
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
SARS-CoV-2 N-protein induces the formation of composite α-synuclein/N-protein fibrils that transform into a strain of α-synuclein fibrils†
Slav A.
Semerdzhiev
 a,
Ine
Segers-Nolten
a,
Paul
van der Schoot
b,
Christian
Blum
*a and
Mireille M. A. E.
Claessens
*a
a,
Ine
Segers-Nolten
a,
Paul
van der Schoot
b,
Christian
Blum
*a and
Mireille M. A. E.
Claessens
*a
aNanobiophysics, Faculty of Science and Technology, MESA + Institute for Nanotechnology and, Technical Medical Centre, University of Twente, P.O. Box 217, 7500 AE Enschede, The Netherlands. E-mail: m.m.a.e.claessens@utwente.nl; c.blum@utwente.nl
bSoft Matter and Biological Physics, Department of Applied Physics and Science Education, Eindhoven University of Technology, P.O. Box 513, 5600 MB Eindhoven, The Netherlands
First published on 18th October 2023
Abstract
The presence of deposits of alpha-synuclein (αS) fibrils in the cells of the brain is a hallmark of several α-synucleinopathies, including Parkinson's disease. As most disease cases are not familial, it is likely that external factors play a role in the disease onset. One of the external factors that may influence the disease onset is viral infection. It has recently been shown in in vitro assays that in the presence of SARS-Cov-2 N-protein, αS fibril formation is faster and proceeds in an unusual two-step aggregation process. Here, we show that faster fibril formation is not due to the SARS-CoV-2 N-protein-catalysed formation of an aggregation-prone nucleus. Instead, aggregation starts with the formation of a population of mixed αS/N-protein fibrils with low affinity for αS. Mixed amyloid fibrils, composed of two different proteins, have not been observed before. After the depletion of N-protein, fibril formation comes to a halt, until a slow transformation into fibrils with characteristics of a pure αS fibril strain occurs. This transformation into a strain of αS fibrils subsequently results in a second phase of fibril growth until a new equilibrium is reached. We hypothesize that this fibril strain transformation may be of relevance in the cell-to-cell spread of the αS pathology and disease onset.
Introduction
The presence of deposits of alpha-synuclein (αS) fibrils in the cells of the brain is a hallmark of α-synucleinopathies, including Parkinson's disease, dementia with Lewy bodies and multiple system atrophy. αS is a 140 amino acid intrinsically disordered protein. It is abundantly present in the brain, where it plays a role in membrane remodeling and membrane trafficking.1–6 For reasons that are not well understood, αS loses its physiological function and self-assembles into amyloid fibrils. These fibrils form deposits in disease-specific brain cells and regions. It has been suggested that, similar to prion diseases, the different pathologies are the result of αS amyloid fibrils with different structural characteristics, also referred to as fibril strains.7–9In the cell-to-cell transmission of the amyloid fold and the progression of the disease, the intrinsic architecture of the amyloid fold is replicated and preserved between cells.7 Which αS fibril strain is found in brain cells therefore likely depends on how the aggregation starts. Various experiments have shown that different external factors, including the presence of (poly)cations, virus proteins, lysozyme fibrils, and fine particulate matter, affect αS aggregation.10–14 Since most cases of α-synucleinopathies are not familial, external factors and/or changes in internal factors due to aging likely play a dominant role in disease onset.
One of the external factors that may influence disease onset is virus infection. For some viruses, this is well established in case studies or on an epidemiological level.15–19 In the context of the COVID pandemic, the first cases that connect SARS-CoV-2 infections to the development of Parkinson's disease (PD) have been reported.20–23 We have recently presented results that point toward direct interactions between the N-protein of SARS-CoV-2 and α-synuclein as a molecular basis for the observed relationship between SARS-CoV-2 infections and Parkinsonism. In the presence of N-protein, αS fibril formation is not only much faster, it also proceeds in an unusual two-step aggregation process in which two different fibril strains are formed.11 However, the specific mechanisms by which N-protein accelerates this aggregation, and whether N-protein becomes a permanent component of the resulting fibril, are still unknown. The question if N-protein is essential for fibril growth has significant implications, especially in the context of the transmission of the amyloid fold to cells that lack N-protein.
Here, we show that the observed aggregation kinetics results from the initial formation of a population of mixed αS/N-protein fibrils with low affinity for αS. After the depletion of N-protein, fibril formation grinds to a halt until a slow transformation to a pure αS fibril strain occurs. This pure αS fibril strain has a higher affinity for αS and dominates the fibril population at later times. Upon addition of monomeric αS, the first fibril strain only slowly elongates or requires the presence of additional N-protein. The second fibril strain readily seeds αS aggregation. In view of the cell-to-cell spread of a virus-induced αS pathology, we therefore hypothesize that this fibril strain conversion is required for the transmission of the amyloid fold between cells.
Results and discussion
Aggregation assays show that the αS fibril formation proceeds considerably faster in the presence of the SARS-CoV-2 N-protein. Moreover, a two-step aggregation process is observed (Fig. 1a).11 In the presence of the amyloid reporter dye thioflavin T (ThT), we observe an increase of ThT emission after a lag time, t0, to a first plateau, p1. After some period of time in which the ThT intensity remains constant, the ThT emission increases again to a final plateau, p2 (Fig. 1a). The nature of the two plateaus and that of the transition is unknown. To characterize the fibrils formed at the plateaus p1 and p2 and those during the transition from p1 to p2, we recorded AFM images at different times. From these AFM images we obtained the helical pitch via a discrete Fourier transform. We observe that going from p1 to p2, the helical pitch of the fibrils changes. Initially, in p1, fibrils with a helical pitch of approximately 86 nm dominate. In due course, an increasing number of fibrils with a pitch of 126 nm appear. Finally, at p2, fibrils with a pitch of approximately 126 nm dominate (Fig. 1b and c). In addition, we observe a change in the height of the fibrils going from p1 to p2 (Fig. 1d). At p1, the mean fibril height is 10.0 ± 1.8 nm, while at p2 the mean height is 8.5 ± 1.7 nm. The different fibril characteristics suggest that these are two different fibril strains. We refer to the fibrils with a pitch of 86 nm and a height of 10 nm as strain 1 (s1) fibrils and those with a pitch of 126 nm and a height of 8.5 nm as strain 2 (s2) fibrils.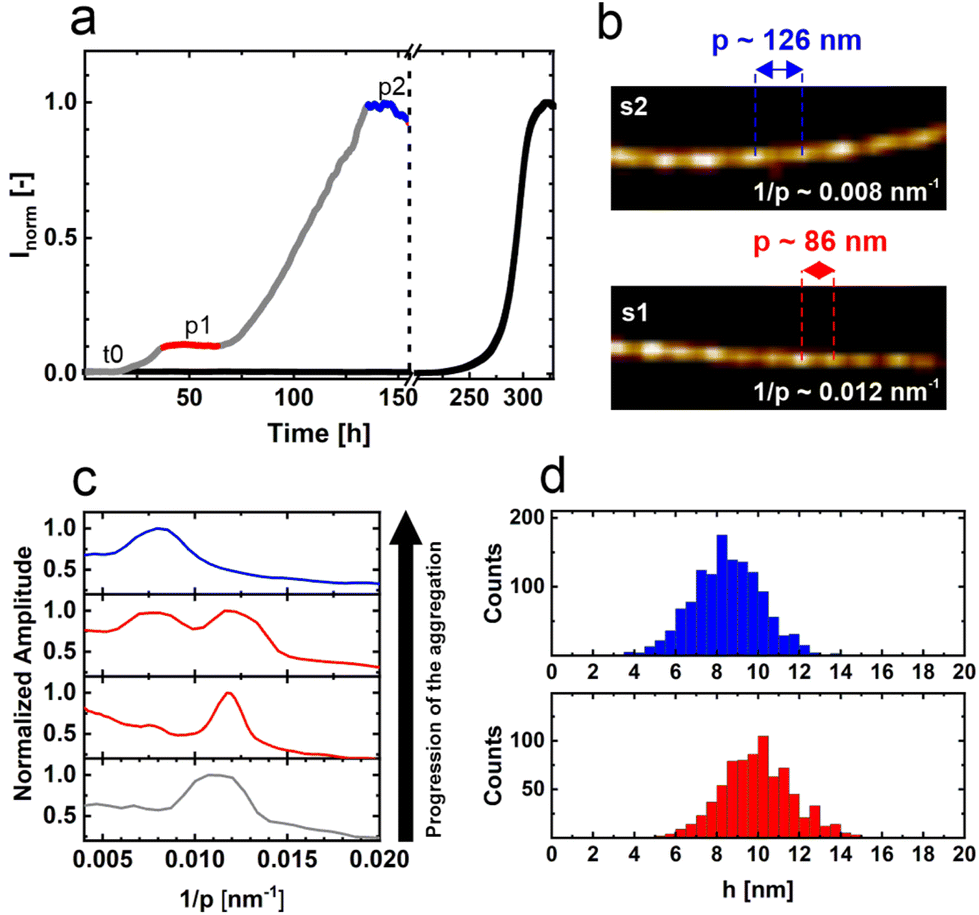 | ||
| Fig. 1 Aggregation of αS in the presence of N-protein. (a) Aggregation assay of 50 μM αS in the absence (black) and presence (grey) of 1 μM SARS-CoV-2 N-protein. The aggregation process is followed by recording the fluorescence of the amyloid-binding dye ThT. The aggregation of αS in the presence of N-protein is much faster compared to the aggregation in the absence of the N-protein. In the presence of N-protein, aggregation proceeds in two steps. After a lag time, t0, a plateau, p1 (red) is reached, and aggregation resumes after some time lag to a second plateau, p2 (blue). The dashed line marks the break in the x-axis. (b) AFM images of fibrils obtained from the stages p1 and p2. At p1 and p2, two different fibril strains s1 and s2 are observed. The mean periodicity p of s1 is 86 nm and that of s2 is 126 nm. (c) Discrete Fourier analysis showing the distribution of 1/p values obtained for fibrils during the aggregation from p1 to p2. The colors of the curves relate to the different phases of fibril growth shown in a. Initially, s1 fibrils with a periodicity of 86 nm dominate. In time, s2 fibrils appear, which are visible as a growing peak at 1/p = 0.008 nm−1. In stage p2, the s2 fibril strain dominates. (d) Height distributions and the mean heights of s1 and s2 fibrils. For s1 fibrils, the height is 10.0 ± 1.8 nm; for s2 fibrils, the height is 8.5 ± 1.7 nm. | ||
It is not clear what the origin of the two different ThT intensity plateaus is. It is well known that the fluorescence intensity of ThT bound to amyloid fibrils depends not only on the fibril mass but also on the fibril morphology.24 The two plateaus could thus just represent the two different fibril strains. To directly determine the fibril mass, we measured the residual αS monomer concentration in solution. To be able to discriminate between αS and N-protein, we used a fraction of fluorescently labelled αS in the aggregation. Samples were taken from the p1 and p2 plateaus, and the formed amyloid fibrils were spun down via high-speed centrifugation. The residual monomer concentration in the supernatant was determined by measuring the absorbance of the fluorescently labelled αS-AF647 at 650 nm and using the Lambert–Beer relation (Fig. 2a). In stage p1, we find an αS residual monomer concentration of 42 μM, whilst in stage p2 we measure a concentration of 26 μM. In p1, 8 μM of αS has aggregated into fibrils while in p2 the amount of αS aggregated increases by a factor three, to 24 μM. The fibril mass clearly increases going from p1 to p2, showing that an increase in the total fibril mass accompanies the two-step aggregation. However, the increase in ThT intensity is not proportional to the increase in fibril mass, which also supports the finding that p1 and p2 contain different fibril strains. The residual αS monomer concentrations at p1 and p2 show that the affinity for αS monomer addition is different for the two fibril strains. First, a fibril strain with lower αS affinity is formed; subsequently a fibril species with higher αS affinity appears. The plateaus in the ThT intensity trace represent the apparent equilibrium between amyloid fibrils and monomers.
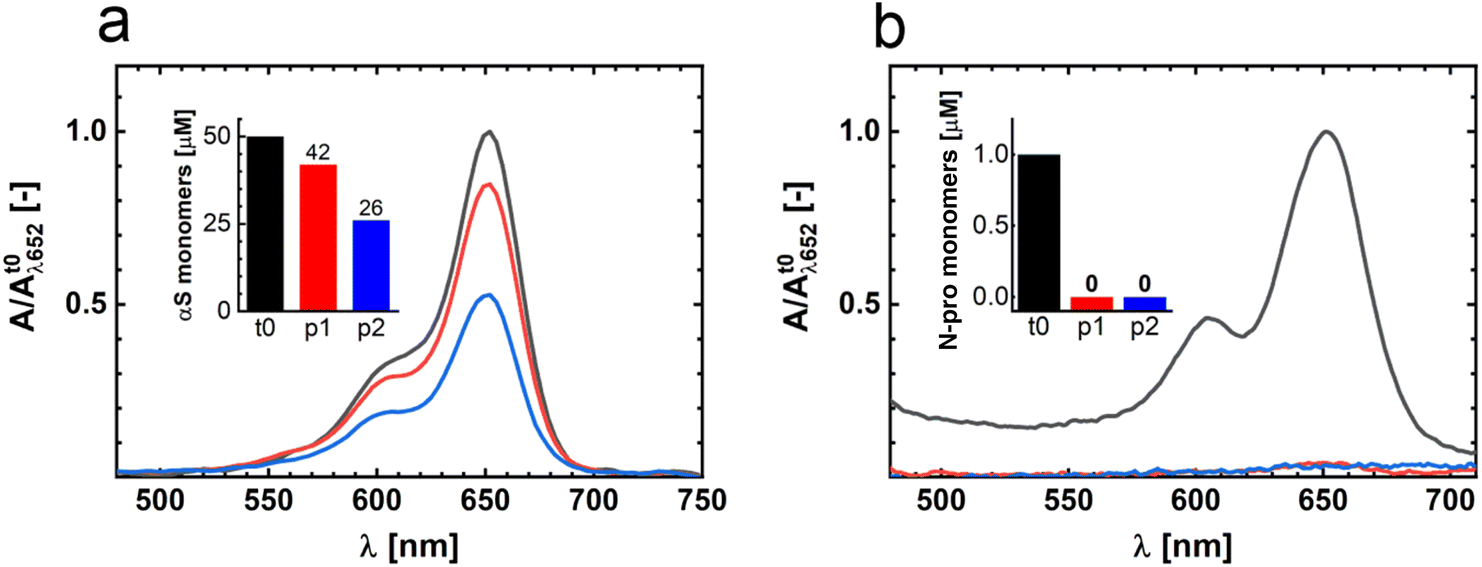 | ||
| Fig. 2 Residual monomer αS and N-protein concentrations in solution. (a) Absorbance spectra of αS-AF647 in the supernatant after spinning down the fibrils. Data are shown for samples obtained in the stages t0 (black), p1 (red) and p2 (blue). The data are peak-normalized to the t0-stage absorbance. The inset shows the derived total residual αS monomer concentrations at different stages of the aggregation process. At t0, the solution contained 50 μM αS and 1 μM N-protein. (b) The experiment was repeated with N-protein-AT647. At t0, the solution contained 50 μM αS and 1 μM N-protein. The absorbance spectra are recorded from the supernatant after spinning down the fibrils. Data are shown for samples obtained in the stages t0 (black), p1 (red) and p2 (blue). The data are peak-normalized to t0 absorbance. The inset shows the derived total residual N-protein concentrations at the different stages of aggregation. | ||
The N-protein induces the aggregation of αS. It has been shown that this proceeds via the formation of αS/N-protein complexes.11 Whether the N-protein acts as a catalyst that triggers aggregation, or plays a direct role in the formation of fibrils, remained unclear. To determine whether the N-protein is consumed in fibril formation, we use the same approach outlined above and determined the residual N-protein concentration in p1 and p2. In p1 and p2, we observe a depletion of the N-protein in the supernatant (Fig. 2b). We therefore conclude that the N-protein does not act as a catalyst, as it is not released after inducing αS aggregation.
Interestingly, after the transfer of the p1 fibrils to a high ionic strength solution, we find some N-protein back in solution (Fig. S1†). This indicates that a small fraction of the net positively charged N-protein binds to the amyloid fibrils that contain the net negatively charged αS. The charge–charge interaction between N-protein and pure preformed αS fibrils is further confirmed by direct observation (Fig. S2†). However, this adsorption of N-protein to the amyloids cannot explain the change in helical pitch observed going from s1 to s2. This, together with the small fraction of N-protein that we find adsorbed to fibrils, suggests that (part of) the N-protein is an integral part of the s1 fibrils. N-protein is a reaction partner in the formation of s1 fibrils.
In p1, αS and N-protein have been consumed in a ratio of approximately 8 to 1, and the supernatant is depleted of the N-protein, while αS is still present in excess. If N-protein is a reaction partner in fibril formation, it should therefore be possible to restart the growth of s1 fibrils. The addition of N-protein in p1 indeed results in a restart of fibril formation as evidenced by an immediate increase in the ThT intensity (Fig. 3a). The increase in the ThT signal levels off with time, as observed for the initial increase to p1. Fibril formation continues until N-protein is depleted from solution again. The newly formed fibril mass scales with the added amount of N-protein. Using fluorescently labelled N-protein in the aggregation confirms that N-protein is part of the formed fibrils. We observe a colocalization of fluorescence from the fluorescently labelled N-protein and ThT (Fig. 3b). To exclude the possibility that N-protein forms amyloid fibrils by itself, we isolated the s1 fibrils, removed the residual αS and subsequently added N-protein. The isolated s1 fibrils do not seed the formation of pure N-protein fibrils (Fig. 3c). Combined, these results show that both N-protein and αS are required for the formation of s1 fibrils. At p1, composite fibrils containing αS and N-protein in an overall ratio of approximately 8 to 1 have been formed. These composite fibrils have a periodicity of 86 nm and an approximate height of 10 nm. While coaggregation of different proteins has been observed in the past, these instances have typically involved heterologous seeding through secondary nucleation, resulting in separate fibrils formed from distinct proteins.10 However, the formation of mixed fibrils, which are composed of two different proteins, is a novel discovery to the best of our knowledge. We cannot resolve how the N-protein is distributed within the amyloid fibrils. We observe both N-protein and αS along the length of the s1 fibril but the optical resolution does not suffice to resolve if the distribution of N-protein and αS is blocky, periodic, statistical, or alternating. In solution, the N-protein dimerizes via the N-terminal dimerization domain. It is not unlikely that the dimeric structure of the N-protein is preserved in the fibril and that the low-complexity domain of the N-protein is part of the fibril core. The formation of composite fibrils containing N-protein is in line with reports on the formation of amyloid-like fibrils of N-protein in the presence of viral RNA.25 It has been observed that the interaction between the low-complexity domain of the N-protein and viral RNA facilitates fibril formation. The net negatively charged disordered αS may act in a similar way, resulting in composite αS/N-protein fibrils.
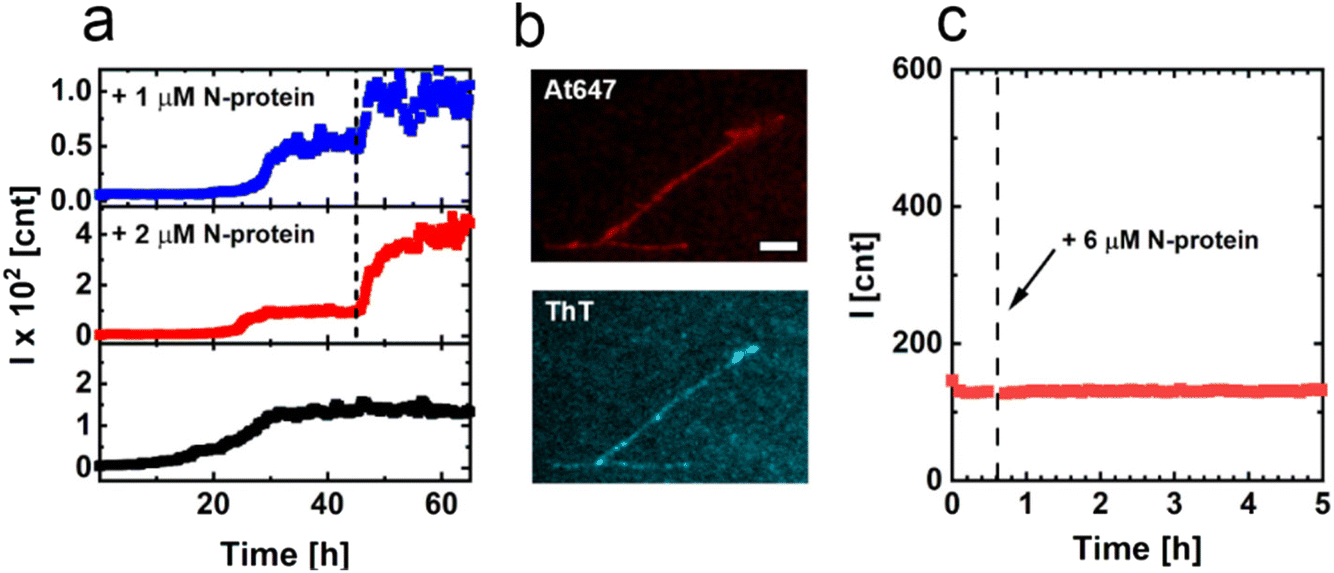 | ||
| Fig. 3 Seeded aggregations and fluorescence images of s1 fibrils. (a) ThT aggregation assay to monitor the restart of fibril formation upon the addition of N-protein. After reaching the plateau p1, N-protein was added at the time-point indicated by the dashed line (1 μM N-protein added (blue) and 2 μM N-protein added (red)). The black curve shows the aggregation to p1, and no additional N-protein was added. The addition of N-protein immediately restarts the aggregation. The increase in ThT emission reflects the added amount of N-protein. (b) Fluorescence microscopy images of fibrils formed in the presence of the At647-labelled N-protein. The top panel shows the emission from the At647-labelled N-protein. The bottom panel shows the ThT emission and confirms that the visualized structures are amyloid fibrils. (c) ThT assay of the resuspended isolated s1 fibrils. At the time-point indicated by the dashed line, N-protein was added. The addition of N-protein did not restart aggregation in the absence of αS. | ||
Since N-protein has been depleted in p1, while αS was still present in excess, the s2 fibrils must predominantly contain αS. Indeed, the characteristics of the s2 fibrils resemble the characteristics of αS fibrils formed in the absence of N-protein under comparable conditions. The fibril periodicity, height and the residual monomer concentration matches the values found for pure αS fibrils.26,27 In agreement with this, we observe that s2 fibrils can seed αS aggregation. The addition of 50 μM αS to s2 fibrils under quiescent conditions resulted in an immediate start and linear increase of the ThT intensity with time (Fig. S3†). The linear increase in ThT intensity with time evidences that the increase in fibril mass is dominated by the addition of αS monomers as expected under these conditions. We conclude that the different height and periodicity observed for s1 fibrils results from the incorporation of N-protein.
The depletion of N-protein is the reason for the observed plateau p1. Next, we investigated the mechanism that underlies the restart of the aggregation and the formation of s2 fibrils. The formation of two different fibril strains in a two-step aggregation process has been observed before. The aggregation of a mixture of the amyloid peptides Aβ40 and Aβ42 results in an apparent two-step aggregation behavior due to the formation of two different fibril strains, which independently form following different lag times.28 However, in our experiments, the aggregation towards p2 is fast compared to the de novo formation of αS fibrils (Fig. 1a). We therefore rule out that the two-step aggregation process is the result of two independent nucleation processes of composite αS /N-protein fibrils and pure αS fibrils. Another mechanism that could account for the observed two-step aggregation process is surface-catalyzed secondary nucleation (SCSN) of amyloids, where the surface of the composite fibrils nucleates the formation of pure αS fibrils.29,30 To test if SCSN is responsible for the observed behavior, we performed the aggregation of αS under quiescent conditions in the presence of s1 fibrils. The experiments were performed with an excess of αS at different αS concentrations. At the start of the experiment, we observed not a concave but a linear increase of the ThT signal for all αS concentrations tested (Fig. 4). Upon the depletion of αS, the curves level off, and the plateau ThT intensity scales with the αS concentration added. The plateau ThT intensity hence reflects the total mass of the fibrils. The fact that we observe a linear and not a concave increase in ThT intensity with time points to growth of the fibrils from the fibril ends. If the aggregation kinetics is governed by the rate of addition of monomers to the fibril ends, the aggregation can be treated as a bimolecular reaction of the first order.27 When we plot the initial slope of the ThT curves as a function of the added αS monomer concentration, we indeed observe a linear relationship (Fig. 4b). This linear increase of the initial growth rate with the added αS monomer concentration signifies that the addition of αS to s1 fibrils results in a bimolecular reaction of the first order. In this case, the increase in ThT intensity with time can also be described as end-growth/evaporation kinetics.31 Fitting the increase of the ThT intensity to the end-growth/evaporation model (see Materials and methods section), we obtain a constant intrinsic relaxation time of 10.2 ± 1.4 hours over all five experiments, which confirms that growth occurs from the fibril ends. We therefore conclude that the transition into the second plateau is not caused by the nucleation of new fibrils via SCSN.
 | ||
| Fig. 4 Aggregation of αS in the presence of fibrils from p1. (a) Increase in ThT emission as a function of time for increasing concentrations of added monomeric αS, 333 μM (black), 267 μM (red), 200 μM (blue), 133 μM (green), 67 μM (purple) and a constant concentration (∼4 μM equivalent monomer) of p1 fibrils. (b) Initial slope of the ThT emission increase observed for the different added αS monomer concentrations. The color coding is the same as shown in (a). | ||
In the absence of new nucleation events, the fibrils themselves have to transform, and the obtained data suggest that s1 fibrils elongate and transform into the s2 phenotype. The observation of the two fibril strains and the transformation from one to the other is reminiscent of the sergeant–soldier mechanism observed in the amplification of chirality in supramolecular polymers.32–34 At p1, the presence of small amounts of N-protein (sergeants) dictates how αS (soldiers) are incorporated into the s1 fibrils. With the depletion of the N-protein from solution upon the growth of the s1 fibrils, this structural dictate weakens. In the absence of the N-protein, fibril growth will be determined by αS, which eventually results in s2 fibrils with a structural characteristic that resembles more that of the pure αS fibrils (Fig. 1c).
To directly visualize this fibril growth, we started the aggregation of αS in the presence of N-protein with a fraction of αS labelled with αS-AF488 under mild agitation. After reaching p1, αS conjugated with αS-AF647 was added and aggregation was allowed to proceed (Fig. 5 schematic). Fluorescence microscopy images of the resulting s2 fibrils are shown in Fig. 5 and Fig. S4.† The images reveal the presence of segments of different colors, where the initial s1 fibrils (green) are elongated by the later added αS (red). Besides fibrils that contain segments of different colors, we also observe fibrils of only one color (ESI†). We attribute the presence of one-color fibrils to fibril breaking. The presence of segments of different colors within one fibril is expected since in the absence of secondary nucleation processes growth of amyloid fibrils occurs from fibril ends. New αS-AF647 add to the ends of the αS-AF488-labelled fibrils. For amyloid fibrils, monomer dissociation is typically slow, which limits the exchange of the N-protein and αS. Once the N-protein is incorporated into the composite fibril, it is likely to stay there. The s1 fibril may retain its structure and elongate into s2 fibrils with different characteristics. We were, however, not able to unequivocally identify s1 domains in s2 fibrils in the AFM images. While it is possible to globally discriminate the s1 and s2 morphologies, the contrast may not be large enough to identify differences in periodicity and height within short segments. Alternatively, the stresses that are created in the growing fibrils due to the absence of N-protein (sergeants) may cause the s1 fibrils to deform and reorganize into a mixed fibril with s2 characteristics. This fibril strain transformation is a chance process; the increase in fibril mass is initially very slow, resulting in a near constant ThT signal at p1. With the occurrence of a fibril strain transformation, the fibrils elongate up to a point where sample agitation causes the elongated fibrils to break, creating more fibrils. The increase in the number of ends results in an exponential increase of the fibril mass towards p2.
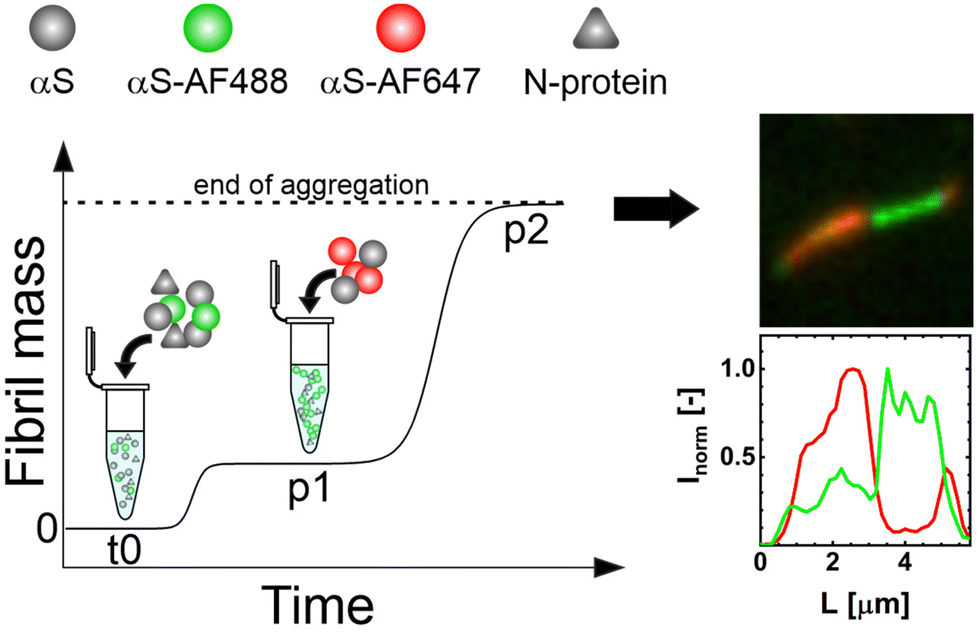 | ||
| Fig. 5 Visualizing the two steps of αS aggregation in the presence of N-protein. The starting αS/N-protein aggregation mixture in stage t0 contains a fraction of αS-AF488 (green). Once the first plateau p1 is reached, αS-AF647 is added (red). The resulting fibrils were imaged using TIRF microscopy after p2 was reached. The resulting fibrils clearly show segments of different colors. This evidences that the s1 fibril strain extends and converts into the s2 fibril strain. | ||
Our study underscores a potential link between SARS-CoV-2 infection and the development of synucleopathies through the interaction between the viral N-protein and αS. Initially, N-protein and αS form mixed fibrils, which require N-protein for propagation, rendering them irrelevant for disease onset in uninfected cells. However, these composite fibrils can transform into an αS fibril strain that no longer needs N-protein and is likely able to propagate in cells irrespective of SARS-CoV-2 infection. Notably, the transformation into the αS strain is faster than the de novo αS fibril formation, suggesting that viral infection may accelerate synucleinopathy development. We hypothesize that fibril strain conversion is crucial for the cell-to-cell transmission of virus-induced αS pathology. This conversion effectively erases the viral signature, making it difficult to identify viral infection as the initial trigger for disease onset. After strain conversion, the structural characteristics of the new fibrils become indistinguishable from those commonly associated with various synucleinopathies, further complicating the traceability of external factors in disease development.
Conclusions
In summary, the SARS-CoV-2 N-protein does not induce αS aggregation by catalysing the formation of an aggregation-prone nucleus. Instead, the N-protein and αS co-aggregate resulting in the formation of a composite αS/N-protein fibril of which the N-protein is an integral part. Although in the fibril the N-protein/αS ratio is small, the presence of N-protein dictates a different fibril morphology compared to that of pure αS fibrils. The observation that a few N-proteins can direct the organization of a large number of αS proteins has not been observed before for amyloid fibrils but is reminiscent of the sergeant-and-soldier principle known from the amplification of chirality in supramolecular chemistry.33–36 The composite fibrils have relatively low affinity for αS and, therefore, do not deplete the solution of αS. After reaching the equilibrium between αS, N-protein and the composite fibril, the increase in fibril mass comes to an apparent stop. However, with the depletion of N-protein, the morphology of the fibrils changes. At the fibril ends, lengthening of the composite fibril by the homo-aggregation of αS results in a slow fibril strain conversion to s2 fibrils. This transformation into a strain of αS fibrils subsequently results in a second phase of fibril growth until a new equilibrium is reached. The fibrils of the second fibril strain resemble pure αS fibrils and have a much higher affinity for the αS monomers. Our study reveals a potentially significant process where the rapid formation of a composite fibril strain, requiring SARS-CoV-2's N-protein for propagation, transforms into an αS fibril strain, which can propagate without N-protein, even in uninfected cells. This transformation, being faster than the de novo αS fibril formation, potentially triggers and accelerates synucleinopathy development.Materials and methods
Preparation and labelling of αS monomers
The expression of the human αS wild-type (αS), and the αSA140C and αSA18C mutants with single alanine to cysteine substitutions, was performed in E. coli B121 (DE3) using the pT7-7-based expression system. Details on the purification procedure for αS and αS140C are described elsewhere.37 SDS-Page gels of the purified αS protein can be found in the ESI (Fig. S5†). The αSA140C and αSA18C were conjugated with the maleimide derivative of AlexaFluor 568 (AF568), AlexaFluor 488 (AF488) or AlexaFluor 647 (AF647) (Thermo Fisher Scientific, USA), targeting the cysteine residues. The following fluorescent αS conjugates were used in this work: αSA140C-AF488, αSA18C-AF647 and αSA18C-AF568. We will refer to the labelled proteins with the shorthand notations αS-AF488, αS-AF647 and αS-AF568, respectively. For the labeling, each of the αS mutants was first incubated with 2 mM DTT to ensure the reduction of any present disulfide bridges. The DTT was removed using a 2 ml ZebaSpin MWCO 7 kDa desalting column (Thermo Fisher Scientific, USA). The column was pre-equilibrated by washing it with 1 ml of 10 mM TRIS buffer at pH 7.5 for three consecutive times by spinning them at 1000×g for 2 minutes for each washing step. The protein solution containing the respective αS mutants was placed on the equilibrated column and was spun down for 2 minutes at 1000×g. The DTT-free eluate containing the αS mutant of interest was collected. 100 μM of αSA140C or αSA18C was incubated with 2-fold excess of the corresponding AlexaFluor dye at room temperature, in the dark and under mild agitation for the duration of two hours. To separate the labeled protein from the free dye, the reaction mixture was transferred to a 2 ml ZebaSpin MWCO 7 kDa (Thermo Fisher Scientific, USA) column that was washed in advance 3 times with 10 mM TRIS buffer at pH 7.5. The column with the dye–protein reaction mixture was spun down for 2 minutes at 1000×g and the eluate containing the labeled protein was collected for further use and characterization. The degree of labeling (DOL) was determined by means of UV-vis absorbance spectroscopy. The DOL was approximately 0.5, 1 and 1 for the αS-AF647, αS-AF488 and αS-AF568, respectively.Preparation and labelling of the SARS-CoV-2 N-protein
The SARS-CoV-2 N-protein was recombinantly produced in our laboratory using plasmid nr. #157867 (Addgene, USA), following the procedure described in detail elsewhere.38 SDS-Page gels of the purified N-protein can be found in the ESI (Fig. S5†). In all experiments, we used N-protein in the concentration range that shows a strong decrease in the αS aggregation lag time.11 The purified N-protein was labeled using Atto 647 NHS – ester (Atto-Tec, Germany) targeting the accessible amino groups of the protein. 40 μM N-protein in 20 mM Hepes, pH = 8, and 100 mM NaCl were incubated with 3-fold excess of Atto 647N NHS at room temperature, under gentle agitation, for 2 hours in the dark. The reaction mixture was then transferred to a 2 ml ZebaSpin MWCO 7 kDa desalting column (Thermo Fisher Scientific, USA) to separate the labeled protein from the free dye. The columns were pre-equilibrated by washing them with 20 mM Hepes, pH = 8, 100 mM NaCl for three consecutive times by spinning them at 1000×g for 2 minutes for each washing step. The reaction mixture was spun down for 2 minutes at 1000×g and the eluate containing the labeled protein was collected for further characterization. The degree of labeling (DOL) of ∼1.8 was determined by means of UV-vis absorbance spectroscopy. We will refer to the labelled protein as N-protein-At647.THT aggregation assays
In the aggregation assays, a solution of 50 μM αS and 1 μM N-protein in 20 mM Tris buffer (Sigma-Aldrich, UK), pH = 7.4, 5 μM ThT (Fluka, Sigma-Aldrich, UK), and 10 mM NaCl (Sigma-Aldrich, USA) was placed in the wells of a 96-well half-area clear flat-bottomed polystyrene NBS (low bind) microplate (3881, Corning, USA). Samples were prepared in 3 replicates of 50 μL. The aggregation proceeded while shaking at 500 rpm at 37 °C. To follow the aggregation, the increase in ThT fluorescence was monitored using a plate reader (Infinite 200 Pro, Tecan Ltd, Switzerland). The ThT dye was excited at 446 nm and the fluorescence signal was measured at 485 nm every 10 min. Both the N-protein and αS have been reported to phase-separate into liquid condensates at high concentrations.38–40 In phase contrast microscopy, we observe no signs of phase separation at the protein concentrations and buffer conditions used here.Atomic force microscopy
The samples from the aggregation mixture (50 μM αS, 1 μM N-protein) were diluted five times, deposited onto freshly cleaved mica (Muscovite mica, V-1 quality, EMS, USA), and left to rest for 5 minutes. The sample was carefully washed 3 times with 20 μL of demineralized water (Milli-Q) and gently dried under flow (low flow rate) of nitrogen gas. AFM images were obtained using a BioScope Catalyst (Bruker, USA) in soft tapping mode using a silicon probe, NSC36 tip B with a force constant of 1.75 N m−1 (MikroMasch, Bulgaria). Images were captured with a resolution of 512 × 512 (10 μm × 10 μm) pixels per image at a scan rate of 0.2 to 0.5 Hz. AFM images were processed using the Scanning Probe Image Processor (SPIP, Image Metrology, Denmark) and Nanoscope Analysis (Bruker, USA) packages. The fibril length and periodicity morphology were analyzed using a custom fibril analysis MATLAB script and the FiberApp package.41Determination of the residual monomer concentration
The residual αS monomer concentration was determined by first aggregating 49 μM αS, 1 μM αS-AF647 (DOL = 0.5) and 1 μM N-protein in conditions identical to those described in the Aggregation assays section. Samples were taken out of the well plate at either p1 or p2 and were spun down for 1 hour at 18 000×g. The absorbance of the supernatant was measured at λ = 650 nm using the Nanodrop ND-1000 (ThermoFisher Scientific, USA) or the UV-2401 (Shimadzu, Japan) UV-vis spectrophotometer. The concentration of αS-AF674 was determined spectroscopically and from this the total residual αS monomer concentration was derived by considering the ratio between labelled and unlabelled αS.The residual N-protein concentration was determined by first aggregating 50 μM αS, 0.5 μM N-protein and 0.5 μM N-protein-At647 (DOL = 1.8) at conditions identical to those described in the Aggregation assays section. Samples were taken out of the well plate at either p1 or p2 and the residual N-protein concentration was determined in an identical manner as for αS (see above). An additional set of samples from p1 and p2 was incubated at high-ionic strength conditions (0.5 M NaCl) for 1 hour to minimize adsorption of the net positively charged N-protein to the fibrils, which are mainly composed of net negatively charged αS. After that the residual N-protein monomer concentration was determined in the same way as it was done for αS and N-protein without the high-salt incubation step (Fig. S1†).
Aggregation in the presence of fibrils from p1 and p2
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000×g. The pellet with p1 fibrils was resuspended in 20 mM Tris buffer and 10 mM NaCl. The solution of resuspended p1 fibrils was placed in a well and the well plate was returned to the plate reader. After ∼45 min, the N-protein concentration was increased by 6 μM. After the addition of extra N-protein, the measurement was continued.
000×g. The pellet with p1 fibrils was resuspended in 20 mM Tris buffer and 10 mM NaCl. The solution of resuspended p1 fibrils was placed in a well and the well plate was returned to the plate reader. After ∼45 min, the N-protein concentration was increased by 6 μM. After the addition of extra N-protein, the measurement was continued.
In this equation, Iinf represents the plateau value of the thioflavin T (ThT) fluorescence intensity, I0 is the initial intensity at the beginning of the experiment, and τ stands for the intrinsic relaxation time.

Microscopy
Visualizing the two steps of αS aggregation in the presence of N-protein
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to thank Kirsten A. van Leijenhorst-Groener for the production of the recombinant proteins. We are grateful to the Dutch Parkinson's disease foundation “Stichting ParkinsonFonds” for financial support.References
- D. Snead and D. Eliezer, Alpha-synuclein function and dysfunction on cellular membranes, Exp. Neurobiol., 2014, 23, 292–313 CrossRef PubMed.
- J. Lautenschlager, C. F. Kaminski and G. S. Kaminski Schierle, alpha-Synuclein - Regulator of Exocytosis, Endocytosis, or Both?, Trends Cell Biol., 2017, 27, 468–479 CrossRef CAS PubMed.
- J. Burre, M. Sharma, T. Tsetsenis, V. Buchman, M. R. Etherton and T. C. Sudhof, Alpha-synuclein promotes SNARE-complex assembly in vivo and in vitro, Science, 2010, 329, 1663–1667 CrossRef CAS PubMed.
- T. Logan, J. Bendor, C. Toupin, K. Thorn and R. H. Edwards, alpha-Synuclein promotes dilation of the exocytotic fusion pore, Nat. Neurosci., 2017, 20, 681–689 CrossRef CAS PubMed.
- M. A. Fakhree, N. Zijlstra, C. C. Raiss, C. J. Siero, H. Grabmayr, A. R. Bausch, C. Blum and M. M. Claessens, The number of alpha-synuclein proteins per vesicle gives insights into its physiological function, Sci. Rep., 2016, 6, 30658 CrossRef CAS PubMed.
- M. A. A. Fakhree, I. B. M. Konings, J. Kole, A. Cambi, C. Blum and M. Claessens, The Localization of Alpha-synuclein in the Endocytic Pathway, Neuroscience, 2021, 457, 186–195 CrossRef CAS PubMed.
- L. Bousset, L. Pieri, G. Ruiz-Arlandis, J. Gath, P. H. Jensen, B. Habenstein, K. Madiona, V. Olieric, A. Bockmann, B. H. Meier and R. Melki, Structural and functional characterization of two alpha-synuclein strains, Nat. Commun., 2013, 4, 2575 CrossRef PubMed.
- W. Peelaerts, L. Bousset, A. Van der Perren, A. Moskalyuk, R. Pulizzi, M. Giugliano, C. Van den Haute, R. Melki and V. Baekelandt, alpha-Synuclein strains cause distinct synucleinopathies after local and systemic administration, Nature, 2015, 522, 340–344 CrossRef CAS PubMed.
- Y. Yang, Y. Shi, M. Schweighauser, X. Zhang, A. Kotecha, A. G. Murzin, H. J. Garringer, P. W. Cullinane, Y. Saito, T. Foroud, T. T. Warner, K. Hasegawa, R. Vidal, S. Murayama, T. Revesz, B. Ghetti, M. Hasegawa, T. Lashley, S. H. W. Scheres and M. Goedert, Structures of α-synuclein filaments from human brains with Lewy pathology, Nature, 2022, 610, 791–795 CrossRef CAS PubMed.
- J. Vaneyck, I. Segers-Nolten, K. Broersen and M. Claessens, Cross-seeding of alpha-synuclein aggregation by amyloid fibrils of food proteins, J. Biol. Chem., 2021, 296, 100358 CrossRef CAS PubMed.
- S. A. Semerdzhiev, M. A. A. Fakhree, I. Segers-Nolten, C. Blum and M. Claessens, Interactions between SARS-CoV-2 N-Protein and alpha-Synuclein Accelerate Amyloid Formation, ACS Chem. Neurosci., 2022, 13, 143–150 CrossRef CAS PubMed.
- T. Antony, W. Hoyer, D. Cherny, G. Heim, T. M. Jovin and V. Subramaniam, Cellular polyamines promote the aggregation of alpha-synuclein, J. Biol. Chem., 2003, 278, 3235–3240 CrossRef CAS PubMed.
- X. Yuan, Y. Yang, C. Liu, Y. Tian, D. Xia, Z. Liu, L. Pan, M. Xiong, J. Xiong, L. Meng, Z. Zhang, K. Ye, H. Jiang and Z. Zhang, Fine Particulate Matter Triggers alpha-Synuclein Fibrillization and Parkinson-like Neurodegeneration, Mov. Disord., 2022, 37, 1817–1830 CrossRef CAS PubMed.
- A. Binolfi, R. M. Rasia, C. W. Bertoncini, M. Ceolin, M. Zweckstetter, C. Griesinger, T. M. Jovin and C. O. Fernandez, Interaction of alpha-synuclein with divalent metal ions reveals key differences: a link between structure, binding specificity and fibrillation enhancement, J. Am. Chem. Soc., 2006, 128, 9893–9901 CrossRef CAS PubMed.
- H. Vlajinac, E. Dzoljic, J. Maksimovic, J. Marinkovic, S. Sipetic and V. Kostic, Infections as a risk factor for Parkinson's disease: a case-control study, Int. J. Neurosci., 2013, 123, 329–332 CrossRef PubMed.
- C. P. Maurizi, Why was the 1918 influenza pandemic so lethal? The possible role of a neurovirulent neuraminidase, Med. Hypotheses, 1985, 16, 1–5 CrossRef CAS PubMed.
- R. T. Ravenholt and W. H. Foege, 1918 influenza, encephalitis lethargica, parkinsonism, Lancet, 1982, 2, 860–864 CrossRef CAS PubMed.
- M. A. Harris, J. K. Tsui, S. A. Marion, H. Shen and K. Teschke, Association of Parkinson's disease with infections and occupational exposure to possible vectors, Mov. Disord., 2012, 27, 1111–1117 CrossRef PubMed.
- A. Calculli, T. Bocci, M. Porcino, M. Avenali, C. Casellato, S. Arceri, S. Regalbuto, A. Priori and A. Pisani, Parkinson disease following COVID-19: Report of six cases, Eur. J. Neurol., 2023, 30, 1272–1280 CrossRef PubMed.
- M. Merello, K. P. Bhatia and J. A. Obeso, SARS-CoV-2 and the risk of Parkinson's disease: facts and fantasy, Lancet Neurol., 2021, 20, 94–95 CrossRef CAS PubMed.
- M. E. Cohen, R. Eichel, B. Steiner-Birmanns, A. Janah, M. Ioshpa, R. Bar-Shalom, J. J. Paul, H. Gaber, V. Skrahina, N. M. Bornstein and G. Yahalom, A case of probable Parkinson's disease after SARS-CoV-2 infection, Lancet Neurol., 2020, 19, 804–805 CrossRef CAS PubMed.
- A. Mendez-Guerrero, M. I. Laespada-Garcia, A. Gomez-Grande, M. Ruiz-Ortiz, V. A. Blanco-Palmero, F. J. Azcarate-Diaz, P. Rabano-Suarez, E. Alvarez-Torres, C. P. de Fuenmayor-Fernández de la Hoz, D. Vega Pérez, R. Rodriguez-Montalban, A. Perez-Rivilla, J. Sayas Catalán, A. Ramos-Gonzalez and J. González de la Aleja, Acute hypokinetic-rigid syndrome following SARS-CoV-2 infection, Neurology, 2020, 95, e2109–e2118 CrossRef CAS PubMed.
- I. Faber, P. R. P. Brandão, F. Menegatti, D. D. de Carvalho Bispo, F. B. Maluf and F. Cardoso, Coronavirus Disease 2019 and Parkinsonism: A Non-post-encephalitic Case, Mov. Disord., 2020, 35, 1721–1722 CrossRef CAS PubMed.
- A. Sidhu, J. Vaneyck, C. Blum, I. Segers-Nolten and V. Subramaniam, Polymorph-specific distribution of binding sites determines thioflavin-T fluorescence intensity in alpha-synuclein fibrils, Amyloid, 2018, 25, 189–196 CrossRef CAS PubMed.
- E. Tayeb-Fligelman, X. Cheng, C. Tai, J. T. Bowler, S. Griner, M. R. Sawaya, P. M. Seidler, Y. X. Jiang, J. Lu, G. M. Rosenberg, L. Salwinski, R. Abskharon, C. T. Zee, K. Hou, Y. Li, D. R. Boyer, K. A. Murray, G. Falcon, D. H. Anderson, D. Cascio, L. Saelices, R. Damoiseaux, F. Guo and D. S. Eisenberg, Inhibition of amyloid formation of the Nucleoprotein of SARS-CoV-2, bioRxiv, 2021, 2021.03.05.434000 Search PubMed.
- A. Sidhu, I. Segers-Nolten, V. Raussens, M. M. Claessens and V. Subramaniam, Distinct Mechanisms Determine alpha-Synuclein Fibril Morphology during Growth and Maturation, Eur. J. Neurol., 2017, 8, 538–547 CAS.
- V. V. Shvadchak, M. M. Claessens and V. Subramaniam, Fibril breaking accelerates alpha-synuclein fibrillization, J. Phys. Chem. B, 2015, 119, 1912–1918 CrossRef CAS PubMed.
- R. Cukalevski, X. Yang, G. Meisl, U. Weininger, K. Bernfur, B. Frohm, T. P. J. Knowles and S. Linse, The Abeta40 and Abeta42 peptides self-assemble into separate homomolecular fibrils in binary mixtures but cross-react during primary nucleation, Chem. Sci., 2015, 6, 4215–4233 RSC.
- R. Gaspar, G. Meisl, A. K. Buell, L. Young, C. F. Kaminski, T. P. J. Knowles, E. Sparr and S. Linse, Secondary nucleation of monomers on fibril surface dominates alpha-synuclein aggregation and provides autocatalytic amyloid amplification, Q. Rev. Biophys., 2017, 50, e6 CrossRef PubMed.
- M. Tornquist, T. C. T. Michaels, K. Sanagavarapu, X. Yang, G. Meisl, S. I. A. Cohen, T. P. J. Knowles and S. Linse, Secondary nucleation in amyloid formation, Chem. Commun., 2018, 54, 8667–8684 RSC.
- A. N. Semenov and I. A. Nyrkova, End-growth/evaporation living polymerization kinetics revisited, J. Chem. Phys., 2011, 134, 114902 CrossRef CAS PubMed.
- J. van Gestel, P. van der Schoot and M. A. J. Michels, Amplification of Chirality in Helical Supramolecular Polymers, Macromolecules, 2003, 36, 6668–6673 CrossRef CAS.
- M. M. Green, M. P. Reidy, R. D. Johnson, G. Darling, D. J. O'Leary and G. Willson, Macromolecular stereochemistry: the out-of-proportion influence of optically active comonomers on the conformational characteristics of polyisocyanates. The sergeants and soldiers experiment, J. Am. Chem. Soc., 1989, 111, 6452–6454 CrossRef.
- L. J. Prins, J. Huskens, F. de Jong, P. Timmerman and D. N. Reinhoudt, Complete asymmetric induction of supramolecular chirality in a hydrogen-bonded assembly, Nature, 1999, 398, 498–502 CrossRef CAS.
- J. van Gestel, P. van der Schoot and M. A. J. Michels, in Molecular Gels: Materials with Self-Assembled Fibrillar Networks, ed. R. G. Weiss and P. Terech, Springer, Netherlands, 2006, pp. 79–97 Search PubMed.
- M. M. Smulders, I. A. Filot, J. M. Leenders, P. van der Schoot, A. R. Palmans, A. P. Schenning and E. W. Meijer, Tuning the extent of chiral amplification by temperature in a dynamic supramolecular polymer, J. Am. Chem. Soc., 2010, 132, 611–619 CrossRef CAS PubMed.
- S. A. Semerdzhiev, D. R. Dekker, V. Subramaniam and M. M. Claessens, Self-assembly of protein fibrils into suprafibrillar aggregates: bridging the nano- and mesoscale, ACS Nano, 2014, 8, 5543–5551 CrossRef CAS PubMed.
- T. M. Perdikari, A. C. Murthy, V. H. Ryan, S. Watters, M. T. Naik and N. L. Fawzi, SARS-CoV-2 nucleocapsid protein phase-separates with RNA and with human hnRNPs, EMBO J., 2020, 39, e106478 CrossRef CAS PubMed.
- H. Chen, Y. Cui, X. Han, W. Hu, M. Sun, Y. Zhang, P.-H. Wang, G. Song, W. Chen and J. Lou, Liquid–liquid phase separation by SARS-CoV-2 nucleocapsid protein and RNA, Cell Res., 2020, 30, 1143–1145 CrossRef CAS PubMed.
- A. Savastano, A. Ibáñez de Opakua, M. Rankovic and M. Zweckstetter, Nucleocapsid protein of SARS-CoV-2 phase separates into RNA-rich polymerase-containing condensates, Nat. Commun., 2020, 11, 6041 CrossRef CAS PubMed.
- I. Usov and R. Mezzenga, FiberApp: An Open-Source Software for Tracking and Analyzing Polymers, Filaments, Biomacromolecules, and Fibrous Objects, Macromolecules, 2015, 48, 1269–1280 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3nr03556e |
| This journal is © The Royal Society of Chemistry 2023 |