
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOne-step electrodeposition of binder-containing Cu nanocube catalyst layers for carbon dioxide reduction†
Andrea
Serfőző
,
Gábor András
Csík
,
Attila
Kormányos
 ,
Ádám
Balog
,
Csaba
Janáky
and
Balázs
Endrődi
*
,
Ádám
Balog
,
Csaba
Janáky
and
Balázs
Endrődi
*
Department of Physical Chemistry and Materials Science, University of Szeged, Rerrich Square 1, Szeged, H-6720 Hungary. E-mail: endrodib@chem.u-szeged.hu
First published on 21st September 2023
Abstract
To reach industrially relevant current densities in the electrochemical reduction of carbon dioxide, this process must be performed in continuous-flow electrolyzer cells, applying gas diffusion electrodes. Beyond the chemical composition of the catalyst, both its morphology and the overall structure of the catalyst layer are decisive in terms of reaction rate and product selectivity. We present an electrodeposition method for preparing coherent copper nanocube catalyst layers on hydrophobic carbon paper, hence forming gas diffusion electrodes with high coverage in a single step. This was enabled by the appropriate wetting of the carbon paper (controlled by the composition of the electrodeposition solution) and the use of a custom-designed 3D-printed electrolyzer cell, which allowed the deposition of copper nanocubes selectively on the microporous side of the carbon paper substrate. Furthermore, a polymeric binder (Capstone ST-110) was successfully incorporated into the catalyst layer during electrodeposition. The high electrode coverage and the binder content together result in an increased ethylene production rate during CO2 reduction, compared to catalyst layers prepared from simple aqueous solutions.
Introduction
Electrochemical reduction of carbon dioxide (CO2RR) has undoubtedly been among the most intensively studied electrochemical processes in recent years.1,2 The large number of researchers working in the field made notable progress in understanding the reaction mechanisms, identifying active and selective catalysts, and developing efficient electrolyzer cells and stacks. The latter becomes more and more important as the industrial implementation of CO2RR seems to be feasible in the near future.3–6The maximum rate of CO2RR in aqueous solutions is limited by the solubility of CO2, which is around 30 mM at room temperature. The mass transport of CO2 in such solutions limits the maximum current density to a few tens of mA cm−2 for 2-electron products (i.e., carbon monoxide, formate).7 Furthermore, a large cell voltage develops due to the large distance between the electrodes in the cells typically used in laboratory experiments (e.g., H-cells). Finally, product separation from the solution (in the case of formate) in batch reactors is challenging. These obstacles can be overcome by using continuous-flow electrolyzer cells.8 Here, CO2 is fed to the cathode catalyst through a porous substrate, the gas diffusion layer (GDL), hence decreasing the diffusion layer thickness by several orders of magnitude.9 The distance between the electrodes is minimized, and they are typically separated by only a membrane (zero-gap electrolyzer cells), a thin liquid electrolyte (microfluidic electrolyzer cells), or two liquid electrolytes and a membrane (hybrid electrolyzer cells).10
GDLs are typically formed of two layers: a macroporous layer, with larger pore size for gas transport, and a microporous layer. The GDL with a catalyst-coated microporous layer is called the gas diffusion electrode (GDE), which is the central piece of electrolyzer cells.11 The structure of this GDE assures the proper reactant and product transport. It is mechanically stable and electrically conductive, resulting in low cell resistance. A further important role of the GDE is to separate the gas and liquid phases, hence avoiding gas breakthrough or electrode flooding.12,13 For this reason, the typically applied GDLs are impregnated with hydrophobic compounds, such as polytetrafluoroethylene (PTFE). GDEs are usually formed by physical means, immobilizing nanoparticles on GDLs by spray coating, drop-casting, sputtering, or other alternative techniques.11,14 Electrodeposition is another possibility for GDE preparation. In this case, the catalyst layer is directly formed on the microporous side of the GDL, hence a low contact resistance, and the formation of a strongly adhering layer is expected. Furthermore, electrochemical methods allow precise tuning of the deposited catalyst amount by simply controlling the deposition time, charge, or cycle number (in the case of dynamic electrodeposition methods). Also, electrodeposition conditions dictate the morphology, crystallinity, and size of the forming catalyst particles and layers.15–18 Finally, electrodeposition is relatively easy to scale up to form large-area electrodes.19
Copper is one of the most frequently studied catalysts for CO2RR, rooted in its unique capability to form C2+ products from CO2 in a single step. The selectivity is highly dependent on the catalyst morphology and the exposed crystal facets.20–22 Among other morphologies, copper nanocubes (Cu NCs) offer high selectivity for ethylene production in CO2RR.23–28 The selectivity is affected by the size of the catalyst particles that can be tuned by varying the synthesis parameters. Electrodeposition is a versatile method in this regard, as the reaction conditions (e.g., precursor concentration, overpotential) were proven to control the forming catalyst layer and, consequently, the reaction rate and selectivity.29–31 We note that the main drawback of using Cu NCs for CO2RR is the morphological change of the catalyst during the reaction. Different mitigation strategies are being pursued by multiple research groups to avoid this, including the deposition of organic or inorganic protective coatings on the nanoparticles.32 Stabilization of these particles is, however, beyond the scope of this contribution. Here, we focus on the electrochemical formation of GDEs and apply Cu NCs as a model system.
Performing electrodeposition from aqueous electrolyte solutions is not straightforward on the typically used GDLs due to their hydrophobicity. It is not even trivial to immerse such substrates in water-based solutions. Using apolar organic solvents, which fully wet the GDL, on the other hand, leads to the penetration of the electrolyte solution into deeper pores, where electrodeposition might also occur. In this case, the pore structure of the GDL can be distorted, which should be avoided. Here, we demonstrate, using the example of Cu NC catalysts, how the microporous side of a hydrophobic GDL can be selectively and fully coated via electrodeposition by tailoring the precursor solution composition and using a simple 3D-printed electrodeposition cell. Furthermore, we show that a binder material (Capstone ST-110 polymer in this case) can also be incorporated into the catalyst layer in the same single electrodeposition step. The electrocatalytic activity of the formed layers are compared based on CO2RR experiments in a continuous-flow microfluidic electrolyzer cell. Overall, the approach presented here to form fully covered binder-containing GDEs is expected to be generally applicable for different GDL–catalyst systems.
Experimental section
Materials
All chemicals used in this study were purchased from Sigma-Aldrich or VWR International. Chemicals of high purity (at least ACS reagent grade) were purchased and used without further purification. Ultrapure water (18.2 MΩ cm) was used for the experiments, freshly produced using a Millipore Direct Q3 UV instrument. 4.5 purity CO2 gas was employed in the CO2RR studies.Catalyst electrodeposition on gas diffusion layers (GDLs)
Electrodeposition of the catalyst layers was performed in a custom-designed, 3D-printed electrolyzer cell (see Fig. 1). A Raise3D N2-type 3D printer was used to manufacture the cell components. The cells were printed from an acrylonitrile butadiene styrene polymer (ABS), with 100% filling ratio to ensure the liquid and gas tightness of the cell. A regular three-electrode arrangement was applied for the electrodeposition, in which an Ag/AgCl/3 M NaCl electrode and a copper mesh served as the reference and counter electrodes, respectively, while a Freudenberg H23C6-type GDL was used as the working electrode. Importantly, only the microporous side of the GDL is in direct contact with the electrolyte solution, hence enabling to selectively deposit the catalyst layer on that side. The anode electrodes were made by spray coating a suspension of commercial Ir powder (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 isopropanol/water mixture as the solvent, 15 wt% Nafion ionomer content with reference to the total Ir + binder amount, and an Ir concentration of 20 mg cm−3) on a Freudenberg H23C6 GDL, with a catalyst loading of 1 mg cm−2. Electrodeposition experiments were carried out using a Metrohm Autolab 204-type instrument.
:1 isopropanol/water mixture as the solvent, 15 wt% Nafion ionomer content with reference to the total Ir + binder amount, and an Ir concentration of 20 mg cm−3) on a Freudenberg H23C6 GDL, with a catalyst loading of 1 mg cm−2. Electrodeposition experiments were carried out using a Metrohm Autolab 204-type instrument.
 | ||
| Fig. 1 Schematic drawing of the 3D-printed electrolyzer cell used for the electrodeposition of Cu NC catalyst layers on the microporous side of hydrophobic GDLs. The opening where the GDL surface is in contact with the electrolyte solution is 3.5 cm × 3.5 cm large. | ||
Physical characterization of the samples
A Thermo Scientific Apreo 2 scanning electron microscope (SEM) was employed to collect information on the morphology of the formed electrodes. A Krüss EasyDrop instrument was used to measure the wetting properties (that is, contact angles) of different solvent mixtures on the microporous side of the Freudenberg H23C6 GDL. A droplet of the solvent mixture was formed on a plate using a syringe. Using the CCD camera of a goniometer, the drop contour of the captured photographs was analyzed. The pH of the solutions was measured using a Mettler Toledo FiveEasy Plus FP20 pH meter.Measurements in a continuous-flow electrolyzer cell
Continuous-flow electrolysis experiments were performed in a two-electrode setup employing a microfluidic cell designed based on the work of the Kenis research group.33,34 This consisted of two stainless steel electrode contacts, separated by a single poly(methyl methacrylate) (PMMA) flow channel (d = 2 mm thickness). 3 mm deep cavities were formed on the metal electrodes to serve as gas flow channels. An inlet and an outlet port were added to the cathode current collector for CO2 transport, while a single outlet port was formed on the anode for the evolving O2. A 2 cm × 0.5 cm large opening was created in the middle of the PMMA separator, defining the A = 1 cm2 electrolysis geometric area. ϕ = 1 mm holes were drilled in two opposite sides of this plastic element, going through the middle of the formed opening, for the transport of the electrolyte solution. The connection for the liquid pump was established by mounting 1 mm needles in the holes. The cathode GDE, together with a PTFE gasket around it, was mounted between the cathode electrode and the plastic flow element. The anode was mounted in the cell similarly. CO2 gas was fed to the cathode in a flow-by mode at a rate of u = 20 sccm, while 1 M KOH electrolyte was directed between the two electrodes at a flow rate of 0.5 cm3 min−1. A Bronkhorst EL-FLOW Select F-201CV mass flow controller and a KF Technology NE-300 syringe pump were used for regulating the gas and the liquid flow rate, respectively. Electrochemical measurements were controlled using a Biologic VMP300 type instrument. The CO2RR products were monitored during electrolysis using a Shimadzu GC-2030 Plus gas chromatograph (operated with 6.0 He carrier gas), equipped with a barrier discharge ionization (BID) detector and an automatic 6-way valve injection system. The faradaic efficiency of the CO2 electrolysis was calculated from the GC results and the measured gas flow rate (Agilent ADM flow meter). Importantly, the pressure increase in the gas line – which could lead to electrode flooding – was avoided by applying a small vacuum pump to fill the sample loop of the injector, sampling the main gas stream.Results and discussion
An electrodeposition cell was designed to ensure that electrodeposition of the catalyst layer occurs only on the microporous side of the GDL (Fig. 1). This is achieved by mounting the GDL between the plastic backplate of the cell and a gasket, on which an opening defines the electrode area that is in contact with the liquid electrolyte. Importantly, if the solution is not wetting the GDL fully, any electrochemical (Faradaic) process can only occur on the surface. This cell design is based on the cells typically applied in anodization studies,35,36 and it was prepared by 3D printing. This allowed us to scale the electrolyzer cell to the desired electrode size rapidly.Electrodeposition of Cu nanocube (Cu NC) layers was based on previous pioneering studies.29–31 As the first step, we used aqueous CuSO4/KCl solutions and successfully implemented their deposition protocol, leading to the formation of Cu NC catalysts. We found, however, that the coverage of the GDL was low, approximately 15–20% of the total surface area. This is related to the hydrophobicity of the GDL's microporous layer, which is not wetted by the solution.
To overcome this challenge, based on our earlier experience,37 we used isopropanol:water (IPA:H2O) solvent mixtures instead of pure water. Increasing the IPA content of the solution led to improved surface wetting (i.e., decreased contact angle) of the microporous surface of the GDL (Fig. 2). We note that there is a fine balance here: while appropriate wetting of the surface is a prerequisite for fully coated GDE preparation, the electrolyte solution should not penetrate the deeper pores of the GDL, as this would lead to catalyst deposition throughout the GDL. The latter could distort the gas transport and affect the flooding properties of the GDE during CO2RR, the targeted application for these electrodes. The amount of the deposition material was regulated by the number of cycles. The deposition was performed from a solution containing CuSO4 and KCl at the same concentration.
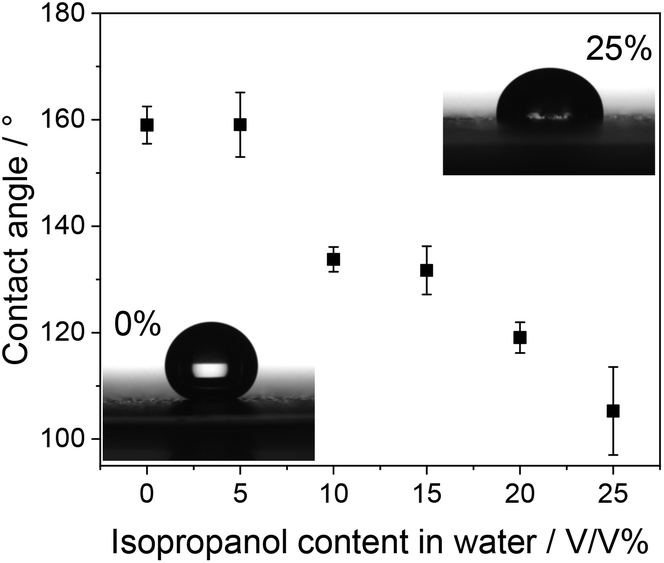 | ||
| Fig. 2 Contact angles of different water/isopropanol solvent mixtures on the microporous side of a Freudenberg H23C6 GDL. | ||
Electrodeposition was performed via a potentiodynamic method, applying the same potential limits as in ref. 30 (between +0.55 V and +0.22 V, both vs. RHE) (Fig. 3A). Low deposition currents were observed at very low precursor concentrations (1 mM), related to the mass transfer limitations arising in the quiescent solution. The deposition rate increases with increasing precursor concentration, caused by the increased mass transport rate and decreased solution resistance. As an illustrative example, we compare here the morphology of the layers formed from 5 mM CuSO4/KCl solution in pure water and in 15 V/V% IPA containing aqueous solutions, applying the same number of deposition cycles (Fig. 3B and C, respectively). In agreement with former reports,29–31 we confirmed the deposition of Cu NCs using pure water as the solvent (Fig. 3B). The formation of densely covered and “empty” regions was observed in the SEM images, and the surface coverage was estimated to be around 15–20%. In stark contrast, an almost fully covered surface was witnessed when the solvent mixture contained 15 V/V% IPA (Fig. 3C and Fig. S1†). Importantly, the cube morphology was observed in this case as well. This proves our hypothesis, namely that by tailoring the solvent composition, the surface coverage of the electrode can be tuned during electrodeposition. As for the effect of varying solvent composition, we performed studies with different IPA:H2O mixtures at a constant precursor concentration. In agreement with the contact angle measurements (Fig. 2), an increasing surface coverage was observed, up to 15% IPA content. At higher IPA contents, however, a gradually decreasing amount of catalyst deposition was observed on the GDL surface, and the cube morphology was also distorted; the formation of platelets was seen (Fig. S2†). This is caused by the penetration of the solution in the pores of the GDL, and hence deposition occurs there as well. A high capacitive current appeared in this case due to the large inner surface area of the porous substrate (Fig. S3†).38 Under the applied potentiodynamic conditions, only a small fraction of the total charge is consumed in a Faradaic process (i.e., the deposition of copper nanoparticles). Furthermore, a gradual decrease in the particle size was also witnessed with increasing IPA ratio (Fig. S4†). This causes an increase in the surface area of the catalyst particles and hence leads to a larger number of exposed crystal edges that can result in higher CO2RR activity and selectivity.
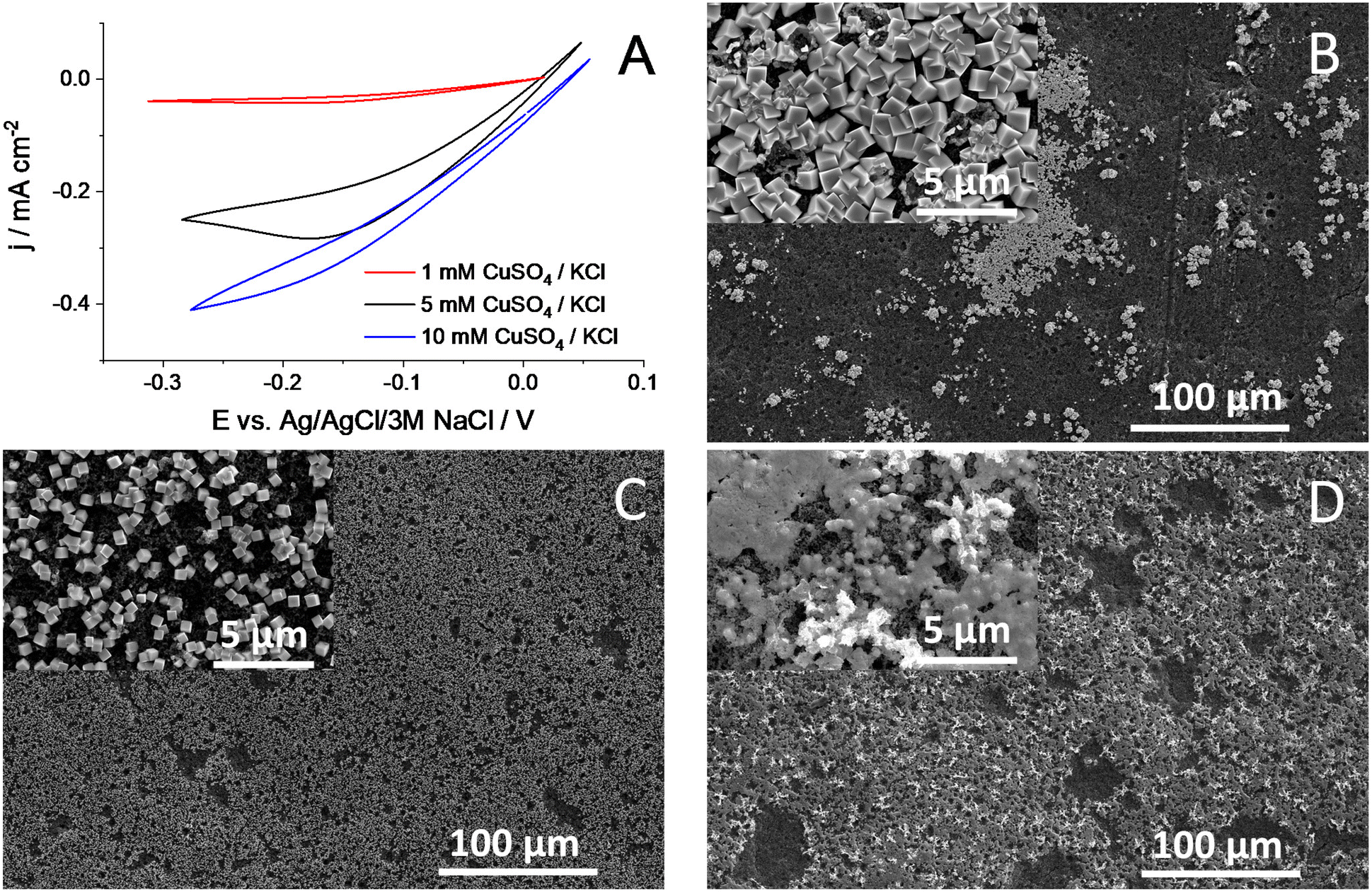 | ||
| Fig. 3 (A) Representative potentiodynamic curves recorded for Cu NC synthesis on a Freudenberg H23C6 GDL. The sweep rate was ν = 100 mV s−1, and the measurements were performed at room temperature in 15 v/v% isopropanol-containing aqueous solutions. SEM images taken at two different magnifications (see the insets) of layers deposited from 5 mM CuSO4/KCl solutions; in (B) pure water, (C) 15 v/v% isopropanol-containing solvent mixture, and (D) a solution identical to (C), but also containing 100 mg l−1 Capstone ST-110. 500 electrodeposition cycles were applied for (B), (C) and (D). The integrated charge during the depositions varied within the 20% range. | ||
Furthermore, this also implies the decrease of the layer thickness, as under the applied conditions, a “monolayer” of Cu NCs forms on the GDLs. We also mention that the formation of larger copper aggregates is typically seen on the layers formed from pure water, while these structures are absent when a solvent mixture that wets the GDL fully is used (Fig. S5†). All further measurements were performed using pure water or 15% IPA-containing solutions with 5 or 10 mM CuSO4 concentration. The Cu NC catalyst containing GDEs were tested in CO2RR in a microfluidic electrolyzer cell,39,40 in galvanostatic measurements (Fig. 4, Table S1†). Comparing the cell voltage stability using the GDEs formed from pure water and in the 15% IPA:water mixture, a striking difference is seen, even without analyzing the formed products. While stable cell voltages were recorded in the latter case, the GDE formed from pure water failed rapidly at the highest studied current density (200 mA cm−2), signaled by a large decrease (from ca. 3.2 V to ca. 2.6 V) of the cell voltage (Fig. 4A). This is caused by the flooding of the GDE, and the consequent dominance of the cathodic HER, which proceeds at a less negative potential compared to CO2RR. We believe that the rapid cell failure is rooted in the high local current densities at the catalyst-covered parts of the GDE; note that the current density is normalized by the geometric surface area of the GDE. However, as only 15–20% of the surface is coated with Cu NCs, the 200 mA cm−2 geometric area normalized current density translates to 1–1.3 A cm−2 catalyst geometric area normalized current density. This can cause rapid catalyst degradation, increased local temperature, and electrowetting, which together lead to the flooding of the GDE.
 | ||
| Fig. 4 (A) Typical galvanostatic CO2RR measurements with Cu NC catalyst layers formed from solutions of identical precursor concentration, using pure water or a 15 v/v% IPA and water mixture as the solvent. Product distribution during galvanostatic CO2RR experiments on Cu NC catalyst layers electrodeposited (B) from pure water or 15 v/v% IPA-containing solution and (C) from solutions of 15 v/v% IPA in water, also containing different amounts of the Capstone ST110 ionomer. The experiments in (B) and (C) were performed at both 5 and 10 mM CuSO4/KCl concentrations. All measurements were performed at room temperature in a microfluidic electrolyzer cell, with a cathodic CO2 feed of 20 cm3 min−1 and with a 1 M KOH solution flowing between the electrodes at a rate of 0.5 cm3 min−1. All GDEs were formed by 500 times, repeating the potentiodynamic deposition protocol shown in Fig. 3A. | ||
Irrespective of the solvent used during the electrodeposition of Cu NCs, ethylene was the dominant CO2RR product in the gas phase, while methane and carbon monoxide were detected at small concentrations on all GDEs (Fig. 4B). As expected from the low surface coverage, a relatively low CO2RR rate was measured for the GDEs formed using pure water as the solvent. At 200 mA cm−2 current density, the HER was the dominant electrode process, proceeding with ca. 75% Faradaic efficiency (FE), independent of the CuSO4/KCl concentration.
The HER was efficiently suppressed on the GDEs deposited from IPA:water mixtures. The FE(H2) was between 10 and 15% at all studied current densities. In parallel to this, the FE(C2H4) was between 45 and 50%, with slightly lower values measured for the samples prepared at lower precursor concentrations. At higher current densities, notable ethanol (FE ∼5–6%) and acetate (FE ∼3–4%) formation rates were witnessed for these GDEs.
To increase the stability of the electrodeposited layers, we aimed to incorporate a binder into the catalyst layer during electrodeposition. Therefore, the electrodeposition of the catalyst layers was repeated from 15 V/V% IPA-containing precursor solutions, containing different amounts of Capstone ST-110, a commercial pore sealer.40 This resulted in the formation of a coherent layer between the Cu NCs, implying the incorporation of the polymer (Fig. 3D and Fig. S6, S7†).
The polymer layer thickness increases with the binder concentration in the precursor solution but has no apparent effect on the crystal size of the forming particles, suggested by the almost identical XRD patterns (Fig. S7†). When testing these layers in CO2RR (Fig. 4C), a further increased ethylene (FE(C2H4) above 55%) and ethanol (FE(EtOH) between 10 and 15%) formation selectivity was witnessed, and the HER was further suppressed (FE(H2) ≤ 10%). Interestingly, a lower polymer content was found to be beneficial for the layers deposited from higher concentration precursor solutions, while a higher CST concentration resulted in better selectivity for the layers formed at lower CuSO4 concentration. We attribute this trend to the differences in the particle size of the Cu NCs (Fig. S8†). Importantly, the measured selectivity greatly exceeds that obtained with commercial Cu nanopowder (FE(C2H4) ≈ 35%) under identical conditions (Fig. S9†).
The stability of the catalyst layers formed from pure water solvent and from IPA- and CST-containing solutions was compared in constant current electrolysis experiments at j = 200 mA cm−2 (Fig. 5A and B, Table S1†). In these studies, almost instantaneous flooding of the former was experienced, while a constant ethylene formation rate with the FE(C2H4) between 57 and 60% was observed on the polymer-containing GDE for ca. 45 minutes when flooding of the layer occurred, as shown by the rapid cell voltage decrease.
 | ||
| Fig. 5 Constant current electrolysis experiments at j = 200 mA cm−2 using GDEs formed from 5 mM CuSO4/KCl-containing solutions in (A) pure water and (B) 100 mg l−1 CST- and 15 V/V% IPA-containing aqueous solution with 500 deposition cycles. All measurements were performed at room temperature in a microfluidic electrolyzer cell, with a cathodic CO2 feed of 20 cm3 min−1 and with a 1 M KOH solution flowing between the electrodes at a rate of 0.5 cm3 min−1. All GDEs were formed by 500 times repeating the potentiodynamic deposition protocol shown in Fig. 3A. (C) SEM images of the GDE deposited from IPA- and CST-containing solutions before and after the electrochemical measurements shown in (B). | ||
When these experiments were repeated a few times, very similar conclusions were obtained, although with slightly varying times until flooding. We attribute the eventual flooding to the degradation of the Cu NCs. According to our SEM studies, the initial structure in which both the cube morphology and polymer coverage can be observed changes during the reaction, resulting in the damage of the coherent catalyst layer, exposing a larger area of the GDL (Fig. 5C). Although we expect that by further tuning the layer thickness and the deposition solution composition, the lifetime of such catalyst layers could be extended. However, the degradation of Cu NCs will eventually occur,41 and therefore, we did not attempt to extend this study in this direction.
Conclusions
By tailoring the solvent mixture composition, ideal wetting of the surface of a hydrophobic GDL can be achieved. Performing electrodeposition from such a solution mixture leads to high and homogeneous surface coverage, as demonstrated on the example of copper nanocubes (Cu NCs). Importantly, when performing the same electrodeposition from pure water, only 15–20% of the substrate was coated with the catalyst, while using a fully wetting solvent, the electrodeposition occurred within the GDL, blocking the pores that are pivotal for proper gas transfer and distorting the structure of the substrate. Increasing the surface coverage resulted in better performance in CO2RR in a continuous-flow microfluidic electrolyzer cell, which was demonstrated by the better tolerance against flooding and the higher formation rate of C2+ products (ethylene in particular). By performing the electrodeposition with a polymeric binder added to the precursor solution, Capstone ST-110 binder-containing catalyst layers could be deposited. A slightly increased C2+ selectivity was witnessed when using these catalyst layers for CO2RR. More importantly, the stability of the layers increased due to the homogeneous GDL coverage and binder incorporation. Although a specific example is shown here to highlight the benefits of electrochemically forming GDEs from solutions with a tailored composition, we believe that this strategy can be extended to other systems as well. Electrodeposition is a highly controllable and fairly easily scalable method, and hence it can offer an alternative to prepare large-area GDEs for CO2 electrolyzers while also avoiding any physical catalyst immobilization steps (e.g., spray coating). This offers faster GDE preparation and fully circumvents any possible contamination originating from the physical deposition of the catalyst.Author contributions
Andrea Serfőző: investigation and visualization. Gábor András Csík: investigation and methodology. Attila Kormányos: investigation, visualization, and data curation. Ádám Balog: investigation and methodology. Csaba Janáky: resources, writing – review and editing, supervision, and funding acquisition. Balázs Endrődi: conceptualization, data curation, writing – original draft, supervision, and funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project has received funding under the European Union's Horizon 2020 research and innovation program from the FlowPhotoChem project (Grant Agreement No. 862453). The research was supported by the National Research, Development and Innovation Office (NKFIH) through the FK-132564 project. B. E. and A. K. also acknowledge the financial support from the János Bolyai Research Scholarship of the Hungarian Academy of Sciences and the ÚNKP-22-5 New National Excellence Program of the Ministry for Innovation and Technology from the National Research, Development and Innovation Fund. Project No. RRF-2.3.1-21-2022-00009, titled the National Laboratory for Renewable Energy, has been implemented with the support provided by the Recovery and Resilience Facility of the European Union within the framework of Programme Széchenyi Plan Plus”.References
- J. Yu, J. Wang, Y. Ma, J. Zhou, Y. Wang, P. Lu, J. Yin, R. Ye, Z. Zhu and Z. Fan, Adv. Funct. Mater., 2021, 31, 2102151 CrossRef CAS.
- A. Liu, M. Gao, X. Ren, F. Meng, Y. Yang, L. Gao, Q. Yang and T. Ma, J. Mater. Chem. A, 2020, 8, 3541–3562 RSC.
- H. Shin, K. U. Hansen and F. Jiao, Nat. Sustainability, 2021, 1–10 Search PubMed.
- S. W. Sheehan and R. Buonsanti, Chem. Catal., 2021, 1, 751–753 CrossRef CAS.
- A. Somoza-Tornos, O. J. Guerra, A. M. Crow, W. A. Smith and B. M. Hodge, iScience, 2021, 24, 102813 CrossRef CAS PubMed.
- I. E. L. Stephens, K. Chan, A. Bagger, S. W. Boettcher, J. Bonin, E. Boutin, A. Buckley, R. Buonsanti, E. Cave, X. Chang, S. W. Chee, A. H. M. da Silva, P. de Luna, O. Einsle, B. Endrődi, M. E. Escribano, J. V. Ferreira de Araujo, M. C. Figueiredo, C. Hahn, K. U. Hansen, S. Haussener, S. Hunegnaw, Z. Huo, Y. J. Hwang, C. Janáky, B. S. Jayathilake, F. Jiao, Z. P. Jovanov, P. Karimi, M. T. M. Koper, K. Kuhl, W. H. Lee, Z. Liang, X. Liu, S. Ma, M. Ma, H.-S. Oh, M. Robert, B. R. Cuenya, J. Rossmeisl, C. Roy, M. P. Ryan, E. H. Sargent, P. Sebastián-Pascual, B. Seger, L. Steier, P. Strasser, A. S. Varela, R. E. Vos, X. Wang, B. Xu, H. Yadegari and Y. Zhou, J. Phys.: Energy, 2022, 4, 042003 Search PubMed.
- B. Endrődi, G. Bencsik, F. Darvas, R. Jones, K. Rajeshwar and C. Janáky, Prog. Energy Combust. Sci., 2017, 62, 133–154 CrossRef.
- D. Wakerley, S. Lamaison, J. Wicks, A. Clemens, J. Feaster, D. Corral, S. A. Jaffer, A. Sarkar, M. Fontecave, E. B. Duoss, S. Baker, E. H. Sargent, T. F. Jaramillo and C. Hahn, Nat. Energy, 2022, 7, 130–143 CrossRef CAS.
- T. Burdyny and W. A. Smith, Energy Environ. Sci., 2019, 12, 1442–1453 RSC.
- Á. Vass, A. Kormányos, Z. Kószó, B. Endrődi and C. Janáky, ACS Catal., 2022, 12, 1037–1051 CrossRef PubMed.
- H. Rabiee, L. Ge, X. Zhang, S. Hu, M. Li and Z. Yuan, Energy Environ. Sci., 2021, 14, 1959–2008 RSC.
- B. De Mot, M. Ramdin, J. Hereijgers, T. J. H. Vlugt and T. Breugelmans, ChemElectroChem, 2020, 7, 3839–3843 CrossRef CAS.
- K. Yang, R. Kas, W. A. Smith and T. Burdyny, ACS Energy Lett., 2021, 6, 33–40 CrossRef CAS.
- H. R. Q. Jhong, F. R. Brushett and P. J. A. Kenis, Adv. Energy Mater., 2013, 3, 589–599 CrossRef CAS.
- R. L. Machunda, H. Ju and J. Lee, Curr. Appl. Phys., 2011, 11, 986–988 CrossRef.
- H. Rabiee, X. Zhang, L. Ge, S. Hu, M. Li, S. Smart, Z. Zhu and Z. Yuan, ACS Appl. Mater. Interfaces, 2020, 12, 21670–21681 CrossRef CAS PubMed.
- H. Rabiee, L. Ge, X. Zhang, S. Hu, M. Li, S. Smart, Z. Zhu and Z. Yuan, Appl. Catal., B, 2021, 286, 119945 CrossRef CAS.
- R. L. Machunda, J. Lee and J. Lee, Surf. Interface Anal., 2010, 42, 564–567 CrossRef CAS.
- S. Oh, H. Park, H. Kim, Y. S. Park, M. G. Ha, J. H. Jang and S.-K. Kim, Coatings, 2020, 10, 341 CrossRef CAS.
- G. Zhang, Z.-J. Zhao, D. Cheng, H. Li, J. Yu, Q. Wang, H. Gao, J. Guo, H. Wang, G. A. Ozin, T. Wang and J. Gong, Nat. Commun., 2021, 12, 5745 CrossRef CAS PubMed.
- A. Conte, M. Baron, S. Bonacchi, S. Antonello and A. Aliprandi, Nanoscale, 2023, 15, 3693–3703 RSC.
- S. T. Ahn, S. Sen and G. T. R. Palmore, Nanoscale, 2022, 14, 13132–13140 RSC.
- P. De Luna, R. Quintero-Bermudez, C.-T. Dinh, M. B. Ross, O. S. Bushuyev, P. Todorović, T. Regier, S. O. Kelley, P. Yang and E. H. Sargent, Nat. Catal., 2018, 1, 103–110 CrossRef CAS.
- S. Nitopi, E. Bertheussen, S. B. Scott, X. Liu, A. K. Engstfeld, S. Horch, B. Seger, I. E. L. Stephens, K. Chan, C. Hahn, J. K. Nørskov, T. F. Jaramillo and I. Chorkendorff, Chem. Rev., 2019, 119, 7610–7672 CrossRef CAS PubMed.
- W. Tang, A. A. Peterson, A. S. Varela, Z. P. Jovanov, L. Bech, W. J. Durand, S. Dahl, J. K. Nørskov and I. Chorkendorff, Phys. Chem. Chem. Phys., 2012, 14, 76–81 RSC.
- F. S. Roberts, K. P. Kuhl and A. Nilsson, Angew. Chem., 2015, 127, 5268–5271 CrossRef.
- G. O. Larrazábal, V. Okatenko, I. Chorkendorff, R. Buonsanti and B. Seger, ACS Appl. Mater. Interfaces, 2022, 14, 7779–7787 CrossRef PubMed.
- W. Ye, X. Guo and T. Ma, Chem. Eng. J., 2021, 414, 128825 CrossRef CAS.
- T. Möller, F. Scholten, T. N. Thanh, I. Sinev, J. Timoshenko, X. Wang, Z. Jovanov, M. Gliech, B. Roldan Cuenya, A. S. Varela and P. Strasser, Angew. Chem., 2020, 132, 18130–18139 CrossRef.
- P. Grosse, D. Gao, F. Scholten, I. Sinev, H. Mistry and B. Roldan Cuenya, Angew. Chem., Int. Ed., 2018, 57, 6192–6197 CrossRef CAS PubMed.
- P. Grosse, A. Yoon, C. Rettenmaier, A. Herzog, S. W. Chee and B. Roldan Cuenya, Nat. Commun., 2021, 12, 6736 CrossRef CAS PubMed.
- S. Popović, M. Smiljanić, P. Jovanovič, J. Vavra, R. Buonsanti and N. Hodnik, Angew. Chem., 2020, 132, 14844–14854 CrossRef.
- D. T. Whipple, E. C. Finke and P. J. A. Kenis, Electrochem. Solid-State Lett., 2010, 13, B109 CrossRef CAS.
- B. Kim, S. Ma, H.-R. Molly Jhong and P. J. A. Kenis, Electrochim. Acta, 2015, 166, 271–276 CrossRef CAS.
- N. Kumar, G. D. Varma, R. Nath and A. K. Srivastava, Appl. Phys. A, 2011, 104, 1169–1174 CrossRef CAS.
- G. F. Samu, C. Visy, K. Rajeshwar, S. Sarker, V. R. Subramanian and C. Janáky, Electrochim. Acta, 2015, 151, 467–476 CrossRef CAS.
- B. Endrődi, A. Samu, E. Kecsenovity, T. Halmágyi, D. Sebők and C. Janáky, Nat. Energy, 2021, 6, 439–448 CrossRef.
- B. Endrődi, G. F. Samu, D. Fejes, Z. Németh, E. Horváth, A. Pisoni, P. K. Matus, K. Hernádi, C. Visy, L. Forró and C. Janáky, J. Polym. Sci., Part B: Polym. Phys., 2015, 53, 1507–1518 CrossRef.
- A. A. Samu, A. Kormányos, E. Kecsenovity, N. Szilágyi, B. Endrődi and C. Janáky, ACS Energy Lett., 2022, 7, 1859–1861 CrossRef CAS PubMed.
- A. Kormányos, B. Endrődi, Z. Zhang, A. Samu, L. Mérai, G. F. Samu, L. Janovák and C. Janáky, EES Catal., 2023, 1, 263–273 RSC.
- R. M. Arán-Ais, R. Rizo, P. Grosse, G. Algara-Siller, K. Dembélé, M. Plodinec, T. Lunkenbein, S. W. Chee and B. R. Cuenya, Nat. Commun., 2020, 11, 1–8 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Further characterization of the catalyst layers. See DOI: https://doi.org/10.1039/d3nr03834c |
| This journal is © The Royal Society of Chemistry 2023 |