of sEVs secreted by MSCs isolated from the Wharton Jelly or by human umbilical cord blood MNCs (hUC-MNCs) in endothelial cells, microglia and neurons as well as their effect in the modulation of microglia polarization. We further investigated the interaction of sEVs with different cell types after stereotaxic injection in the brain and assessed the degree of neuroprotection, angiogenesis and modulation of microglia polarization 3 days after treatment with sEVs. sEVs isolated from cell sources available in cell banks (i.e. MNCs and MSCs) have been used in the current study (Scheme 1). Due to their immunomodulatory properties, MSCs have been one of the sources usually used in preclinical models of ischemic stroke.11,18 The regenerative properties of sEVs secreted by hUCB-MNCs have been reported by us,23 but their bioactivity in the context of stroke has not been explored yet. Nonetheless, it has been shown that hUCB-MNCs have neuroprotective activity and improve motor function in animal models of ischemic stroke,24,25 and thus are a relevant sEV source for clinical applications in this context. In order to minimize donor-related heterogeneity, cells from different donors have been used in this study (WJ-MSCs from 3 different donors and HUCB-MNCs from 6 donors). HUCB-MNCs expressed the hematopoietic markers CD45 and HLA-DR while WJ-MSCs expressed CD73, CD90 and CD105 and were negative for CD45 and HLA-DR (ESI Fig. 1†). sEVs were isolated by differential centrifugation, purified by size exclusion chromatography and characterized by nanoparticle tracking analysis (NTA), transmission electron microscopy (TEM) and flow cytometry (ESI Fig. 2†). NTA analyses showed that the majority of sEVs had a size in the range of 100–200 nm. TEM analyses revealed the presence of cup-shaped structures, typical of sEVs. As for the purity of our samples, MNC-sEVs and MSC-sEVs showed averages of 2.29 × 109 parts per μg and 3.30 × 109 parts per μg of protein, respectively. To ensure that our preparations were enriched in sEVs, we performed western blot and flow cytometry analyses to detect common sEV markers (ESI Fig. 2†). Our results showed that sEVs derived from both cell sources expressed the surface markers CD9 and CD63 and cytosolic marker GAPDH, while they were negative for calnexin, an endoplasmic reticulum marker present in cells but not in sEVs. Overall, our results showed that our samples were enriched in sEVs.
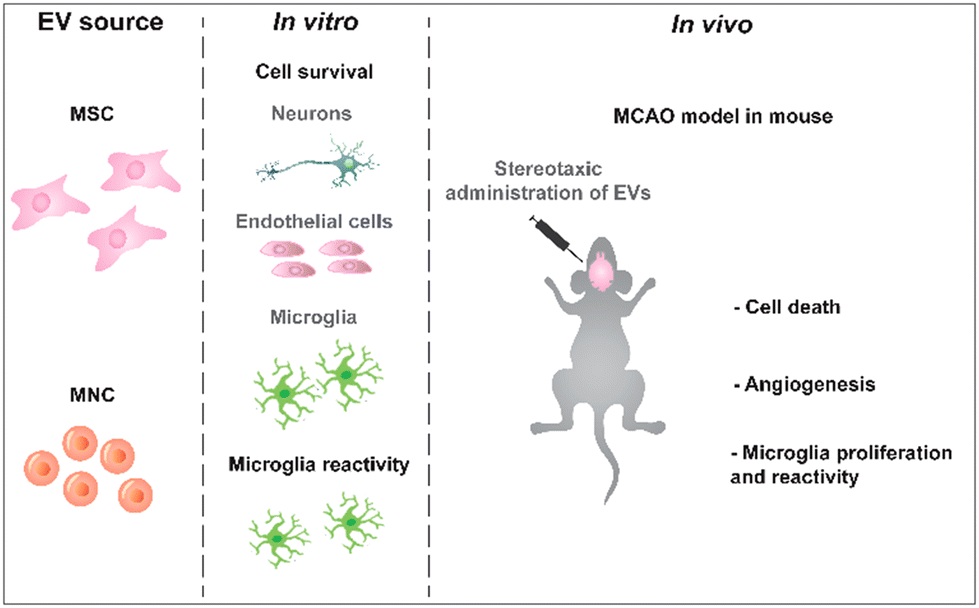 | ||
| Scheme 1 Experimental design. sEVs were isolated from MSCs collected from human Wharton jelly and mononuclear cells from umbilical cord blood. The bioactivity of these sEVs was tested in vitro in different brain cells and then in vivo in a MCAO mouse model. | ||
Initially, the bioactivity of sEVs in brain cells (endothelial cells, cortical neurons and microglia) was evaluated (Scheme 1). Cells were first characterized by immunofluorescence for endothelial, microglial and neuronal markers (ESI Fig. 3†). Endothelial cells and cortical neurons were incubated for 2 h under glucose and oxygen deprivation (OGD) to mimic a stroke situation and then incubated with different concentrations of sEVs for 24 h or 48 h, after which the number of live cells was quantified by high-content microscopy (Fig. 1A1). To test whether cell death was induced by nutrient deprivation, positive control cells were incubated with glucose-containing medium under normoxic conditions (sham) during the 2 h. Both sEVs protected endothelial cells and microglia (Fig. 1A2 and A4) from death, with enhanced bioactivity directly related to the concentration of sEVs in the case of endothelial cells; however, only sEVs from MNCs protected neurons from OGD-induced cell death (Fig. 1A3). Next, we evaluated whether sEVs had an effect in suppressing microglia activation, which is exacerbated after acute stroke.26 For this purpose, TSPO, a protein highly expressed by M1 activated microglia in the ischemic brain,27 was assessed by immunofluorescence (Fig. 1B1). After 6 h of OGD, the expression of TSPO increased (by almost 40% relative to cells kept under normoxia). MNC-sEVs (at 3 × 109 particles per mL and 4.5 × 109 particles per mL), but not MSC-sEVs, were successful in decreasing the expression of TSPO to levels comparable to those of the control (Fig. 1B2 and B3). In line with these results, MNC-sEVs increased the expression of CD206, a typical M2 microglia marker28 (ESI Fig. 4†). This means that these sEVs can modulate microglia polarization. Indeed, these results agree with previous studies showing that MNCs (for sEVs was not demonstrated) may modulate microglia reactivity both in vitro29 and in vivo.30
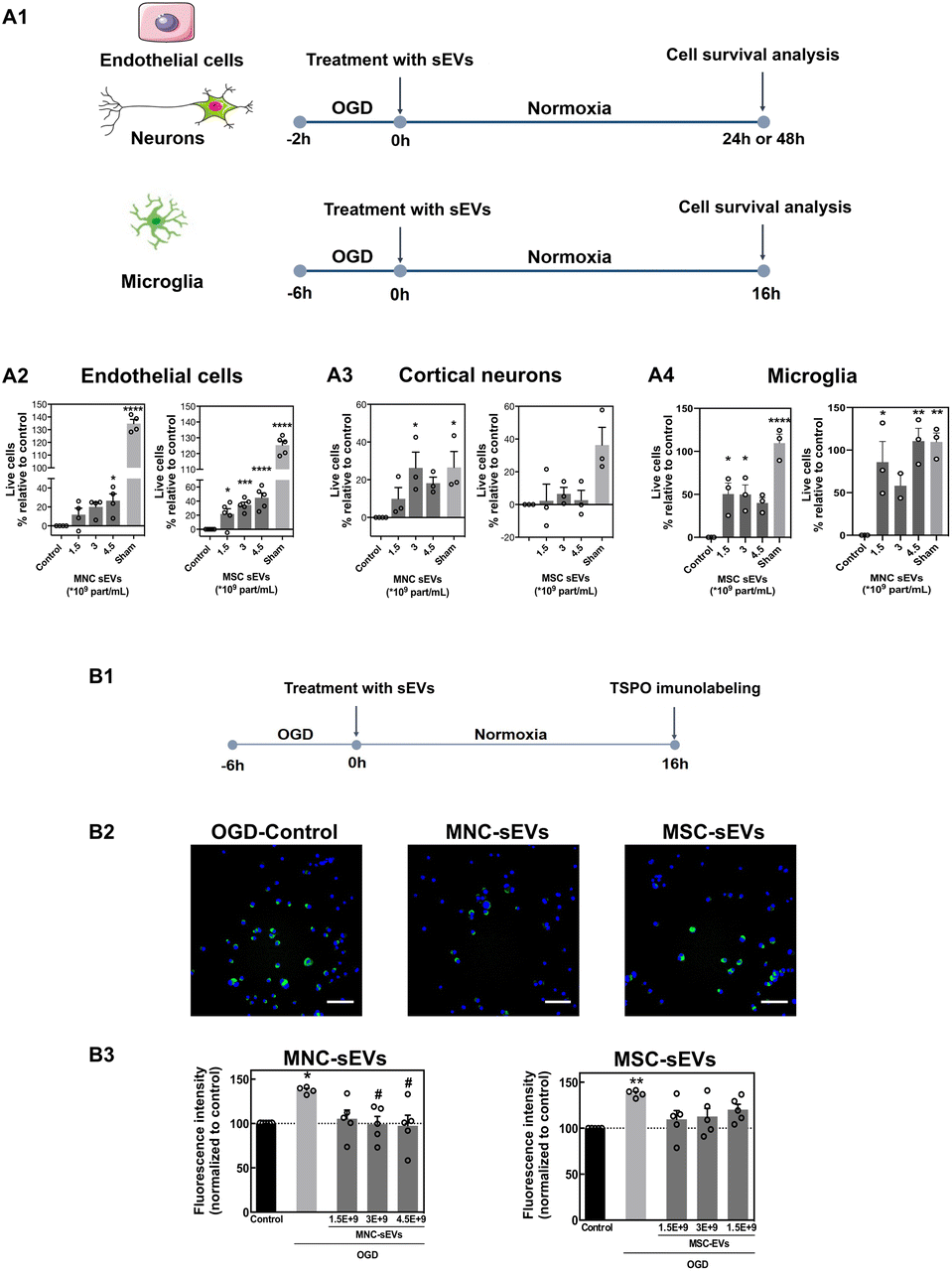 | ||
| Fig. 1 sEV bioactivity in endothelial cells, cortical neurons and microglia. (A1) HUVEC, cortical neurons and microglia were cultured under oxygen and glucose deprivation and immediately treated with different concentrations of MNC-sEVs or MSC-sEVs. (A2 and A3) The cell number was quantified at 48 h (endothelial cells), at 24 h (neurons) or at 16 h (microglia) by DAPI staining using high-content microscopy. Control refers to cells cultured under OGD without any treatment. The results are expressed as the percentage of live cells over the control. Results are expressed as mean ± SEM (n = 3–5 independent experiments, 3 technical replicates per experiment), **, ***, and **** denote statistical significance (p < 0.01; p < 0.001; and p < 0.0001) assessed by one-way ANOVA followed by Tukey's post-hoc test. (B1) Microglia reactivity evaluated by the expression of translocator protein (TSPO). Microglia cells were cultured under hypoxia for 6 h and then treated with different concentrations of MNC-EVs or MSC-EVs under normoxic conditions. (B2) The expression of TSPO in microglia cells as monitored by immunofluorescence. Scale bar corresponds to 50 μm. (B3) Quantification of TSPO levels in microglia cells treated or not with sEVs. Cells were stained for TSPO and images were acquired using a high-content microscope for quantification of fluorescence. Results are expressed as mean ± SEM (n = 5 independent experiments, 2 technical replicates, 15 image fields per replicate). * and ** denote statistical significance (p < 0.05 and p < 0.01) in relation to the control group (normoxia) assessed by one-way ANOVA. # denotes statistical significance (p < 0.05) in relation to control (OGD) assessed by one-way ANOVA. | ||
Because the bioactivity of sEVs from different cell sources may be related to differences in their cellular internalization, we studied the sEV uptake kinetics by high-content fluorescence microscopy (ESI Fig. 5†). In endothelial cells and cortical neurons, both fluorescently-labelled sEVs followed the same uptake kinetics whereas in microglia MNC-sEVs were preferentially internalized as compared to MSC-sEVs. To confirm the internalization of sEVs, we evaluated by confocal microscopy the colocalization of endolysosomes with sEVs in endothelial cells and microglia (ESI Fig. 6A†). In microglia cells, the colocalization of endolysosomes with MNC-sEVs is higher than the colocalization with MSC-sEVs (ESI Fig. 6B†), which is consistent with a higher number of cells internalizing MNC-sEVs in the uptake kinetics experiment (ESI Fig. 5†). It is possible that the presence of specific proteins on the MNC-sEV surface may mediate their highest internalization by microglia cells. We focused our attention on the glycoprotein HLA-DR, which is present on MNC-sEVs but not on MSC-sEVs (ESI Fig. 2†). Since HLA-DR is a molecule associated with antigen presentation via specific interaction with CD4 receptor, we evaluated by flow cytometry the presence of CD4 in microglia cells (ESI Fig. 7†), being expressed in the entire cell population. To further validate if MNC-sEVs uptake is governed by HLA-DR and CD4 interaction, MNC-EVs were pre-incubated with an antibody against HLA-DR in order to block the binding to CD4 present on the cell surface. This pre-incubation was able to block specifically the uptake of MNC-sEVs but not MSC-sEVs (ESI Fig. 7†). Nevertheless, the blockage was not fully effective in inhibiting MNC-sEV internalization, indicating that the uptake of these sEVs may be mediated by other molecules besides HLA-DR. Overall, our results indicated that MNC-sEVs have higher bioactivity in cortical neurons and microglia than MSC-sEVs and thus they were subsequently selected for in vivo tests. To study the effect of MNC-sEVs in the brain, without having the contribution of immunomodulatory systemic effects, sEVs were locally delivered by stereotaxic injection right after MCAO using a transient intraluminal filament (Scheme 1). The MCAO mouse model is one of the models that most closely simulates ischemic stroke, exhibiting a penumbra region similar to what happens in human ischemic stroke.31 To select the sEV dose for this procedure, we first quantified the accumulation of radiolabeled sEVs (2.5 × 1010) in the brain 1 h after intravenous administration in the tail vein (ESI Fig. 8†). The accumulation of sEVs was below 1% of the injected dose. Thus, for stereotaxic administration, the amount of injected sEVs was 4.5 × 109 particles (equivalent to approximately 2 μg in terms of the protein content). This value represents approximately 1% of the amount delivered by intravenous and intra-arterial injection in previous studies.11,12,18 We focused first on the study of the interaction with different cell types 6 h after injection and then on the evaluation of the neuroprotective and immunomodulatory effects of sEVs at 3 days after administration. To track sEVs in the brain by confocal microscopy, sEVs were labelled with a fluorescent dye (DiD). Mice with brain ischemic lesions assessed by magnetic resonance imaging (MRI) and impaired neurological and locomotor function evaluated using the Clark's score32 (ESI Fig. 9†) received sEVs by stereotaxic administration in the penumbra region (Fig. 2A and B). At 6 h post administration, sEVs were preferentially internalized by microglial cells (33% of sEVs per field colocalized with microglia), followed by neurons and endothelial cells (approximately 25% of sEVs) and finally astrocytes (16.8% of sEVs) (Fig. 2C and D). The number of sEVs in the contralateral hemisphere was negligible.
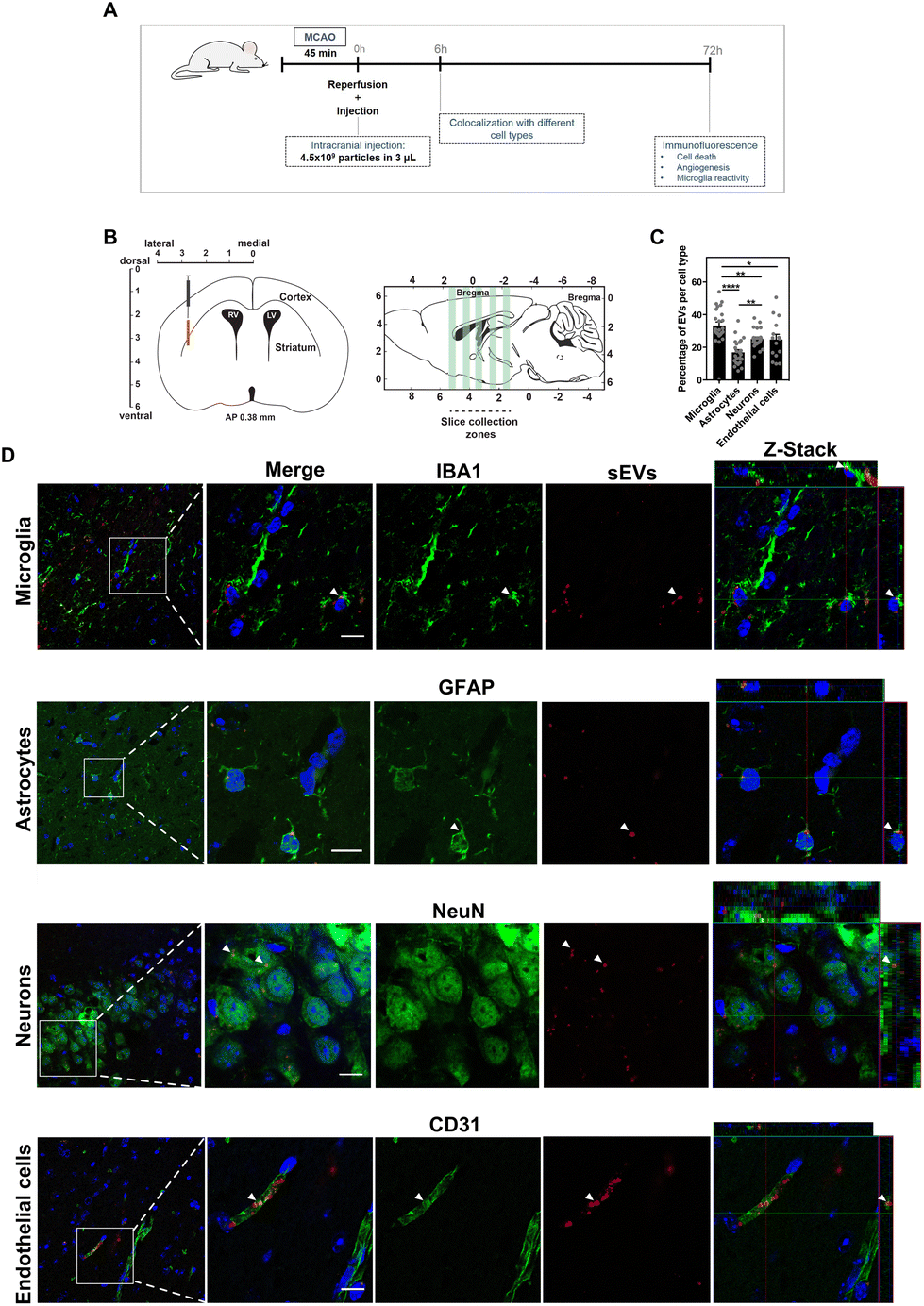 | ||
| Fig. 2 Uptake of sEVs by brain cells. (A) Experimental setup. Mice were subjected to MCAO for 45 min, after which sEVs were administered by stereotaxic injection in the penumbra region. A subset of animals was injected with fluorescent sEVs and sacrificed at 6 h after injection for biodistribution studies and the other subset was sacrificed 72 h after injection for the evaluation of neuroprotection, angiogenesis and microglia activation by immunofluorescence. (B) For colocalization studies, different regions of the brain were sliced and stained for IBA-1 (microglia), GFAP (astrocytes), NeuN (neurons) and CD31 (endothelial cells). Cryosections with 50 μm in the coronal plane were obtained from the 5 different regions of the brain ranging from anteroposterior (AP) +1.54 to AP −2.18. (C) The percentage of colocalization of sEVs with different cell types. Results are expressed as mean ± SEM (n = 5 animals, 3–5 sections per brain were analysed). (D) Representative fluorescence images showing colocalization with microglia, astrocytes, neurons and endothelial cells. White arrows indicate cells colocalizing with sEVs. Images were acquired using a high-content microscope and analysed using the INCell analyser developer toolbox. Scale bars correspond to 50 μm. | ||
To evaluate neuroprotection caused by sEVs, we measured cell apoptosis (TUNEL staining) and endothelial cell proliferation (CD31/Ki67 staining), 3 days after treatment. Analysis of TUNEL staining revealed that treatment with MNC-sEVs decreased cell apoptosis in the ipsilateral hemisphere compared to the control group (Fig. 3A1 and A2). Endothelial cell proliferation was quantified by staining brain slices for CD31 (endothelial cell marker) and Ki67 (cell proliferation marker). Compared to the control group, MNC-sEVs were able to increase significantly (2.4-fold) the proliferation of endothelial cells in the ipsilateral hemisphere relative to the contralateral hemisphere (Fig. 3B1 and B2). These results are in agreement with the pro-angiogenic effect of these sEVs previously reported by us in the context of wound healing.23 Indeed, angiogenesis has been considered a potential therapeutic target to improve the outcome after stroke, namely through a coupling between angiogenesis and neuronal remodelling.33 To evaluate the immunomodulatory properties of sEVs, the number of microglia cells and their polarization were quantified by immunofluorescence. No significant differences were observed between the control and sEV groups regarding microglia proliferation (Fig. 3C1 and C2); however, in agreement with the in vitro studies, microglia activation was significantly attenuated in the ipsilateral hemisphere in the group treated with MNC-sEVs (Fig. 3D1 and D2).
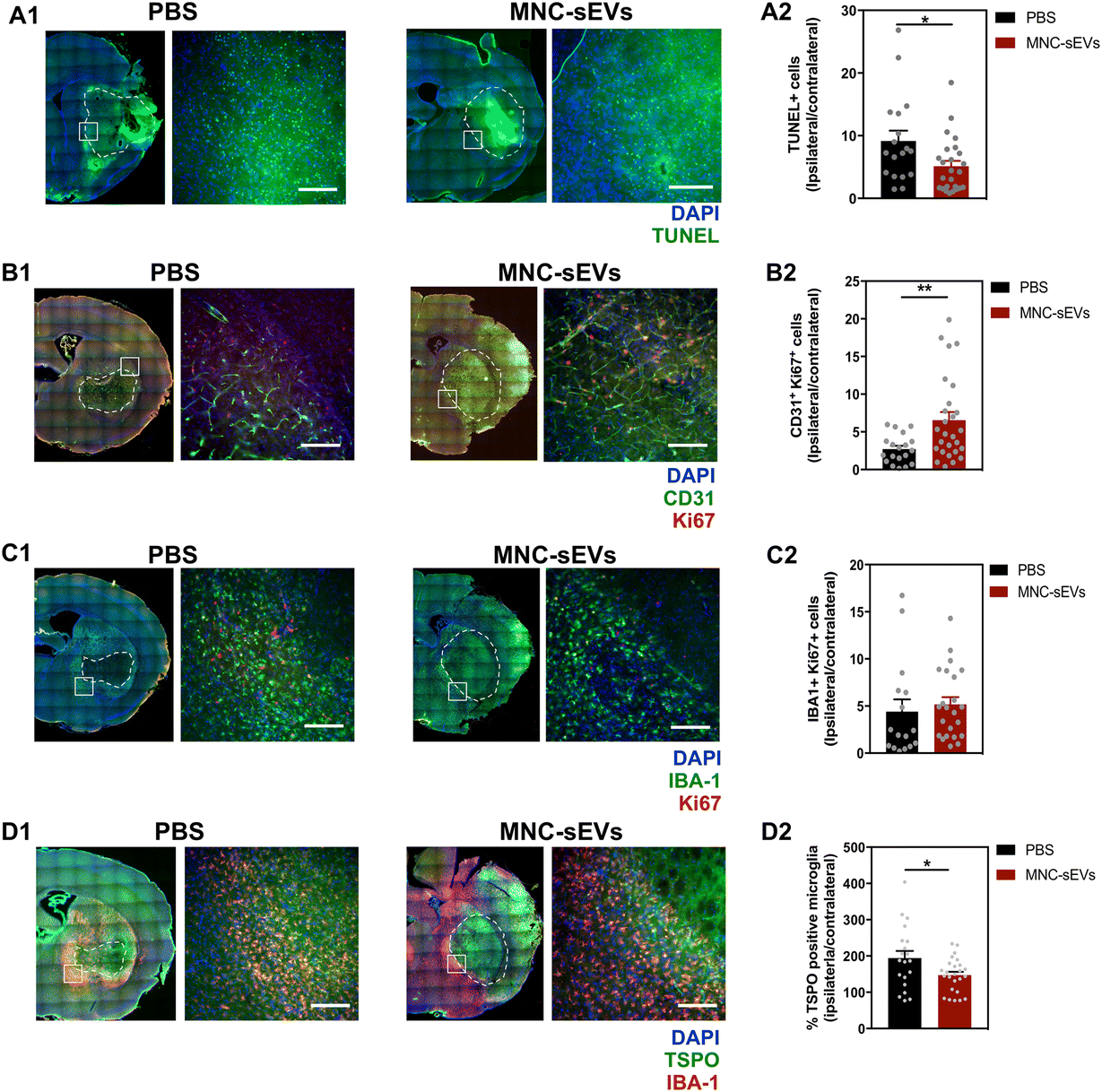 | ||
| Fig. 3 MNC-sEVs decrease cell death and induce angiogenesis at 3 days post-administration. (A) TUNEL staining images and (A2) quantification of TUNEL positive cells (ratio between the ipsilateral and the contralateral hemisphere). Results are expressed as mean ± SEM (n = 18–25 brain sections; PBS: 4 animals, MNC-sEVs: 5 animals). Scale bar corresponds to 150 μm. (B1) Representative fluorescence images of the ipsilateral hemisphere stained for CD31 and Ki67. Scale bar corresponds to 150 μm. (B2) Fold change of double-stained cells in the ipsilateral hemisphere relative to the contralateral hemisphere. Results are expressed as mean ± SEM (n = 19–26 brain sections, PBS: 4 animals, MNC-sEVs: 6 animals). (C1) Representative fluorescence images of the ipsilateral hemisphere stained for IBA-1 and Ki67. Scale bar corresponds to 150 μm. (C2) Fold change of double-stained cells in the ipsilateral hemisphere relative to the contralateral hemisphere. Results are expressed as mean ± SEM (n = 20–26 brain sections, PBS: 4 animals, MNC-sEVs: 6 animals). (D1) Representative fluorescence images of the ipsilateral hemisphere stained for IBA-1 and TSPO. (D2) Fold change of double-stained cells in the ipsilateral hemisphere relative to the contralateral hemisphere. Results are expressed as mean ± SEM (n = 20–24 brain sections, PBS: 4 animals, MNC-sEVs: 6 animals). Scale bar corresponds to 150 μm. The ischemic lesion is delimited with a dashed white line. White squares in A2, B2, C2 and D2, results are expressed as mean ± SEM (n = 4–6 animals, 5 sections per animal). * and ** denote statistical significance (p < 0.05 and p < 0.01) assessed by the unpaired t-test. | ||
Previous studies have shown that MSC-sEVs or lipopolysaccharide-stimulated macrophages sEVs administered by intracardiac (hypoxia-ischemic injury in neonatal mice)34 or intravenous (in a MCAO rat model)35 administration, respectively, were able to decrease the number of Iba-1+ cells34 and polarize the cells to an M2 phenotype.34,35 However, it was unclear whether the microglia polarization effect was due to the direct internalization of sEVs by the microglia cells or other effects. The results presented here show that MNC-sEVs administered locally in the brain promoted in vivo neuroprotection and attenuated microglia reactivity in an animal model of cerebral ischemia. Taking in account that the attenuation of microglia reactivity has been reported to promote brain repair and regeneration,36 our results show that the accumulation of sEVs in the brain might be required to maximize their therapeutic effect.
Conclusions
In summary, under the experimental conditions tested in this work, our results indicate that the neuroprotection and microglia modulation depend on the sEV source. sEVs increased the survival of endothelial cells, microglia and neurons and decreased microglia reactivity after cell culture in OGD conditions, this effect being higher for sEVs isolated from MNCs than for the ones isolated from MSC-sEVs (at least for neurons and microglia). Part of the differences might be due to the differences in sEV internalization. We showed for the first time that sEV internalization in microglia is mediated, at least in part, by the axis HLA-DR (sEVs): CD4 (microglia cells). Our results further show that neuroprotection and microglia modulation can be mediated directly by sEVs, without the contribution of indirect systemic effects. The brain cells that are more proactive in the uptake of MNC-sEVs are microglia cells. Thus, it is likely that platforms that promote the accumulation of sEVs in the brain after stroke may increase the neuroprotection/immunomodulation programs and ultimately brain regeneration.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was partially supported by FCT through project EXPL/BTM-ORG/1348/2021, project “NeuroAtlantic”, Ref: EAPA_791/2018, which is co-funded by Program Interreg Atlantic Space through the European fund for Regional Development, and the project 0624_2IQBIONEURO_6_E, co-funded by the European Regional Development Fund (ERDF), through the program INTERREG V-A España Portugal (POCTEP). Project “Agendas/Alianças mobilizadoras para a reindustrialização” (Ref: C630926586-00465198) funded by PRR.References
- V. Saini, L. Guada and D. R. Yavagal, Neurology, 2021, 97, S6–S16 CrossRef PubMed.
- G. Tsivgoulis, A. H. Katsanos and A. V. Alexandrov, Front. Neurol., 2014, 5, 215 Search PubMed.
- W. J. Powers, A. A. Rabinstein, T. Ackerson, O. M. Adeoye, N. C. Bambakidis, K. Becker, J. Biller, M. Brown, B. M. Demaerschalk, B. Hoh, E. C. Jauch, C. S. Kidwell, T. M. Leslie-Mazwi, B. Ovbiagele, P. A. Scott, K. N. Sheth, A. M. Southerland, D. V. Summers and D. L. Tirschwell, Stroke, 2019, 50, e344–e418 CrossRef PubMed.
- M. Tymianski, Stroke, 2013, 44, 2942–2950 CrossRef PubMed.
- X. Y. Xiong, L. Liu and Q. W. Yang, Front. Neurol., 2018, 9, 249 CrossRef PubMed.
- L. Chen, G. Zhang, Y. Gu and X. Guo, Sci. Rep., 2016, 6, 32291 CrossRef CAS PubMed.
- A. Kumar, M. Prasad, V. P. Jali, A. K. Pandit, S. Misra, P. Kumar, K. Chakravarty, P. Kathuria and A. Gulati, Acta Neurol. Scand., 2017, 135, 496–506 CrossRef CAS PubMed.
- Q. Vu, K. Xie, M. Eckert, W. Zhao and S. C. Cramer, Neurology, 2014, 82, 1277–1286 CrossRef PubMed.
- M. M. Lino, S. Simoes, F. Tomatis, I. Albino, A. Barrera, D. Vivien, T. Sobrino and L. Ferreira, J. Controlled Release, 2021, 338, 472–485 CrossRef CAS PubMed.
- T. R. Doeppner, J. Herz, A. Gorgens, J. Schlechter, A. K. Ludwig, S. Radtke, K. de Miroschedji, P. A. Horn, B. Giebel and D. M. Hermann, Stem Cells Transl. Med., 2015, 4, 1131–1143 CrossRef CAS PubMed.
- K.-H. Chen, C.-H. Chen, C. G. Wallace, C.-M. Yuen, G.-S. Kao, Y.-L. Chen, P.-L. Shao, Y.-L. Chen, H.-T. Chai, K.-C. Lin, C.-F. Liu, H.-W. Chang, M. S. Lee and H.-K. Yip, Oncotarget, 2016, 7, 74537–74556 CrossRef PubMed.
- J. Y. Lee, E. Kim, S.-M. Choi, D.-W. Kim, K. P. Kim, I. Lee and H.-S. Kim, Sci. Rep., 2016, 6, 33038–33038 CrossRef CAS PubMed.
- L. Otero-Ortega, F. Laso-García, M. D. C. Gómez-de Frutos, B. Rodríguez-Frutos, J. Pascual-Guerra, B. Fuentes, E. Díez-Tejedor and M. Gutiérrez-Fernández, Sci. Rep., 2017, 7, 44433–44433 CrossRef CAS PubMed.
- M. Safakheil and H. Safakheil, J. Mol. Neurosci., 2020, 70, 724–737 CrossRef CAS PubMed.
- X. Sun, J.-H. Jung, O. Arvola, M. R. Santoso, R. G. Giffard, P. C. Yang and C. M. Stary, Front. Cell. Neurosci., 2019, 13, 394 CrossRef CAS PubMed.
- R. L. Webb, E. E. Kaiser, B. J. Jurgielewicz, S. Spellicy, S. L. Scoville, T. A. Thompson, R. L. Swetenburg, D. C. Hess, F. D. West and S. L. Stice, Stroke, 2018, 49, 1248–1256 CrossRef PubMed.
- R. L. Webb, E. E. Kaiser, S. L. Scoville, T. A. Thompson, S. Fatima, C. Pandya, K. Sriram, R. L. Swetenburg, K. Vaibhav, A. S. Arbab, B. Baban, K. M. Dhandapani, D. C. Hess, M. N. Hoda and S. L. Stice, Transl. Stroke Res., 2018, 9, 530–539 CrossRef CAS PubMed.
- H. Xin, Y. Li, Y. Cui, J. J. Yang, Z. G. Zhang and M. Chopp, J. Cereb. Blood Flow Metab., 2013, 33, 1711–1715 CrossRef CAS PubMed.
- C. Wang, V. Borger, M. Sardari, F. Murke, J. Skuljec, R. Pul, N. Hagemann, E. Dzyubenko, R. Dittrich, J. Gregorius, M. Hasenberg, C. Kleinschnitz, A. Popa-Wagner, T. R. Doeppner, M. Gunzer, B. Giebel and D. M. Hermann, Stroke, 2020, 51, 1825–1834 CrossRef CAS PubMed.
- G. Morad, C. V. Carman, E. J. Hagedorn, J. R. Perlin, L. I. Zon, N. Mustafaoglu, T. E. Park, D. E. Ingber, C. C. Daisy and M. A. Moses, ACS Nano, 2019, 13, 13853–13865 CrossRef CAS PubMed.
- A. Banerjee, V. Alves, T. Rondao, J. Sereno, A. Neves, M. Lino, A. Ribeiro, A. J. Abrunhosa and L. S. Ferreira, Nanoscale, 2019, 11, 13243–13248 RSC.
- B. W. McColl, S. M. Allan and N. J. Rothwell, Neuroscience, 2009, 158, 1049–1061 CrossRef CAS PubMed.
- H. Henriques-Antunes, R. M. S. Cardoso, A. Zonari, J. Correia, E. C. Leal, A. Jimenez-Balsa, M. M. Lino, A. Barradas, I. Kostic, C. Gomes, J. M. Karp, E. Carvalho and L. Ferreira, ACS Nano, 2019, 13, 8694–8707 CrossRef CAS PubMed.
- L. Huang, Y. Liu, J. Lu, B. Cerqueira, V. Misra and T. Q. Duong, Stem Cell Res. Ther., 2017, 8, 74 CrossRef PubMed.
- J. Boltze, D. M. Reich, S. Hau, K. G. Reymann, M. Strassburger, D. Lobsien, D. C. Wagner, M. Kamprad and T. Stahl, Cell Transplant., 2012, 21, 723–737 Search PubMed.
- M. A. Yenari, T. M. Kauppinen and R. A. Swanson, Neurotherapeutics, 2010, 7, 378–391 CrossRef CAS PubMed.
- B. Gulyas, M. Toth, M. Schain, A. Airaksinen, A. Vas, K. Kostulas, P. Lindstrom, J. Hillert and C. Halldin, J. Neurol. Sci., 2012, 320, 110–117 CrossRef CAS PubMed.
- J. Suenaga, X. Hu, H. Pu, Y. Shi, S. H. Hassan, M. Xu, R. K. Leak, R. A. Stetler, Y. Gao and J. Chen, Exp. Neurol., 2015, 272, 109–119 CrossRef CAS PubMed.
- S. Sharma, B. Yang, R. Strong, X. Xi, M. Brenneman, J. C. Grotta, J. Aronowski and S. I. Savitz, J. Neurosci. Res., 2010, 88, 2869–2876 CAS.
- S. S. Bedi, P. A. Walker, S. K. Shah, F. Jimenez, C. P. Thomas, P. Smith, R. A. Hetz, H. Xue, S. Pati, P. K. Dash and C. S. Cox, Jr., J. Trauma Acute Care Surg., 2013, 75, 410–416 CrossRef CAS PubMed.
- F. Fluri, M. K. Schuhmann and C. Kleinschnitz, Drug Des., Dev. Ther., 2015, 9, 3445–3454 CAS.
- W. M. Clark, N. S. Lessov, M. P. Dixon and F. Eckenstein, Neurol. Res., 1997, 19, 641–648 CrossRef CAS PubMed.
- L. Ruan, B. Wang, Q. ZhuGe and K. Jin, Brain Res., 2015, 1623, 166–173 CrossRef CAS PubMed.
- D. Xin, T. Li, X. Chu, H. Ke, Z. Yu, L. Cao, X. Bai, D. Liu and Z. Wang, Acta Biomater., 2020, 113, 597–613 CrossRef CAS PubMed.
- Y. Zheng, R. He, P. Wang, Y. Shi, L. Zhao and J. Liang, Biomater. Sci., 2019, 7, 2037–2049 RSC.
- J. Lyu, D. Xie, T. N. Bhatia, R. K. Leak, X. Hu and X. Jiang, CNS Neurosci. Ther., 2021, 27, 515–527 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3nr03861k |
| This journal is © The Royal Society of Chemistry 2023 |