
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBiochemical transformations of inorganic nanomedicines in buffers, cell cultures and organisms
Anna L.
Neuer
 ab,
Inge K.
Herrmann
ab and
Alexander
Gogos
*ab
ab,
Inge K.
Herrmann
ab and
Alexander
Gogos
*ab
aLaboratory for Particles-Biology Interactions, Department of Materials Meet Life, Swiss Federal Laboratories for Materials Science and Technology (Empa), Lerchenfeldstrasse 5, 9014 St. Gallen, Switzerland. E-mail: alexander.gogos@empa.ch; Tel: +41 (0)58 765 78 05
bNanoparticle Systems Engineering Laboratory, Institute of Process Engineering, Department of Mechanical and Process Engineering, ETH Zurich, Sonneggstrasse 3, 8092 Zurich, Switzerland
First published on 30th October 2023
Abstract
The field of nanomedicine is rapidly evolving, with new materials and formulations being reported almost daily. In this respect, inorganic and inorganic–organic composite nanomaterials have gained significant attention. However, the use of new materials in clinical trials and their final approval as drugs has been hampered by several challenges, one of which is the complex and difficult to control nanomaterial chemistry that takes place within the body. Several reviews have summarized investigations on inorganic nanomaterial stability in model body fluids, cell cultures, and organisms, focusing on their degradation as well as the influence of corona formation. However, in addition to these aspects, various chemical reactions of nanomaterials, including phase transformation and/or the formation of new/secondary nanomaterials, have been reported. In this review, we discuss recent advances in our understanding of biochemical transformations of medically relevant inorganic (composite) nanomaterials in environments related to their applications. We provide a refined terminology for the primary reaction mechanisms involved to bridge the gaps between different disciplines involved in this research. Furthermore, we highlight suitable analytical techniques that can be harnessed to explore the described reactions. Finally, we highlight opportunities to utilize them for diagnostic and therapeutic purposes and discuss current challenges and research priorities.
Introduction
Over the past 25 years, nanotechnology has received increasing attention in the biomedical field. The design,1,2 tunability,3,4 biomedical efficacy,5 and characterization6,7 of nanomaterials (NMs) for biomedical applications have been widely studied. In addition to studies on organic nanomaterials, such as liposomes and micelles, detailed studies on inorganic and organic–inorganic composite NMs have been conducted.8,9 Such materials have been used extensively in applications, as imaging agents,10–13 in drug delivery systems,14,15 in radio-enhancement therapy,16–19 and in wound healing.20–22 They have also been used as theranostic agents (combining diagnostics and therapy)9,23 due their exclusive and tailorable properties.Nonetheless, compared to the abundance of nanomedical research, with around 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 publications per year, the clinical use of nanomedicines is greatly limited, with only about 200 organic and inorganic nanomedical drugs having entered clinical phases to date.24 Clinical translation remains challenging25 due to the oftentimes limited understanding and control over the nanomaterial chemistry during all stages of material life-cycle from stable storage to applications, covering bioavailability and fate within the body, metabolism and ultimately excretion/elimination processes. While many of these aspects have been well-characterized for organic nanomaterials (liposomes and micelles),26,27 research on the physical and chemical modification and transformation of inorganic NMs within the body is still scarce.28 Physical and chemical changes in NMs are related to modifications in NM composition and structure, and therefore to modified efficacy and safety profiles. Those factors need to be strictly controlled if NMs are intended for medical use, as NMs might undergo physical and chemical transformations upon interaction with the different chemical environments within the body, such as variations in pH, ion composition, and redox potential. In this context, research on NM stability has mostly focused on NM decomposition and degradation (often monitored as a loss of function over time),29 as well as on corona formation;30,31i.e., the formation of secondary (bio-)molecule layers on the NM surface. However, in biomedical research, there is a gap between the number of studies reporting the design of new NMs and those assessing NM fate, including primary reactions and secondary effects. Primary transformations may result in alterations in elemental speciation, crystal structure, material composition and/or morphology of the NM, as well as the generation of secondary nanoparticles. These alterations can have a major impact on the in vivo efficacy, toxicity, and fate. The occurrence and impact of such NM transformations have been widely acknowledged and studied in various fields, such as nano-environmental health and safety (nanoEHS),32–34 catalysis35 and energy applications.36–38 However, over the past several years, the importance and relevance of NM transformation have become also recognized by the biomedical field.39
000 publications per year, the clinical use of nanomedicines is greatly limited, with only about 200 organic and inorganic nanomedical drugs having entered clinical phases to date.24 Clinical translation remains challenging25 due to the oftentimes limited understanding and control over the nanomaterial chemistry during all stages of material life-cycle from stable storage to applications, covering bioavailability and fate within the body, metabolism and ultimately excretion/elimination processes. While many of these aspects have been well-characterized for organic nanomaterials (liposomes and micelles),26,27 research on the physical and chemical modification and transformation of inorganic NMs within the body is still scarce.28 Physical and chemical changes in NMs are related to modifications in NM composition and structure, and therefore to modified efficacy and safety profiles. Those factors need to be strictly controlled if NMs are intended for medical use, as NMs might undergo physical and chemical transformations upon interaction with the different chemical environments within the body, such as variations in pH, ion composition, and redox potential. In this context, research on NM stability has mostly focused on NM decomposition and degradation (often monitored as a loss of function over time),29 as well as on corona formation;30,31i.e., the formation of secondary (bio-)molecule layers on the NM surface. However, in biomedical research, there is a gap between the number of studies reporting the design of new NMs and those assessing NM fate, including primary reactions and secondary effects. Primary transformations may result in alterations in elemental speciation, crystal structure, material composition and/or morphology of the NM, as well as the generation of secondary nanoparticles. These alterations can have a major impact on the in vivo efficacy, toxicity, and fate. The occurrence and impact of such NM transformations have been widely acknowledged and studied in various fields, such as nano-environmental health and safety (nanoEHS),32–34 catalysis35 and energy applications.36–38 However, over the past several years, the importance and relevance of NM transformation have become also recognized by the biomedical field.39
In this review, we highlight studies on the transformation of inorganic and organic–inorganic composite NMs (metals, metal-oxides, and metal organic frameworks (MOFs)) that are relevant for various biomedical applications; such applications are realized under cell-free, in vitro, ex vivo, and in vivo conditions. Hereafter, “NMs” refers to inorganic and inorganic–organic composite materials and will be assessed with a focus on chemical transformation, including changes in element speciation, crystal structure, material composition and morphology,40 as well as the formation of secondary nanoscale phases from the parent material. Studies that solely investigate dissolution are excluded since they have been extensively reviewed elsewhere.29–31 Additionally, we point to successful measures that protect clinically relevant inorganic NMs from transformation, as well as specific transformations that might be harnessed to endow NMs with additional functionality. Moreover, we provide a comprehensive overview of suitable analytical techniques that can be leveraged to gain insights into the underlying reaction mechanisms for material transformations. Ultimately, a better understanding of potential transformation reactions allows for better safe-by-design approaches and tunable transformation for therapeutic, diagnostic, and elimination purposes.
Application routes for inorganic nanomedicines and associated biological environments
Application routes for inorganic nanomedicines in vivo can be divided into four principal categories: (i) oral administration,41 (ii) inhalation/tracheal administration,41 (iii) injection (which can further be divided into (iii.a) intravascular41–46 (IV) (iii.b) direct site47 and (iii.c) retroorbital48 injection (which is solely found in rodent studies)), as well as (iv) subcutaneous and transdermal49–52 application (see Fig. 1A). The route of administration is highly dependent on the intended medical application, the bioavailability, as well as the NM surface chemistry and size.53 The size and surface chemistry of NM are of paramount importance. As the size decreases, the proportion of surface atoms becomes increasingly pronounced, which in turn significantly influences the long-term fate of NM, especially under varying pH values and ionic strengths of the environment. The surface chemistry, along with any coatings, can serve dual roles: they can act as protective layers, stabilizing the NM, or they can actively modulate interactions with the surrounding environment. This surface chemistry determines the charge, hydrophobicity, and affinity of the NM, all of which play pivotal roles in how these materials interact with biological systems.54 The application route defines the local fate of the NM as well as the chemical environment the NM is first exposed to. After oral administration, studied typically with oral gavage in rodents,41 the NM is exposed to the milieus of the gastrointestinal (GI) tract starting from the stomach, all the way to the small intestine, and then to the large intestine (SI/LI). This involves drastic changes in pH, from pH 1.0–3.0 (stomach) to pH 5.4–7.5 (SI) and pH 6.6–7.4 (LI)55 (Fig. 1B). Additionally, digestive enzymes may be present, which, again, is strongly dependent on the exact anatomical location. The entire gastrointestinal system is lined by a mucosal layer, separating the cells of the gastrointestinal tract from its contents. In a healthy GI tract, the mucosal layer shields the intestinal cells, to a certain extent, from interactions with ingested nanomaterials. However, disruption of this barrier, e.g., due to inflammatory bowel disease (IBD), has shown altered exposure of the GI to ingested NMs, hence indicating a strong dependence of NM fate on the barrier being intact.56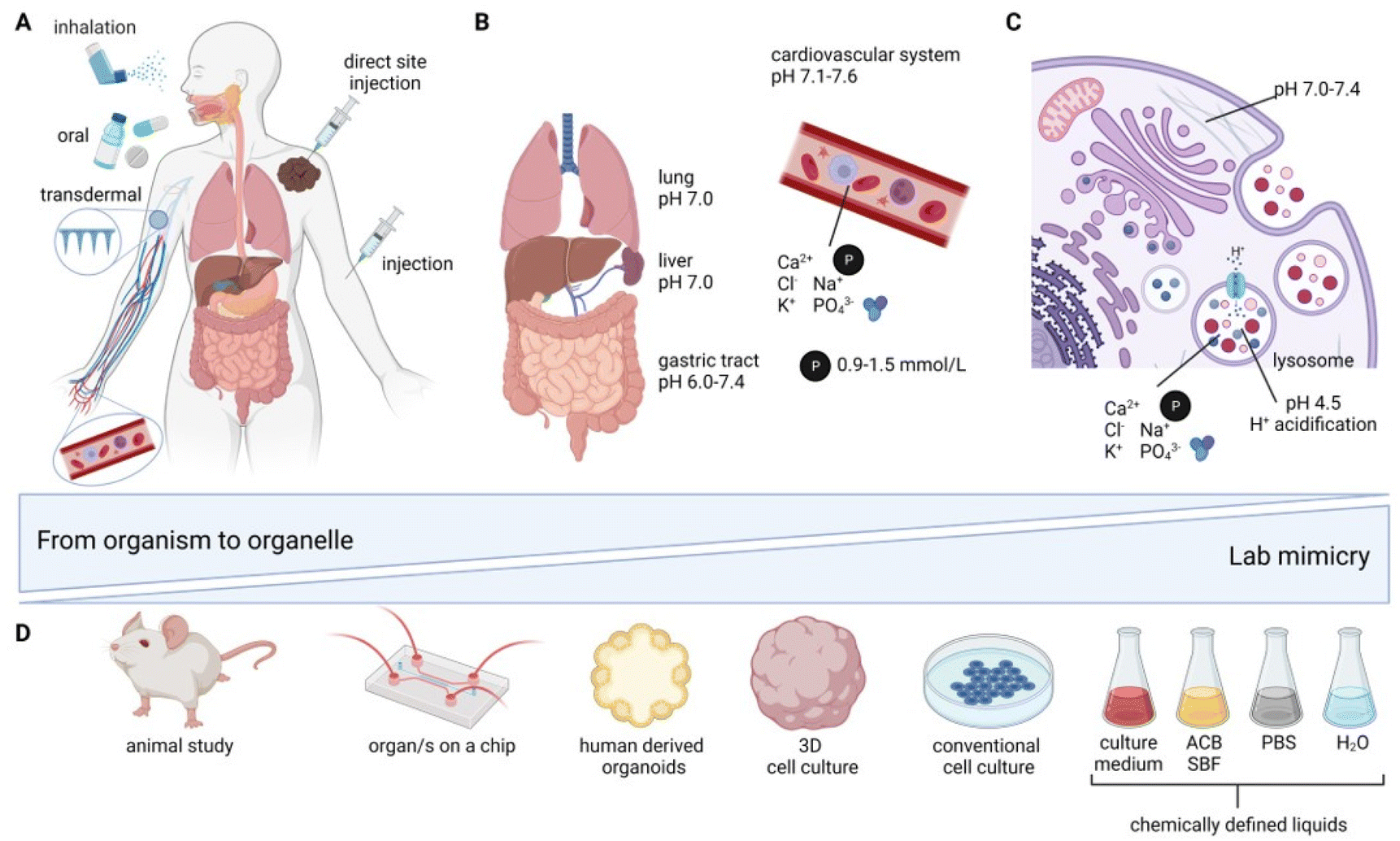 | ||
| Fig. 1 Application routes for inorganic nanomedicines and associated biological environments (A) in a whole organism, (B) at the organ level with tissue-specific pH values and ion/macromolecule compositions, and (C) in the endosomal–lysosomal cellular uptake mechanism of NMs, with lysosomal-specific composition of ions, macromolecules, phosphate, and pH values. The organism to organelle compartments are juxtaposed to (D) lab mimicries with bar “Lab mimicry” indicating restricted availability, high throughput and simplicity. Ranging from chemically defined liquids (phosphate buffered saline (PBS), artificial compartment buffer (ACB), simulated body fluids (SBF)), in vitro systems to in vivo. | ||
In contrast to oral NM administration, the conditions encountered by NMs via inhalation or tracheal administration41 are distinctly different; the first organ of contact is the lung, with a neutral pH of around 7.0, coated with a mucosal layer that protects it from the external environment. For inhalation or intratracheal exposure, NM uptake into alveolar macrophages and epithelial cells has been reported, with further translocation of NM to the vascular system.57
A benefit of injection into blood either intravenously41–46 (or retroorbitally in rodents48), which is favored by most studies, is direct systemic distribution with exposure to a near-neutral pH of 7.1–7.6.58 Nonetheless, blood (Fig. 1B) is a very complex environment, containing phosphates (0.9–1.5 mmol l−1 (ref. 59)), macromolecules (60–80 g l−1 (ref. 60)), and a variety of ions (e.g., Ca2+, Cl−, K+, Na+). Unless the GI tract or the lung is the primary target organ for the clinical application of nanomedicine, injections are typically preferable. Intravenous administration gives access to circulation in the blood stream and, therefore, a systemic distribution; however NM fate is heavily influenced by their interaction with the reticuloendothelial system (RES), including the liver, kidneys, and/or spleen, where NMs preferentially accumulate.42,43,45,48 Depending on the target organ and therapy involved, a direct site injection may have advantages over intravenous administration, achieving a highly defined and concentrated local NM distribution.47 This is crucial for several therapies, such as NM-enhanced radiotherapy (Hensify®, NBTXR3)61 and magnetic hyperthermia (MagForce AG, NanoTherm® Therapy)62,63 of solid tumors.
Irrespective of the route of administration, the therapeutic efficacy of almost all NMs relies on the NM uptake by cells.64 The most relevant cellular uptake mechanism for NM is via endocytosis (Fig. 1C).64 The NM is taken up by cells from the extracellular space into endosomes with an initial pH of 6.0–6.5.65 Over the course of endosomal maturation and V-ATPase acidification (H+ pump), the endosomes mature to late endosomes with a pH value of 5.0–6.0.65 Further maturation and acidification fully converts the endosomes into lysosomes with a pH of 4.5–5.0,65–68 corresponding approximately to a 25 μM H+ concentration, which is roughly 500 times higher than the cytosolic H+ concentration.62 Lysosomes (Fig. 1C) are vesicles found in most eukaryotic cells containing a wide variety of enzymes relevant for (bio-) macromolecule degradation. Additionally, lysosomes play a role in cellular processes, such as the secretion of proteins, cell signaling, and metabolic processes.69 The size of lysosomes varies from 0.1–0.6 μm, and they are containing a diverse set of enzymes (e.g., phosphatases, lipases, proteases, and sulfatases, responsible for different degradation and digestion processes) and ions (0.5 mM Ca2+, 20–140 mM Na+, 2–50 mM K+, 60–80 mM Cl−).62 All of the aforementioned factors, including phosphates, macromolecules, and ions (Ca2+, Cl−, K+, Na+, H+), and various combinations of these, add complexity to the biochemical environment that the NM is exposed to. The major challenges when studying transformation in whole organisms include: (i) highly dynamic effects, (ii) multifactorial reactions, as well as (iii) observer bias (i.e., “you only see what you look for”). Even though there is increased focus on the chemical and physical changes inherent to NM research for biomedical applications, most studies continue to focus on degradation and loss of function and not on primary environment-specific transformation reactions. Therefore, the interaction between complex in vitro or in vivo environments and NMs with associated transformations can currently be considered a “black-box”, with native NMs as the input and transformed NMs as the output. However, the underlying reactions are potentially a series of primary/elementary reactions/processes that result in secondary processes, such as degradation, amorphization, and recrystallization. To understand these fundamental processes, it is often essential to carefully simplify the environment to better discern the reaction mechanisms at play. However, such simplified studies should always be complemented by research in more advanced in vitro human models, as well as ex vivo or in vivo models. This approach verifies the occurrence of significant transformations. For example, morphological changes to CuS/CuS-iron oxide NM were first observed in vitro and afterwards confirmed in vivo.70 Drawing correlations between findings in intricate in vivo scenarios and those in more basic systems will contribute to the safe and sustainable translation of NMs towards clinical trials. As systematic in vivo investigations are time consuming, laborious, and resource intensive, adequate alternatives are needed, not only to reduce the amount of animals sacrificed, but also to unravel primary reaction mechanisms in less complex environments (Fig. 1D). The complexity of the lab systems need to be as defined as possible, with greatly reduced numbers of unknown variables that mimic single compartments and biological environments, such as lysosomes, blood, and tissue differences. Gradual complexity reduction, from whole in vivo organisms to cell-free systems, might be obtained using: (i) ex vivo71,72 and organ-on-a-chip approaches,73 (ii) mono- or co-cultured 3D in vitro models,74,75 (iii) conventional monolayer in vitro cell cultures,75,76 (iv) cell-free buffer systems, and (v) in silico modeling approaches77 (Fig. 1D).
Nanomaterial transformation: a cascade of primary and secondary reactions
Most studies on NMs for biomedical applications discuss endpoints, such as degradation or loss of function. These endpoints are secondary effects rather than primary/elementary reactions, on which the focus will be placed in the following.In classical chemical reaction theory, a reaction mechanism has one or more elementary reactions. These reactions consist of only one reaction step and one transition state involving one or more reactants. As previously discussed, the reactions that NMs undergo in model systems are intrinsically complex, not only due to the complexity of the model system and environments the NM's are facing, but also the NM's own complex structure (e.g., MOFs). Here we break down the transformation processes into two main classes: primary and secondary transformation processes. Our aim is to elucidate the black box of NM transformation, and to clarify transformation reaction terminology to produce a starting point in unifying the scientific language of NM transformation. We consider primary processes to be the very first steps in initiating a transformation of the parent material, i.e., a deviation from the material's original chemical, morphological, and/or structural states. Primary processes can be easily differentiated from secondary processes, as secondary processes are the result of one or more sequentially or simultaneously occurring primary processes (primary–secondary-reaction-cascade). We systematically analyzed studies (Table 1) in which primary and secondary processes that involved NMs for biomedical applications were determinable. To give a short example of such a primary–secondary-reaction cascade, the transformation of ZIF-8, a Zn-based MOF, in phosphate buffered saline (PBS) is described as a corrosion reaction of the Zn2+ centers with free phosphate groups.78 The gradual formation of zinc phosphate leads to a release of the organic linker, gradual amorphization, and the loss of the original structure as secondary processes.78
| NM | Chemical formula (MOFs only linker) | Environment | Transformation type | Details of reaction mechanism | Kinetics: max. reaction time | Analytical technique(s) |
|---|---|---|---|---|---|---|
| a Definition of corrosion in terms of chemistry according to Ahmad 2006:102 “Corrosion is the result of interaction between a metal and environments which results in its gradual destruction” or “Corrosion is the destructive attack of a metal by chemical or electrochemical reaction with the environment” (examples are rust formation, sulfidation of metallic Ag etc.). For example, the reaction of Fe2+ with water under oxygenated conditions to give 2Fe3O3·xH2O. | ||||||
| Metals | ||||||
| AgNP79 | Ag0 | In vitro, cell culture medium | Corrosiona (Ag2S formation), complexation (Ag-cystein, -histidine, -acetate) | NA | 24 h | ICP-MS synchrotron XAS (XANES) |
| AgNP80 | Ag0 | In vitro (THP-1) | Oxidation (Ag2O) | Ag0 → Ag+ and AgO–R → AgS–R | 48 h | Synchrotron (X-ray microscopy, XANES) |
| Complexation (AgO–R, AgS–R) | Complexation with macromolecules | |||||
| AgNP81 | Ag0 | In vitro | Dissolution (Ag+) | Ag0 → Ag+ (50% within 1 h) → AgO–R (after 12 h) → AgS–R (stable) | 24 h | XANES |
| Oxidation (Ag2O) | ||||||
| Complexation (AgO–R, AgS–R) | ||||||
| AuNP82 | Au | In vitro primary fibroblasts, lysosomes | Redox reaction: dissolution and recrystallization | 1st stage: Fenton/OH˙83 (mediated by NADPH oxidase) → release of ionic Au | 6 months | Electron microscopy (HR-TEM, SAED, STEM, EDX) |
| Secondary NP generation (nano leaves) | 2nd stage: recrystallization of ionic Au (ev. mediated by metalthioneines) | |||||
| Metal oxides | ||||||
| CeO2, TiO284 |
CeO2, TiO2 | In vivo mice – intra-venous & intra-tracheal administration | Redox reaction: dissolution, generation of secondary NP | Suggested transformation (oxidation) route published by Graham et al.85 | 180 d | (sp)IC-PMS |
| CeO285 |
CeO2 | In vivo rat liver | Redox reaction: dissolution and recrystallization | Redox reaction (CeIV → CeIII), partial dissolution of high energy edge sites and recrystallization to generate ultra-small secondary NP (1–3 nm), associated changes in morphology | 90 d | Electron microscopy (TEM, SAED, EELS, EDX) |
| CeO242 |
CeO2 | In vivo rat (liver and spleen) | Corrosion: dissolution, recrystallization | Differences in transformation between liver and spleen; liver: similar to Graham 2014,85 spleen: lesser degree of “bioprocessing”, formation of Ce-phosphates | 90 d | Electron microscopy (HR-TEM, STEM SAED, EELS, EDX) |
| CdSe@ZnS86 | CdSe@ZnS core shell QDs | In vivo mice (balb/c, liver, spleen, lung, blood, kidney, intestine, urine, faeces) | Dissolution (Zn2+) Reaction with phosphorous (Zn3(PO4)2) | Organ specific transformation of all elements: Zn from the ZnS shell (e.g., to Zn3(PO4)2 and different other species (e.g. Zn-cysteine) in blood and quick (∼10 d) transformation to mainly Zn3(PO4)2 in liver, spleen and lung), integration of Zn from ZnS to biomolecules similar to Cao et al.87 | 1, 10, 30, 90, 150, 365 d | XANES |
| Slow transformation of Se from CdSe to endogenous Se forms (mainly selenomethionine, selenocysteine). Slow transformation of Cd from CdSe to Cd-cysteine and CdS | ||||||
| Corrosion: formation of CdS from CdSe | ||||||
| Complexation: Zn-/Cd-/Se-cysteine, Zn-histidine, Zn-citrate, Se-methionine | ||||||
| Hollow CuS NP/iron oxide nanoflower hybrids70 | Cu2−xS, Fe-oxide (maghemite)@CuS | In vitro human mesenchyma cells and Glioblastoma cells U87 | Corrosion: dissolution, recrystallization (Fe) | Transformation of maghemite-Fe to ferrihydrite-Fe, possibly stored within the ferritin protein. Although no phase change was observed for sulfide-Cu, the initial CuS particle size and structure was not preserved per TEM imaging | 3 d, 21 d | XANES, electron microscopy (TEM) |
| γ-Fe2O3 (maghemite)48 | Fe2O3 | In vivo mice (lysosomes) – retro-orbital injection, spleen and liver | Chelation: dissolution and amorphization | Transformation to amorphous, non-magnetic Fe in spleen and liver – potentially in lysosomes, formation of ferritin-Fe | 44 d | FMR, SQUID, electron microscopy (TEM, SAED, EDX) |
| γ-Fe2O3 (maghemite)88 | Fe2O3 | In vitro mesenchymal stem cells (endosomes) 2D and 3D | Dissolution-recrystallization | Degradation by stem cells, generation of secondary NP with poorer crystallinity that remagnetized, ferrihydrite generation | NP uptake 30 min, 21 d | Electron microscopy (TEM, SAED) |
| Fe-oxides (magnetite) with PEG shell coating44 | In vivo rats | Dissolution, degradation | Transformation of magnetite-NP to ferritin-iron (Fe-hydroxide/oxide bound to a Ferritin complex), mainly in the liver | 24 h, 7 d, 30 d | Electron microscopy (TEM) | |
| Fe-oxides (maghemite), Fe2O3 citrate-coated89 | Fe2O3 | Endosomal fluid pH 4.7 with citrate | Chelation: dissolution | 27 d | TEM | |
| In vitro MSC spheroids (3D) | Magnetometry | |||||
| Fe3O4 (FDA approv.) (maghemite)citrate and phosphonoacedic coated90 | Fe3O4 | Artificial lysosomal fluid (ALF) containing citrate, pH 2.5, 3.5, 4.5, RT/37 °C | Chelation: dissolution, loss of crystallinity, secondary species generation | Iron ion release, iron citrate complex | 24 h | Magnetometry |
| Size reduction – coating dependent | ICP-OES | |||||
| Decrease of saturation magnetization | PXRD | |||||
| DLS | ||||||
| TEM | ||||||
| Fe-oxides (maghemite), multi-core nanoflowers91 | Fe2O3 | Artificial lysosomal fluid (ALF) containing citrate, pH 4.7, 37 °C | Chelation and corrosion: dissolution | Fe-chelate formation with citrate, transformation of multi-core into single-core magnetite-NP | 1–90 d//23 d | Magnetometry (FMR – ferromagnetic resonance spectroscopy), SAR (specific absorption rate), electron microscopy (HR-TEM, EFTEM, EDX) |
| Damage in atomic structure after 30 min, formation of holes in the atomic structure | ||||||
| InP QDs (core/core–shell)92 | Core: InP | Hydra vulgaris (eukariotic) | Ligand exchange | In–O bonds get replaced by In–P/In–S bonds (1 h after treatment) | 1, 3, 24, 48 h//3 h | μXANES, XRF |
| 1st shell: ZnSexS1−x | High affinity of COO− for In3+ | |||||
| 2nd shell: ZnS | 1. Phase transition (ligand exchange) | |||||
| 2. Degradation in absence of protective shell | ||||||
| MoS2 2D nanodots87 | MoS2 conjugated to human serum albumin (HSA) | In vivo C57BL/6 mice, in vitro embryonic murine hepatocyte cell line BNL CL.2 and murine monocyte/macrophage cell line J774A.1 | Redox reaction | Formation of protein corona, oxidation of Mo(IV) to Mo(VI) in the liver mediated by phase I oxidases, formation of (MoO4)2−, Mo from MoS2 used for biosynthesis of Mo-enzymes such as aldehyde oxidase and xanthine oxidoreductase, which are able to generate factors for tumor progression | 6 h, 1 d, 3 d, 7 d, 14 d, 60 d | XANES, XPS |
| Rare earth oxide NPs93 | E. coli | Corrosion: dissolution | Formation of Gd-phosphates and carboxylates after NP-cell contact | 2 h | XAFS | |
| Metal organic frameworks (MOFs) | ||||||
| A diverse set of MOFs (e.g., Mn-MOF-74)94 | Cell culture medium 10% FBS, 37 °C | Corrosion: pot. ligand exchange, amorphization | n/a/phase transformation (generation of new rod-like MnCO3 particles) | 24 h | PXRD, ICP-OES, electron microscopy (TEM, SAED) | |
| Mg-gallate95 | C6H2(OH)3COOH | Water, RPMI cell culture medium, 37 °C | Corrosion: ligand exchange | n/a/formation of Mg-phosphate. Complexation with phosphate is suggested | 24 h | PXRD, 13C/1H NMR, HPLC |
| Gallic acid (H4gal) | Gallic-acid release was faster in P rich medium | |||||
| MIL-100 (Fe)96 | C6H3(CO2H)3 | Simulated body fluids, PBS (different P concentrations), 37 °C | Corrosion: ligand exchange, amorphization | Replacement of trimesate linker by phosphate ions (ligand-exchange) independent of P content, limited by diffusion of phosphate into and linker out of the MOF → formation of amorphous Fe-phosphate shell | 1–8 d (microMOFs) | PXRD, Raman microscopy, Mössbauer spectrometry, electron microscopy (TEM) |
| Trimesic acid | ||||||
| MIL-100 (Fe), MIL-53 (Fe)97 | MIL-100: C6H3(CO2H)3 | Water (100 °C) | Hydrolysis | Formation of amorphous ferrihydrite from MOF hydrolysis at pH 7: | 2 h, 24 h | PXRD, electron microscopy (TEM, SEM) |
| Trimesic acid | Water (RT, pH 7) | (a) 2[Fe3O(OH)(H2O)2](BTC)2 + 7H2O → 3{Fe2O3·2H2O} + 4H3BTC | ||||
| MIL-53: C6H4(CO2H)2 | (b) 2Fe(OH)BDC + 3H2O → {Fe2O3·2H2O} + 2H2BDC | |||||
| Terephtalic acid | BDC = 1,4-benzenedicarboxylate (MIL-53) | |||||
| BTC = 1,3,5-benzenetricarboxylate (MIL-100) | ||||||
| MIL-100 (Fe)45 | C6H3(CO2H)3 | In vivo wistar rats | Corrosion: dissolution, ligand exchange | BTC/trimesate and Fe release by replacement with phosphates | 15 min–1 h after administration | HPLC |
| Trimesic acid | ||||||
| MIL-100 (Fe)98 | MIL-100: C6H3(CO2H)3 | Water, PBS, PBS + BSA 37 °C | Hydrolysis: ligand exchange, degradation | Linker (BTC/trimesate) release into medium | Up to 24 h | HPLC, XRPD |
| Trimesic acid | HCL pH 1.2, lis-SIF, lis-SIF + pancreatin, lis-SIF + mucine 37 °C | |||||
| MIL-88A (Fe), MIL-100 (Fe)46 | MIL-88A: HO2CCH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHCO2H CHCO2H |
Simulated physiological conditions | Ligand exchange, amorphization | Linker release and degradation of nano MOF | In vitro 8 d | HPLC, SEM |
| Fumaric acid | In vitro mouse macrophages | In vivo 3 months | ||||
| In vivo wistar rats | ||||||
| MIL-100: C6H3(CO2H)3 | ||||||
| Trimesic acid | ||||||
| PCN-223 (Zr)99 | C9H18Cl3O4P | Water, PBS, cell culture medium | Degradation | Up to 7 d | ||
| Tris-(2-chlorisopropyl)-phosphat | ||||||
| UiO-66/UiO-66-PEG (Zr)100 | C6F4-1,4-(CO2H) | PBS | Ligand release/exchange, degradation, amorphization | Linker release speed dependent on surface modification and pH | Up to 24 h | UV-Vis, PXRD |
| 1,4-Benzenedicarboxylic acid | ||||||
| ZIF-8 (Zn), Comparison micro- and nano-sized78 | C4H6N2 | PBS pH 7.4, 37 °C | Corrosion: ligand exchange | Competition of phosphate for metallic centers, linker release, conversion of rhombic dodecahedron ZIF-8(Zn) to spherical Zn-phosphate NP | Main changes in 1st hour//1, 3, 6, 24 h | PXRD, IR, electron microscopy (SEM, EDX), AFM, GC-MS |
| 2-Methylimidazol | ||||||
| ZIF-8 (Zn)101 | C4H6N2 | Solution with biomacromolecules (proteins, DNA, enzymes) | Hydrolysis: decomposition | Change in acidity from pH 7.4 to 6 37 °C disintegrates ZIF-8 layer | PXRD, electron microscopy (SEM) | |
| 2-Methylimidazol | ||||||
Primary chemical reaction mechanisms dependent on the chemical environment
Every biological environment, such as tissue, blood, and cell organelles, can be translated to an environment that drives chemical reactions. From the studies analyzed in Table 1, four main types are identified: hydrolysis, redox reactions, corrosion and complexation. Their relative importance is heavily dependent on the environment and intended application: pH, (mineral) ion composition and concentration, protein and macromolecule composition, as well as irradiation as an external parameter (Fig. 2). All of these parameters initiate specific reaction mechanisms, hereafter termed “primary reactions”.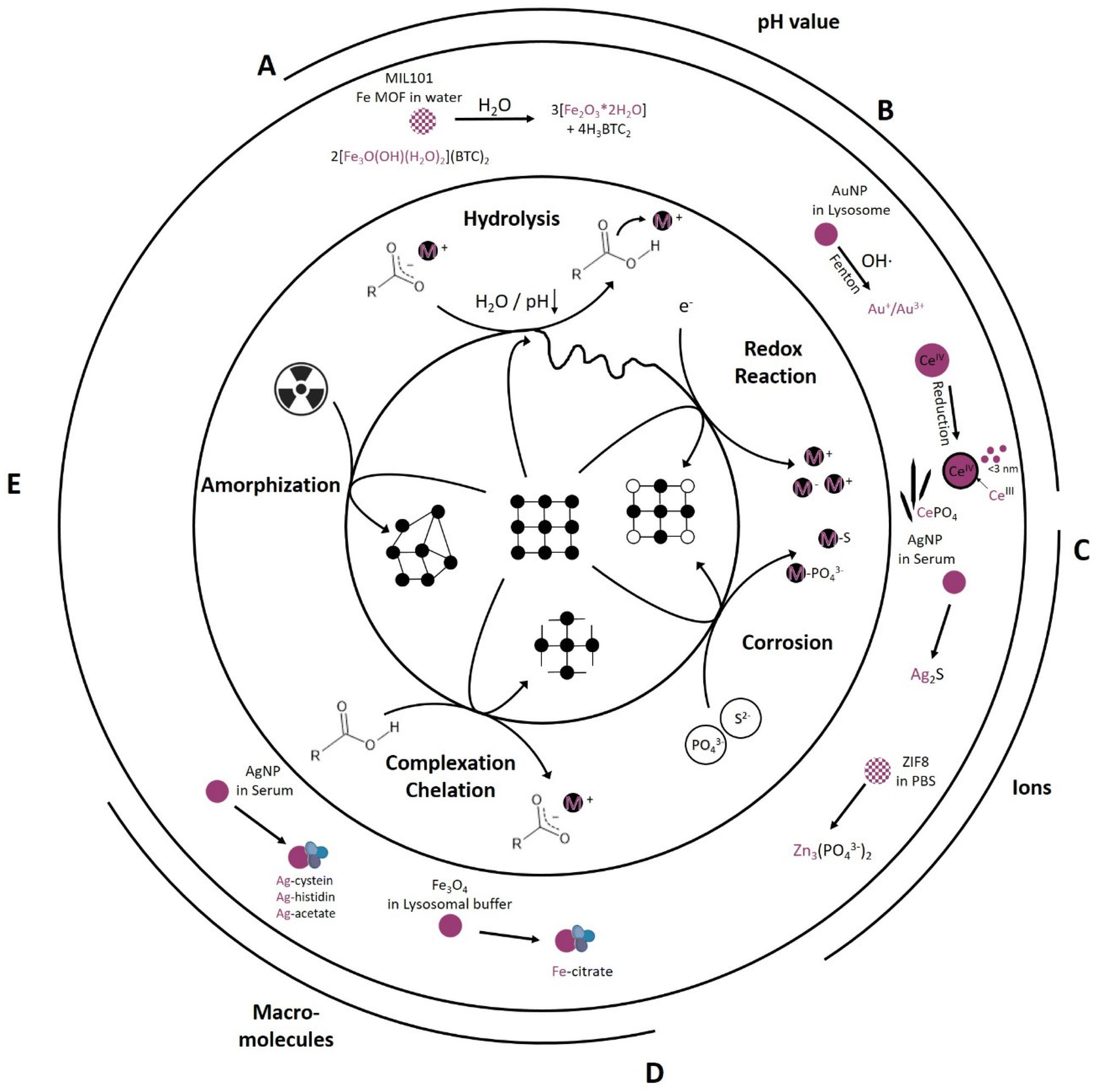 | ||
| Fig. 2 Primary reactions initiate transformation of inorganic NM dependent on the chemical environment including (A) hydrolysis reactions (e.g., MIL-101 transformation into an amorphous ferrihydrite97), (B) redox reactions (e.g., AuNP transformation into Au+/Au3+ (ref. 82) and formation of CeIII surface sites on CeO2 as well as formation of Ce-phosphate needles in vivo42,43), (C) corrosion reaction (e.g., AgNP transformation into Ag2S79 and ZIF-8 transformation into Zinc phosphate78), (D) complexation and chelation reactions (e.g., Fe3O4 transformation into Fe-citrate90 and AgNP transformation into Ag-cystein/-histidin/-acetate79), and (E) radiation induced amorphization reaction. | ||
One of the simplest and most widely studied primary reactions is hydrolysis. This reaction is mediated by water and its dissociation products, OH− and H+, which react with the original molecule, causing it to break down into smaller molecules. Hydrolysis is an integral reaction in many biological processes, such as food digestion and metabolism, also known as chemical digestion.103,104 This reaction can be accelerated under basic or acidic conditions and is temperature dependent; however, near-neutral to acidic conditions dominate under normal human physiological conditions, with individual variations in temperature around 37 ± 0.5 °C (Fig. 1).105 For example, Fe-MIL MOFs, mainly investigated for drug delivery applications, hydrolyzed in water, resulting in cleavage of the metal clusters and the linker into amorphous ferrihydrite (Fe2O3·2H2O) and its respective linker molecule97 (Fig. 2A). This reaction was more pronounced in acidic environments compared to neutral environments.97 Taken together, increased temperature and acidic environments can enhance hydrolysis. Generally, MOFs exhibit a lower chemical stability than their metal oxide counterparts due to the covalent binding of MOF metal clusters and organic linkers that makes them more prone to hydrolysis. However, this “Achilles heel” holds the potential to be used in acidic environments as a stimulus for drug release. An example for this is given by Liang et al.,101 where a PBS pH reduction from 7.4 to 6 was sufficient to release an encapsulated macromolecule (bovine serum albumin (BSA)) from a zinc-based ZIF-8 MOF. ZIF-8 MOFs are a widely studied material for drug delivery applications.106 Iron oxide nanoparticles are also prone to hydrolysis, undergoing degradation to ferrihydrite and losing their magnetic properties.88
The pH, however, not only significantly influences hydrolysis, but also drives reduction–oxidation (redox) reactions (Fig. 2B). In redox reactions, electrons are generally transferred between a NM and its chemical environment. The pH plays a pivotal role in this respect as it influences the redox potential and thereby the capability of the NM to acquire or loose electrons. For example, an increased transformation of Au0 NP into ionic gold, in which Au+ seems to be more stable in biological environments than Au3+ (Fig. 2B), occurred in acidic (pH 2) and neutral (pH 7) saline solutions (NaCl), whereas under basic conditions (pH 12), this reaction was prevented. This shows that pH is a driving force in redox reactions, with the constraint that the associated experiments were performed under a focused electron beam, which might have had its own influence on the transformation.82,107 Additionally, the oxidation state of the target element can be altered via electron transfer, e.g., by the transfer of free electrons or highly reactive radicals, such as reactive oxygen species (ROS). In physiological environments, in addition to purely chemically driven redox reactions including Fenton-like108 reactions also enzyme-mediated reactions can occur. Enzymes, such as superoxide dismutase (SOD) or catalase (CAT), maintain the redox balance in physiological environments to support cellular health and functionality. SOD catalyzes oxidation reactions, converting highly reactive oxygen (superoxide, O2−) to hydrogen peroxide (H2O2) and oxygen (O2), whereas CAT catalyzes reduction reactions, in which the H2O2 is further converted to water and oxygen. Therefore, CAT and SOD play pivotal roles in cellular oxidative stress protection.109–112 An example of enzyme-mediated oxidation is one involving 4 nm gold nanoparticles (Au0 NPs), which are generally believed to be chemically inert. Balfourier et al. proposed a complex biotransformation reaction, where in a first stage the NOX complex (membrane associated protein complex: NADPH oxidase) converted O2 into ˙O2−, followed by its conversion into H2O2via SOD. Hydrogen peroxide then oxidized the Au0 NP into ionic gold via a Fenton reaction, resulting in either Au+ or Au3+ (Fig. 2B).82 Besides SOD mediated H2O2 generation also a spontaneous dismutation is conceivable, most likely due to the reduction of O2 to ˙O2− in the mitochondrial respiration pathway.113 Therefore, depending on the source of the ROS, the transformation is either a bio- or chemical transformation. Another example of a redox reaction is the in vivo reduction of CeIVO2 particles. As-synthesized 30 nm cubic CeIVO2 particles were analyzed 90 days after IV injection in rat liver and spleen. In vivo dissolution of high energy edges occurred, resulting in a rounded morphology with increased CeIII levels in the shell region. The transformation rate in the liver, which possessed a higher acidity, was higher than that of the spleen. These findings were consistent with previous work that showed that the transformation rate of ceria was dependent on pH and electrochemical potential. The in vivo dissolution of these CeIVO2 particles resulted in the formation of CeIII enriched clouds as well as the recrystallization of ultra-small 1–3 nm CeO2−x particles; these particles had a significantly increased free radical scavenging potential (antioxidant effect) (Fig. 2B).43,114 Following the CeIII ion release, Graham et al. observed tissue-dependent phosphatization and recrystallization in form of CeIIIPO4 nanoneedles, with an individual length of about 20 nm being part of a greater network.42 Generally, metals, such as cerium, iron, chromium and nickel are known to catalyze Fenton-like reactions in vitro and in vivo.108,115,116 Also, some metals or trace elements are essential for the body and were recently shown to be converted to active biological molecules, thus over time exerting an effect on the metabolism.86,87 For example, Mo was shown to be integrated into Mo-cofactors in the liver after enzyme-mediated oxidation of Mo(IV) to Mo(VI) from MoS2 particles. This in turn led to an increased activity of the main molybdoflavoenzymes (aldehyde oxidase and xanthine oxidoreductase) which can generate nitric oxide, involved in tumor progression.87 Thus, metals should be closely monitored over their entire in vitro or in vivo life cycle if considered for biomedical applications to ensure their efficacy and safety.
Like hydrogen and oxygen, sulfur and phosphorous are essential and abundant elements in physiological environments. Sulfur is present in different forms, such as sulfate (SO42−), sulfides (S2−), and thiol groups (–SH), in proteins and cellular structures. Phosphorous is an integral component of biological molecules, nucleic acids (DNA and RNA), adenosine triphosphate (ATP), and phospholipids, the building blocks of the cell and cell organelle membranes. In their different forms, depending on the composition and affinity, those ions can interact with NMs. Here, we term the underlying chemical reaction “corrosion”, which leads to the gradual destruction of an NM as the ion reacts with the metal, resulting in the formation of metal-sulfides or metal-phosphates in amorphous or crystalline forms (Fig. 2C). The metal-sulfide or metal-phosphate can also still be bound to the macromolecule and therefore forming metal-sulfide- or metal-phosphate-complexes.80 The corrosion of the chalcophile silver (Ag) (-NP) with e.g. H2S into Ag2S is widely known and has been described for different systems were sulfur-bearing species are prevalent, significantly impacting behavior and fate of the material in the different systems.117–120 In biomedical applications, silver nanoparticles (AgNPs) are studied for their antimicrobial properties,121 advanced wound healing properties,122,123 implant coating properties,124 and applicability in drug delivery systems.125 In in vitro lymphocyte experiments, Ag either stayed intact as metallic silver or was transformed into Ag2S in a corrosion reaction or complexed as Ag-cystein.79 The transformation and fractionation into intact AgNPs, Ag2S, and Ag-cystein was highly dependent on the original particle size. The smaller the as-synthesized particle, the higher the uptake and intracellular sulfidation into Ag2S in contrast to a lower AgNP uptake and intracellular sulfidation for bigger as-synthesized particles (Fig. 2C).79 Jiang et al. and Wang et al. proposed a multiple-step intracellular transformation that saw the dissolution of Ag0 NP to Ag+, followed by an oxidation to Ag–O species after 12 h and a sulfidation to Ag2S after 24 h, resulting in a more stable intracellular form compared to the intermediate states.80,81 Also, the phosphatization of zinc-based ZIF-8 MOFs has been found to exhibit particle-size-dependent transformation kinetics. Phosphates in PBS were found to compete with the linker of the framework structures, resulting in a release of the linker and a conversion of the rhombic, dodecahedron ZIF-8 particles into spherical, amorphous Zinc phosphate particles (Fig. 2C).78 This process was highly particle-size-dependent, with a faster transformation for smaller nano-sized ZIF-8 particles.78 Similarly, also Zn from ZnS shells was shown to transform (amongst others) into Zn3(PO4)2.86 Also other elements with a high affinity towards specific S- or P-bearing species are likely to be subject to corrosion (e.g. Cu126 or Cd86). This transformation will alter biocompatibility and nanomedical application efficiency. A better understanding of the underlying corrosion mechanisms can help to improve stability and reduce undesired side effects, already during the NM design and synthesis. The synthesis could be augmented by e.g., mesoporous silica shells127 or organic polymer coatings, such as poly ethylene glycol (PEG)128 coatings, to protect the material from corrosion. For semi-conductor NM, ZnS is sometimes proposed as a protective shell. However, recently, Chen et al. showed that a ZnS shell can be rapidly degraded within 10 days in an in vivo setting.86 Ultimately, whether a more protective or a more labile shell is required will depend largely on the needs of the final application and/or on the clearance pathways of the respective NM and its constituent elements.
So far, only interactions between ions and small molecules have been discussed. However, physiological environments also contain larger and complex (macro) molecules, such as proteins, amino acids, and other organic molecules. These molecules have key functional moieties, such as carboxyl- (–COOH), amino- (–NH2), aldehyde- (–C(O)H), and thiol (–SH) moieties, which can interact and coordinate NM-related elements/ions. Generally, metal coordination, complexation, and chelation processes (Fig. 2D) play a key role in cellular metabolism and signaling, e.g., in metalloenzymes such as peptidases, the zinc finger-proteins of transcription factors, oxygen transport via hemoglobin and excretion. As chelation and complexation are systemically present, and many protein functional groups can function as chelators, they influence NM cellular fate. AgNPs have been shown to transform apart from the main product Ag2S also into Ag-cysteine, Ag-histidine, and Ag-acetate complexes (Fig. 2D).79 The proportional distribution of Ag-cysteine, Ag-histidine, and Ag-acetate complexation was surface-functionalization and particle-size dependent; for example, no Ag-cysteine complexes were found for differently sized citrate-capped AgNP, whereas the opposite was the case for polyethylenimine-(PEI)-coated AgNPs.79 Gutiérrez et al. also proved the impact of surface functionalization on chelation kinetics.90 This study used citric acid (CIT) and phosphonoacetic acid (PAA) on magnetic Fe-oxides (Fe3O4) in 20 mM citric acid lysosomal mimicking buffer (pH 2.5, 3.5, and 4.5).90 The transformation was faster for PAA-functionalized particles than for CIT-functionalized particles, with a size reduction and faster transformation in strong acidic environments (pH 2.5). They compared the temperature (RT and 37 °C) dependence at pH 4.5 and found a tremendous impact on transformation kinetics for both coatings, with a significantly faster transformation for PAA-functionalized Fe-oxides (Fig. 2D).90 Surface functionalization and coatings potentially protect NMs from chelation and complexation; i.e., the metal centers are shielded from the chelators and complexing agents by the coating layer. Therefore, a denser and thicker coating might improve the protection, but at the same time hamper applications relying on core material activity.
So far, all primary transformation reactions discussed were induced by the chemical environment, i.e., the concentration and combination of ions, the pH (basic, neutral, acidic conditions), or presence of macromolecules and proteins. However, external factors, such as radiation, are known to induce transformation (Fig. 2E). To our knowledge this has not yet been studied for biomedically relevant nanomaterials. Except for Lin et al., who has introduced the first radio-enhancing Hf-based MOF, RiMO-301, which is currently under clinical trials (Phase I: NCT03444714). They did not find any phase transformation upon 16 Gy X-ray (250 kVp, 15 mA, 1 mm Cu filter) irradiation.129 However, since radio-enhancement applications of nanomaterials have found increasing potential, phase transformation must be carefully considered if ionizing radiation is applied.
In other fields, radiation-induced transformations have been observed; for example in waste management, hydroxyapatite nanoparticles (Ca10(PO4)6(OH)2) are used for the disposal of actinides and fission products.130 These particles amorphize under 1 MeV Kr2+ ions in a size-dependent manner (20 to 280 nm); the smaller the particles, the faster the amorphization process. This suggests a surface-to-volume enhanced transformation process due to the fast accumulation of oxygen vacancies. Interestingly, after amorphization, they observed a transmission electron microscope-induced (electron energy of 200 keV) recrystallization, again size dependent, with a significantly faster recrystallization process into smaller hydroxyapatite nanoparticles, respective to the native particle size.130 Furthermore, influence of particle size on radiation-induced amorphization has been studied. Smaller CePO4 particles (20 nm) underwent faster amorphization at RT, than larger particles (40 nm) under 1 MeV Kr2+ irradiation. It was proposed that for smaller NP a proportionally higher fraction of amorphization induced swelling and deformation which led to a faster complete loss of crystallinity. This effect was further increased for both particle sizes in higher temperature conditions (100 °C).131 However, there are also the opposite indications, that small single crystals/grains can enhance radiation tolerance, i.e. the resistance to phase transitions.132,133 The mechanisms governing this dependence remain yet to be determined.
Harnessing analytics to unravel (bio-) transformation processes
Generally, the analysis of (bio-)transformations of inorganic nanomaterials in a near native manner and based only on minimal perturbation of the system is key. However, high-resolution chemical and structural analysis relies on adequate sample preparation and analysis settings. These often necessitate analysis under vacuum conditions and might involve other modifications to the system, such as introducing labels. In particular, the introduction of widely used (fluorescence)-labels, e.g., for the visualization of metal ions or entire nanoparticles has a high potential for critical alteration of the system. Similarly, also the use of high-vacuum analytical techniques, such as (analytical) electron microscopy or X-ray photoelectron spectroscopy, which require sample drying, and sometimes even chemical fixation, heavy metal staining and/or embedding into resin can bear considerable source for sample preparation artifacts. In addition, also measurement induced changes need to be carefully considered, including damage induced by the electron beam of the electron microscope (e.g. through knock-on displacement, radiolysis or electrostatic charging and heating), potential re-crystallization of amorphous materials under the electron beam or changes induced by sample heating when using lasers, e.g. in vibrational spectroscopy. However, many challenges, such as sampling artifacts, sample heating, and other beam-induced damages, can be mitigated by using cryogenic conditions (typically characterized as temperatures below −150 °C (ref. 134)). By employing these methods along with other meticulous sampling strategies that prevent further reactions of the analytes, “snapshots” of the processes can be obtained.Hence, all analytical investigations need to be carefully designed to include appropriate controls that help to identify potential errors. Also, the use of as many as possible complementary and/or orthogonal techniques135 is highly advisable in order to reduce uncertainty and increase consistency of the data and hence their trustworthiness. This becomes even more important, as in some cases observing no change in one parameter does not mean that there is no change also in another – as shown for CuS particles that showed no phase change in X-ray absorption spectroscopy but significant morphological changes in electron microscopy.70
In the following, analytical techniques suitable to give insights into the respective previously discussed reaction mechanisms (Fig. 3) will be systematically discussed.
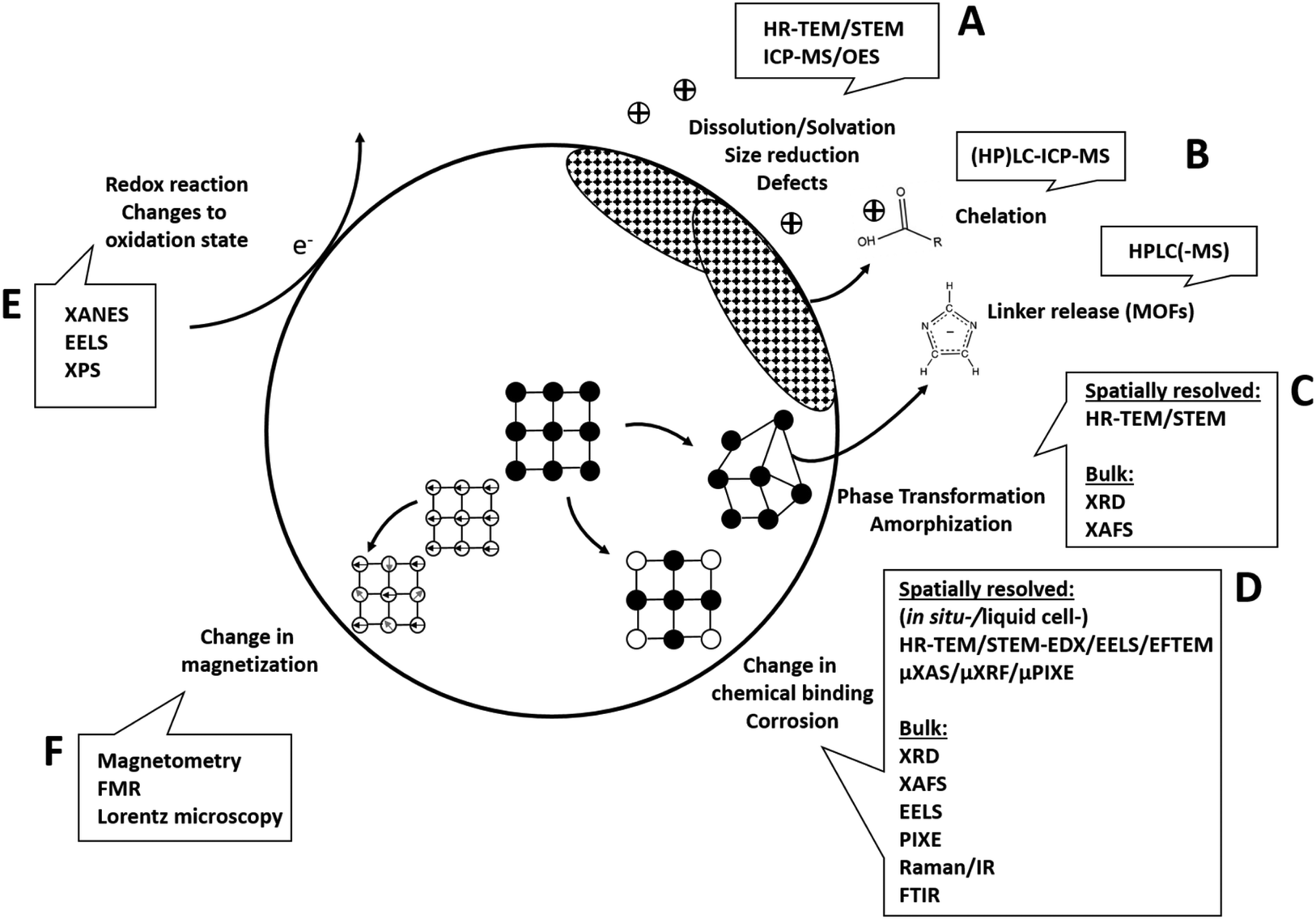 | ||
| Fig. 3 Analytical techniques to unravel underlying transformation processes. (A) Structural changes and accompanying ion release can be analyzed using a combination of high-resolution (scanning) transmission electron microscopy (HR-TEM/STEM) and inductively coupled plasma mass spectroscopy or optical emission spectroscopy (ICP-MS/OES). (B) Organic components (e.g. MOF linkers, surface molecules) released from the solid material can be analyzed with high performance liquid chromatography (HPLC). (C) Phase transformations including amorphization can be analyzed with HR-TEM/STEM, bulk X-ray diffraction (XRD) or X-ray absorption fine structure/extended fine structure spectroscopy (XANES/EXAFS). (D) Corrosion, complexation and changes in chemical binding can either be analyzed with high spatial resolution using HR-TEM/STEM combined with energy-dispersive X-ray spectroscopy (EDX) or electron energy loss spectroscopy (EELS, EFTEM) as well as X-ray micro/bulk-analysis, e.g., (micro-)X-ray absorption spectroscopy (XANES/EXAFS, μXAS), micro-X-ray fluorescence (μXRF), micro-particle induced X-ray emission (μPIXE). (E) For analysis of changes in oxidation state EELS, X-ray absorption near edge spectroscopy (XANES) or X-ray photoelectron spectroscopy (XPS) are the techniques of choice. (F) Magnetic materials as well as changes in magnetic potential is analyzed using magnetometry, ferromagnetic resonance (FMR) or Lorentz microscopy. | ||
Changes to the material that involve structural changes together with release of components (e.g. hydrolysis, dissolution in general) require a combined approach including electron microscopy and elemental analysis, eventually together with chromatographic techniques (Fig. 3A and B). For example, imaging techniques such as high-resolution (scanning) transmission electron microscopy (HR-(S)TEM) in combination with EDXS can be employed to study structural and compositional changes to the solid phase (dry or cryogenically preserved), while the fractionation between solid and dissolved components can be studied using inductively coupled plasma techniques (ICP-OES or -MS, liquid samples (aqueous or organic)) together with suitable separation techniques (e.g. filtration, centrifugation). In addition, single particle ICP-MS gives access to particle size distribution (see e.g. Modrzynska et al.41). Here, transient signals of highly diluted particle dispersions are being recorded, where single particle events can be distinguished from a dissolved background. The intensity distribution of these events can then be converted to a particle size distribution, where the specific geometry has to be known or assumed. Detection limits in terms of particles size mostly depend on the constituent element and have been evaluated.136 Size detection limits are lowest for the more heavy elements, for example therapeutically relevant Hf or Ce-oxides can be determined down to ≈10 nm, whereas most of the other relevant elements such as Zn or Fe are detectable roughly down to 30–50 nm or even >100 nm (Si, Ca). Conventional quadrupole ICP-MS systems are only able to measure one element at a time, so for materials where multiple elements have to be monitored, a more costly time-of-flight (TOF) analyzer has to be used. Such an analyzer is capable of simultaneous detection across almost the entire mass range between 7 and 275 amu.137 This additionally gives access to monitoring compositional changes to the individual NPs (e.g., making use of isotope labelling), which makes this technique very powerful for biotransformation studies.138
For organo-metallic materials, such as MOFs, it is necessary to additionally monitor the release of organic building blocks and/or metabolites. In the absence of interfering absorbers, this can be achieved using rather straightforward techniques such as UV-Vis spectroscopy100 or, if more detail and higher sensitivity is required, more advanced techniques such as high performance liquid chromatography (HPLC) with coupling to UV detection46 and/or mass spectrometry (MS, Fig. 3B, transfer of the analytes to compatible solvents might be necessary). However, some biogenic organic molecules can also act as chelating agents, able to complex metals and act as promotors of dissolution within the cell/body. To study chelation processes or generally binding of organic molecules to inorganic ions, a combination of HPLC and ICP-MS is usually employed.139 For inorganic complexes, ion chromatography in combination with ICP-OES/MS is a potent technique combination.
Changes in bulk phase and crystallinity are commonly assessed using X-ray diffraction (XRD, see e.g. ref. 78). However, this often requires the availability of larger sample amounts in the order of mg to g,140 depending on the sample (usually dry powders). While these amounts are mostly available for the initial materials, this is not the case after experiments conducted with relevant dosing. Also, XRD shows significant peak broadening with decreasing particle size, making the characterization of very small (<10 nm) particles challenging.141 Here, complementary to bulk XRD, high resolution (HR) – (scanning) transmission electron microscopy ((S)TEM) in combination with electron diffraction can be used on single particles to determine their crystal structure, requiring only minor sample amounts. Furthermore, due to the size-related peak broadening in XRD, the comparison of (S)TEM-determined particle size to XRD-determined particle size can allow conclusions regarding the dominating size fraction in the sample.141 In addition, most microscopes capable of analyzing electron diffraction are equipped with an energy dispersive X-ray spectrometer (EDXS), which allows to determine the elemental composition of the particle. This classic combination of XRD and electron microscopy has been used with success in many of the reviewed studies. For example, Graham et al.42 used a combination of electron diffraction and EDX elemental mapping to follow the transformation of nano-ceria in an in vivo setting.
However, new developments in terms of automation in electron microscopy might revolutionize the way in which this set of techniques can be applied to study particles even in complex environments such as cells. For example, pieces of software are already available that allow a fully automated analysis workflow consisting of image acquisition, particle identification, particle compositional analysis using EDX and particle size and morphological characterization.142 Also, more and more free software is available that aids image based particle analysis (see for example the Particle-Sizer plugin for ImageJ143). Such automated routines give access to much better statistics, as thousands of particles can be analyzed in a rather short time. As the development of artificial intelligence is progressing rapidly, our capabilities of automatically recognizing and analyzing particles in complex environments will increase drastically in the future (see e.g. ref. 144), potentially making electron microscopy a key technique for biotransformation studies. Also in terms of changes to the oxidation state of atoms, electron microscopy plays an important role, as this parameter is accessible from electron energy loss spectroscopy (EELS, Fig. 3E) in specialized TEMs. With EELS, the electronic structure and chemical bonding can be analyzed at the nanoscale as the energy distribution of the electrons is analyzed after their interaction with the sample. The combination of STEM and EELS allows for a very detailed characterization of complex NMs, apart from element identification and oxidation state also yielding valuable information such as element molar ratios or identification of specific carbon bonds of organic coatings.145 Apart from analysis of single particles/pure materials, EELS can also be applied to (embedded) cells/tissues.146 Here, EELS can especially serve to collect complementary biological information, such as mapping the distribution of proteins or water in the cell by low-energy loss EELS. Also, studies are available that analyzed NM transformation in tissues using EELS, e.g. CeO2 in the liver of rats.85 Here, the high spatial resolution of STEM-EELS allowed to discriminate between CeIII on the surface of single particles as opposed to CeIV in their center. However, generally, for successful EELS measurements, sample quality is absolutely critical. This includes the absence of carbon contamination build-up, appropriate sample thickness and analyte mass fraction. Measurement temperatures ≤LN2 as well as low-dose techniques are helpful in this regard.
Apart from EELS and X-ray spectroscopy in the EM, X-ray based spectroscopy in general holds a multitude of possibilities for elemental analysis as well as for determination of chemical states and structure. One of the most powerful techniques in this regard is synchrotron X-ray absorption spectroscopy (X-ray absorption near-edge (XANES) and extended X-ray absorption fine structure (EXAFS) spectroscopy), giving access to element oxidation state and local coordination geometry. It can be performed <LN2 temperatures, which allows to resolve material changes in frozen time series with minimal sample alteration. Samples for investigation include cell free systems but can be extended to cells, cell cultures and tissues.86,147,148 Some beamlines additionally offer the possibility of a micro-focused or even nano-focused (≈100 nm2)149,150 X-ray beam, adding spatial resolution and resulting in hyperspectral data-cubes.151 However, either high intracellular analyte element concentrations or high local concentrations of it within the cell are necessary to record interpretable data.149 In addition, especially with micro/nano-focused beams, potential influences of the X-ray beam on the sample during the measurement have to be taken into account. For cells and/or tissues, sample preparation methods have been mostly adapted from electron microscopy, including classical embedding and sectioning or fully cryogenic preparation.150
As a more available, lab-scale technique, X-ray photoelectron spectroscopy (XPS) also allows to access oxidation and binding states as well as general elemental composition (all elements except H and He152). Here, mono-energetic soft X-rays are being used to extract electrons from the surface in ultrahigh-vacuum via the photo-electric effect, whose energies are subsequently analyzed. This then gives access to the electron binding energies, which inform about the elements chemical state. However, as photoelectrons generally can only escape from a depth roughly below 10 nm, XPS is only sensitive to the sample surface.152 This makes the technique ideal to study changes to surface terminations and/or coatings. For example, after incubation with glutathione – an important peptide in the body – a reduction of disulfide bonds to thiol groups in the outer layer of organo-silica NPs was observed using XPS.153 When determining oxidation states however, care has to be taken to account for potential reduction by the X-ray beam. If reduction is observed, determination of the reduction rate law can still allow to determine the true sample composition by extrapolation to zero X-ray exposure time.154 To address questions regarding the relevance of XPS to hydrated biological environments, freeze hydration or cryo-XPS was developed almost 30 years ago.152,155 Here, the sample is fast-frozen in the XPS entry chamber, where the subsequent exposure to ultrahigh-vacuum sublimates the ice. However, it is estimated that during the following XPS analysis conducted below −120 °C about 2–3 layers of water molecules still remain at the surface together with structural water inside the cells.156 Conducting the analysis at or below cryogenic temperatures then also has been shown to reduce thermal induced beam damage, similar to electron microscopy or other X-ray based techniques.156
Finally, changes to magnetic properties can be accessed using magnetometry approaches, including electron paramagnetic spectroscopy (EPR), ferromagnetic resonance spectroscopy (FMR), vibrating sample magnetometry (VSM), Alternating Gradient Field Magnetometer (AGFM)157 and Superconducting Quantum Interference Device (SQUID)158 measurements. Also magnetic resonance imaging (MRI), and especially quantitative parameters R2, R2* and QSM have been shown to be sensitive to changes in iron oxidation state.159
Conclusions and outlook
A solid understanding of NM (bio-)transformations and their mechanisms opens up entirely new avenues of research. Especially, it increases our ability to predict and explore crucial safety aspects of these materials, which will in turn pave the way for the safe-by-design development of new nanomedicines. Furthermore, (bio-) transformation may be harnessed for diagnostic and therapeutic purposes, giving access to multistage diagnostics or therapeutics. Such materials would undergo chemical changes over time and exert multiple functions. For example, they could provide feedback after initiation of the transformation reaction that could be read out externally, e.g., through changes in medical image contrast. Such feedback could include information either on the tissue environment faced by the nanotherapeutic, or on the status of the treatment (e.g., change in contrast indicating the release of a therapeutic agent). To fully harness these potentials, the community needs to adapt a rigorous testing process that involves in vivo experiments complemented by experiments at carefully reduced complexity to unravel underlying mechanisms, all accompanied by multi-scale and multi-modal complementary analytics to fully describe the fate of the materials in the body.Author contributions
AN: investigation, data curation, writing – original draft; IKH: funding acquisition, resources, conceptualization, writing – review and editing; AG: conceptualization, data curation, investigation, supervision, writing – review and editing. All authors agreed to the final version of the manuscript.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
We thank Sebastian Habermann for fruitful discussions and proof reading. We also like to thank the Swiss National Science Foundation (SNF, Grant No. 181290) for funding.References
- B. Buesser and S. E. Pratsinis, Annu. Rev. Chem. Biomol. Eng., 2012, 3, 103–127 CrossRef CAS PubMed.
- L. Yang, W. Li, M. Kirberger, W. Liao and J. Ren, Biomater. Sci., 2016, 4, 785–802 RSC.
- Q. Tang, Z. Zhou and Z. Chen, Nanoscale, 2013, 5, 4541 RSC.
- L. Jing, W. Zhou, G. Tian and H. Fu, Chem. Soc. Rev., 2013, 42, 9509 RSC.
- P. Mukhopadhyay, R. Mishra, D. Rana and P. P. Kundu, Prog. Polym. Sci., 2012, 37, 1457–1475 CrossRef CAS.
- P. Sanderson, J. M. Delgado-Saborit and R. M. Harrison, Atmos. Environ., 2014, 94, 353–365 CrossRef CAS.
- S. Anu Mary Ealia and M. P. Saravanakumar, IOP Conf. Ser.: Mater. Sci. Eng., 2017, 263, 032019 Search PubMed.
- K. Zarschler, L. Rocks, N. Licciardello, L. Boselli, E. Polo, K. P. Garcia, L. De Cola, H. Stephan and K. A. Dawson, Nanomedicine, 2016, 12, 1663–1701 CrossRef CAS PubMed.
- J. Xie, S. Lee and X. Chen, Adv. Drug Delivery Rev., 2010, 62, 1064–1079 CrossRef CAS PubMed.
- D. Anderson, T. Anderson and F. Fahmi, Phys. Status Solidi A, 2019, 216, 1801008 CrossRef.
- S. Caspani, R. Magalhães, J. P. Araújo and C. T. Sousa, Materials, 2020, 13, 2586 CrossRef CAS PubMed.
- Z. R. Stephen, F. M. Kievit and M. Zhang, Mater. Today, 2011, 14, 330–338 CrossRef CAS PubMed.
- C. Sun, J. S. H. Lee and M. Zhang, Adv. Drug Delivery Rev., 2008, 60, 1252–1265 CrossRef CAS PubMed.
- W.-H. Chen, W.-C. Liao, Y. S. Sohn, M. Fadeev, A. Cecconello, R. Nechushtai and I. Willner, Adv. Funct. Mater., 2018, 28, 1705137 CrossRef.
- R. Singh and J. W. Lillard, Exp. Mol. Pathol., 2009, 86, 215–223 CrossRef CAS PubMed.
- A. Guerreiro, N. Chatterton, E. M. Crabb and J. P. Golding, Cancer Nanotechnol., 2019, 10, 10 CrossRef CAS.
- K. T. Butterworth, S. J. McMahon, F. J. Currell and K. M. Prise, Nanoscale, 2012, 4, 4830 RSC.
- A. L. Neuer, A. Jessernig, L. R. H. Gerken, A. Gogos, F. H. L. Starsich, A. H. C. Anthis and I. K. Herrmann, Biomater. Sci., 2022, 10(22), 6558–6569 RSC.
- Z. Kuncic and S. Lacombe, Phys. Med. Biol., 2018, 63, 02TR01 CrossRef PubMed.
- S. Kumar Nethi, S. Das, C. Ranjan Patra and S. Mukherjee, Biomater. Sci., 2019, 7, 2652–2674 RSC.
- M. M. Mihai, M. B. Dima, B. Dima and A. M. Holban, Materials, 2019, 12, 2176 CrossRef CAS PubMed.
- A. Barroso, H. Mestre, A. Ascenso, S. Simões and C. Reis, Nano Sel., 2020, 1, 443–460 CrossRef.
- H. Zhao, C. Serre, E. Dumas and N. Steunou, in Metal-Organic Frameworks for Biomedical Applications, ed. M. Mozafari, Woodhead Publishing, 2020, pp. 397–423 Search PubMed.
- H. Huang, W. Feng, Y. Chen and J. Shi, Nano Today, 2020, 35, 100972 CrossRef CAS.
- J. M. Metselaar and T. Lammers, Drug Delivery Transl. Res., 2020, 10, 721–725 CrossRef PubMed.
- L. van der Koog, T. B. Gandek and A. Nagelkerke, Adv. Healthcare Mater., 2022, 11, 2100639 CrossRef CAS PubMed.
- P. Liu, G. Chen and J. Zhang, Molecules, 2022, 27, 1372 CrossRef CAS PubMed.
- H. Bouwmeester, I. Lynch, H. J. P. Marvin, K. A. Dawson, M. Berges, D. Braguer, H. J. Byrne, A. Casey, G. Chambers, M. J. D. Clift, G. Elia, T. F. Fernandes, L. B. Fjellsbø, P. Hatto, L. Juillerat, C. Klein, W. G. Kreyling, C. Nickel, M. Riediker and V. Stone, Nanotoxicology, 2011, 5, 1–11 CrossRef CAS PubMed.
- T. L. Moore, L. Rodriguez-Lorenzo, V. Hirsch, S. Balog, D. Urban, C. Jud, B. Rothen-Rutishauser, M. Lattuada and A. Petri-Fink, Chem. Soc. Rev., 2015, 44, 6287–6305 RSC.
- M. Hadjidemetriou and K. Kostarelos, Nat. Nanotechnol., 2017, 12, 288–290 CrossRef CAS PubMed.
- M. P. Monopoli, C. Åberg, A. Salvati and K. A. Dawson, Nat. Nanotechnol., 2012, 7, 779–786 CrossRef CAS PubMed.
- G. V. Lowry, K. B. Gregory, S. C. Apte and J. R. Lead, Environ. Sci. Technol., 2012, 46, 6893–6899 CrossRef CAS PubMed.
- G. Pulido-Reyes, F. Leganes, F. Fernández-Piñas and R. Rosal, Environ. Toxicol. Chem., 2017, 36, 3181–3193 CrossRef CAS PubMed.
- J. Lv, P. Christie and S. Zhang, Environ. Sci. Nano, 2019, 6, 41–59 RSC.
- M. Ganesh and J. Ramakrishna, Asian J. Org. Chem., 2020, 9, 1341–1376 CrossRef CAS.
- M. Fayette and R. D. Robinson, J. Mater. Chem. A, 2014, 2, 5965–5978 RSC.
- Y. Xu, K. Ren, T. Ren, M. Wang, S. Yu, Z. Wang, X. Li, L. Wang and H. Wang, J. Mater. Chem. A, 2020, 8, 19873–19878 RSC.
- C. Zhang, W. Liu, C. Chen, P. Ni, B. Wang, Y. Jiang and Y. Lu, Nanoscale, 2022, 14, 2915–2942 RSC.
- H. Zhou, J. Ge, Q. Miao, R. Zhu, L. Wen, J. Zeng and M. Gao, Bioconjugate Chem., 2020, 31, 315–331 CrossRef CAS PubMed.
- D. A. Porter and K. E. Easterling, Phase Transformations in Metals and Alloys (Revised Reprint), CRC Press, 2009 Search PubMed.
- J. Modrzynska, T. Berthing, G. Ravn-Haren, K. Kling, A. Mortensen, R. R. Rasmussen, E. H. Larsen, A. T. Saber, U. Vogel and K. Loeschner, PLoS One, 2018, 13, e0202477 CrossRef PubMed.
- U. M. Graham, R. A. Yokel, A. K. Dozier, L. Drummy, K. Mahalingam, M. T. Tseng, E. Birch and J. Fernback, Toxicol. Pathol., 2018, 46, 47–61 CrossRef CAS PubMed.
- U. M. Graham, M. T. Tseng, J. B. Jasinski, R. A. Yokel, J. M. Unrine, B. H. Davis, A. K. Dozier, S. S. Hardas, R. Sultana, E. A. Grulke and D. A. Butterfield, ChemPlusChem, 2014, 79, 1083–1088 CrossRef CAS PubMed.
- A. Ruiz, L. Gutiérrez, P. R. Cáceres-Vélez, D. Santos, S. B. Chaves, M. L. Fascineli, M. P. Garcia, R. B. Azevedo and M. P. Morales, Nanoscale, 2015, 7, 16321–16329 RSC.
- T. Simon-Yarza, T. Baati, F. Neffati, L. Njim, P. Couvreur, C. Serre, R. Gref, M. F. Najjar, A. Zakhama and P. Horcajada, Int. J. Pharm., 2016, 511, 1042–1047 CrossRef CAS PubMed.
- P. Horcajada, T. Chalati, C. Serre, B. Gillet, C. Sebrie, T. Baati, J. F. Eubank, D. Heurtaux, P. Clayette, C. Kreuz, J.-S. Chang, Y. K. Hwang, V. Marsaud, P.-N. Bories, L. Cynober, S. Gil, G. Férey, P. Couvreur and R. Gref, Nat. Mater., 2010, 9, 172–178 CrossRef CAS PubMed.
- F. Boateng and W. Ngwa, Int. J. Mol. Sci., 2020, 21, 273 CrossRef CAS PubMed.
- M. Levy, N. Luciani, D. Alloyeau, D. Elgrabli, V. Deveaux, C. Pechoux, S. Chat, G. Wang, N. Vats, F. Gendron, C. Factor, S. Lotersztajn, A. Luciani, C. Wilhelm and F. Gazeau, Biomaterials, 2011, 32, 3988–3999 CrossRef CAS PubMed.
- D. K. Mishra, V. Pandey, R. Maheshwari, P. Ghode and R. K. Tekade, in Basic Fundamentals of Drug Delivery, ed. R. K. Tekade, Academic Press, 2019, pp. 595–650 Search PubMed.
- H. Marwah, T. Garg, A. K. Goyal and G. Rath, Drug Delivery, 2016, 23, 564–578 CrossRef CAS PubMed.
- N. Davies, D. Hovdal, N. Edmunds, P. Nordberg, A. Dahlén, A. Dabkowska, M. Y. Arteta, A. Radulescu, T. Kjellman, A. Höijer, F. Seeliger, E. Holmedal, E. Andihn, N. Bergenhem, A.-S. Sandinge, C. Johansson, L. Hultin, M. Johansson, J. Lindqvist, L. Björsson, Y. Jing, S. Bartesaghi, L. Lindfors and S. Andersson, Mol. Ther.–Nucleic Acids, 2021, 24, 369–384 CrossRef CAS PubMed.
- T. W. Prow, J. E. Grice, L. L. Lin, R. Faye, M. Butler, W. Becker, E. M. T. Wurm, C. Yoong, T. A. Robertson, H. P. Soyer and M. S. Roberts, Adv. Drug Delivery Rev., 2011, 63, 470–491 CrossRef CAS PubMed.
- M. de J. Velásquez-Hernández, M. Linares-Moreau, E. Astria, F. Carraro, M. Z. Alyami, N. M. Khashab, C. J. Sumby, C. J. Doonan and P. Falcaro, Coord. Chem. Rev., 2021, 429, 213651 CrossRef.
- J. Schubert and M. Chanana, Curr. Med. Chem., 2019, 25, 4556–4586 Search PubMed.
- E. Lu, S. Li and Z. Wang, Asian J. Pharm. Sci., 2017, 12, 9–20 CrossRef PubMed.
- I. L. Bergin and F. A. Witzmann, Int. J. Biomed. Nanosci. Nanotechnol., 2013, 3 DOI:10.1504/IJBNN.2013.054515.
- N. R. Yacobi, F. Fazllolahi, Y. H. Kim, A. Sipos, Z. Borok, K.-J. Kim and E. D. Crandall, Air Qual., Atmos. Health, 2011, 4, 65–78 CrossRef PubMed.
- H. G. Preuss, Clin. Lab. Med., 1993, 13, 103–116 CrossRef CAS PubMed.
- E. Moutzouri, E. N. Liberopoulos and M. Elisaf, Arch. Med. Sci., 2011, 7, 736–739 CrossRef CAS PubMed.
- K. E. Barrett, S. Boitano and S. M. Barman, Ganong's Review of Medical Physiology, McGraw-Hill Professional Publishing, New York, USA, 23rd edn, 2010 Search PubMed.
- S. Bonvalot, P. L. Rutkowski, J. Thariat, S. Carrère, A. Ducassou, M.-P. Sunyach, P. Agoston, A. Hong, A. Mervoyer, M. Rastrelli, V. Moreno, R. K. Li, B. Tiangco, A. C. Herraez, A. Gronchi, L. Mangel, T. Sy-Ortin, P. Hohenberger, T. de Baère, A. Le Cesne, S. Helfre, E. Saada-Bouzid, A. Borkowska, R. Anghel, A. Co, M. Gebhart, G. Kantor, A. Montero, H. H. Loong, R. Vergés, L. Lapeire, S. Dema, G. Kacso, L. Austen, L. Moureau-Zabotto, V. Servois, E. Wardelmann, P. Terrier, A. J. Lazar, J. V. M. G. Bovée, C. Le Péchoux and Z. Papai, Lancet Oncol., 2019, 20, 1148–1159 CrossRef CAS PubMed.
- P. C. Trivedi, J. J. Bartlett and T. Pulinilkunnil, Cells, 2020, 9, 1131 CrossRef CAS PubMed.
- M. Johannsen, B. Thiesen, P. Wust and A. Jordan, Int. J. Hyperth. Off. J. Eur. Soc. Hyperthermic Oncol. North Am. Hyperth. Group, 2010, 26, 790–795 CrossRef PubMed.
- H. Kettiger, A. Schipanski, P. Wick and J. Huwyler, Int. J. Nanomed., 2013, 8, 3255–3269 Search PubMed.
- D. Schmaljohann, Adv. Drug Delivery Rev., 2006, 58, 1655–1670 CrossRef CAS PubMed.
- S. Ohkuma and B. Poole, Proc. Natl. Acad. Sci. U. S. A., 1978, 75, 3327–3331 CrossRef CAS PubMed.
- Y.-B. Hu, E. B. Dammer, R.-J. Ren and G. Wang, Transl. Neurodegener., 2015, 4, 18 CrossRef PubMed.
- P. Watson, A. T. Jones and D. J. Stephens, Adv. Drug Delivery Rev., 2005, 57, 43–61 CrossRef CAS PubMed.
- C. Settembre, A. Fraldi, D. L. Medina and A. Ballabio, Nat. Rev. Mol. Cell Biol., 2013, 14, 283–296 CrossRef CAS PubMed.
- A. Curcio, A. V. de Walle, E. Benassai, A. Serrano, N. Luciani, N. Menguy, B. B. Manshian, A. Sargsian, S. Soenen, A. Espinosa, A. Abou-Hassan and C. Wilhelm, ACS Nano, 2021, 15, 9782–9795 CrossRef CAS PubMed.
- T. G. Meijer, K. A. Naipal, A. Jager and D. C. van Gent, Future Sci. OA, 2017, 3, FSO190 CrossRef CAS PubMed.
- P. Wick, S. Chortarea, O. T. Guenat, M. Roesslein, J. D. Stucki, S. Hirn, A. Petri-Fink and B. Rothen-Rutishauser, Eur. J. Nanomed., 2015, 7, 169–179 Search PubMed.
- C. M. Leung, P. de Haan, K. Ronaldson-Bouchard, G.-A. Kim, J. Ko, H. S. Rho, Z. Chen, P. Habibovic, N. L. Jeon, S. Takayama, M. L. Shuler, G. Vunjak-Novakovic, O. Frey, E. Verpoorte and Y.-C. Toh, Nat. Rev. Methods Primers, 2022, 2, 1–29 CrossRef.
- C. Jubelin, J. Muñoz-Garcia, L. Griscom, D. Cochonneau, E. Ollivier, M.-F. Heymann, F. M. Vallette, L. Oliver and D. Heymann, Cell Biosci., 2022, 12, 155 CrossRef PubMed.
- M. Kapałczyńska, T. Kolenda, W. Przybyła, M. Zajączkowska, A. Teresiak, V. Filas, M. Ibbs, R. Bliźniak, Ł. Łuczewski and K. Lamperska, Arch. Med. Sci., 2018, 14, 910–919 Search PubMed.
- Y. Imamura, T. Mukohara, Y. Shimono, Y. Funakoshi, N. Chayahara, M. Toyoda, N. Kiyota, S. Takao, S. Kono, T. Nakatsura and H. Minami, Oncol. Rep., 2015, 33, 1837–1843 CrossRef CAS PubMed.
- M. Viceconti, F. Pappalardo, B. Rodriguez, M. Horner, J. Bischoff and F. Musuamba Tshinanu, Methods, 2021, 185, 120–127 CrossRef CAS PubMed.
- M. de J. Velásquez-Hernández, R. Ricco, F. Carraro, F. Ted Limpoco, M. Linares-Moreau, E. Leitner, H. Wiltsche, J. Rattenberger, H. Schröttner, P. Frühwirt, E. M. Stadler, G. Gescheidt, H. Amenitsch, C. J. Doonan and P. Falcaro, CrystEngComm, 2019, 21, 4538–4544 RSC.
- A. Malysheva, A. Ivask, C. L. Doolette, N. H. Voelcker and E. Lombi, Nat. Nanotechnol., 2021, 16, 926–932 CrossRef CAS PubMed.
- L. Wang, T. Zhang, P. Li, W. Huang, J. Tang, P. Wang, J. Liu, Q. Yuan, R. Bai, B. Li, K. Zhang, Y. Zhao and C. Chen, ACS Nano, 2015, 9, 6532–6547 CrossRef CAS PubMed.
- X. Jiang, T. Miclăuş, L. Wang, R. Foldbjerg, D. S. Sutherland, H. Autrup, C. Chen and C. Beer, Nanotoxicology, 2015, 9, 181–189 CrossRef CAS PubMed.
- A. Balfourier, N. Luciani, G. Wang, G. Lelong, O. Ersen, A. Khelfa, D. Alloyeau, F. Gazeau and F. Carn, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 103–113 CrossRef CAS PubMed.
- A. M. Nowicka, U. Hasse, M. Hermes and F. Scholz, Angew. Chem., Int. Ed., 2010, 49, 1061–1063 CrossRef CAS PubMed.
- J. Modrzynska, T. Berthing, G. Ravn-Haren, K. Kling, A. Mortensen, R. R. Rasmussen, E. H. Larsen, A. T. Saber, U. Vogel and K. Loeschner, PLoS One, 2018, 13, e0202477 CrossRef PubMed.
- U. M. Graham, M. T. Tseng, J. B. Jasinski, R. A. Yokel, J. M. Unrine, B. H. Davis, A. K. Dozier, S. S. Hardas, R. Sultana, E. A. Grulke and D. A. Butterfield, ChemPlusChem, 2014, 79, 1083–1088 CrossRef CAS PubMed.
- G. Chen, Y. Zhang, D. Huang, Y. Liu, C. Li and Q. Wang, Nano Today, 2022, 44, 101504 CrossRef CAS.
- M. Cao, R. Cai, L. Zhao, M. Guo, L. Wang, Y. Wang, L. Zhang, X. Wang, H. Yao, C. Xie, Y. Cong, Y. Guan, X. Tao, Y. Wang, S. Xu, Y. Liu, Y. Zhao and C. Chen, Nat. Nanotechnol., 2021, 16, 708–716 CrossRef CAS PubMed.
- A. V. de Walle, A. P. Sangnier, A. Abou-Hassan, A. Curcio, M. Hémadi, N. Menguy, Y. Lalatonne, N. Luciani and C. Wilhelm, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 4044–4053 CrossRef PubMed.
- F. Mazuel, A. Espinosa, N. Luciani, M. Reffay, R. Le Borgne, L. Motte, K. Desboeufs, A. Michel, T. Pellegrino, Y. Lalatonne and C. Wilhelm, ACS Nano, 2016, 10, 7627–7638 CrossRef CAS PubMed.
- L. Gutiérrez, S. Romero, G. B. da Silva, R. Costo, M. D. Vargas, C. M. Ronconi, C. J. Serna, S. Veintemillas-Verdaguer and M. del Puerto Morales, Biomed. Eng. Biomed. Tech., 2015, 60, 417–425 Search PubMed.
- Y. Javed, L. Lartigue, P. Hugounenq, Q. L. Vuong, Y. Gossuin, R. Bazzi, C. Wilhelm, C. Ricolleau, F. Gazeau and D. Alloyeau, Small, 2014, 10, 3325–3337 CrossRef CAS PubMed.
- G. Veronesi, M. Moros, H. Castillo-Michel, L. Mattera, G. Onorato, K. D. Wegner, W. L. Ling, P. Reiss and C. Tortiglione, ACS Appl. Mater. Interfaces, 2019, 11, 35630–35640 CrossRef CAS PubMed.
- X. He, Y. Pan, J. Zhang, Y. Li, Y. Ma, P. Zhang, Y. Ding, J. Zhang, Z. Wu, Y. Zhao, Z. Chai and Z. Zhang, Environ. Pollut., 2015, 196, 194–200 CrossRef CAS PubMed.
- À. Ruyra, A. Yazdi, J. Espín, A. Carné-Sánchez, N. Roher, J. Lorenzo, I. Imaz and D. Maspoch, Chem. – Eur. J., 2015, 21, 2508–2518 CrossRef PubMed.
- L. Cooper, T. Hidalgo, M. Gorman, T. Lozano-Fernández, R. Simón-Vázquez, C. Olivier, N. Guillou, C. Serre, C. Martineau, F. Taulelle, D. Damasceno-Borges, G. Maurin, Á. González-Fernández, P. Horcajada and T. Devic, Chem. Commun., 2015, 51, 5848–5851 RSC.
- X. Li, L. Lachmanski, S. Safi, S. Sene, C. Serre, J. M. Grenèche, J. Zhang and R. Gref, Sci. Rep., 2017, 7, 1–11 CrossRef PubMed.
- I. Bezverkhyy, G. Weber and J.-P. Bellat, Microporous Mesoporous Mater., 2016, 219, 117–124 CrossRef CAS.
- E. Bellido, M. Guillevic, T. Hidalgo, M. J. Santander-Ortega, C. Serre and P. Horcajada, Langmuir, 2014, 30, 5911–5920 CrossRef CAS PubMed.
- J. Yang, X. Chen, Y. Li, Q. Zhuang, P. Liu and J. Gu, Chem. Mater., 2017, 29, 4580–4589 CrossRef CAS.
- I. Abánades Lázaro, S. Haddad, S. Sacca, C. Orellana-Tavra, D. Fairen-Jimenez and R. S. Forgan, Chem, 2017, 2, 561–578 Search PubMed.
- K. Liang, R. Ricco, C. M. Doherty, M. J. Styles, S. Bell, N. Kirby, S. Mudie, D. Haylock, A. J. Hill, C. J. Doonan and P. Falcaro, Nat. Commun., 2015, 6, 1–8 Search PubMed.
- Z. Ahmad, Principles of Corrosion Engineering and Corrosion Control, Elsevier, 2006 Search PubMed.
- J. J. Patricia and A. S. Dhamoon, in StatPearls, StatPearls Publishing, Treasure Island, FL, 2022 Search PubMed.
- S. Phang-Lyn and V. A. Llerena, in StatPearls, StatPearls Publishing, Treasure Island, FL, 2022 Search PubMed.
- V. E. Del Bene, in Clinical Methods: The History, Physical, and Laboratory Examinations, ed. H. K. Walker, W. D. Hall and J. W. Hurst, Butterworths, Boston, 3rd edn, 1990 Search PubMed.
- S. Feng, X. Zhang, D. Shi and Z. Wang, Front. Chem. Sci. Eng., 2021, 15, 221–237 CrossRef CAS.
- J. Hermannsdörfer, N. de Jonge and A. Verch, Chem. Commun., 2015, 51, 16393–16396 RSC.
- E. G. Heckert, S. Seal and W. T. Self, Environ. Sci. Technol., 2008, 42, 5014–5019 CrossRef CAS PubMed.
- T. Finkel and N. J. Holbrook, Nature, 2000, 408, 239–247 CrossRef CAS PubMed.
- J. M. McCord and I. Fridovich, J. Biol. Chem., 1969, 244, 6049–6055 CrossRef CAS PubMed.
- A. Nandi, L.-J. Yan, C. K. Jana and N. Das, Oxid. Med. Cell. Longevity, 2019, 2019, e9613090 Search PubMed.
- A. Deisseroth and A. L. Dounce, Physiol. Rev., 1970, 50, 319–375 CrossRef CAS PubMed.
- C. M. C. Andrés, J. M. Pérez de la Lastra, C. A. Juan, F. J. Plou and E. Pérez-Lebeña, Stresses, 2022, 2, 256–274 CrossRef.
- R. W. Tarnuzzer, J. Colon, S. Patil and S. Seal, Nano Lett., 2005, 5, 2573–2577 CrossRef CAS PubMed.
- J. A. Imlay, S. M. Chin and S. Linn, Science, 1988, 240, 640–642 CrossRef CAS PubMed.
- K. D. Sugden, R. D. Geer and S. J. Rogers, Biochemistry, 1992, 31, 11626–11631 CrossRef CAS PubMed.
- J. L. Elechiguerra, L. Larios-Lopez, C. Liu, D. Garcia-Gutierrez, A. Camacho-Bragado and M. J. Yacaman, Chem. Mater., 2005, 17, 6042–6052 CrossRef CAS.
- R. Kaegi, A. Voegelin, C. Ort, B. Sinnet, B. Thalmann, J. Krismer, H. Hagendorfer, M. Elumelu and E. Mueller, Water Res., 2013, 47, 3866–3877 CrossRef CAS PubMed.
- A. Wamucho, J. M. Unrine, T. J. Kieran, T. C. Glenn, C. L. Schultz, M. Farman, C. Svendsen, D. J. Spurgeon and O. V. Tsyusko, Environ. Pollut., 2019, 254, 113078 CrossRef CAS PubMed.
- M. Li, J. Li, J. Sun, Y. He, P. Chen and C. Zhang, Environ. Sci. Nano, 2019, 6, 3611–3624 RSC.
- W.-R. Li, T.-L. Sun, S.-L. Zhou, Y.-K. Ma, Q.-S. Shi, X.-B. Xie and X.-M. Huang, Int. Biodeterior. Biodegrad., 2017, 123, 304–310 CrossRef CAS.
- Y. Y. Hong Hu, J. Microb. Biochem. Technol., 2015, 7, 228–233 Search PubMed.
- F. F. Larese, F. D'Agostin, M. Crosera, G. Adami, N. Renzi, M. Bovenzi and G. Maina, Toxicology, 2009, 255, 33–37 CrossRef CAS PubMed.
- Y. Qing, L. Cheng, R. Li, G. Liu, Y. Zhang, X. Tang, J. Wang, H. Liu and Y. Qin, Int. J. Nanomed., 2018, 13, 3311–3327 CrossRef CAS PubMed.
- M. Rai, A. P. Ingle, I. Gupta and A. Brandelli, Int. J. Pharm., 2015, 496, 159–172 CrossRef CAS PubMed.
- A. Gogos, B. Thalmann, A. Voegelin and R. Kaegi, Environ. Sci. Nano, 2017, 4, 1733–1741 RSC.
- D. Borisova, H. Möhwald and D. G. Shchukin, ACS Nano, 2011, 5, 1939–1946 CrossRef CAS PubMed.
- T. W. Quadri, L. O. Olasunkanmi, O. E. Fayemi, M. M. Solomon and E. E. Ebenso, ACS Omega, 2017, 2, 8421–8437 CrossRef CAS PubMed.
- K. Ni, G. Lan, C. Chan, B. Quigley, K. Lu, T. Aung, N. Guo, P. La Riviere, R. R. Weichselbaum and W. Lin, Nat. Commun., 2018, 9, 2351 CrossRef PubMed.
- J. Zhou, M. Kirk, P. Baldo and F. Lu, Materialia, 2021, 18, 101154 CrossRef CAS.
- F. Lu, Y. Shen, X. Sun, Z. Dong, R. C. Ewing and J. Lian, Acta Mater., 2013, 61, 2984–2992 CrossRef CAS.
- T. D. Shen, Nucl. Instrum. Methods Phys. Res., Sect. B, 2008, 266, 921–925 CrossRef CAS.
- G. M. Cheng, W. Z. Xu, Y. Q. Wang, A. Misra and Y. T. Zhu, Scr. Mater., 2016, 123, 90–94 CrossRef CAS.
- K. D. Timmerhaus and T. M. Flynn, Cryogenic Process Engineering, Springer Science & Business Media, 2013 Search PubMed.
- C. G. Simon, S. E. Borgos, L. Calzolai, B. C. Nelson, J. Parot, E. J. Petersen, M. Roesslein, X. Xu and F. Caputo, J. Controlled Release, 2023, 354, 120–127 CrossRef CAS PubMed.
- S. Lee, X. Bi, R. B. Reed, J. F. Ranville, P. Herckes and P. Westerhoff, Environ. Sci. Technol., 2014, 48, 10291–10300 CrossRef CAS PubMed.
- L. Hendriks, A. Gundlach-Graham, B. Hattendorf and D. Günther, J. Anal. At. Spectrom., 2017, 32, 548–561 RSC.
- X. Tian, H. Jiang, L. Hu, M. Wang, W. Cui, J. Shi, G. Liu, Y. Yin, Y. Cai and G. Jiang, TrAC, Trends Anal. Chem., 2022, 157, 116746 CrossRef CAS.
- H. M. Neu, S. A. Alexishin, J. E. P. Brandis, A. M. C. Williams, W. Li, D. Sun, N. Zheng, W. Jiang, A. Zimrin, J. C. Fink, J. E. Polli, M. A. Kane and S. L. J. Michel, Mol. Pharm., 2019, 16, 1272–1281 CrossRef CAS PubMed.
- A. A. Bunaciu, E. gabriela Udriştioiu and H. Y. Aboul-Enein, Crit. Rev. Anal. Chem., 2015, 45, 289–299 CrossRef CAS PubMed.
- C. F. Holder and R. E. Schaak, ACS Nano, 2019, 13, 7359–7365 CrossRef CAS PubMed.
- T. Uusimaeki, T. Wagner, H. G. Lipinski and R. Kaegi, J. Nanopart. Res., 2019, 21, 1–11 CrossRef.
- T. Wagner and J. Eglinger, thorstenwagner/ij-particlesizer Zenodo 2017 Search PubMed.
- M. Botifoll, I. Pinto-Huguet and J. Arbiol, Nanoscale Horiz., 2022, 7, 1427–1477 RSC.
- M. M. van Schooneveld, A. Gloter, O. Stephan, L. F. Zagonel, R. Koole, A. Meijerink, W. J. M. Mulder and F. M. F. de Groot, Nat. Nanotechnol., 2010, 5, 538–544 CrossRef CAS PubMed.
- M. A. Aronova and R. D. Leapman, MRS Bull., 2012, 37, 53–62 CrossRef CAS PubMed.
- M. Podgórczyk, W. M. Kwiatek, W. M. Zaj
![[a with combining cedilla]](https://www.rsc.org/images/entities/char_0061_0327.gif) c, J. Dulińska-Litewka, E. Welter and D. Grolimund, X-Ray Spectrom., 2009, 38, 557–562 CrossRef.
c, J. Dulińska-Litewka, E. Welter and D. Grolimund, X-Ray Spectrom., 2009, 38, 557–562 CrossRef. - A. Levina, A. I. McLeod, A. Pulte, J. B. Aitken and P. A. Lay, Inorg. Chem., 2015, 54, 6707–6718 CrossRef CAS PubMed.
- C. Sanchez-Cano, D. Gianolio, I. Romero-Canelon, R. Tucoulou and P. J. Sadler, Chem. Commun., 2019, 55, 7065–7068 RSC.
- H. A. Castillo-Michel, C. Larue, A. E. Pradas del Real, M. Cotte and G. Sarret, Plant Physiol. Biochem., 2017, 110, 13–32 CrossRef CAS PubMed.
- M. Podgórczyk, W. M. Kwiatek, D. Grolimund and C. Borca, Radiat. Phys. Chem., 2009, 78, S53–S57 CrossRef.
- S. L. McArthur, G. Mishra and C. D. Easton, in Surface Analysis and Techniques in Biology, ed. V. S. Smentkowski, Springer International Publishing, Cham, 2014, pp. 9–36 Search PubMed.
- H. Mekaru, A. Yoshigoe, M. Nakamura, T. Doura and F. Tamanoi, ACS Appl. Nano Mater., 2019, 2, 479–488 CrossRef CAS.
- C.-K. Wu, M. Yin, S. O'Brien and J. T. Koberstein, Chem. Mater., 2006, 18, 6054–6058 CrossRef CAS.
- K. B. Lewis and B. D. Ratner, J. Colloid Interface Sci., 1993, 159, 77–85 CrossRef CAS.
- M. Kjærvik, M. Ramstedt, K. Schwibbert, P. M. Dietrich and W. E. S. Unger, Front. Chem., 2021, 9, 666161 CrossRef PubMed.
- A. L. Urbano-Bojorge, N. Félix-González, T. Fernández, F. del Pozo-Guerrero, M. Ramos and J. J. Serrano-Olmedo, J. Nano Res., 2015, 31, 129–137 CAS.
- M. Škrátek, A. Dvurečenskij, M. Kluknavský, A. Barta, P. Bališ, A. Mičurová, A. Cigáň, A. Eckstein-Andicsová, J. Maňka and I. Bernátová, Nanomaterials, 2020, 10, 1993 CrossRef PubMed.
- C. Birkl, A. M. Birkl-Toeglhofer, C. Kames, W. Goessler, J. Haybaeck, F. Fazekas, S. Ropele and A. Rauscher, NeuroImage, 2020, 220, 117080 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2023 |