
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Probing physical properties of single amyloid fibrils using nanofluidic channels†
Nima
Sasanian
a,
Rajhans
Sharma
 a,
Quentin
Lubart
a,
Sriram
KK
a,
Marziyeh
Ghaeidamini
a,
Kevin D.
Dorfman
b,
Elin K.
Esbjörner
*a and
Fredrik
Westerlund
*a
a,
Quentin
Lubart
a,
Sriram
KK
a,
Marziyeh
Ghaeidamini
a,
Kevin D.
Dorfman
b,
Elin K.
Esbjörner
*a and
Fredrik
Westerlund
*a
aDivision of Chemical Biology, Department of Life Sciences, Chalmers University of Technology, Kemivägen 10, 412 96 Gothenburg, Sweden. E-mail: eline@chalmers.se; fredrikw@chalmers.se
bDepartment of Chemical Engineering and Materials Science, University of Minnesota-Twin Cities, 421 Washington Ave SE, Minneapolis, Minnesota 55455, USA
First published on 13th November 2023
Abstract
Amyloid fibril formation is central to the pathology of many diseases, including neurodegenerative disorders such as Alzheimer's and Parkinson's disease. Amyloid fibrils can also have functional and scaffolding roles, for example in bacterial biofilms, and have also been exploited as useful biomaterials. Despite being linear protein homopolymers, amyloid fibrils can exhibit significant structural and morphological polymorphism, making it relevant to study them on the single fibril level. We here introduce the concept of nanofluidic channel analysis to the study of single, fluorescently-labeled amyloid fibrils in solution, monitoring the extension and emission intensity of individual fibrils confined in nanochannels with a depth of 300 nm and a width that gradually increases from 300 to 3000 nm. The change in fibril extension with channel width permitted accurate determination of the persistence length of individual fibrils using Odijk's theory for strongly confined polymers. The technique was applied to amyloid fibrils prepared from the Alzheimer's related peptide amyloid-β(1–42) and the Parkinson's related protein α-synuclein, obtaining mean persistence lengths of 5.9 ± 4.5 μm and 3.0 ± 1.6 μm, respectively. The broad distributions of fibril persistence lengths indicate that amyloid fibril polymorphism can manifest in their physical properties. Interestingly, the α-synuclein fibrils had lower persistence lengths than the amyloid-β(1–42) fibrils, despite being thicker. Furthermore, there was no obvious within-sample correlation between the fluorescence emission intensity per unit length of the labelled fibrils and their persistence lengths, suggesting that stiffness may not be proportional to thickness. We foresee that the nanofluidics methodology established here will be a useful tool to study amyloid fibrils on the single fibril level to gain information on heterogeneity in their physical properties and interactions.
Introduction
Protein aggregation and the formation of amyloid fibrils is associated with more than 20 incurable disorders,1 including severe neurodegenerative conditions such as Alzheimer's and Parkinson's disease, where the process of amyloid fibril formation has been shown to generate neurotoxicity.2 Besides being involved in the pathological development of disease, amyloid fibrils can also have functional biological roles.2–4 For example, amyloid fibrils formed by the human Pmel17 protein are important in the catalysis of melanin formation and protection against UV-mediated damage of the skin.5 Amyloid fibrils can also act as structural scaffolds, for example in bacterial biofilms,6 as well as in novel biomaterials.7,8To understand the role of amyloid fibrils in a biological context, and to be able to tune their properties to be used as functional biomaterials,9,10 it is important not only to solve their structures, but also to understand their mechanical and physical properties. Single fibril level technologies are important in this respect as they can account for polymorphism within samples. Various techniques, such as atomic force microscopy (AFM),11,12 transmission electron microscopy (TEM),13 cryoEM,14,15 total internal reflection microscopy (TIRF)16 and super-resolution fluorescence microscopy,17 have been used to study the morphological, structural and physical properties of amyloid fibrils. However, these techniques typically rely on the immobilization of the fibrils to a surface and, in particular for AFM, drying of the immobilized sample. In recent years, microfluidic and nanofluidic technologies have introduced new and powerful platforms to explore biomolecules, including polypeptides, on a single molecule level and in solution. Microfluidic chip-based methods have for example been used to study properties of amyloid structure and their interactions with other proteins and compounds.18–21 The dimensions of nanofluidic devices confine biopolymers in an extended conformation, which facilitates their visualization and analysis. Furthermore, the confinement in a nanochannel keeps the object of interest in focus, which means that standard epi-fluorescence microscopy can be used for visualization with minimal background. Nanochannel devices have been extensively used by us and others to study basic polymer physics of single DNA molecules, as well as their interactions with DNA-binding proteins.22 The degree of stretching of a polymer in a nanochannel is mainly governed by its persistence length and the dimensions of the channels. Theoretical predictions of the extension of biopolymers in different regimes were pioneered by de Gennes23 and Odijk.24 Persson et al. have previously established a nanofluidic device with funnel-like nanochannels that allows single biopolymers to be studied at several different confinements as they are moved along the nanochannel using pressure-driven flow.25 The device has been used to study DNA, and a similar geometry was later implemented by us to determine the persistence length of RecA filaments formed on dsDNA.26 Similar approaches to study physical properties of biopolymers have been demonstrated for actin27–29 and vimentin fibrils.30
In this study, we extend the application of nanofluidic channels with funnel-like dimensions to demonstrate that amyloid fibrils can be confined in nanochannels and studied at the single fibril level free in solution. We furthermore address the physical properties of fibrils by studying their extension as a function of degree of confinement, as well as the mean fluorescence intensities per unit length. These data allow us to probe the concept of polymorphism on the single fibril level. We also compare amyloid fibrils prepared from two different amyloidogenic proteins of different sizes and function; the ∼4 kDa Alzheimer's related peptide Aβ(1–42) and the ∼14 kDa Parkinson's related protein α-synuclein. We envision that this methodological platform can be extended to study amyloid formation mechanisms and amyloid interactions on the single fibril level in a semi high-throughput manner that, moreover, requires very low sample volumes, opening possibilities to study both in vitro prepared amyloid material and samples retrieved from cells.
Materials and methods
Recombinant protein production
Wild-type (wt) Aβ(1–42) and α-syn were produced by recombinant expression in E. coli. Aβ(1–42) was expressed as a fusion protein to the His tag-labelled NT solubility tag31 and purified by affinity chromatography, TEV (tobacco etch virus) enzymatic cleavage and size exclusion chromatography as described in ref. 32. The peptide was obtained in a 20 mM sodium phosphate buffer at pH 8.0 and freeze dried in small aliquots that were stored at −20 °C until further use. α-syn was purified by consecutive steps of ion exchange and size exclusion chromatography into 20 mM Tris-HCl buffer pH 7.4 as described in ref. 33. The protein was aliquoted, snap-frozen in liquid nitrogen and stored at −80 °C until further use.Preparation and handling of HF555-Aβ(1–42) peptides
Synthetic N-terminal labelled HF555-Aβ(1–42) peptides were bought from Anaspec Inc. as lyophilized powders and dissolved in hexafluoro-2-isopropanol to disrupt aggregates and monomerize the peptide. The solutions were vortexed briefly and aliquoted at 4 °C. The solvent was removed by rotary evaporation. The resulting peptide films were snap frozen in liquid nitrogen and kept at −80 °C until further use. The concentration of HF555-Aβ(1–42) in the aliquots was determined by dissolving one peptide film in 1% ammonium hydroxide (v/v) followed by absorption measurements on a Cary 4000 UV-Vis Spectrophotometer (Agilent Technologies). An extinction coefficient of 150![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 M−1 cm−1 at 551 nm was used for the HF555 dye. Prior to each experiment, a new peptide film was dissolved in a small volume of 1% ammonium hydroxide (v/v) and thereafter diluted with 20 mM sodium phosphate buffer pH 8.0.
000 M−1 cm−1 at 551 nm was used for the HF555 dye. Prior to each experiment, a new peptide film was dissolved in a small volume of 1% ammonium hydroxide (v/v) and thereafter diluted with 20 mM sodium phosphate buffer pH 8.0.
Labelling of α-syn
α-syn monomers were labeled with ATTO488 (ATTO Technology Inc.) using NHS-ester chemistry according to the manufacturer's protocol. The recombinant protein was transferred into a PBS buffer with 100 mM NaCl using Amicon Ultra 0.5 mL 10 kDa cut-off centrifugal filters. 5 mg ml−1 of protein was incubated for 1 h at room temperature with a 2-fold molar excess of the ATTO-488 NHS-ester reagent. The α-syn protein was separated from unreacted dye using a Sephadex G-25 gel filtration spin column at 5000g for 5 min. The labeling ratio was determined by absorbance (Fig. S4†) and was approximately 90% (that is, on average 90% of the α-syn molecules carried one dye molecule). The labelled protein was flash-frozen on dry ice and stored at −80 °C.Amyloid fibril formation
Aβ(1–42) amyloid fibrils were prepared by incubation of 90 mol% of recombinant Aβ(1–42) with 10 mol% HF555-Aβ(1–42) in quiescent condition at 37 °C for 48 h. α-syn amyloid fibrils were prepared by seeding using so-called PFFs (pre-formed fibrils) that were prepared according to the Michael J. Fox Foundation protocol.34 Briefly, α-syn was assembled in PBS at 5 mg mL−1 by incubation for 7 days at 37 °C under constant agitation (1000 rpm, Benchmark Incu-Shaker Mini). The resulting PFFs were flash-frozen on dry ice and stored at −80 °C until further use. The labelled α-syn fibrils were prepared by seed amplification in a 100 μM solution containing 90% unlabeled and 10% labeled monomeric α-syn and 5% of PFFs. The samples were incubated in a Thermoshaker for three days at 37 °C at 600 rpm to elongate the seeds and obtain sufficiently long fibrils for the nanochannel analysis.Nanofluidic device
The nanofluidic devices used in this work were fabricated using traditional semiconductor processing methods that are described in detail elsewhere.22,35 Briefly, the design consists of 80 nanochannels, that run between two microchannels, each having two loading reservoirs. The nanochannels are 500 μm long and 300 nm deep. The width of the nanochannels increases in steps from 300 nm at one end via 600 nm, 900 nm, 1500 nm, 2400 nm and finally 3000 nm at the other end (Fig. S1†). Each of the above regions spans over 80 μm with a smooth transition in between consecutive widths.Lipid passivation
The nanochannels were passivated by creation of a supported lipid bilayer (SLB) as described by Persson et al.36 Large unilamellar vesicles with 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) and doped with 1% Marina Blue 1,2-dihexadecanoyl-sn-glycero-3-phosphoethanolamine (Marina Blue DHPE) for visualization purposes were used. To create the SLB, 5 μL of a 1 mM solution of large unilamellar vesicles were added to all 4 reservoirs. When the vesicles land at a sufficient density they rupture and the SLB is formed. Remaining vesicles can be flushed out and removed.DNA preparation
60 μM of T4-DNA (Nippon gene) was stained with YOYO-1 dye (1:5 dye:bp ratio) in 0.5× TBE buffer (10 μl total volume) and incubated at 50 °C for 30 minutes. Then, 88 μl of MilliQ water and 2 μl β-mercaptoethanol were added to a total volume of 100 μl. 15 μl of this solution was added into one of the loading reservoirs and the other three reservoirs were filled with 0.05× TBE buffer.
Data acquisition
The amyloid fibrils or the T4-DNA molecules were loaded into the nanofluidic chips. The sample was driven from the reservoir to the microchannel by applying pressure to the inlet containing the sample. The sample was then introduced into the nanochannel array by applying pressure to both inlets connecting that microchannel. The samples were imaged using an epi-configured fluorescence microscope (Zeiss AxioObserver.Z1), equipped with 100× TIRF oil immersion objective (Zeiss, NA = 1.46), a Colibri 7 LED light source (Zeiss) and an EMCCD camera (Andor Ixon). YOYO-1 (for visualisation of DNA) was excited in the 469/38 nm wavelength band and emission was collected at 530/25 nm. ATTO488 (for visualisation of α-syn fibrils) was excited in the 469/38 nm wavelength band and HF555 (for visualisation of Aβ(1-42) fibrils) in the 555/30 nm wavelength band and emission from both fluorophores were collected through a 90 HE multi bandpass emission filter (Zeiss). The images were analyzed with ImageJ and a custom written Matlab code.Results and discussion
We prepared fluorescent Aβ(1–42) amyloid fibrils from a 9:1 mix of recombinant wild-type (wt) and synthetic N-terminally labelled HF555-Aβ(1–42) peptides (as described in Material and methods). The fibrils were first introduced into the microchannels of the nanofluidic device (Fig. 1a), where free-flowing fibrils could be visualized with high contrast using epi-fluorescence timelapse imaging (Fig. 1b, left and Video S1†), thanks to the shallow channel depth (300 nm in nanochannel and 1.2 μm in microchannels), which effectively reduces out-of-focus fluorescence. As comparison, we also analyzed a sample of T4-DNA (166000 base pairs; contour length ∼56 μm37) stained with YOYO-1 (1:5 dye:base pairs) (Fig. 1b, right and Video S2†). Significant differences in the physical properties of the Aβ(1–42) amyloid fibrils and the T4-DNA were observed, suggesting that the amyloid fibrils are very stiff biopolymers, compared to the much softer DNA. The Aβ(1–42) amyloid fibrils were thereafter injected into the nanochannels of the device (Fig. 1a) by applying pressure to both inlets of one of the microfluidic channels (Video S3†). The nanochannels have a constant depth of 300 nm and a step-wise increase in width from 300 to 3000 nm, as schematically depicted in Fig. 1c. The change in the width of the nanochannels alters the level of confinement of the amyloid fibrils and in turn their extension, as shown for Aβ(1–42) amyloid fibrils in Fig. 1d and Videos S4–S7;† YOYO-1 stained T4-DNA was included for comparison. The difference in the change in extension when comparing the amyloid fibrils and the T4-DNA at 300 nm and 3000 nm is apparent and supports that amyloid fibrils are much stiffer than the DNA, even though the images also confirm that the Aβ(1–42) amyloid fibrils are flexible enough to be affected by the level of confinement.
 | ||
| Fig. 1 (a) Schematic illustration of the nanofluidic device, depicting four loading reservoirs (grey circles), feeding into two horizontal microchannels (light blue), which are connected by multiple vertical nanochannels with widths ranging from 300 nm to 3000 nm, see also (c). (b) Snap shot epifluorescence images from time-lapse videos of HF555-labelled Aβ(1–42) amyloid fibrils (left) or YOYO-1 stained T4-DNA molecules (right) residing in one of the microchannels of the device. (c) Close-up schematic illustration of the nanochannels, illustrating the principle of polymer stretching as function of degree of confinement. (d) Epifluorescence images of a single Aβ(1–42) amyloid fibril and a single T4-DNA molecule captured in the narrow (300 nm width) and wide (3000 nm width) end of a single nanochannel, respectively. All scale bars represent 4 μm. | ||
To determine the variation in fibril extension at different degrees of confinement, we recorded time-lapse movies of the Aβ(1–42) fibrils in the nanochannels (Videos S4–S7†) and translated the data into kymographs, where each image is represented by one row, and the rows are stacked on top of each other to represent the time dimension. Fig. 2a (left panels) shows representative kymographs at 300 nm and 1500 nm nanochannel width for the Aβ(1–42) fibrils. To extend the study, we included analysis of fluorescently labelled α-synuclein (α-syn) amyloid fibrils (Fig. 2a, middle panels and Videos S8–S11†), prepared as described in the Methods section to have a similar labelling density as the Aβ(1–42) fibrils. We also, again, compared the amyloid fibrils to the T4-DNA (Fig. 2a, right panels). It is again obvious that the amyloid fibrils are much stiffer than DNA. Plotting the average extension of each of the single polymers as function of the degree of confinement (nanochannel width) further supports this observation (Fig. 2b). The data for the Aβ(1–42) and α-syn amyloid fibrils have some resemblance to that of filaments of the bacterial DNA-repair protein RecA, which have a persistence length of ∼1 μm,26 whereas the persistence length of double stranded DNA is around 50 nm.38
 | ||
| Fig. 2 Extension and persistence lengths of single amyloid fibrils and DNA molecules. (a) Kymographs showing the polymer extension as function of time for single Aβ(1–42) and α-syn fibrils and a T4-DNA molecule confined in, respectively, the 300 nm and 1500 nm wide regions of single nanochannels. All three kymographs are 3 seconds long. All scale bars correspond to 4 μm. (b) Polymer extension as a function of channel width for the Aβ(1–42) fibril, α-syn fibril, and T4-DNA molecule shown in (a). (c and d) Fits of the data in (b) using the Odijk theory (eqn (1)) for the Aβ(1–42) fibril (c) and α-syn fibril (d), respectively. (e) Persistence lengths of individual Aβ(1–42) fibrils (n = 82) and α-syn fibrils (n = 46). | ||
Since the amyloid fibrils were much stiffer than DNA, the extension of the amyloid filaments at the different nanochannel geometries should follow the scaling proposed by Odijk24 for a biopolymer in a confined environment, as we have previously reported for RecA filaments.26 For rectangular channels of dimensions that are smaller than the polymer persistence length, and where the polymer is thus fluctuating between the channel walls, Odijk's theory predicts:
 | (1) |
Using the above-described approach, we then determined the persistence lengths of 82 individual Aβ(1–42) fibrils and 46 individual α-syn fibrils (Fig. 2e). The persistence length for the Aβ(1–42) fibrils varied from 1.6 μm to 16.4 μm, with an average of 5.9 ± 4.5 μm, whereas the persistence length for the α-syn fibrils varied from 1.5 μm to 7.0 μm, with an average of 3.0 ± 1.6 μm. The differences between the two fibril types are statistically significant (t test, p < 0.0001), and indicate that Aβ(1–42) fibrils are stiffer than α-syn fibrils. Moreover, the obtained values are in the same range as previously reported persistence lengths for amyloid fibrils formed by for example β-lactoglobulin,12 but considerably longer than the persistence lengths of protofibrils formed by for example β-lactoglobulin12 or the N-terminal domain of the hydrogenase maturation factor HypF.40 The data can, furthermore, be compared to reported persistence lengths of other protein polymers such as RecA filaments (Lp ∼ 1 μm26), actin filaments (Lp ∼ 17 μm41) and microtubules (Lp > 5000 μm42).
Next, we compared the persistence length data that were obtained using the nanofluidic device and analysis of single amyloid fibrils in solution to a more conventional method based on atomic force microscopy (AFM) topographical images of amyloid fibrils deposited onto mica and thereafter dried. Representative images of the Aβ(1–42) and α-syn fibrils are shown in Fig. 3a and b, confirming amyloid appearance and typical unbranched filaments. Average persistence lengths of 4.9 ± 2.2 μm for Aβ(1–42) fibrils and 2.1 ± 1.5 μm for α-syn fibrils were determined using the Easyworm software43,44 which is based on worm-like chain theory for semi-flexible polymers and uses contour lengths and polymer end-to-end distances obtained from the AFM images. The polymer flexibility was also depicted by aligning the analysed fibril fragments in contour plots (Fig. 3c and d). The average persistence lengths, as determined by Easyworm, are in good agreement with those obtained by single fibril analysis using our nanochannel-based method and Odijk theory (Fig. 2). The results furthermore support the above conclusion that the α-syn fibrils are less rigid than the Aβ(1–42) fibrils (the Lp difference as measured by AFM is also statistically significant (paired Student's t-test; p < 0.0001)). However, the AFM-based analysis may be prone to artifacts related to difficulties in preparing samples such that the conformation, and hence end-to-end distance, of each filament on the AFM substrate is entirely equilibrated,45,46 and one can, at best, only obtain an ensemble-averaged persistence length. Our solution-based nanofluidic method avoids this problem and thereby provides single fibril data. As will be further discussed below, this makes it possible to analyse heterogeneity within fibril samples.
 | ||
| Fig. 3 AFM analysis of amyloid fibril morphology and persistence length. (a and b) Representative AFM image of Aβ(1–42) (a) and α-syn (b) amyloid fibrils. The scale bars are 5 μm. (c and d) Contours of Aβ(1–42) (n = 310) (c) and α-syn (n = 341) (d) amyloid fibrils with their initial tangents aligned. The average persistence lengths of depicted fibrils, calculated using the Easyworm software, are indicated in the figure. (e and f) Histograms of fibril height distributions for the Aβ(1–42) (e) and α-syn (f) amyloid fibril samples, the numbers of analyzed individual fibrils are indicated in each graph. | ||
Since AFM probes the morphology of a surface with high (sub nanometer) resolution in the Z direction, we also used the AFM images to measure fibril height distributions (Fig. 3e and f). The data in Fig. 3e show a broad distribution of Aβ(1–42) amyloid fibril heights with a mode value of around 2.5 nm and a smaller shoulder around 4.2 nm, consistent with previously reported fibril thicknesses.47,48 There also appears to be a minor fibril population with a height of 6.5 nm. This digitation of thickness could indicate co-existence of fibril polymorphs with different numbers of protofilaments as observed by Meinhardt et al.49 for Aβ(1–40) fibrils. The α-syn amyloid fibrils were more homogenous with respect to height, with a major population at 6.3 ± 0.9 nm and a minor population at around 3.0 nm (Fig. 3f), possibly corresponding to fibrils consisting of two and one protofilaments respectively.
In the final part of the study, we took advantage of the fact that the confinement of amyloid fibrils into the nanochannels in our setup makes it possible to determine, in addition to persistence lengths, both amyloid fibril contour lengths and their mean fluorescence intensity per unit length (IPL); here defined on a per pixel basis, and hence explore correlations between different fibril properties on the single fibril level (Fig. 4). The ‘IPL’ should correlate to the fibril mass per unit length (MPL), assuming an even distribution of fluorophores along the fibril extension (which is reasonable given the kymographs in Fig. 2a). MPL is related to fibril thickness and a common descriptor of protofilament packing,47,48 and has been reported to vary between amyloid fibrils formed from different proteins as well as between amyloid fibrils from a single protein, reflecting their polymorphism.15,50 The IPL values (Fig. 4c and d, y-axis) of the different fibrils are not directly comparable as different fluorophores were used to label Aβ(1–42) and α-syn, but the observed variations in IPL within samples are still important. The contour lengths of the 82 Aβ(1–42) amyloid fibrils varied from 6.9 μm to 17.6 μm, with an average of 11.8 ± 2.6 μm (Fig. 4a, y-axis), whereas the contour lengths of the 46 α-syn fibrils varied from 6.1 to 9.3 μm, with an average of 7.3 ± 0.7 μm (Fig. 4b, y-axis histogram) and with no detected fibrils longer than 10 μm.
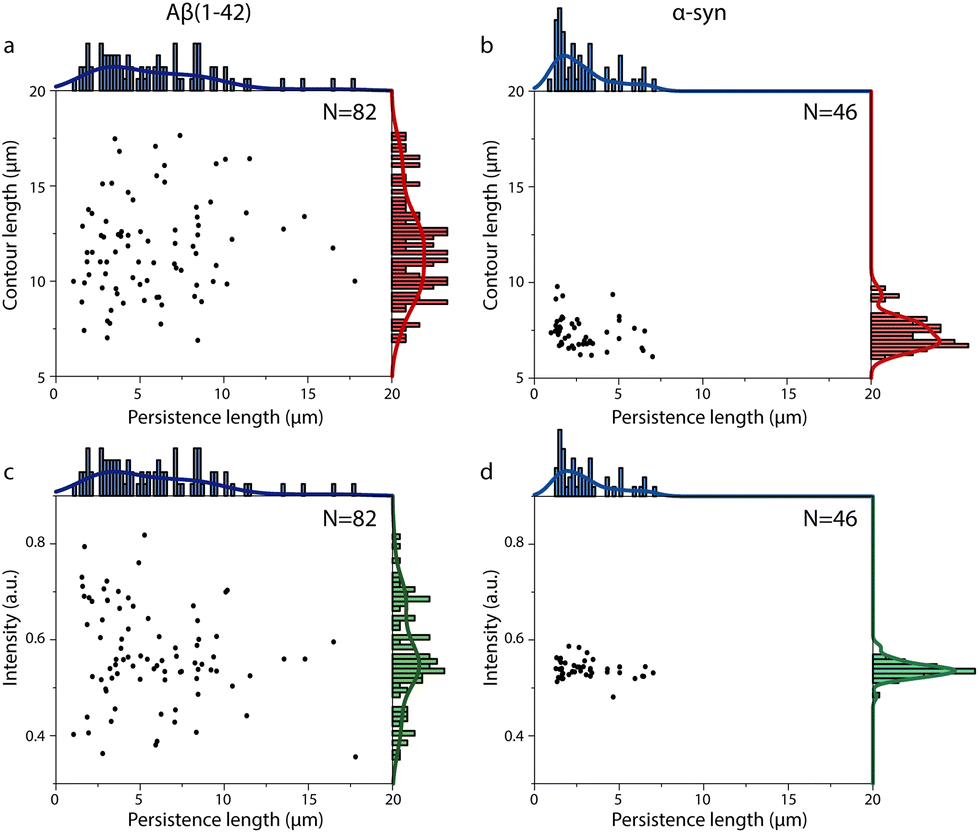 | ||
| Fig. 4 Relationships between persistence length, contour length, and fluorescence intensity per length unit for single amyloid fibrils. (a and c) Scatter plots of contour length versus persistence length for Aβ(1–42) (a) and α-syn (c) amyloid fibrils. (b and d) Scatter plots of fluorescence intensity per length unit (intensity) versus persistence length for Aβ(1–42) (b) and α-syn (d) amyloid fibril. | ||
Fig. 4 indicates that both the Aβ(1–42) and α-syn fibril samples are heterogeneous with respect to persistence lengths and IPL (intensity). A comparison of IPL (intensity) distributions and fibril height distributions suggests that the parameters are correlated (Fig. S5†) and that IPL can thus be used as a reasonable proxy of fibril thickness in the common discussion of the results. The Aβ(1–42) fibrils (Fig. 4a and c) appear to be more heterogeneous than the α-syn fibrils (Fig. 4b and d), and we even observed a few fibrils with very long persistence lengths of ∼15 μm, suggesting that our method can report on fibril polymorphism with respect to fibril stiffness. The observation of polymorphism is in agreement with previous reports on the heterogeneity of Aβ(1–42) amyloid fibrils.50
In Fig. 4 we further explored the relationship between persistence lengths and contour lengths. It should be noted that we only included fibrils that were longer than 6 μm in the analysis to be able to accurately record the effect of the change of nanochannel confinements on their extensions. We therefore cannot exclude that short fibrils could have different mechanical properties, even though the similarity in mean persistence lengths estimated from the nanochannel recordings (Fig. 4) and based on AFM analysis (Fig. 3, no restriction of fibril length) are very similar. Starting with the Aβ(1–42) amyloid fibrils (Fig. 4a), there was no apparent correlation between persistence length and contour length. There was also no correlation between IPL and contour length (Fig. S6†). This is consistent with a model of templated elongation, and hence perseverance of structure, as the underlying mechanism of the growth of individual amyloid fibrils. However, it would also be possible that structurally different amyloid polymorphs elongate at different rates and thus give rise to populations that differ in contour lengths. We therefore further explored the relation between persistence lengths of the Aβ(1–42) and their IPL (Fig. 4c). Considering the significant heterogeneity in both measures and the fact that polymer thickness in homogenous polymer materials correlates to stiffness, it is a bit surprising that our data suggest the two parameters to be uncorrelated. A possible explanation to this observation is that the persistence length variation is mainly related to differences in the arrangement of the β-sheet core of the fibrils (often reported to consist of dimers)47,50–53 and less dependent on the number of protofilaments in the fibril (hence its thickness).
The distribution of α-syn fibril persistence lengths (Fig. 4b, top axis histogram) is, as mentioned above, less heterogeneous than that of the Aβ(1–42) fibrils. There is also clearer support for the existence of a main population of fibrils with an average persistence length of 2.2 ± 0.7 μm and a smaller proportion of fibrils with longer persistence lengths. This could potentially reflect the co-existence of two polymorphs with different filament packing as observed in recent cryo-EM studies.15,54 As for the Aβ(1–42) fibrils, there was no correlation between persistence length and contour length (Fig. 4b) or IPL (Fig. 4d) or between contour length and IPL (Fig. S6b†). Furthermore, the IPL of the α-syn fibrils was significantly more uniform than for the Aβ(1–42) fibrils, with one narrow single mode distribution (Fig. 4d, y-axis histogram). This is consistent with the observation of a narrow height distribution in AFM (Fig. 3d) and suggests that heterogeneity in the α-syn samples studied here mainly manifests in relation to their rigidity, which, again, appears to not be directly correlated to fibril thickness (IPL) (Fig. 4d), but presumably more to filament packing.
Comparing the data for the Aβ(1–42) and α-syn fibrils displayed in Fig. 2 and 4, we conclude that there is a clear difference in their mean persistence length (Fig. 2e and 4a, b). We note that α-syn is a considerably larger protein than Aβ(1–42) (14.5 kDa vs. 4.2 kDa) and that it has a distinctly different fibril fold with a more complex topography15,47,53,55 compared to Aβ(1–42) fibrils.47,53 This is also reflected in the AFM data, showing that the α-syn fibrils are thicker (Fig. 3e and f). This may allow for greater conformational flexibility between the monomer ‘layers’ in the fibril, despite that the α-syn fibrils are likely to have a more extensive network of cross-β core hydrogen bonds. Indeed, it has been reported that there are additional constraints in packing of larger proteins into an amyloid core that might destabilize their fibril structure and make the fibrils softer43 and an inverse correlation between axial stiffness and cross-sectional area of amyloid fibrils formed by different proteins have been reported.56 The observation that the distributions of IPL and fibril thicknesses of the α-syn fibrils (Fig. 4d and 3d) were much narrower than those for the Aβ(1–42) fibrils (Fig. 4c and 3c) may be related to the former being amplified by seeding, thus conserving to a larger extent a pre-defined packing.
Conclusion
We have demonstrated that nanofluidic channels can be used for multiparametric analysis of single fluorescent amyloid fibrils in solution. We have determined the extensions, emission intensities (related to the mass per unit length of the fibrils) and persistence lengths for fibrils formed by Aβ(1–42) and α-syn. As expected, the persistence lengths for fibrils formed by both proteins are in the μm regime and considerably larger than the persistence length of DNA. The Aβ(1–42) fibrils are significantly stiffer than the α-syn fibrils, which may relate to that they are formed from a shorter polypeptide chain and hence have a more compact fibril core packing. Interestingly, we observe that both Aβ(1–42) and α-syn fibril samples are heterogeneous and that there is a significant variation in the persistence lengths of individual fibrils within each sample. An interesting observation is that we did not observe any correlation between IPL (which can be interpreted as a proxy of fibril thickness). This suggests that the amyloid core assembly and filament packing may be more decisive for the stiffness of an amyloid fibril than its actual thickness (number of filaments). Furthermore, the observations of persistence length heterogeneity provide a complementary perspective to that of solving amyloid structures or using amyloid sensitive dyes.57 It is also important to note that heterogeneity manifests in the mesoscopic morphological characteristics (persistence length) of the amyloid polymer and that this can be detected and quantified using our nanofluidic-based method. We foresee that nanofluidic channels can be a useful future tool for analyzing single amyloid fibrils in solution and to reveal how the principles that affect their physical properties may relate to their pathological potential in a disease context.Author contributions
E. K. E. and F. W. conceived the idea of the project. N. S. designed and performed a majority of the experiments and analysed the data. R. S. and Q. L. contributed to experimental design and initial experiments. S. K. K. fabricated the nanofluidic devices and performed the experiments on DNA. M. G. prepared and characterized the α-syn fibrils. K. D. D. contributed with the theoretical framework to determine persistence lengths. N. S., K. D. D., E. K. E., and F. W. interpreted the data and wrote the paper. All authors read and commented on the final version.Conflicts of interest
There are no conflicts to declare.Acknowledgements
N. S. is funded by the Chalmers Area of Advance Nano Excellence PhD student program. E. K. E. acknowledges funding from the Knut and Alice Wallenberg foundation's academy fellow program, the Swedish Research Council (2016-03902 and 2020-05303) and the Wenner-Gren foundations. F. W. acknowledges funding from the European Research Council, in the form of an ERC consolidator grant (nanoDNArepair, no. 866238), and the Swedish Research Council (2020-03400). The Matlab software for analyzing kymographs was developed by the group of Prof. Tobias Ambjörnsson at Lund University. The nanofluidic device fabrication was done at MyFab at Chalmers University of Technology.References
- J. D. Sipe, M. D. Benson, J. N. Buxbaum, S.-i. Ikeda, G. Merlini, M. J. Saraiva and P. Westermark, Amyloid, 2016, 23, 209–213 CrossRef CAS PubMed.
- F. Chiti and C. M. Dobson, Annu. Rev. Biochem., 2006, 75, 333–366 CrossRef CAS PubMed.
- D. M. Fowler, A. V. Koulov, W. E. Balch and J. W. Kelly, Trends Biochem. Sci., 2007, 32, 217–224 CrossRef CAS.
- M. Tanaka, S. R. Collins, B. H. Toyama and J. S. Weissman, Nature, 2006, 442, 585–589 CrossRef CAS.
- D. M. Fowler, A. V. Koulov, C. Alory-Jost, M. S. Marks, W. E. Balch and J. W. Kelly, PLoS Biol., 2006, 4, e6 CrossRef.
- D. Otzen and P. H. Nielsen, Cell. Mol. Life Sci., 2008, 65, 910–927 CrossRef CAS PubMed.
- C. Lendel and N. Solin, RSC Adv., 2021, 11, 39188–39215 RSC.
- X. Ye, K. Junel, M. Gällstedt, M. Langton, X.-F. Wei, C. Lendel and M. S. Hedenqvist, ACS Sustainable Chem. Eng., 2018, 6, 5462–5469 CrossRef CAS.
- D. Otzen and R. Riek, Cold Spring Harbor Perspect. Biol., 2019, 11, a033860 CrossRef CAS.
- T. Scheibel, R. Parthasarathy, G. Sawicki, X.-M. Lin, H. Jaeger and S. L. Lindquist, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 4527–4532 CrossRef CAS.
- F. S. Ruggeri, F. Benedetti, T. P. Knowles, H. A. Lashuel, S. Sekatskii and G. Dietler, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 7230–7235 CrossRef CAS PubMed.
- C. C. vandenAkker, M. F. Engel, K. P. Velikov, M. Bonn and G. H. Koenderink, J. Am. Chem. Soc., 2011, 133, 18030–18033 CrossRef CAS PubMed.
- L. M. Sagis, C. Veerman and E. van der Linden, Langmuir, 2004, 20, 924–927 CrossRef CAS PubMed.
- M. Kollmer, W. Close, L. Funk, J. Rasmussen, A. Bsoul, A. Schierhorn, M. Schmidt, C. J. Sigurdson, M. Jucker and M. Fändrich, Nat. Commun., 2019, 10, 1–8 CrossRef CAS.
- B. Li, P. Ge, K. A. Murray, P. Sheth, M. Zhang, G. Nair, M. R. Sawaya, W. S. Shin, D. R. Boyer and S. Ye, Nat. Commun., 2018, 9, 1–10 CrossRef PubMed.
- L. J. Young, G. S. K. Schierle and C. F. Kaminski, Phys. Chem. Chem. Phys., 2017, 19, 27987–27996 RSC.
- C. F. Kaminski and G. S. K. Schierle, Neurophotonics, 2016, 3, 041807 CrossRef PubMed.
- P. Arosio, T. Müller, L. Rajah, E. V. Yates, F. A. Aprile, Y. Zhang, S. I. Cohen, D. A. White, T. W. Herling and E. J. De Genst, ACS Nano, 2016, 10, 333–341 CrossRef CAS PubMed.
- K. L. Saar, Y. Zhang, T. Müller, C. P. Kumar, S. Devenish, A. Lynn, U. Łapińska, X. Yang, S. Linse and T. P. Knowles, Lab Chip, 2018, 18, 162–170 RSC.
- M. Wolff, J. J. Mittag, T. W. Herling, E. D. Genst, C. M. Dobson, T. P. Knowles, D. Braun and A. K. Buell, Sci. Rep., 2016, 6, 1–10 CrossRef.
- E. V. Yates, T. Müller, L. Rajah, E. J. De Genst, P. Arosio, S. Linse, M. Vendruscolo, C. M. Dobson and T. P. Knowles, Nat. Chem., 2015, 7, 802–809 CrossRef CAS PubMed.
- K. Frykholm, V. Müller, K. Sriram, K. D. Dorfman and F. Westerlund, Q. Rev. Biophys., 2022, 55, e12 CrossRef.
- P.-G. de Gennes, Scaling concepts in polymer physics, Cornell university press, 1979 Search PubMed.
- T. Odijk, Macromolecules, 1983, 16, 1340–1344 CrossRef CAS.
- F. Persson, P. Utko, W. Reisner, N. B. Larsen and A. Kristensen, Nano Lett., 2009, 9, 1382–1385 CrossRef CAS PubMed.
- K. Frykholm, M. Alizadehheidari, J. Fritzsche, J. Wigenius, M. Modesti, F. Persson and F. Westerlund, Small, 2014, 10, 884–887 CrossRef CAS PubMed.
- S. Köster, J. Kierfeld and T. Pfohl, Eur. Phys. J. E: Soft Matter Biol. Phys., 2008, 25, 439–449 CrossRef PubMed.
- S. Köster, H. Stark, T. Pfohl and J. Kierfeld, Biophys. Rev. Lett., 2007, 2, 155–166 CrossRef.
- S. Köster, D. Steinhauser and T. Pfohl, J. Phys.: Condens. Matter, 2005, 17, S4091 CrossRef.
- S. Köster, D. A. Weitz, R. D. Goldman, U. Aebi and H. Herrmann, Curr. Opin. Cell Biol., 2015, 32, 82–91 CrossRef PubMed.
- A. Abelein, G. Chen, K. Kitoka, R. Aleksis, F. Oleskovs, M. Sarr, M. Landreh, J. Pahnke, K. Nordling and N. Kronqvist, Sci. Rep., 2020, 10, 1–10 CrossRef PubMed.
- J. E. Tropea, S. Cherry and D. S. Waugh, High throughput protein expression and purification: methods and protocols, 2009, pp. 297–307 Search PubMed.
- A. Abelein, G. Chen, K. Kitoka, R. Aleksis, F. Oleskovs, M. Sarr, M. Landreh, J. Pahnke, K. Nordling and N. Kronqvist, Sci. Rep., 2020, 10, 235 CrossRef CAS PubMed.
- N. K. Polinski, L. A. Volpicelli-Daley, C. E. Sortwell, K. C. Luk, N. Cremades, L. M. Gottler, J. Froula, M. F. Duffy, V. M. Lee and T. N. Martinez, J. Parkinson's Dis., 2018, 8, 303–322 Search PubMed.
- E. S. Kang, K. Sriram, I. Jeon, J. Kim, S. Chen, K. H. Kim, K. H. Kim, H. S. Lee, F. Westerlund and M. P. Jonsson, Adv. Sci., 2022, 9, 2201907 CrossRef CAS PubMed.
- F. Persson, J. Fritzsche, K. U. Mir, M. Modesti, F. Westerlund and J. O. Tegenfeldt, Nano Lett., 2012, 12, 2260–2265 CrossRef CAS PubMed.
- M. Ueda, K. Yoshikawa and M. Doi, Polym. J., 1997, 29, 1040 CrossRef CAS.
- C. G. Baumann, S. B. Smith, V. A. Bloomfield and C. Bustamante, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 6185–6190 CrossRef CAS PubMed.
- K. Jo, D. M. Dhingra, T. Odijk, J. J. de Pablo, M. D. Graham, R. Runnheim, D. Forrest and D. C. Schwartz, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 2673–2678 CrossRef CAS PubMed.
- A. Relini, S. Torrassa, R. Ferrando, R. Rolandi, S. Campioni, F. Chiti and A. Gliozzi, Biophys. J., 2010, 98, 1277–1284 CrossRef CAS PubMed.
- H. Isambert, P. Venier, A. C. Maggs, A. Fattoum, R. Kassab, D. Pantaloni and M.-F. Carlier, J. Biol. Chem., 1995, 270, 11437–11444 CrossRef CAS PubMed.
- M. E. Janson and M. Dogterom, Biophys. J., 2004, 87, 2723–2736 CrossRef CAS PubMed.
- T. P. Knowles, A. W. Fitzpatrick, S. Meehan, H. R. Mott, M. Vendruscolo, C. M. Dobson and M. E. Welland, science, 2007, 318, 1900–1903 CrossRef CAS.
- G. Lamour, J. B. Kirkegaard, H. Li, T. P. Knowles and J. Gsponer, Source Code Biol. Med., 2014, 9, 1–6 CrossRef PubMed.
- S. Mantelli, P. Muller, S. Harlepp and M. Maaloum, Soft Matter, 2011, 7, 3412–3416 RSC.
- C. Rivetti, M. Guthold and C. Bustamante, J. Mol. Biol., 1996, 264, 919–932 CrossRef CAS PubMed.
- M. T. Colvin, R. Silvers, Q. Z. Ni, T. V. Can, I. Sergeyev, M. Rosay, K. J. Donovan, B. Michael, J. Wall and S. Linse, J. Am. Chem. Soc., 2016, 138, 9663–9674 CrossRef CAS PubMed.
- R. Cukalevski, X. Yang, G. Meisl, U. Weininger, K. Bernfur, B. Frohm, T. P. Knowles and S. Linse, Chem. Sci., 2015, 6, 4215–4233 RSC.
- J. Meinhardt, C. Sachse, P. Hortschansky, N. Grigorieff and M. Fändrich, J. Mol. Biol., 2009, 386, 869–877 CrossRef CAS PubMed.
- M. Fändrich, S. Nyström, K. P. R. Nilsson, A. Böckmann, H. LeVine III and P. Hammarström, J. Intern. Med., 2018, 283, 218–237 CrossRef PubMed.
- L. Gremer, D. Schölzel, C. Schenk, E. Reinartz, J. Labahn, R. B. Ravelli, M. Tusche, C. Lopez-Iglesias, W. Hoyer and H. Heise, Science, 2017, 358, 116–119 CrossRef CAS.
- M. Schmidt, A. Rohou, K. Lasker, J. K. Yadav, C. Schiene-Fischer, M. Fändrich and N. Grigorieff, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 11858–11863 CrossRef CAS.
- M. A. Wälti, F. Ravotti, H. Arai, C. G. Glabe, J. S. Wall, A. Böckmann, P. Güntert, B. H. Meier and R. Riek, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, E4976–E4984 CrossRef.
- R. Guerrero-Ferreira, N. M. Taylor, A.-A. Arteni, P. Kumari, D. Mona, P. Ringler, M. Britschgi, M. E. Lauer, A. Makky and J. Verasdonck, eLife, 2019, 8, e48907 CrossRef CAS PubMed.
- M. D. Tuttle, G. Comellas, A. J. Nieuwkoop, D. J. Covell, D. A. Berthold, K. D. Kloepper, J. M. Courtney, J. K. Kim, A. M. Barclay and A. Kendall, Nat. Struct. Mol. Biol., 2016, 23, 409–415 CrossRef CAS PubMed.
- R. Nassar, E. Wong, J. r. Gsponer and G. Lamour, J. Am. Chem. Soc., 2018, 141, 58–61 CrossRef PubMed.
- D. J. Lindberg, M. S. Wranne, M. G. Gatty, F. Westerlund and E. K. Esbjörner, Biochem. Biophys. Res. Commun., 2015, 458, 418–423 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3nr02740f |
| This journal is © The Royal Society of Chemistry 2023 |