
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRuthenium nanoparticles stabilized by 1,2,3-triazolylidene ligands in the hydrogen isotope exchange of E–H bonds (E = B, Si, Ge, Sn) using deuterium gas†‡
Pablo
Molinillo
a,
Maxime
Puyo
a,
Florencia
Vattier
b,
Bertrand
Lacroix
 c,
Nuria
Rendón
*a,
Patricia
Lara
*a and
Andrés
Suárez
*a
c,
Nuria
Rendón
*a,
Patricia
Lara
*a and
Andrés
Suárez
*a
aInstituto de Investigaciones Químicas (IIQ), Departamento de Química Inorgánica, and Centro de Innovación en Química Avanzada (ORFEO-CINQA). CSIC and Universidad de Sevilla, Avda. Américo Vespucio, 49, 41092 Sevilla, Spain. E-mail: nuria@iiq.csic.es; patricia@iiq.csic.es; andres.suarez@iiq.csic.es
bInstituto de Ciencia de Materiales de Sevilla. CSIC-Universidad de Sevilla, Avda. Américo Vespucio 49, 41092 Sevilla, Spain
cDepartamento de Física Aplicada I, Escuela Politécnica Superior, Universidad de Sevilla, Virgen de África 7, 41011 Sevilla, Spain
First published on 9th August 2023
Abstract
A series of ruthenium nanoparticles (Ru·MIC) stabilized with different mesoionic 1,2,3-triazolylidene (MIC) ligands were prepared by decomposition of the Ru(COD)(COT) (COD = 1,5-cyclooctadiene; COT = 1,3,5-cyclooctatriene) precursor with H2 (3 bar) in the presence of substoichiometric amounts of the stabilizer (0.1–0.2 equiv.). Small and monodisperse nanoparticles exhibiting mean sizes between 1.1 and 1.2 nm were obtained, whose characterization was carried out by means of transmission electron microscopy (TEM), including high resolution TEM (HRTEM), inductively coupled plasma (ICP) analysis and X-ray photoelectron spectroscopy (XPS). In particular, XPS measurements confirmed the presence of MIC ligands on the surfaces of the nanoparticles. The Ru·MIC nanoparticles were used in the isotopic H/D exchange of different hydrosilanes, hydroboranes, hydrogermananes and hydrostannanes using deuterium gas under mild conditions (1.0 mol% Ru, 1 bar D2, 55 °C). Selective labelling of the E–H (E = B, Si, Ge, Sn) bond in these derivatives, with high levels of deuterium incorporation, was observed.
Introduction
Hydrogen isotope exchange (HIE) reactions have drawn significant attention due to the important role that isotopically labelled compounds have in different fields. For example, molecules containing heavier isotopes of hydrogen (i.e. deuterium and tritium) are used in drug discovery and development as internal mass spectrometry standards, for the realization of biochemical mechanistic studies or in the preparation of pharmaceutical derivatives with improved properties.1–5 Moreover, deuterated reagents are important tools for the investigation of the mechanisms of chemical reactions.6,7The synthesis of deuterium labelled organic molecules is more conveniently achieved through direct H/D exchange involving catalytic C–H bond activation at a late synthetic stage.8,9 By using a directing group on the molecule, transition metal catalysts selectively activate C–H bonds and endow the corresponding labelled derivatives with high levels of deuterium incorporation. Since the seminal studies of Chaudret, Rousseau, Pieters et al.,10 based on the initial observations from the Finke group,11 the use of mono- and bimetallic nanoparticles (NPs) in HIE has been growing due to their high activity and selectivity in the deuteration of functionalized molecules using readily available deuterium gas.12–28 This observed reactivity is attributable to the fact that metal nanoparticles readily activate C–H and H–H bonds under mild conditions, therefore lending suitable catalytic systems for H/D exchange.29
Hydrosilanes, hydroboranes and hydrides of elements heavier than silicon (Ge and Sn) are widely employed reagents in synthetic organic chemistry, particularly due to their ability to catalytically add to unsaturated C–C, C–O, and C–N bonds30 and reduce carbon–halogen bonds.31 H/D exchange in these metalloid hydrides might find application in deuterium labelling of complex organic molecules using well-developed catalytic protocols, as well as in the mechanistic study of these transformations. Selective deuteration of hydrosilanes using D2 has been effected using different transition metal complexes32–43 and heterogeneous metal catalysts.44 Borane deuteration based on H/D exchange with deuterium gas has also been accomplished through the use of similar catalysts.41,42,45 Meanwhile, to our knowledge, only one catalytic system has been reported for H/D exchange in germananes and stannanes.41 Development of catalytic systems applicable to all these main-group element hydrides remains an interesting challenge.
In addition to H–H and C–H bonds, metal nanoparticles have also been shown to activate Si–H46 and B–H bonds.47 However, their use in H/D exchange in hydrosilanes, as well as in other hydrides of main group elements (B, Sn, Ge), has not been reported. Herein, we describe the synthesis and characterization of a series of small and monodisperse Ru nanoparticles stabilized by mesoionic 1,2,3-triazolylidene ligands (MICs)48 and their application in the deuteration of metalloid hydrides under mild conditions using deuterium gas.49 Use of ligands for metal nanoparticle stabilization allows a facile tuning of the size, shape and surface properties of the nanomaterials through easy modification of the structure and amount of the capping ligand.50 N-Heterocyclic carbenes (NHCs) are a structurally ample family of ligands widely employed in coordination chemistry and catalysis.51 Straightforward synthesis and manipulation of these derivatives, along with their structural and steric variability and strong donor properties, make them ideal molecules to be used as stabilizing agents for the synthesis of nanoparticles for catalytic applications.52 Examples of NHC-stabilized metal nanoparticles are mainly limited to the use of imidazole-2-ylidene (uNHC)53,54 and imidazolidin-2-ylidene (sNHC)55 ligands as capping agents (Fig. 1). Mesoionic 1,2,3-triazolylidene ligands possess stronger electron donor properties than more traditional imidazole-2-ylidene and imidazolidin-2-ylidene derivatives, which makes them appealing stabilizers for the synthesis of nanoparticles.48 Moreover, precursors of 1,2,3-triazolylidenes (i.e. 1,2,3-triazolium salts) are readily accessible and easily amenable to structural changes in few steps through conventional organic synthetic methodologies.
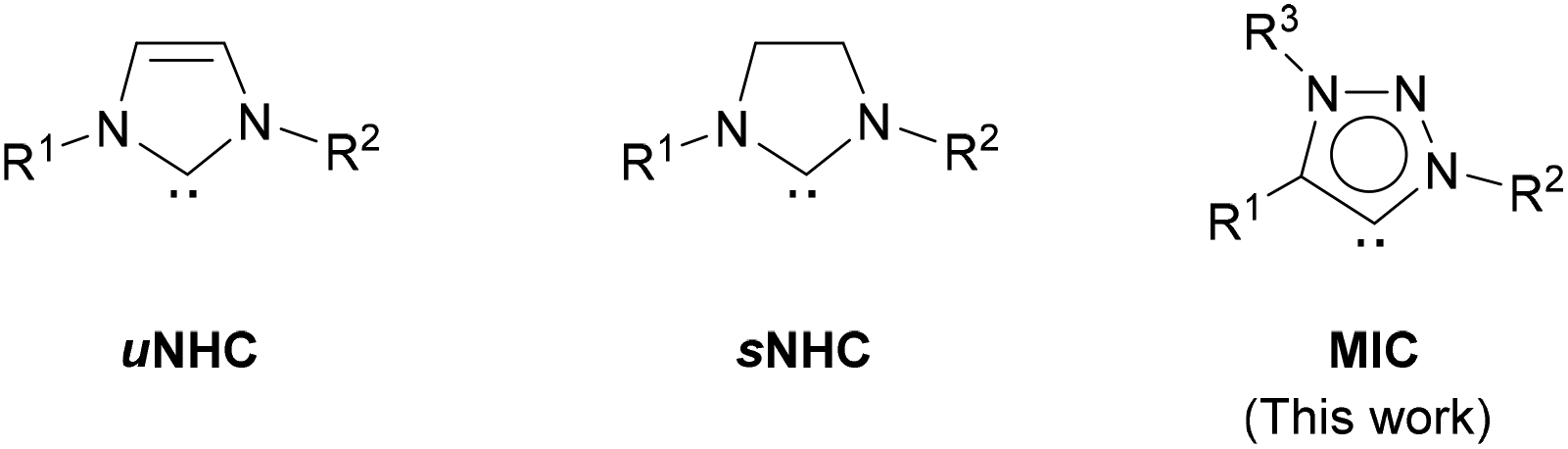 | ||
| Fig. 1 N-Heterocyclic carbene ligands used for nanoparticle stabilization. | ||
Results and discussion
Synthesis and characterization of Ru-MIC nanoparticles
A series of ruthenium nanoparticles stabilized by 1,2,3-triazolylidene ligands (MICs) were synthesized by decomposition of the ruthenium complex Ru(COD)(COT), (1,5-cyclooctadiene)(1,3,5-cyclooctatriene)ruthenium(0), dissolved in THF with H2 (3 bar), in the presence of substoichiometric amounts of the corresponding MIC ligand (MIC/Ru = 0.1 or 0.2 equiv.) (Scheme 1).56 In turn, the triazolylidene ligands were generated in situ by treatment of the corresponding 1,2,3-triazolium tetrafluoroborate salts with potassium bis(trimethylsilyl)amide, KHMDS. Three different carbene ligands MIC1–MIC3, using MIC/Ru ratios of 0.2 equiv., were initially examined. As determined by TEM analysis, in all cases, small (1.1–1.2 nm mean sizes) and monodisperse nanoparticles were obtained (Table 1 and Fig. 2). When a lower amount of the MIC1 ligand was employed in the synthetic procedure, superstructures formed of individual nanoparticles of a mean size of 1.8 (0.5) nm were observed, which is an indication of slightly poorer nanoparticle stabilization (Fig. 2). Metal contents of the obtained nanomaterials, which are stable both in solution and in the solid state under an inert atmosphere, were determined by ICP analysis of the purified samples (Table 1). Finally, control experiments aimed at the preparation of nanoparticles using directly the 1,2,3-triazolium salts in the absence of a base led to the formation of bulk metal, therefore ruling out the possibility of nanoparticle stabilization by the triazolium salts. | ||
| Scheme 1 Synthesis of Ru·MIC nanoparticles and 1,2,3-triazolylidene ligands used. | ||
 | ||
| Fig. 2 TEM images with size distribution histograms of Ru·MIC10.2, Ru·MIC10.1, Ru·MIC20.2 and Ru·MIC30.2 (from top to bottom). | ||
High-resolution transmission electron microscopy (HRTEM) images were recorded in order to assess the crystal structure of the Ru·MIC10.2 and Ru·MIC30.2 nanoparticles, as representative examples (Fig. 3, left and Fig. S1‡). The structure of these materials matches the hexagonal close-packed (hcp) structure expected for Ru nanoparticles.53 Moreover, the ruthenium composition of the nanoparticles was confirmed by STEM-EDS analysis (Fig. 3, right).
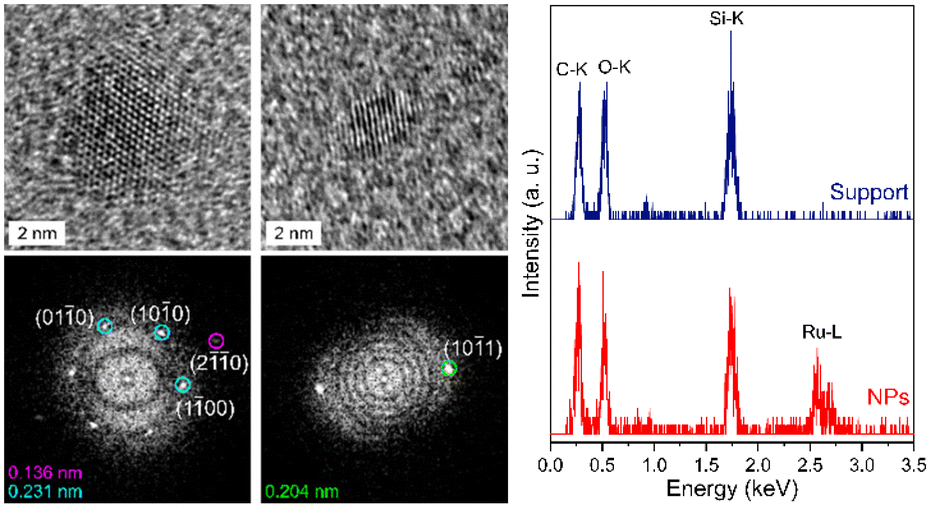 | ||
| Fig. 3 HRTEM images (left, top) of the Ru·MIC10.2 nanoparticles. The corresponding fast Fourier transform analyses of spatial frequencies (left, bottom) show that the lattice fringes observed on the HRTEM images correspond to the planes of the hexagonal close-packed (hcp) structure. STEM-EDS analysis (right) of the Ru·MIC10.2 nanoparticles. | ||
The composition and chemical state of the surfaces of the nanoparticles were analysed by X-ray photoelectron spectroscopy (XPS). Since the Ru 3d signal partially overlaps with the C 1s peak, the Ru 3p photoemission signal was preferred for XPS analysis.57Fig. 4(a) shows the high resolution Ru 3p region for the Ru·MIC10.2 (a-1), Ru·MIC20.2 (a-2) and Ru·MIC30.2 (a-3) nanoparticles. The Ru 3p region shows two peaks centred at 484.1 and 461.9 eV binding energy (BE) that correspond to the 3p1/2 and 3p3/2 peaks, respectively. The Ru 3p3/2 region is well fitted into two components separated by 3.4 eV, corresponding to the Ru(0) and Ru(IV) oxidation states, respectively.57,58 In all cases, the percentage of ruthenium oxidized is ca. 7–9% of the surface metal, which is very similar to the Ru(0)/Ru(IV) ratio observed for other Ru nanoparticles.59
 | ||
| Fig. 4 X-ray photoelectron spectroscopy (XPS) peaks of (a) Ru 3p regions of the Ru·MIC10.2 (a-1), Ru·MIC20.2 (a-2) and Ru·MIC30.2 (a-3) nanoparticles (experimental and fitted spectra) and (b) N 1s regions of Ru·MIC10.2 (b-1) (experimental and fitted spectra) and the triazolium salt precursor of MIC1 (b-2) (experimental spectrum). | ||
The presence of the MIC ligands on the surfaces of the nanoparticles was confirmed by X-ray photoelectron spectroscopy (XPS). The spectra of the high resolution N 1s region of the Ru·MIC10.2 nanoparticles and the MIC1 precursor are shown in Fig. 4(b-1) and (b-2), respectively. In the case of the triazolium salt of MIC1 (b-2), a signal at 402.5 eV BE (fwhm = 2.20 eV) was observed, which corresponds to the three nitrogen atoms in the molecule.60 The N 1s photoemission peak of the Ru·MIC10.2 nanoparticles consists of a broad signal centred at 401.2 eV, which can be deconvoluted into two components. The lowest BE peak at 399.4 eV (fwhm = 2.50 eV) belongs to the N atom adjacent to the carbene C atom. The major peak observed at a higher BE, 401.4 eV (fwhm = 2.77 eV), can be assigned to the other two N atoms in the ligand that are further away from the carbenic carbon atom. As can be seen in Fig. 4(b-1), the peak area ratio of the two components corresponds to the expected 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 ratio of the nitrogen atoms in the ligand. The N 1s signal for the Ru·MIC10.2 nanoparticles was broader and appeared at a lower BE than that observed for the same peak of the MIC1 precursor. While the coordination of the MIC ligand to the Ru nanoparticles should produce a loss of charge density on the N atom adjacent to the carbenic carbon and give rise to a peak appearing at a higher BE value than in the case of the free carbene ligand, this could not be corroborated since the MIC ligand was not isolable. However, as shown in the case of metal nanoparticles stabilized by other NHC ligands, the position of the N 1s peak for the Ru·MIC nanoparticles appears at a lower BE than that of the cationic ligand precursor.16,61 Therefore, the latter observation is in line with a direct coordination of the carbene ligand to the surface metal atoms. Similar results have been found for the corresponding triazolium salts of the MIC2 and MIC3 ligands and their corresponding Ru·MIC20.2 and Ru·MIC30.2 nanoparticles (Fig. S3 and S4‡).
:2 ratio of the nitrogen atoms in the ligand. The N 1s signal for the Ru·MIC10.2 nanoparticles was broader and appeared at a lower BE than that observed for the same peak of the MIC1 precursor. While the coordination of the MIC ligand to the Ru nanoparticles should produce a loss of charge density on the N atom adjacent to the carbenic carbon and give rise to a peak appearing at a higher BE value than in the case of the free carbene ligand, this could not be corroborated since the MIC ligand was not isolable. However, as shown in the case of metal nanoparticles stabilized by other NHC ligands, the position of the N 1s peak for the Ru·MIC nanoparticles appears at a lower BE than that of the cationic ligand precursor.16,61 Therefore, the latter observation is in line with a direct coordination of the carbene ligand to the surface metal atoms. Similar results have been found for the corresponding triazolium salts of the MIC2 and MIC3 ligands and their corresponding Ru·MIC20.2 and Ru·MIC30.2 nanoparticles (Fig. S3 and S4‡).
Catalytic H/D exchange
The new Ru·MIC nanoparticles were initially tested in H/D exchange in hydrosilanes. A comparison of the catalytic activity of the prepared nanoparticles was performed using PhMe2SiH (Si-1) as the selected model substrate and D2 (1 bar, ca. 0.4 equiv.) in THF-d8 at 55 °C, with 1.0 mol% Ru catalyst loading (Table 2; see the Experimental section for details). All the Ru·MIC0.2 nanoparticles were found to be active in the reaction, selectively yielding the product deuterated at the SiH position (entries 1–3), with the Ru·MIC10.2 catalyst leading to the highest deuterium incorporation. A lower catalytic activity was observed in the case of Ru·MIC10.1 (entry 4). Interestingly, the catalytic activity achieved with the Ru·MIC nanoparticles was found to be higher than that provided by Ru·IMes0.2 (IMes = 1,3-dimesitylimidazol-2-ylidene) nanoparticles (entry 5), which were prepared following a previously reported method (1.5 (0.3) nm mean size, 66 wt% Ru).53| Entry | Ru·MIC | D incorporation (%) |
|---|---|---|
| a Reaction conditions: 1.0 mol% Ru, 1 bar D2 (ca. 0.4 equiv. D2), 55 °C, THF-d8. [S] = 1.2 M. Reaction time: 19 h. A single D2 loading was used. Deuterium incorporation as determined by 1H NMR spectroscopy. | ||
| 1 | Ru·MIC10.2 | 60 |
| 2 | Ru·MIC20.2 | 53 |
| 3 | Ru·MIC30.2 | 54 |
| 4 | Ru·MIC10.1 | 49 |
| 5 | Ru·IMes0.2 | 45 |
Having determined that Ru·MIC10.2 is the most active catalyst of the series, these nanoparticles were examined in the deuteration of other silanes (Chart 1). To ensure high deuterium incorporation, a modification of the experimental protocol was carried out consisting of the use of two charges of D2 (see the Experimental section for details). Under these conditions, PhMe2SiH (Si-1) showed a conversion of 96% into the corresponding deuterated isotopologue. Other silanes having alkyl and aryl substituents (Si-2, Si-3, Si-4, Si-5, Si-6 and Si-7) were also selectively deuterated at the Si–H position with conversions higher than 85%. In the reaction of tripropylsilane (Si-5), however, low deuterium incorporation (ca. 5%) at the α positions of the Si atoms was also observed. Similarly, selective labelling of an alkoxysilane (Si-8) was carried out with high deuterium incorporation (>99%), whereas deuteration of dimethylchlorosilane (Si-9) was accomplished with 73% conversion. The reactions of diphenyl- (Si-10) and phenylsilane (Si-11), however, required higher catalyst loadings (2.0 mol% Ru), leading to 72% and 42% deuterium incorporation, respectively. The reduced reactivity of (polyhydro)silanes can be ascribed to the formation of ![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) SiR2 and
SiR2 and ![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) SiR silyl groups that strongly interact with the nanoparticle surface. In this respect, it should be noted that ruthenium nanoparticles stabilized by a silane (octylsilane) have been reported and that the formation of SiR silyl groups, probably linked to several ruthenium atoms, has been attested.62 TEM analysis of Ru·MIC10.2 after the catalytic reaction of Si-5 showed only a slight increase in the size of the nanoparticles (mean size: 2.2 (0.4) nm; Fig. S2‡), confirming the nanomaterial integrity after the catalysis.
SiR silyl groups that strongly interact with the nanoparticle surface. In this respect, it should be noted that ruthenium nanoparticles stabilized by a silane (octylsilane) have been reported and that the formation of SiR silyl groups, probably linked to several ruthenium atoms, has been attested.62 TEM analysis of Ru·MIC10.2 after the catalytic reaction of Si-5 showed only a slight increase in the size of the nanoparticles (mean size: 2.2 (0.4) nm; Fig. S2‡), confirming the nanomaterial integrity after the catalysis.
 | ||
| Chart 1 H/D exchange in hydrides of group 14 elements and boranes catalysed by Ru·MIC10.2. a Reaction conditions, unless otherwise noted: 1.0 mol% Ru, 1 bar D2 (ca. 1.4 equiv.), 55 °C, THF. [S] = 1.2 M. Reaction time: 21 h. Two loadings of D2 were employed. Deuterium incorporation as determined by 1H NMR spectroscopy. H/D exchange selectivity determined by 2H NMR spectroscopy. aTHF-d8. b2.0 mol% Ru. Three loadings of D2 were employed. cIn the absence of light. dC6D6. | ||
Since the Ru·MIC10.2 nanomaterial was found to be a rather general catalyst for hydrosilane deuteration, H/D exchange in hydrides of heavier group 14 elements and boranes was also tested. Deuterium labelling at the Ge–H position of triethyl- and triphenylgermanium (Ge-1 and Ge-2) was achieved with >99 and 70% deuterium incorporation, respectively. Similarly, the tributylstannane deuterium isotopologue (Sn-1) was obtained with complete conversion. It should be noted that the formation of tin-decorated nanoparticles by the reaction of ruthenium nanoparticles with tri-n-butyltin hydride has been reported.63 Finally, the B–H bonds of pinacolborane (B-1) and catecholborane (B-2) were selectively deuterated with conversions higher than 99%.
Although detailed mechanistic studies are required, and precise details may depend on the specific substrate, we hypothesise that the catalytic H/D exchange reactions should involve initial E–H activation by the surface Ru(0) atoms,62,63 as previously proposed for the C–H activation with metal nanoparticles.10 Breaking of the E–H bond could be preceded by a weak E–H σ-coordination to the metal surface.20 In fact, partial deuteration at the α position to the Si atom in Si-5 could be explained considering that C–H activation assisted by σ-Si–H coordination to the metal surface takes place. Then, as a consequence of the fast diffusion of hydrides and deuterides (resulting from the previous activation of D2) on the metal surface, H/D exchange should occur, leading to the labelled product after a formal reductive elimination step. Overall, the application of the Ru·MIC nanoparticles in selective H/D exchange in hydrides of main group elements further demonstrates the potential that metal nanoparticles have as catalysts in processes involving the activation of different heteroatom–H bonds.
Conclusions
In conclusion, we have described for the first time the use of 1,2,3-triazolylidenes as efficient stabilizers for the synthesis of metal nanoparticles. Different ruthenium nanoparticles exhibiting small and narrow-dispersed sizes have been prepared and characterized by TEM, HRTEM, XPS and ICP analysis. More interestingly, these nanomaterials are efficient catalysts for selective hydrogen isotope exchange of E–H (E = B, Si, Ge, Sn) bonds using deuterium gas, leading to the corresponding isotopologues with high deuterium incorporation under relatively mild conditions. Current research in our laboratory is directed towards the use of nanoparticles of other metals incorporating 1,2,3-triazolylidenes as capping agents and their applications in HIE reactions.Experimental procedures
General procedures and nanoparticle characterization techniques
All reactions and manipulations were performed under nitrogen or argon, either in a Braun Labmaster 100 glovebox or using standard Schlenk-type techniques. Solvents were distilled under nitrogen with the following desiccants: sodium benzophenone-ketyl for tetrahydrofuran (THF and THF-d8) and sodium for pentane. D2 (99.8% D) was purchased from Sigma-Aldrich. 1,2,3-Triazolylidene ligand precursors were synthesized using previously reported procedures (see the ESI‡ for details). Ru(COD)(COT) was prepared by a previously reported method.64 All the other chemicals were used as received from commercial suppliers.The morphology and size of the 1,2,3-triazolylidene-stabilized ruthenium nanoparticles (Ru·MIC) were determined by transmission electron microscopy (TEM) in an FEI Talos F200S apparatus working at 200 kV at the Centro de Investigación, Tecnología e Innovación-CITIUS (Universidad de Sevilla). TEM samples were prepared by taking a drop of the crude THF colloidal solution and depositing it over a covered holey copper grid. For the approximation of the mean size of the particles, ca. 300 particles were manually measured employing conventional TEM micrographs enlarged with ImageJ software. High-resolution transmission electron microscopy (HRTEM) images were recorded using a Thermo Scientific Talos F200X microscope in order to assess the crystal structure of the Ru·MIC10.2 and Ru·MIC30.2 nanoparticles. An ABSF filter available within the “HRTEM filter” plugin for Gatan Digital Micrograph software was applied to enhance contrast by reducing the noise due to surrounding amorphous materials.65 To analyse the composition of the nanoparticles, a small, focused electron probe (beam current of about 500 pA) was placed across the nanoparticles using the scanning TEM (STEM) mode and the energy-dispersive X-ray spectroscopy (EDS) signals were recorded through four silicon drift detectors.
ICP analyses were performed at Mikroanalytisched Labor Pascher (Remagen, Germany).
X-Ray photoelectron spectroscopy (XPS) experiments were performed using a PHOIBOS-100 spectrometer with non-monochromatic Mg-Kα radiation (hν = 1235.6 eV) and the power source was 230 W. The hemispherical electron energy analyser was operated in the constant pass energy mode (SPECS PHOIBOS 100DLD). Low resolution survey spectra were obtained with a pass energy of 50 eV, while high energy resolution spectra of the detected elements were obtained with a pass energy of 30 eV. The spectra were analysed using the CASA XPS software, version 2.3.16.Dev52 (Neal Fairly, UK). Shirley type backgrounds were used to determine the areas under the peaks. The Ru 3p3/2 spectra were fitted with two components, corresponding to Ru(0) and Ru(IV), using Gaussian–Lorentzian functions (SGL = 10). N 1s peaks in the spectra of the nanoparticles were fitted with two components with the same shape (SGL = 10) and fixing the fwhm value between 2.5 and 3.0 eV.
Synthesis of ruthenium nanoparticles
Ru(COD)(COT) (0.200 g, 0.65 mmol), the 1,2,3-triazolylidene ligand precursor (0.13 mmol for nanoparticles Ru·MIC10.2, Ru·MIC20.2 and Ru·MIC30.2; 0.06 mmol for Ru·MIC10.1) and KHMDS (0.026 g, 0.13 mmol for Ru·MIC10.1, 0.052 g, 0.26 mmol for Ru·MIC10.2, Ru·MIC20.2 and Ru·MIC30.2) were introduced in a Fisher–Porter vessel and cooled to −50 °C. To the mixture, 70 mL of THF, which was freshly distilled and degassed by N2 bubbling, was added. The Fisher–Porter vessel was pressurized with 3 bar of H2, and the solution was allowed to reach room temperature slowly under vigorous stirring. The homogeneous solution, which turned black after 30 min of reaction, was kept under stirring overnight at room temperature. After this period of time, excess of H2 was carefully released. The initial volume of the solvent was reduced to ca. 10 mL under vacuum; then 30 mL of pentane was added, and the colloidal suspension was cooled down to −30 °C to precipitate the particles. After filtration under argon via a cannula, the black solid powder was washed with pentane (3 × 30 mL) and dried under vacuum. For Ru content determination by ICP and TEM analysis of the nanoparticles, see Table 1.Representative procedure for catalyst comparison using PhMe2SiH (Table 2)
In a glovebox, a J. Young NMR tube was charged with a solution of dimethylphenylsilane (Si-1) (95 μL, 0.62 mmol) in THF-d8 (0.2 mL) and 0.3 mL of a freshly prepared stock suspension of the catalyst Ru·MIC10.2 (0.62 μmol Ru) in THF-d8. The tube was purged three times with D2, and finally pressurized to 1 bar (ca. 0.4 equiv. D2) and heated to 55 °C. After 19 h, the sample was analysed by 1H and 2H NMR spectroscopy.Representative procedure for the selective H/D exchange of E–H (E = Si, Ge, Sn, B) bonds (Chart 1)
In a glovebox, a 25 mL Fisher–Porter vessel was charged with a solution of dimethylphenylsilane (Si-1) (190 μL, 1.24 mmol) in THF (0.4 mL) and 0.6 mL of a freshly prepared stock suspension of the catalyst Ru·MIC10.2 (1.24 μmol Ru) in THF. The reactor was purged three times with D2, and finally pressurized to 1 bar (ca. 1.4 equiv. D2) and heated to 55 °C. After 6 h, the pressure was released, and the reactor was pressurized again with 1 bar D2. After 21 h, the reactor was slowly cooled down to room temperature and depressurized. An aliquot of the reaction mixture was filtered through a short pad of Celite and brought to dryness. The conversion and selectivity were determined by 1H and 2H NMR spectroscopy, respectively.Author contributions
P. Molinillo and M. Puyo: data curation, formal analysis, and investigation; F. Vattier: formal analysis (XPS) and investigation; B. Lacroix: formal analysis (HRTEM and EDS) and investigation; N. Rendón, A. Suárez, and P. Lara: conceptualization, data curation, formal analysis, investigation, methodology, visualization, writing – original draft, and writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The financial support (FEDER contribution) from the Spanish Agencia Estatal de Investigación (PID2019-104159GB-I00/MCIN/AEI/10.13039/501100011033) and Junta de Andalucía (P18-FR-3208 and US-1380604) is gratefully acknowledged. B. Lacroix is also thankful for the support from the University of Sevilla (USE-22972-H contract of the VI-PPITUS) and the EMERGIA program of the Junta de Andalucía (EMC21_00427 contract).Notes and references
- J. Atzrodt, V. Derdau, W. J. Kerr and M. Reid, Angew. Chem., Int. Ed., 2018, 57, 1758–1784 CrossRef CAS PubMed.
- T. G. Gant, J. Med. Chem., 2014, 57, 3595–3611 CrossRef CAS PubMed.
- T. Pirali, M. Serafini, S. Cargnin and A. A. Genazzani, J. Med. Chem., 2019, 62, 5276–5297 CrossRef CAS PubMed.
- L. Konermann, J. Pan and Y.-H. Liu, Chem. Soc. Rev., 2011, 40, 1224–1234 RSC.
- T. Lavold, R. Zubarev and J. Astorga-Wells, Hydrogen-Deuterium Exchange Mass Spectrometry in Drug Discovery - Theory, Practice and Future, in Applied Biophysics for Drug Discovery, ed. D. Huddler and E. R. Zartler, John Wiley & Sons, Inc., 2017. DOI:10.1002/9781119099512.ch4.
- M. Gómez-Gallego and M. A. Sierra, Chem. Rev., 2011, 111, 4857–4963 CrossRef PubMed.
- E. M. Simmons and J. F. Hartwig, Angew. Chem., Int. Ed., 2012, 51, 3066–3072 CrossRef CAS PubMed.
- S. Kopf, F. Bourriquen, W. Li, H. Neumann, K. Junge and M. Beller, Chem. Rev., 2022, 122, 6634–6718 CrossRef CAS PubMed.
- J. Atzrodt, V. Derdau, W. J. Kerr and M. Reid, Angew. Chem., Int. Ed., 2018, 57, 3022–3047 CrossRef CAS PubMed.
- M. Lepron, M. Daniel-Bertrand, G. Mencia, B. Chaudret, S. Feuillastre and G. Pieters, Acc. Chem. Res., 2021, 54, 1465–1480 CrossRef CAS PubMed.
- L. S. Ott, M. L. Cline, M. Deetlefs, K. R. Seddon and R. G. Finke, J. Am. Chem. Soc., 2005, 127, 5758–5759 CrossRef CAS PubMed.
- G. Pieters, C. Taglang, E. Bonnefille, T. Gutmann, C. Puente, J.-C. Berthet, C. Dugave, B. Chaudret and B. Rousseau, Angew. Chem., Int. Ed., 2014, 53, 230–234 CrossRef CAS PubMed.
- C. Taglang, L. M. Martínez-Prieto, I. del Rosal, L. Maron, R. Poteau, K. Philippot, B. Chaudret, S. Perato, A. S. Lone, C. Puente, C. Dugave, B. Rousseau and G. Pieters, Angew. Chem., Int. Ed., 2015, 54, 10474–10477 CrossRef CAS PubMed.
- A. Palazzolo, S. Feuillastre, V. Pfeifer, S. Garcia-Argote, D. Bouzouita, S. Tricard, C. Chollet, E. Marcon, D.-A. Buisson, S. Cholet, F. Fenaille, G. Lippens, B. Chaudret and G. Pieters, Angew. Chem., Int. Ed., 2019, 58, 4891–4895 CrossRef CAS PubMed.
- E. Bresó-Femenia, C. Godard, C. Claver, B. Chaudret and S. Castillón, Chem. Commun., 2015, 51, 16342–16345 RSC.
- L. M. Martínez-Prieto, E. A. Baquero, G. Pieters, J. C. Flores, E. de Jesús, C. Nayral, F. Delpech, P. W. N. M. van Leeuwen, G. Lippens and B. Chaudret, Chem. Commun., 2017, 53, 5850–5853 RSC.
- V. Pfeifer, M. Certiat, D. Bouzouita, A. Palazzolo, S. Garcia-Argote, E. Marcon, D.-A. Buisson, P. Lesot, L. Maron, B. Chaudret, S. Tricard, I. del Rosal, R. Poteau, S. Feuillastre and G. Pieters, Chem. – Eur. J., 2020, 26, 4988–4996 CrossRef CAS PubMed.
- D. Bouzouita, G. Lippens, E. A. Baquero, P. F. Fazzini, G. Pieters, Y. Coppel, P. Lecante, S. Tricard, L. M. Martínez-Prieto and B. Chaudret, Nanoscale, 2019, 11, 16544–16552 RSC.
- A. Palazzolo, T. Naret, M. Daniel-Bertrand, D.-A. Buisson, S. Tricard, P. Lesot, Y. Coppel, B. Chaudret, S. Feuillastre and G. Pieters, Angew. Chem., Int. Ed., 2020, 59, 20879–20884 CrossRef CAS PubMed.
- N. Rothermel, T. Röther, T. Ayvalı, L. M. Martínez-Prieto, K. Philippot, H.-H. Limbach, B. Chaudret, T. Gutmann and G. Buntkowsky, ChemCatChem, 2019, 11, 1465–1471 CrossRef CAS.
- M. Valero, D. Bouzouita, A. Palazzolo, J. Atzrodt, C. Dugave, S. Tricard, S. Feuillastre, G. Pieters, B. Chaudret and V. Derdau, Angew. Chem., Int. Ed., 2020, 59, 3517–3522 CrossRef CAS PubMed.
- M. Daniel-Bertrand, S. Garcia-Argote, A. Palazzolo, I. Mustieles Marin, P.-F. Fazzini, S. Tricard, B. Chaudret, V. Derdau, S. Feuillastre and G. Pieters, Angew. Chem., Int. Ed., 2020, 59, 21114–21120 CrossRef CAS PubMed.
- D. Bouzouita, J. M. Asensio, V. Pfeifer, A. Palazzolo, P. Lecante, G. Pieters, S. Feuillastre, S. Tricard and B. Chaudret, Nanoscale, 2020, 12, 15736–15742 RSC.
- V. Pfeifer, T. Zeltner, C. Fackler, A. Kraemer, J. Thoma, A. Zeller and R. Kiesling, Angew. Chem., Int. Ed., 2021, 60, 26671–26676 CrossRef CAS PubMed.
- A. Zuluaga-Villamil, G. Mencia, J. M. Asensio, P.-F. Fazzini, E. A. Baquero and B. Chaudret, Organometallics, 2022, 41, 3313–3319 CrossRef CAS.
- E. Levernier, K. Tatoueix, S. Garcia-Argote, V. Pfeifer, R. Kiesling, E. Gravel, S. Feuillastre and G. Pieters, JACS Au, 2022, 2, 801–808 CrossRef CAS PubMed.
- O. Suárez-Riaño, G. Mencia, S. Tricard, J. Esvan, P.-F. Fazzini, B. Chaudret and E. A. Baquero, Chem. Commun., 2023, 59, 1062–1065 RSC.
- F. Martinez-Espinar, A. Salom-Català, E. Bresó-Femenia, C. Claver, F. Baletto, J. M. Ricart, B. Chaudret, J. J. Carbó, C. Godard and S. Castillon, Inorg. Chem., 2023, 62, 4570–4580 CrossRef CAS PubMed.
- J. M. Asensio, D. Bouzouita, P. W. N. M. van Leeuwen and B. Chaudret, Chem. Rev., 2020, 120, 1042–1084 CrossRef CAS PubMed.
- (a) B. Marciniec, H. Maciejewski, C. Pietraszuk and P. Pawluc, Hydrosilylation of Alkynes and their derivatives, in Hydrosilylation. A Comprehensive Review of Recent Advances, ed. B. Marciniec, Springer, Berlin, 2009 Search PubMed; (b) B. M. Trost and Z. T. Ball, Synthesis, 2005, 853–887 CrossRef CAS; (c) R. H. Morris, Chem. Soc. Rev., 2009, 38, 2282–2291 RSC; (d) D. Addis, S. Das, K. Junge and M. Beller, Angew. Chem., Int. Ed., 2011, 50, 6004–6011 CrossRef CAS PubMed; (e) C. C. Chong and R. Kinjo, ACS Catal., 2015, 5, 3238–3259 CrossRef CAS; (f) J. V. Obligacion and P. J. Chirik, Nat. Rev. Chem., 2018, 2, 15–34 CrossRef CAS PubMed; (g) M. Alami, A. Hamze and O. Provot, ACS Catal., 2019, 9, 3437–3466 CrossRef CAS; (h) H. Yoshida, Synthesis, 2016, 48, 2540–2552 CrossRef CAS.
- (a) F. Alonso, I. P. Beletskaya and M. Yus, Chem. Rev., 2002, 102, 4009–4092 CrossRef CAS PubMed; (b) M. Aizenberg and D. Milstein, Science, 1994, 265, 359–361 CrossRef CAS PubMed.
- M. D. Fryzuk, L. Rosenberg and S. J. Rettig, Organometallics, 1991, 10, 2537–2539 CrossRef CAS.
- J. Gavenonis and T. D. Tilley, Organometallics, 2002, 21, 5549–5563 CrossRef CAS.
- T. Ayed, J.-C. Barthelat, B. Tangour, C. Pradère, B. Donnadieu, M. Grellier and S. Sabo-Etienne, Organometallics, 2005, 24, 3824–3826 CrossRef CAS.
- J. Campos, A. C. Esqueda, J. López-Serrano, L. Sánchez, F. P. Cossio, A. de Cozar, E. Álvarez, C. Maya and E. Carmona, J. Am. Chem. Soc., 2010, 132, 16765–16767 CrossRef CAS PubMed.
- (a) G. C. Fortman, H. Jacobsen, L. Cavallo and S. P. Nolan, Chem. Commun., 2011, 47, 9723–9725 RSC; (b) J. D. Egbert and S. P. Nolan, Chem. Commun., 2012, 48, 2794–2796 RSC.
- K. A. Smart, E. Mothes-Martin, T. Annaka, M. Grellier and S. Sabo-Etienne, Adv. Synth. Catal., 2014, 356, 759–764 CrossRef CAS.
- G. I. Nikonov, S. F. Vyboishchikov, O. G. Shirobokov and R. Simionescu, Eur. J. Inorg. Chem., 2014, 2896–2901 CrossRef CAS.
- B. A. Connor, J. Rittle, D. VanderVelde and J. C. Peters, Organometallics, 2016, 35, 686–690 CrossRef CAS.
- Y. Kratish, D. Bravo-Zhivotovskii and Y. Apeloig, ACS Omega, 2017, 2, 372–376 CrossRef CAS PubMed.
- M. A. Esteruelas, A. Martínez, M. Oliván and A. Vélez, J. Org. Chem., 2020, 85, 15693–15698 CrossRef CAS PubMed.
- T. G. Linford-Wood, M. F. Mahon, M. N. Grayson and R. L. Webster, ACS Catal., 2022, 12, 2979–2985 CrossRef CAS PubMed.
- L. Denker, D. Wullschläger, J. P. Martinez, S. Swierczewski, B. Trzaskowski, M. Tamm and R. Frank, ACS Catal., 2023, 13, 2586–2600 CrossRef CAS.
- (a) D. I. Bradshaw, R. B. Moyes and P. B. Wells, J. Chem. Soc., Chem. Commun., 1975, 137–138 RSC; (b) M. Bartok and A. J. Molnar, J. Organomet. Chem., 1982, 235, 161–164 CrossRef CAS; (c) L. H. Sommer, J. E. Lyons and H. Fujimoto, J. Am. Chem. Soc., 1969, 91, 7051–7061 CrossRef CAS; (d) D. I. Bradshaw, R. B. Moyes and P. B. Wells, J. Chem. Soc., Faraday Trans. 1, 1980, 76, 979–987 RSC.
- (a) A. W. M. Cummins, S. Li, D. R. Willcox, T. Muilu, J. H. Docherty and S. P. Thomas, Tetrahedron, 2020, 76, 131084 CrossRef CAS; (b) J. V. Obligacion and P. J. Chirik, J. Am. Chem. Soc., 2013, 135, 19107–19110 CrossRef CAS PubMed; (c) S. Bontemps, L. Vendier and S. Sabo-Etienne, Angew. Chem., Int. Ed., 2012, 51, 1671–1674 CrossRef CAS PubMed; (d) D. J. Nelson, J. D. Egbert and S. P. Nolan, Dalton Trans., 2013, 42, 4105–4109 RSC; (e) A. L. Colebatch, B. W. H. Gilder, G. R. Whittell, N. L. Oldroyd, I. Manners and A. S. Weller, Chem. – Eur. J., 2018, 24, 5450–5455 CrossRef CAS PubMed; (f) D. Yoshii, T. Yatabe, T. Yabe and K. Yamaguchi, ACS Catal., 2021, 11, 2150–2155 CrossRef CAS.
- L. M. Martínez-Prieto and B. Chaudret, Acc. Chem. Res., 2018, 51, 376–384 CrossRef PubMed.
- (a) L. C. Moraes, R. C. Figueiredo, J. P. Espinós, F. Vattier, A. Franconetti, C. Jaime, B. Lacroix, J. Rojo, P. Lara and S. Conejero, Nanoscale, 2020, 12, 6821–6831 RSC; (b) Y. Zhu and N. S. Hosmane, Coord. Chem. Rev., 2015, 293–294, 357–367 CrossRef CAS; (c) P. Lara, K. Philippot and A. Suárez, ChemCatChem, 2019, 11, 766–771 CrossRef CAS.
- (a) A. Vivancos, C. Segarra and M. Albrecht, Chem. Rev., 2018, 118, 9493–9586 CrossRef CAS PubMed; (b) R. Maity and B. Sarkar, JACS Au, 2022, 2, 22–57 CrossRef CAS PubMed.
- M. R. Axet and K. Philippot, Chem. Rev., 2020, 120, 1085–1145 CrossRef CAS PubMed.
- P. Lara, K. Philippot and B. Chaudret, ChemCatChem, 2013, 5, 28–45 CrossRef CAS.
- P. Bellotti, M. Koy, M. N. Hopkinson and F. Glorius, Nat. Rev. Chem., 2021, 5, 711–725 CrossRef CAS PubMed.
- (a) M. Koy, P. Bellotti, M. Das and F. Glorius, Nat. Catal., 2021, 4, 352–363 CrossRef CAS; (b) H. Shen, G. Tian, Z. Xu, L. Wang, Q. Wu, Y. Zhang, B. K. Teo and N. Zheng, Coord. Chem. Rev., 2022, 458, 214425 CrossRef CAS; (c) C. Cerezo-Navarrete, P. Lara and L. M. Martínez-Prieto, Catalysts, 2020, 10, 1144 CrossRef CAS; (d) C. A. Smith, M. R. Narouz, P. A. Lummis, I. Singh, A. Nazemi, C.-H. Li and C. M. Crudden, Chem. Rev., 2019, 119, 4986–5056 CrossRef CAS PubMed.
- P. Lara, O. Rivada-Wheelaghan, S. Conejero, R. Poteau, K. Philippot and B. Chaudret, Angew. Chem., Int. Ed., 2011, 50, 12080–12084 CrossRef CAS PubMed.
- For other selected examples, see: (a) P. Lara, L. M. Martínez-Prieto, M. Roselló-Merino, C. Richter, F. Glorius, S. Conejero, K. Philippot and B. Chaudret, Nano-Struct. Nano-Objects, 2016, 6, 39–45 CrossRef CAS; (b) F. Martinez-Espinar, P. Blondeau, P. Nolis, B. Chaudret, C. Claver, S. Castillón and C. Godard, J. Catal., 2017, 354, 113–127 CrossRef CAS; (c) A. M. Ruiz-Varilla, E. A. Baquero, B. Chaudret, E. de Jesús, C. Gonzalez-Arellano and J. C. Flores, Catal. Sci. Technol., 2020, 10, 2874–2881 RSC; (d) E. A. Baquero, S. Tricard, J. C. Flores, E. de Jesús and B. Chaudret, Angew. Chem., Int. Ed., 2014, 53, 13220–13224 CrossRef CAS PubMed; (e) A. Ferry, K. Schaepe, P. Tegeder, C. Richter, K. M. Chepiga, B. J. Ravoo and F. Glorius, ACS Catal., 2015, 5, 5414–5420 CrossRef CAS; (f) P. Lara, A. Suárez, V. Collière, K. Philippot and B. Chaudret, ChemCatChem, 2014, 6, 87–90 CrossRef CAS.
- (a) L. M. Martínez-Prieto, A. Ferry, P. Lara, C. Richter, K. Philippot, F. Glorius and B. Chaudret, Chem. – Eur. J., 2015, 21, 17495–17502 CrossRef PubMed; (b) K. V. S. Ranganath, J. Kloesges, A. H. Schäfer and F. Glorius, Angew. Chem., Int. Ed., 2010, 49, 7786–7789 CrossRef CAS PubMed.
- K. Philippot and B. Chaudret, in Comprehensive Organometallic Chemistry III, ed. R. H. Crabtree & and M. P. Mingos, Elsevier, Applications III: Functional Materials, Environmental and Biological Applications, D. O'Hare (Volume Ed.), 2007, ch. 12–03, vol. 12, pp. 71–99 Search PubMed.
- (a) M. A. Ernst and W. G. Sloof, Surf. Interface Anal., 2008, 40, 334–337 CrossRef CAS; (b) D. J. Morgan, Surf. Interface Anal., 2015, 47, 1072–1079 CrossRef CAS.
- (a) R. Nyholm and N. Martenson, J. Phys. C: Solid State Phys., 1980, 13, L279–L284 CrossRef CAS; (b) N. Chakroune, G. Viau, S. Ammar, L. Poul, D. Veautier, M. M. Chehimi, C. Mangeney, F. Villain and F. Fiévet, Langmuir, 2005, 21, 6788–6796 CrossRef CAS PubMed.
- (a) J. B. Ernst, S. Muratsugu, F. Wang, M. Tada and F. Glorius, J. Am. Chem. Soc., 2016, 138, 10718–10721 CrossRef CAS PubMed; (b) L. M. Martínez-Prieto, M. Puche, C. Cerezo-Navarrete and B. Chaudret, J. Catal., 2019, 337, 429–437 CrossRef; (c) P. Molinillo, B. Lacroix, F. Vattier, N. Rendón, A. Suárez and P. Lara, Chem. Commun., 2022, 58, 7176–7179 RSC.
- (a) P. Finn and W. L. Jolly, Inorg. Chem., 1972, 11, 1434–1435 CrossRef CAS; (b) D. M. Hercules, J. Chem. Educ., 2004, 81, 1751–1766 CrossRef CAS; (c) G. Vilé, G. di Liberto, S. Tosoni, A. Sivo, V. Ruta, M. Nachtegaal, A. H. Clark, S. Agnoli, Y. Zou, A. Savateev, M. Antonietti and G. Pacchiooni, ACS Catal., 2022, 12, 2947–2958 CrossRef.
- (a) A. Rühling, K. Schaepe, L. Rakers, B. Vonhören, P. Tegeder, B. J. Ravoo and F. Glorius, Angew. Chem., Int. Ed., 2016, 55, 5856–5860 CrossRef PubMed; (b) R. W. Y. Man, C.-H. Li, M. W. A. MacLean, O. V. Zenkina, M. T. Zamora, L. N. Saunders, A. Rousina-Webb, M. Mambo and C. M. Crudden, J. Am. Chem. Soc., 2018, 140, 1576–1579 CrossRef CAS PubMed.
- (a) K. Pelzer, B. Laleu, F. Lefebvre, K. Philippot, B. Chaudret, J. P. Candy and J. M. Basset, Chem. Mater., 2004, 16, 4937–4941 CrossRef CAS; (b) K. Pelzer, J. P. Candy, G. Bergeret and J. M. Basset, Eur. Phys. J. D, 2007, 43, 197–200 CrossRef CAS.
- E. Bonnefille, F. Novio, T. Gutmann, R. Poteau, P. Lecante, J.-C. Jumas, K. Philippot and B. Chaudret, Nanoscale, 2014, 6, 9806–9816 RSC.
- C. Pertici, G. Vitulli, W. C. Spink and M. D. Rausch, Inorg. Synth., 1984, 22, 176–181 Search PubMed.
- R. Kilaas, J. Microsc., 1998, 190, 45–51 CrossRef.
Footnotes |
| † Dedicated to Prof. Gregory C. Fu on the occasion of his 60th birthday. |
| ‡ Electronic supplementary information (ESI) available: Characterization of the Ru nanoparticles. NMR data of catalytic reaction products. See DOI: https://doi.org/10.1039/d3nr02637j |
| This journal is © The Royal Society of Chemistry 2023 |