
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Multi-functionalized carbon nanotubes towards green fabrication of heterogeneous catalyst platforms with enhanced catalytic properties under NIR light irradiation†
Xuefeng
Pan‡
ad,
Rongying
Liu‡
b,
Zhilong
Yu
a,
Benedikt
Haas
 c,
Zdravko
Kochovski
a,
Sijia
Cao
ad,
Radwan M.
Sarhan
a,
Guosong
Chen
*b and
Yan
Lu
*ad
c,
Zdravko
Kochovski
a,
Sijia
Cao
ad,
Radwan M.
Sarhan
a,
Guosong
Chen
*b and
Yan
Lu
*ad
aDepartment for Electrochemical Energy Storage, Helmholtz-Zentrum Berlin für Materialien und Energie, Hahn-Meitner-Platz 1, 14109 Berlin, Germany. E-mail: yan.lu@helmholtz-berlin.de
bThe State Key Laboratory of Molecular Engineering of Polymers and Department of Macromolecular Science, Fudan University, Shanghai 200433, China. E-mail: guosong@fudan.edu.cn
cDepartment of Physics & IRIS Adlershof, Humboldt-Universität zu Berlin, Newtonstr. 15, 12489 Berlin, Germany
dInstitute of Chemistry, University of Potsdam, Karl-Liebknecht-Str. 24-25, 14476 Potsdam, Germany
First published on 28th August 2023
Abstract
Metal/carbon nanotubes (CNTs) have been attractive hybrid systems due to their high specific surface area and exceptional catalytic activity, but their challenging synthesis and dispersion impede their extensive applications. Herein, we report a facile and green approach towards the fabrication of metal/CNT composites, which utilizes a versatile glycopeptide (GP) both as a stabilizer for CNTs in water and as a reducing agent for noble metal ions. The abundant hydrogen bonds in GP endow the formed GP-CNTs with excellent plasticity, enabling the availability of polymorphic CNT species from dispersion to viscous paste, gel, and even to dough by increasing their concentration. The GP molecules can reduce metal precursors at room temperature without additional reducing agents, enabling the in situ immobilization of metal nanoparticles (e.g. Au, Ag, Pt, and Pd) on the CNT surface. The combination of the excellent catalytic properties of Pd particles with photothermal conversion capability of CNTs makes the Pd/CNT composite a promising catalyst for the fast degradation of organic pollutants, as demonstrated by a model catalytic reaction using 4-nitrophenol (4-NP). The conversion of 4-NP using the Pd/CNT composite as the catalyst has increased by 1.6-fold under near infrared light illumination, benefiting from the strong light-to-heat conversion effect of CNTs. Our proposed strategy opens a new avenue for the synthesis of CNT composites as a sustainable and versatile catalyst platform.
Introduction
Metal functionalized carbon nanotube (CNT) composites have been proven as excellent heterogeneous catalytic platforms for various reactions, such as hydrogenation, oxidation, and reduction reactions, due to the integral merits from the intrinsic high specific surface area of CNTs and catalytically active metal nanoparticles (NPs).1–4 The rational design of stable and highly active metal/CNT hybrid catalysts has become one of the most prevailing works today. Generally, three critical steps are involved in the fabrication of metal/CNT composites, including effective dispersion of CNTs, reduction/stabilization of metal NPs, and immobilization of metal NPs on the CNTs. The strong van der Waals interactions inherent to CNTs result in their super high propensity to form aggregated bundles in a variety of solvents, particularly in water.5–8 Therefore, a pivotal prerequisite for the production of metal/CNTs is to disperse bulk CNTs via a feasible strategy.9 The most frequently reported strategy for spatially disassembling the CNT bundles is to oxidize CNTs with strong acids, thus chemically etching the inherently hydrophobic CNTs towards the introduction of some hydrophilic oxygen-based functional moieties (e.g. carboxylic, hydroxyl, and phenolic groups) on their surface.10–12 However, the daunting manipulation of the number and type of functional groups still hinders the controllable fabrication of CNT-based composites.13 In addition, irreversible damage to the aromatic rings of CNTs caused by harsh chemical modifications usually impairs their intrinsic conductivity and catalytic activity. Moreover, the use of strong acids in these strategies is not environmentally friendly. Therefore, it is essential to develop a sustainable and eco-friendly approach towards simultaneously dispersing CNTs and preserving their structural integrity.Alternatively, non-covalent physical modification using amphiphilic surfactants, polymers, or biomolecules such as DNA and proteins has been demonstrated to be effective not only in dispersing CNTs in aqueous solution but also in maintaining their structure intact.14–18 In such a scenario, the hydrophobic moieties tend to attach to the surface of CNTs via hydrophobic interactions, while the hydrophilic ones are inclined to actively interact with water molecules, thus efficiently stabilizing CNTs in water.17 Nevertheless, the use of wrapped polymers or surfactants may lead to toxicity, non-degradability, and arduous elimination from CNT dispersion, consequently posing environmental sustainability and green chemistry concerns. Moreover, the intrinsic polydispersity and mono-functionality of naturally occurring biomolecules limit their wide application in such areas. Therefore, environmentally-friendly and versatile dispersants are in great demand for the functionalization of CNTs.
When it comes to the reduction, stabilization, and immobilization of metal NPs on CNTs, direct reduction of metal precursor ions by reducing agents (e.g. sodium borohydride – NaBH4) via wet chemistry often involves toxic chemicals or additional heating.2,19 Meanwhile, amphiphilic surfactants or polymers are commonly employed to control the formation/growth of metal NPs and improve their colloidal stability. Subsequent binding agents are also necessary to immobilize metal NPs on the surface of CNTs, potentially causing additional energy consumption and environmental pollution.20 Conventional synthetic protocols such as hydrothermal and electrochemical deposition, or plasma reduction methods are either time-consuming or equipment-demanding.21–24 Therefore, the development of a novel strategy that could integrally perform these abovementioned three critical steps in a green and sustainable fashion will be essential for the advancement of CNT-based heterogeneous catalysis.
In this work, we prepared an amphiphilic glycopeptide (GP) biomolecule composed of a hydrophilic trisaccharide and a hydrophobic triphenylalanine to efficiently disperse CNTs in aqueous solution (Scheme 1). Furthermore, the GP molecules on the CNT surface could capture various metal ions and subsequently reduce them into catalytic metal NPs at room temperature. The obtained metal NPs were simultaneously stabilized and immobilized on CNTs by the GP molecules, enabling the green fabrication of heterogeneous catalytic platforms. Unexpectedly, the introduction of GP molecules also significantly enhances the plasticity of CNTs and thus provides access to the rational construction of various CNT entities with enhanced processability from paste to gel, and even dough by increasing their concentration. Besides, the hydroxyl groups of GP molecules endow the CNT template with an excellent capability to accommodate a wide range of noble metal NPs, e.g. gold (Au), silver (Ag), platinum (Pt), and palladium (Pd) on its surface under mild conditions. Eventually, the catalytic potential of the fabricated heterogeneous platforms has also been demonstrated via degradation of a typical organic pollutant, 4-nitrophenol (4-NP), in water with the assistance of near-infrared (NIR) light irradiation. The method here opens a new avenue to multi-functionalized CNTs towards the fabrication of heterogeneous catalytic systems through a green approach.
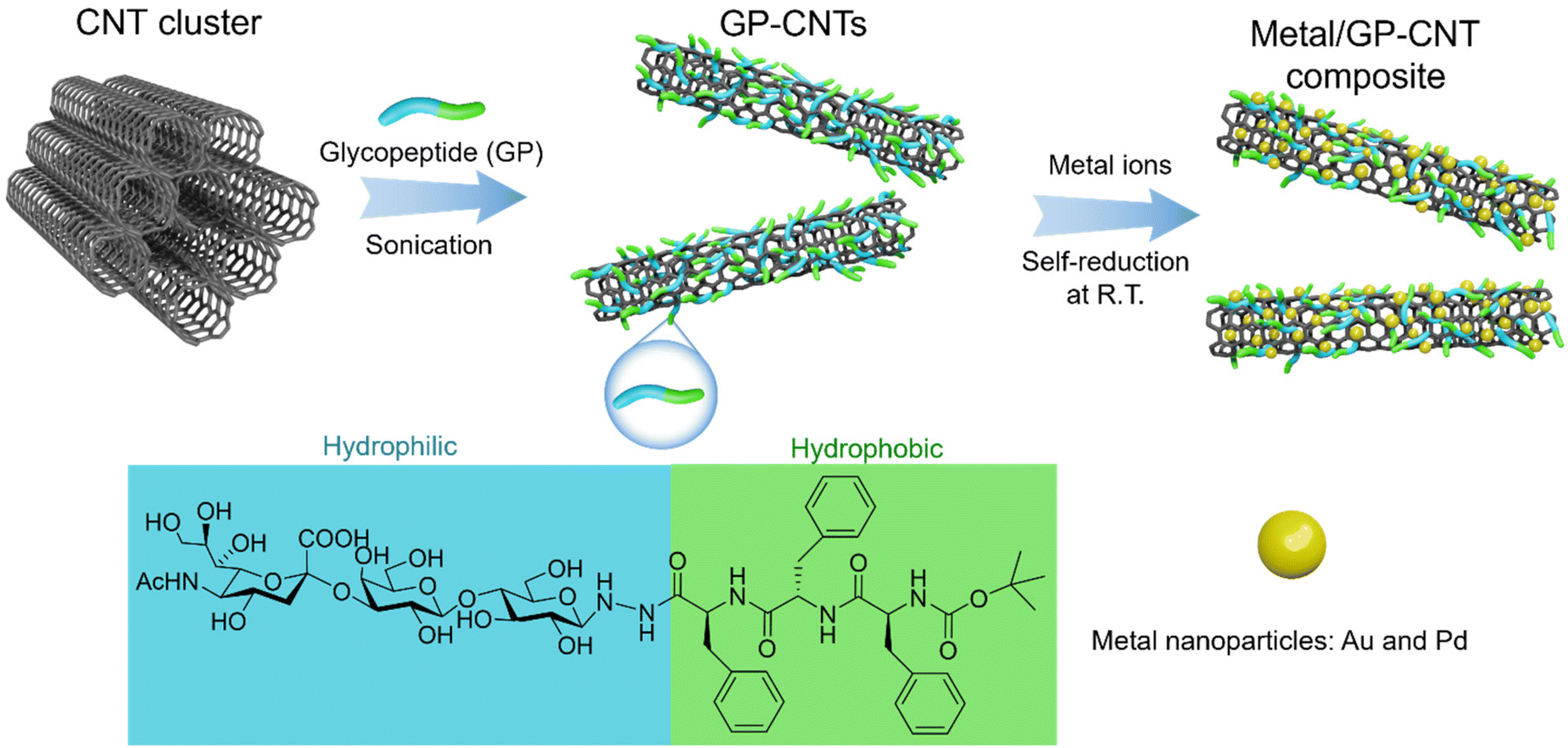 | ||
| Scheme 1 Synthetic strategy for GP-CNTs and their functionalization with metal particles. | ||
Results and discussion
GP-stabilized CNT dispersion
Considering the aromatic scaffold nature of hydrophobic CNTs, amphiphilic molecules containing aromatic groups appear to be ideal dispersant candidates to effectively adhere to the surface of CNTs and thus disperse them in water via aromatic–aromatic interactions between amphiphilic molecules and CNTs. Herein, we prepared an amphiphilic glycopeptide (GP) molecule comprising a hydrophilic moiety (i.e. a trisaccharide with carboxyl and hydroxyl groups) and a hydrophobic part (i.e. three phenylalanine residues) to disperse CNTs in water. The GP molecule was synthesized according to the methods reported in our previous study.25,26 As an initial test, we directly dispersed the as-purchased multi-walled CNT (MWCNT) powders (i.e. pristine CNT cluster, 1 mg) with amphiphilic GP molecules (1 mg) in water (1 mL). A homogeneous CNT dispersion was obtained after simple treatment with sonication and centrifugation (Fig. 1a). A thin layer of GP molecules (∼2 nm) was found to be wrapped on the surface of CNTs via high-resolution transmission electron microscopy (HRTEM), as shown in Fig. 1b. The aromatic phenylalanine moieties of GP molecules are expected to attach to the surface of CNTs via hydrophobic interactions, while the hydrophilic moieties head to the water phase. The presence of carboxyl groups on the head of hydrophilic moieties gives rise to strong electrostatic repulsions among CNTs, thus leading to a homogeneous and stable dispersion of CNTs. As spotted by scanning electron microscopy (SEM), a uniform CNT layer was also achieved by simply drop-casting the GP-CNT dispersion on a silicon wafer and followed by drying under ambient conditions, while the pristine CNT clusters merely showed large aggregates (Fig. 1c and 1f, and S2†). A cryogenic TEM (cryo-TEM) was employed to in situ monitor the native dispersion state of GP-CNTs. The outcome showed that, in the presence of GP molecules, the CNTs were well-dispersed in water without appreciable aggregation, verifying the excellent dispersibility of CNTs enabled by amphiphilic GP molecules (Fig. S3†). In contrast, in the absence of GP molecules, the pristine CNT sample was found to quickly aggregate and precipitate within a few seconds (Fig. 1d and 1e). In addition to MWCNTs, single-walled CNTs (SWCNTs) were also well-dispersed in water with the aid of GP molecules (Fig. S4†). It is worth mentioning that MWCNTs are much more affordable and available compared to SWCNTs, we thereby chose MWCNTs as the model material for practical applications in the subsequent parts of this research. | ||
| Fig. 1 (a and b) TEM and (c) SEM images of the GP-CNT dispersion. (d and e) TEM and (f) SEM images of the pristine CNT cluster dispersion. The insets in (a and d) are the corresponding photos of the CNT dispersions. (g) UV-Vis absorption spectra of the CNT cluster, GP, and GP-CNT dispersion. (h) Raman spectroscopy of the CNT cluster and GP-CNT powder after freeze-drying. (i) UV-Vis absorption spectra of the GP-CNT dispersion at 0.01 mg mL−1 and their corresponding photos (inset) before and after static storage for 2 months. | ||
It has been reported that the uniform dispersion of bundled CNTs will lead to a blue shift in resonant absorption peaks due to the weakened electronic coupling effects among the CNT frameworks.6,9 Accordingly, an appreciable maximum blue shift from 273 nm for pristine CNTs to 260 nm for GP-CNTs in their UV-Vis absorption spectra further confirmed the uniform distribution of CNTs (Fig. 1g). Note that the higher absorption intensity of GP-CNTs compared to the pristine CNT clusters also revealed the superior dispersibility of GP-functionalized CNTs in water.27
To further investigate the electronic interaction between CNTs and GP molecules, both the pristine CNT clusters and GP-CNTs in the solid state were investigated by Raman spectroscopy. As shown in Fig. 1h, three distinct peaks were found in both samples at 1340, 1572, and 2696 cm−1, which respectively refer to the D-band, G-band, and 2D band of MWCNTs. The intensity ratio of the D-band (ID) at 1340 cm−1 to the G-band (IG) at 1572 cm−1 is usually used as an indicator for the evaluation of the defect amount of CNTs. In the current study, the ID/IG of the pristine CNT cluster and the GP-CNTs showed a comparable value of 0.18 and 0.16, respectively, confirming that the aromatic–aromatic interaction between the GP molecules and CNTs does not introduce defects on the sidewall of CNTs, i.e. damage to their surface. This finding confirms that the introduction of GP molecules maintains the structural integrity of CNTs, in contrast to the traditional chemical oxidation strategy.28,29 As shown in the normalized Raman spectra (the inset in Fig. 1h), the red shift from 1572 cm−1 for the pristine CNTs to 1578 cm−1 for the GP-CNTs in the G band verified the occurrence of charge transfer between three aromatic phenylalanine residues of GP molecules and CNT sidewalls via π–π stacking, which will decrease the electron density of CNTs. In addition, the apparent charge transfer observed here could also provide evidence for the attachment of GP molecules to the surface of CNTs.30,31 Overall, we conclude that the strong π–π stacking interaction between CNTs and GP molecules leads to the attachment of GP molecules to CNTs and thus enabled the superior dispersion of CNTs in water.
The colloidal stability of the CNT dispersion is vital for practical applications. Hence, we evaluated the stability of the resulting GP-CNT dispersion against aggregation by storing them statically for 2 months under ambient conditions. The result showed that 2 months of storage did not cause any obvious bundling behaviors of GP-CNTs (inset in Fig. 1i). Besides, similar UV-Vis absorption profiles shared by the GP-CNT dispersion before and after storage verified their long-term stability (Fig. 1i). The HRTEM analysis showed that even after 2 months of storage, a distinct thin layer of GP molecules was still found to be firmly attached to the surface of CNTs (Fig. S5†), accounting for the long-term stability of the GP-CNT dispersion. The good colloidal stability of GP-CNTs could be ascribed to the electrostatically repulsive interaction owing to the deprotonation of carboxyl groups among GP-CNTs. As such, we attempted to manipulate the electrostatically repulsive interaction among GP-CNTs by varying the pH value of the GP-CNT dispersion. The pH of the GP-CNT dispersion was modulated from an initial value of ca. 6.5 to 5 and 10 by adding hydrochloric acid (HCl) and sodium hydroxide (NaOH), respectively. The isolated CNTs in GP-CNT dispersion progressively agglomerated overnight at pH 5, while no obvious change was found for the dispersion at pH 10 (Fig. S6†). Adding HCl to the GP-CNT dispersion is likely to weaken the deprotonation of the carboxyl groups in the GP molecules, thus decreasing the electrostatic repulsion between the neighboring GP molecules.25 Overall, owing to the functionalization with amphiphilic GP molecules, CNTs exhibit outstanding colloidal stability in the water phase.
At a low concentration of GP-CNTs in water, the electrostatic repulsion among the GP molecules wrapped on the CNT surface primarily governs the stable and uniform dispersion of GP-CNTs. However, at high concentrations, another type of non-covalent interaction, namely hydrogen bonding, replaced the role of electrostatic repulsion. This transition occurs because the distance between GP and surrounding CNTs is dramatically decreased when the concentration of GP-CNTs increases, leading to a significant intensification of the inter-CNT saccharide hydrogen bonds. Besides, it might be conceivable that the available inter-CNT saccharide hydrogen bonds will confer excellent plasticity upon the GP-CNTs (Fig. 2a). Herein, as a step forward to explore its plasticity, we further increased the GP-CNT concentration (∼5 to 200 mg mL−1) in water with simple sonication and/or grinding carefully in a mortar. Interestingly, paste, gel, and dough states were continuously achieved from the powder (Fig. 2b–f). Note that a better dispersion of the GP-CNTs in various states can be achieved by concentrating the diluted dispersion through centrifugation. When increasing the GP-CNT concentration to up to ∼5 mg mL−1, a viscous paste was obtained (Fig. 2c), which can be further spread on aluminum foil to form a uniform thin film (Fig. 2d). The GP-CNT film still showed a good dispersion state without obvious aggregation (Fig. S7†). A drastic increase in the GP-CNT concentration to up to ∼70 mg mL−1 led to a self-standing gel (Fig. 2e). Once their concentration was finally elevated up to ∼200 mg mL−1, a playdough-like CNT was produced. To demonstrate its superior processing capability, a typical landmark of Berlin (e.g. United Buddy Bear here) model was directly crafted by hand (Fig. 2f). The outstanding plasticity and shaping capability of versatile GP-CNT materials experimentally demonstrated their enhanced processibility, owing to the abundant inter-CNT saccharide hydrogen bonds. Beyond the conventional approaches that rely on either toxic organic solvents (surfactants) or inevitable surface modifications, the use of eco-friendly and biocompatible GP molecules offers a promising alternative for wide and green applications of pristine CNTs.6,7,32
 | ||
| Fig. 2 (a) Scheme of the formation of processable GP-CNTs. Photos of pristine CNT powders (b) and highly concentrated GP-CNTs in various states: (c and d) paste (∼5 mg mL−1), (e) gel (∼70 mg mL−1), and (f) playdough (∼200 mg mL−1) of a handmade “United Buddy Bear” model with an inset of its corresponding real landmark. | ||
GP-enabled in situ generation of metal NPs on CNTs
Considering numerous hydroxyl groups in the GP molecules, we subsequently employed the GP-CNTs as a versatile template for the preparation of metal/CNT composites, where GP molecules could functionalize CNTs with metal NPs (i.e. Pd, Ag, and Au) through an in situ reducing and stabilizing strategy. Thus, the corresponding metal/GP-CNT composites (i.e. GP-CNTs–Pd, GP-CNTs–Au, GP-CNTs–Pt, and GP-CNTs–Ag) were simply obtained under mild stirring at room temperature. Taking Pd as an example, PdCl2 was used as the metal precursor and directly added to the GP-CNT dispersion without any external reducing agents. Positive Pd2+ ions were electrostatically captured by the negative carboxyl group of GP. As a result, the Pd NPs were in situ reduced by GP and subsequently stably anchored on the surface of CNTs, as revealed in the TEM and SEM images (Fig. 3a and 3b, and S8†). As seen in the photo (inset in Fig. 3a), a uniform GP-CNTs–Pd dispersion was achieved, demonstrating good stability even after functionalization with metal NPs. Moreover, no obvious change was observed in the TEM images of the metal/CNT composites after storing for one month, demonstrating their good stability (Fig. S9†). The lattice spacing was determined from its corresponding HRTEM image to be 0.23 nm, which matches well with the (111) plane of the Pd crystal (Fig. 3c). The obtained Pd NPs show an ultra-small size of 1.8 ± 0.4 nm (Fig. S10†). The tiny Pd NPs and their distributions on CNTs were confirmed by high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) and the corresponding electron energy loss spectroscopy (EELS), as shown in Fig. 3d. The HAADF-STEM image revealed a dense decoration of small Pd NPs on the CNT surface (Fig. 3d1). The presence of Pd was further confirmed through EELS-STEM analysis (Fig. 3d2). As the STEM data are a projection through the sample, the seemingly higher density of Pd NPs at the borders of the GP-CNTs is consistent with those located on the surface of GP-CNTs. Additionally, the C, N, and O species exhibited a homogeneous distribution in the GP-CNTs–Pd composite (Fig. 3d3–5). A distinct hollow core was observed in all the C, N, and O maps, suggesting the successful incorporation of GP molecules and Pd NPs on the outer surface of CNTs. X-ray photoelectron spectroscopy (XPS) was used to study the oxidation state of Pd NPs in the GP-CNTs–Pd composite. The raw and fitted Pd 3d spectra demonstrated the presence of a mixture of Pd0 and Pd2+ (Fig. S11†). The Pd 3d5/2 peak at 335.7 eV and Pd 3d3/2 peak at 340.9 eV correspond to metallic Pd0. In addition, the shoulder peaks at 337.7 and 342.9 eV are assigned to Pd2+, primarily indicating the presence of oxidized Pd (PdO) on the surface of Pd NPs.33 The PdO species mainly resulted from exposure to air during sample preparation. Smaller Pd NPs show higher specific surface areas, indicating more exposed Pd atoms that are easily oxidized. | ||
| Fig. 3 (a–c) TEM and (d1) HAADF-STEM images of GP-CNTs–Pd. (d2–d5) The corresponding STEM-EELS maps (same scale bar) of various chemical elements in the GP-CNTs–Pd composite. TEM images (e–g) of the GP-CNTs–Au composite. The insets in (a and e) are the corresponding photos of the metal/CNT dispersions. (h) UV-Vis absorption spectra of the pristine GP-CNTs, GP-CNTs–Pd, and GP-CNTs–Au dispersion. (i) XRD patterns of the pristine GP-CNTs, GP-CNTs–Au, and GP-CNTs–Pd powder after freeze-drying. | ||
Compared with Pd NPs, the obtained Au NPs on CNTs were relatively larger, with an average size of 5.6 ± 1.3 nm (Fig. 3e–g, S10†). The lattice spacing of 0.22 nm corresponds to the (110) planes of the Au NPs (Fig. 3g).34 The size of Au NPs on the GP-CNTs has been tuned by varying the GP concentration, HAuCl4 concentration, and reaction temperature (Fig. S12†). First of all, when keeping the Au precursor concentration constant, the particle size of Au NPs slightly increased from ∼4 to 5 nm, as the GP concentration was increased from 0.02 to 0.05 mg mL−1. A further increase of the GP concentration to 0.1 mg mL−1 led to the formation of inhomogeneous Au NPs with various morphologies, as indicated with red circles in Fig. S12a–c.† Secondly, when keeping the GP concentration constant, no obvious change in the particle size was observed by increasing the concentration of the Au precursor from 0.05 to 0.15 mM, but the number of the Au NPs deposited on the CNT surface significantly increased (Fig. S12b and S12d†). Further increase of the Au precursor concentration to 0.3 mM results in a sharp increase in the particle size from 5 to 23 nm, with an irregular shape and broad size distribution, as indicated in Fig. S12e.† Finally, elevating the reaction temperature from room temperature to 40 °C led to the formation of irregular morphology and huge aggregates of Au NPs (Fig. S12f†). Therefore, we focused on the sample (Fig. S12b†) prepared with 0.05 mg mL−1 of GP and 0.15 mM of HAuCl4 at room temperature for the following catalysis study.
The successful incorporation of Au NPs into GP-CNTs was further confirmed by UV-Vis absorption spectroscopy. A broad peak at ca. 530 nm was observed for the GP-CNTs–Au dispersion (Fig. 3h), which resulted from the surface plasmon resonance of Au NPs. To further confirm the presence of metal NPs, the obtained metal/GP-CNT composites were characterized by X-ray diffraction (XRD). As shown in Fig. 3i, the distinct peaks located at 25.6, 44.7, 55.1, and 77.6° were observed for all the metal/GP-CNT composites, which respectively refer to the (002), (100), (004), and (110) planes of the MWCNTs (JPCDS no. 96-101-1061). Meanwhile, the peaks at 38.6, 44.3, and 64.5° were observed for GP-CNTs–Au, which can be indexed to the (110), (200), and (220) planes of the face-centered cubic (fcc) phase of the metallic Au crystal (JPCDS no. 00-004-0784), respectively.35 In the case of GP-CNTs–Pd, two peaks at 40.1 and 46.5° were observed, which respectively stand for the (111) and (200) reflections of the (fcc) metallic Pd (JPCDS no. 05-0681).36 Apart from Au and Pd NPs, Ag and Pt NPs can be also successfully incorporated into GP-CNTs (Fig. S13†). As a proof-of-concept, the proposed method demonstrates that our versatile GP-CNTs can be applied for the facile synthesis of various noble metal NPs. The amount of metal loading in the metal/GP-CNT composites was estimated from the aerobic residual in the thermogravimetric analysis (TGA) measurement (Fig. S14†). The metal loadings in GP-CNTs–Au and GP-CNTs–Pd were calculated to be 13.2 and 11.5 wt%, respectively. In control experiments, the pristine CNTs alone were found to be incapable of reducing metal ions to metallic NPs (e.g. Au), as shown in Fig. S15a.† By contrast, directly mixing GP molecules with the Au precursor (e.g. HAuCl4) solution in the absence of the CNT template led to the formation of stably dispersed Au NPs, as demonstrated in Fig. S15b.† These findings confirm that GP molecules indeed function as the reducing agent (and stabilizer) for the generation of metal NPs on CNTs even at room temperature. The noticeable reducibility of GP molecules for the formation of metal NPs is presumably due to their abundant hydroxyl groups in the trisaccharide moieties, which has been reported in our previous work.37 During the reduction process, the hydroxyl groups of the GP molecules are likely to be oxidized into carboxyls by HAuCl4.38 In brief, we demonstrated a straightforward strategy for the green synthesis of water-dispersible metal/CNT composites enabled by amphiphilic GP molecules. Importantly, the small size of metal NPs in situ generated on the CNT surface indicates a high specific surface area, which could provide sufficient active sites for efficient catalysis.
Catalytic reduction of 4-nitrophenol
Noble metal NPs are crucial catalysts for the catalytic degradation of organic pollutants in water via reduction or coupling reactions.39,40 As a typical temperature-dependent and surface reaction, the reduction of 4-nitrophenol (4-NP) to 4-aminophenol (4-AMP) in aqueous solution has been regarded as a benchmark reaction and was utilized to evaluate the catalytic performance of various catalysts.41 Thanks to the outstanding photothermal conversion capability under near-infrared (NIR) light irradiation, CNTs have also found numerous applications including cancer therapy, bio-sensing, and photothermal-assisted catalysis.42,43 In this study, we used the as-prepared metal/GP-CNT composites (GP-CNTs–Pd and GP-CNTs–Au) as the catalyst and NaBH4 as the reducing agent, to investigate their catalytic behavior in the reduction of 4-NP under NIR light irradiation.As an initial test, the catalytic performance of the GP-CNTs–Pd composite in the reduction of 4-NP without NIR light irradiation was assessed in aqueous solution. As the reaction progressed, a characteristic absorption peak at 400 nm gradually faded and disappeared due to the conversion of 4-NP, while a new peak at 290 nm appeared and progressively increased in intensity due to the generation of 4-AMP (Fig. 4a).44 To compare the catalytic performance of various metal/CNT composite catalysts, we fixed the same surface area of the catalytic metal (Pd in GP-CNTs–Pd and Au in GP-CNTs–Au) used for the reaction. The apparent rate constant (kapp) and surface area-normalized rate constant (k1) were determined to quantitatively evaluate the reaction rate of these two catalysts (more details are given in Fig. S16†). As a result, GP-CNTs–Pd displays a fast catalytic conversion of 4-NP, giving a much higher k1 value of 0.062 s−1 m−2 L compared with that of GP-CNTs–Au (k1 of 0.0081 s−1 m−2 L). Note that the as-synthesized GP-CNTs–Pd shows a high rate constant among the reported Pd-based catalysts under similar reaction conditions (Table S1†), demonstrating their good catalytic performance. The particle dimension and electron structure of catalytic metal particles on the surface have been reported to influence the adsorption/desorption behavior of 4-NP molecules.45,46 Hence, the fast conversion of 4-NP could be predominantly explained by the small size as well as the intrinsic catalytic activity of Pd when applied to the reduction of 4-NP. As a reference, acid-oxidized CNTs were also used as the template for the preparation of Pd/CNT composites (denoted as a-CNTs–Pd).2 When used as the catalyst for the reduction of 4-NP with the same concentration as that of GP-CNTs–Pd at 0.02 mg mL−1, a-CNTs–Pd shows a reaction conversion of 32.3% after the reaction for 10 min, which is much lower than that of GP-CNTs–Pd (a conversion of 99.2%) (Fig. S17†). Considering that the type and number of the functional groups (hydroxyl or carboxyl groups) on the CNT surface from strong acid oxidation are not very controllable, this may cause the inhomogeneous distribution and propensity of aggregation of the Pd NPs on CNTs. These factors will lead to the relatively lower catalytic activity of a-CNTs–Pd. Based on these findings, GP-CNTs–Pd was utilized in the subsequent measurements. It was found that an increase in the concentration of the GP-CNTs–Pd catalyst from 0.01 to 0.04 mg mL−1 led to the increase of kapp from 1.81 × 10−3 to 7.21 × 10−3 s−1 (Fig. 4b). Moreover, GP-CNTs–Pd still presented a high conversion above 90% even after 5 consecutive catalytic cycles, demonstrating their excellent catalytic stability (Fig. 4c). No visible change in the morphology of GP-CNTs–Pd was found after the catalytic cycles, confirming its good structural stability (Fig. S18a†). In control experiments, no conversion of 4-NP was observed when only pristine GP-CNTs–Pd or GP molecules were used in the absence of NaBH4, revealing that neither GP molecules nor CNTs alone can reduce 4-NP into 4-AMP (Fig. S18b†).
 | ||
| Fig. 4 (a) UV-Vis absorption spectra of the reduction of 4-NP with the concentration of GP-CNTs–Pd at 0.01 mg mL−1. (b) Plots of ln(c/c0) as a function of reaction time of GP-CNTs–Pd at various concentrations. (c) Catalytic cycles of GP-CNTs–Pd with the concentration of GP-CNTs–Pd at 0.01 mg mL−1, 4-NP at 0.1 mM, and NaBH4 at 20 mM at room temperature. | ||
Next, we tested the photothermal conversion of the as-prepared GP-CNTs–Pd dispersion in a quartz cuvette under NIR light irradiation (a wavelength of 808 nm, 3 W cm−2). As visualized using an IR camera, the temperature of the GP-CNTs–Pd dispersion increased with increasing the irradiation time and concentration of the GP-CNTs–Pd dispersion (Fig. 5a). To precisely track the temperature rise of GP-CNTs–Pd under NIR irradiation, the temperature of the dispersion was further measured and confirmed using a digital thermometer. Notably, the temperature of the GP-CNTs–Pd dispersion with a concentration of 0.08 mg mL−1 increased by ca. 23 °C after irradiation for 15 min (Fig. S19†). By comparison, pure water merely showed a temperature change of ca. 5 °C under the same light intensity, indicating the negligible photothermal conversion effect of water alone. In addition, the photothermal performance of the GP-CNTs and pristine CNT cluster was also evaluated under the same conditions (e.g. concentration, laser power, and irradiation time). GP-CNTs showed a similar temperature variation to GP-CNTs–Pd, while the pristine CNT cluster only showed a mild temperature increase by ca. 8 °C (Fig. 5b). The photothermal conversion efficiency (η) of the GP-CNT dispersion was calculated to be 22.3%, which is higher than that of the pristine CNT cluster dispersion (4.2%), as shown in Fig. S20.† As mentioned above, the pristine CNTs tend to form big aggregates in water owing to the inherent hydrophobicity. In such a scenario, only little surface area can be exposed for light harvest. The majority of incident light will just pass through the water molecules. In comparison, the NIR light can be efficiently captured by the GP-CNTs owing to a high exposed surface area. In addition, a better dispersity of the GP-CNTs improves water diffusion, thus enhancing the heat transport from the surface of CNTs to the surrounding solution. Overall, the GP-modified CNTs show better photothermal conversion performance than the pristine CNTs. Also, the loading of the small size Pd NPs exerts no influence on their photothermal heating activities. Furthermore, GP-CNTs–Pd showed superior photothermal stability, as revealed by its negligible reduction in its photothermal conversion performance after 5 consecutive ON–OFF laser cycles (Fig. 5c).
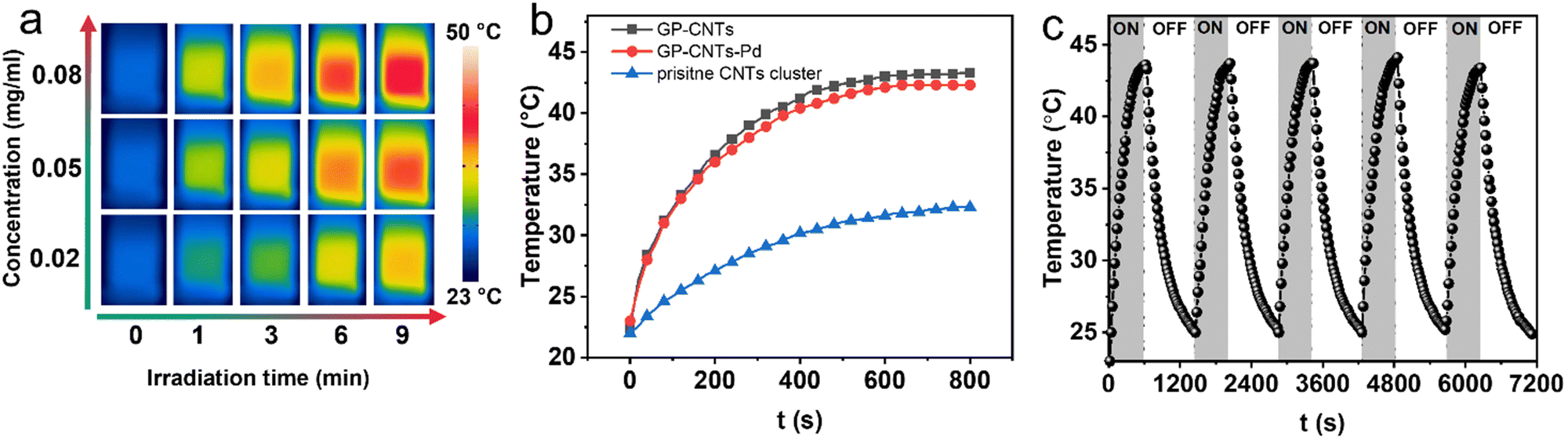 | ||
| Fig. 5 (a) Time-dependent IR thermal photos of the GP-CNTs–Pd dispersion under an 808 nm laser (3 W cm−2) irradiation at various concentrations (0.02, 0.05, and 0.08 mg mL−1). (b) Comparison of the photothermal heating of various dispersions at 0.08 mg mL−1. (c) Heating/cooling profiles of the GP-CNTs–Pd dispersion at 0.08 mg mL−1 under NIR irradiation at 3 W cm−2. | ||
Taking advantage of the photothermal conversion effect of GP-CNTs–Pd, we further explored its catalytic performance under NIR light irradiation (Fig. 6a). To reach an equilibrium temperature (32 °C), the GP-CNTs–Pd dispersion was first continuously stirred under NIR light irradiation (3 W cm−2) for 15 min prior to the reaction, i.e. in the absence of NaBH4. The UV-Vis absorption spectrum showed no 4-NP conversion on this occasion (Fig. S21†), indicating that the NIR light irradiation alone does not affect the reduction process of 4-NP. Afterwards, a fresh NaBH4 solution was rapidly injected into the reaction mixture to trigger the reduction of 4-NP (Fig. 6b). As expected, the reaction under NIR light illumination showed a high conversion of up to ∼91.2% in 5 min, which is 6.8-fold higher than the conversion (∼13.5%) achieved at room temperature without light illumination. Considering that light irradiation leads to a significant increase in the dispersion temperature, the enhanced reaction conversion is thus assigned to the high temperature induced by the photothermal heating of CNTs. To further confirm the role of the photoheating effect, the reduction of 4-NP was conducted at the same reaction temperature (32 °C) in a water bath, in the absence of light illumination. In this case, only ∼35.1% of conversion was achieved, which is much lower than that obtained under NIR light irradiation (Fig. 6c and S22†). As a control experiment, the reduction of 4-NP was conducted under NIR light irradiation at a wavelength of 976 nm, as shown in Fig. S23.† It was found that GP-CNTs–Pd shows conversion of the reduction reaction of ∼88.5% after the reaction for 5 min (solution temperature at 33 °C), which is similar to the reduction reaction conducted at the wavelength of 808 nm (91.2%, solution temperature at ∼32 °C). This indicates that the catalytic performance does not show an obvious difference when exposed to light wavelengths of 808 and 976 nm. As the reduction of 4-NP is a thermally driven reaction, the catalytic performance is primarily controlled by the reaction temperature under NIR illumination. Therefore, the comparable catalytic performance of the GP-CNTs–Pd composite can be attributed to the similar photothermal conversion performance at these two wavelengths, which was confirmed by its similar solution temperature under NIR irradiation at 808 nm (∼32 °C) and 976 nm (∼34 °C), respectively. It has been reported that the hollow architecture of nanomaterials is capable of facilitating light harvesting and subsequent localization, resulting in a relatively higher surface temperature compared to the bulk solution.47,48 In the present work, we hypothesized that the heat induced by light irradiation is relatively higher on the nanotube surface supporting the catalytic Pd NPs than in the solution phase. This localized heat predominantly contributes to the substantial enhancement in the fast conversion of 4-NP, as the reduction of 4-NP is a typical temperature-dependent and surface reaction. Overall, the incorporation of catalytic metal particles into photothermal CNTs realized by the multi-functional GP molecules demonstrates great potential in the efficient degradation of organic pollutants. In addition, a more systematic study will be conducted in the future to gain deeper insights into the dependence of the catalytic performance of the metal/CNT composites on the light wavelength. Considering that Au NPs could also show significant photothermal effects, the combination of photothermal Au NPs and CNTs could be promising for enhancing the catalytic performance. Thus, we will continue the optimization of Au NP synthesis and even investigate Pd–Au nanoalloy particles in our future work.
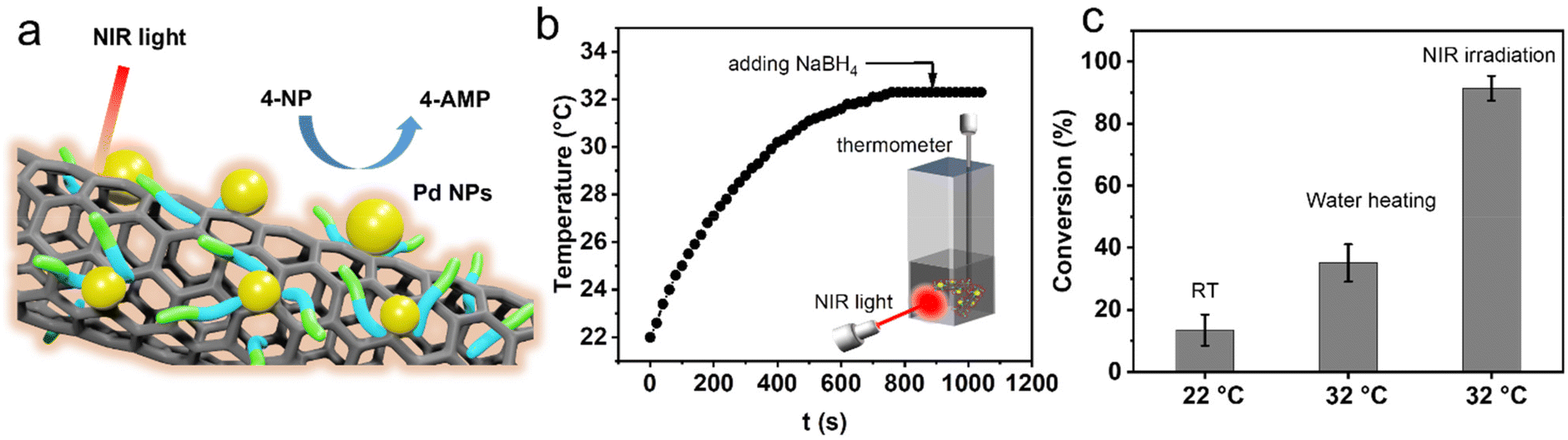 | ||
| Fig. 6 (a) Schematic of the reduction reaction of 4-NP using GP-CNTs–Pd as the catalyst under NIR irradiation. (b) Temperature profile of the reactant under light irradiation. (c) Conversion comparison of reduction of 4-NP under various heating conditions in 5 min with the concentrations of GP-CNTs–Pd at 0.03 mg mL−1, 4-NP at 1 mM, and NaBH4 at 0.2 M. | ||
Conclusions
In summary, we provided a novel strategy for the facile and green synthesis of versatile metal/CNT composites based on amphiphilic glycopeptide (GP) biomolecules. GP was first employed to disassemble CNT clusters in water through non-covalent π–π interactions. In particular, GP-CNTs exhibit an enhanced processibility, forming a paste, gel, and dough at high concentrations. In contrast to conventional single-functional surfactants, the GP molecules also show strong reducibility towards noble metal ions. This unique property enables the in situ generation of various metal NPs including Au, Ag, Pt, and Pd NPs on CNTs at room temperature, which was scarcely reported in the literature. To explore their practical applications, GP-CNTs–Pd with an ultra-small size of Pd NPs (1–3 nm on average) was applied as the catalyst for the reduction reaction of the typical organic pollutant, 4-NP, with the assistance of NIR light irradiation. The enhanced degradation of 4-NP was realized mainly due to the small size of catalytic Pd NPs as well as the localized photothermal heating of the CNT template under NIR light irradiation. The proposed approach provides new opportunities for the integral green synthesis of CNT-based catalytic platforms that are inaccessible to the conventional surfactant strategy.Experimental section/methods
Chemicals
Single-walled carbon nanotubes (SWCNTs, diameter of 1–2 nm, length of 5–30 μm, ≥95%) and multiple-walled carbon nanotubes (MWCNTs, outer diameter of 10–30 nm, inner diameter of 3–5 nm, length of 10–50 μm, ≥99.9%) were obtained from Timesnano. Palladium chloride (PdCl2, 99%), gold chloride trihydrate (HAuCl4·3H2O, ≥99%), chloroplatinic acid hydrate (H2PtCl6), silver nitrate (AgNO3, ≥99.99%), sodium borohydride (NaBH4, 99%), sodium hydroxide (NaOH, ≥97%), hydrochloric acid (HCl, 37%), 4-nitrophenol (4-NP, ≥99%), dimethyl sulfoxide-d6 (DMSO-d6, ≥99%), and tetramethylsilane (TMS, ≥99%) were purchased from Sigma-Aldrich. Ultrapure water with a resistance of 18.2 MΩ cm was purified using a Thermo Scientific Barnstead GenPurex CAD Plus system. All chemicals were used without any further purification.Material synthesis
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) but the total concentration in water increased from ∼70 to 200 mg mL−1, or even higher and followed by simple hand-grinding carefully in a mortar for 30 min.
:1) but the total concentration in water increased from ∼70 to 200 mg mL−1, or even higher and followed by simple hand-grinding carefully in a mortar for 30 min.
Catalytic reduction of 4-nitrophenol
Characterization
X-ray diffraction (XRD) analysis was conducted on a Bruker D8 diffractometer with a monochromatic X-ray beam with Cu Kα radiation at a scan rate of 0.05° min−1. All the samples were collected after freeze-drying overnight and stored in air-tight XRD holders for measurements. The oxidation state of Pd in the GP-CNTs–Pd composite was determined by X-ray photoelectron spectroscopy (XPS) with an ESCA-Lab-220i-XL X-ray photoelectron spectrometer (Thermo Fisher Scientific) with Al Kα sources (hν = 1486.6 eV). Nuclear magnetic resonance (NMR) measurements were performed on AVANCE III HD 400 MHz of Bruker BioSpin International. Dimethyl sulfoxide-d6 (DMSO-d6) was used as a solvent and tetramethylsilane (TMS) was employed as the reference with the peak of chemical shift at 0 ppm.Conventional transmission electron microscopy (TEM), high-resolution TEM (HRTEM), and cryogenic TEM (cryo-TEM) were performed on a JEOL JEM-2100 instrument operated at an accelerating voltage of 200 kV. For conventional TEM and HRTEM, all the samples were prepared by dropping ∼5 μL of the resulting dispersions on continuous carbon-coated copper TEM grids (200 mesh, Science Services). Cryo-TEM specimens were prepared on Lacey carbon-coated copper grids (200 mesh, Science Services) and subsequently plunge-frozen into liquid ethane with an FEI Vitrobot Mark IV at 4 °C with a humidity of 95%. Vitrified grids were either directly transferred to the cryogenic transfer holder (Gatan 914, Gatan, Munich, Germany) or stored in a liquid N2 tank. The imaging process was carried out at ca. −183 °C with a defocus of the objective lens ranging from 2.5 to 3 μm. Cryo-TEM images were recorded at various magnifications using a bottom-mounted 4 k × 4 k CMOS camera (TemCam-F416, TVIPS, Gauting, Germany). The total electron dose for each image was maintained below 20 e− Å−2. Scanning transmission electron microscopy (STEM) for electron energy-loss spectroscopy (EELS) analysis was performed on a Nion HERMES microscope (Nion Co., Kirkland, WA, USA) equipped with a Dectris ELA direct electron detector (Dectris AG, Baden-Daettwil, Switzerland). Similar to the sample preparation of cryo-TEM, ∼5 μL of the GP-CNTs–Pd dispersion was placed on a lacey carbon-coated copper grid (200 mesh, Electron Microscopy Sciences, Hatfield, PA) and followed by drying under mild conditions. Scanning electron microscopy (SEM) images were recorded using a LEO GEMINI 1530 microscope at 5 kV.
Thermogravimetric analysis (TGA) was performed on a Netzch Phoenix F204 instrument with a temperature range of 25–900 °C at a heating rate of 10 °C min−1 under a synthetic air atmosphere. Raman spectra were recorded using an inVia 2R9X81 Raman spectrometer equipped with a detector of Renishaw Centrus 2MYA08 and a HeNe laser (a wavelength of 532 nm). UV-Vis absorption spectra were recorded at room temperature using a PerkinElmer Lambda 650 spectrometer. All the aqueous solutions or dispersions were placed in a clean quartz cuvette (110-QS Hellma, 10 mm) for measurement.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the Deutsche Forschungsgemeinschaft (DFG, project number: 410871749) for financial support. G. C. thanks National Natural Science Foundation of China (NSFC/China, No. 52125303, 51721002, and 21861132012) for financial support. The authors also appreciate the Joint Lab for Structural Research at the Integrative Research Institute for the Sciences (IRIS Adlershof) for the cryo-TEM and STEM measurements.References
- Z. Li, J. Peng and Z. Lin, Giant, 2021, 5, 100048 CrossRef CAS.
- D. H. Kweon, M. S. Okyay, S.-J. Kim, J.-P. Jeon, H.-J. Noh, N. Park, J. Mahmood and J.-B. Baek, Nat. Commun., 2020, 11, 1278 CrossRef CAS PubMed.
- K.-I. Kim, S. Yoon, J. Chang, S. Lee, H. H. Cho, S. H. Jeong, K. Jo and J. H. Lee, Small, 2020, 16, 1905821 CrossRef CAS PubMed.
- B. Zhang, T. Li, L. Huang, Y. Ren, D. Sun, H. Pang, J. Yang, L. Xu and Y. Tang, Nanoscale, 2021, 13, 5400–5409 RSC.
- K. Chiou, S. Byun, J. Kim and J. Huang, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 5703–5708 CrossRef CAS PubMed.
- S. Wang, Z. Huang, W. Shi, D. Lee, Q. Wang, W. Shang, Y. Stein, Y. Shao-Horn, T. Deng, B. L. Wardle and K. Cui, ACS Appl. Mater. Interfaces, 2021, 13, 28583–28592 CrossRef CAS PubMed.
- K. Chiou and J. Huang, Matter, 2020, 3, 302–319 CrossRef.
- T. Premkumar, R. Mezzenga and K. E. Geckeler, Small, 2012, 8, 1299–1313 CrossRef CAS PubMed.
- M. Bystrzejewski, A. Huczko, H. Lange, T. Gemming, B. Büchner and M. H. Rümmeli, J. Colloid Interface Sci., 2010, 345, 138–142 CrossRef CAS PubMed.
- V. A. Davis, A. N. G. Parra-Vasquez, M. J. Green, P. K. Rai, N. Behabtu, V. Prieto, R. D. Booker, J. Schmidt, E. Kesselman, W. Zhou, H. Fan, W. W. Adams, R. H. Hauge, J. E. Fischer, Y. Cohen, Y. Talmon, R. E. Smalley and M. Pasquali, Nat. Nanotechnol., 2009, 4, 830–834 CrossRef CAS PubMed.
- X. Wu, Z. Wang, D. Zhang, Y. Qin, M. Wang, Y. Han, T. Zhan, B. Yang, S. Li, J. Lai and L. Wang, Nat. Commun., 2021, 12, 4018 CrossRef CAS PubMed.
- Z. Ma, C. Lian, D. Niu, L. Shi, S. Hu, X. Zhang and H. Liu, ChemSusChem, 2019, 12, 1724–1731 CrossRef CAS PubMed.
- A. Benko, J. Duch, M. Gajewska, M. Marzec, A. Bernasik, M. Nocuń, W. Piskorz and A. Kotarba, Nanoscale, 2021, 13, 10152–10166 RSC.
- M. Youssry, M. Al-Ruwaidhi, M. Zakeri and M. Zakeri, Emergent Mater., 2020, 3, 25–32 CrossRef CAS.
- A. Ezzeddine, Z. Chen, K. S. Schanze and N. M. Khashab, ACS Appl. Mater. Interfaces, 2015, 7, 12903–12913 CrossRef CAS PubMed.
- A. Star, J. F. Stoddart, D. Steuerman, M. Diehl, A. Boukai, E. W. Wong, X. Yang, S. W. Chung, H. Choi and J. R. Heath, Angew. Chem., 2001, 113, 1771–1775 CrossRef.
- L. Vaisman, H. D. Wagner and G. Marom, Adv. Colloid Interface Sci., 2006, 128–130, 37–46 CrossRef CAS PubMed.
- T. Li, Y. Chen, W. Hu, W. Yuan, Q. Zhao, Y. Yao, B. Zhang, C. Qiu and C. M. Li, Nanoscale, 2021, 13, 4444–4450 RSC.
- J. John, E. Gravel, I. N. N. Namboothiri and E. Doris, Nanotechnol. Rev., 2012, 1, 515–539 CAS.
- S. Chu and A. Majumdar, Nature, 2012, 488, 294–303 CrossRef CAS PubMed.
- Y. Shi, W.-M. Huang, J. Li, Y. Zhou, Z.-Q. Li, Y.-C. Yin and X.-H. Xia, Nat. Commun., 2020, 11, 4558 CrossRef CAS PubMed.
- Y. Liu, X. Li, Q. Zhang, W. Li, Y. Xie, H. Liu, L. Shang, Z. Liu, Z. Chen, L. Gu, Z. Tang, T. Zhang and S. Lu, Angew. Chem., Int. Ed., 2020, 59, 1718–1726 CrossRef CAS.
- D. Wang, Q. Li, C. Han, Q. Lu, Z. Xing and X. Yang, Nat. Commun., 2019, 10, 3899 CrossRef.
- F. Morales-Lara, V. K. Abdelkader-Fernández, M. Melguizo, A. Turco, E. Mazzotta, M. Domingo-García, F. J. López-Garzón and M. Pérez-Mendoza, J. Mater. Chem. A, 2019, 7, 24502–24514 RSC.
- R. Liu, R. Zhang, L. Li, Z. Kochovski, L. Yao, M.-P. Nieh, Y. Lu, T. Shi and G. Chen, J. Am. Chem. Soc., 2021, 143, 6622–6633 CrossRef CAS PubMed.
- R. Liu, L. Li, S. Chen, Z. Yang, Z. Kochovski, S. Mei, Y. Lu, L. Zhang and G. Chen, ACS Nano, 2023, 17, 2245–2256 CrossRef CAS PubMed.
- S. Attal, R. Thiruvengadathan and O. Regev, Anal. Chem., 2006, 78, 8098–8104 CrossRef CAS PubMed.
- S. Costa and E. Borowiak-Palen, Acta Phys. Pol., A, 2009, 116, 32–35 CrossRef CAS.
- B. Shen, W. Zhai, C. Chen, D. Lu, J. Wang and W. Zheng, ACS Appl. Mater. Interfaces, 2011, 3, 3103–3109 CrossRef CAS PubMed.
- Q. Xu and W. Zhang, e-Polymers, 2021, 21, 166–178 CrossRef CAS.
- P. Song, Z. Xu, Y. Wu, Q. Cheng, Q. Guo and H. Wang, Carbon, 2017, 111, 807–812 CrossRef CAS.
- L. Yu, C. Shearer and J. Shapter, Chem. Rev., 2016, 116, 13413–13453 CrossRef CAS PubMed.
- P. J. Cappillino, J. D. Sugar, M. A. Hekmaty, B. W. Jacobs, V. Stavila, P. G. Kotula, J. M. Chames, N. Y. Yang and D. B. Robinson, J. Mater. Chem. A, 2012, 22, 14013–14022 RSC.
- M. Mohammadnezhad, G. S. Selopal, O. Cavuslar, D. Barba, E. G. Durmusoglu, H. Y. Acar, Z. M. Wang, G. P. Lopinski, B. Stansfield, H. Zhao and F. Rosei, Chem. Eng. J., 2021, 421, 127756 CrossRef CAS.
- Q. Yu, J. Li, Y. Zhang, Y. Wang, L. Liu and M. Li, Sci. Rep., 2016, 6, 26667 CrossRef CAS.
- X. Shu, Q. Yang, F. Yao, Y. Zhong, W. Ren, F. Chen, J. Sun, Y. Ma, Z. Fu, D. Wang and X. Li, Chem. Eng. J., 2019, 358, 903–911 CrossRef CAS.
- L. Su, C. Wang, F. Polzer, Y. Lu, G. Chen and M. Jiang, ACS Macro Lett., 2014, 3, 534–539 CrossRef CAS PubMed.
- E. V. Pronina, Y. A. Vorotnikov, T. N. Pozmogova, A. O. Solovieva, S. M. Miroshnichenko, P. E. Plyusnin, D. P. Pishchur, I. V. Eltsov, M. V. Edeleva, M. A. Shestopalov and O. A. Efremova, ACS Sustainable Chem. Eng., 2020, 8, 5371–5379 CrossRef CAS.
- P. Hervés, M. Pérez-Lorenzo, L. M. Liz-Marzán, J. Dzubiella, Y. Lu and M. Ballauff, Chem. Soc. Rev., 2012, 41, 5577–5587 RSC.
- M. Hartings, Nat. Chem., 2012, 4, 764–764 CrossRef CAS PubMed.
- S. Mei, Z. Kochovski, R. Roa, S. Gu, X. Xu, H. Yu, J. Dzubiella, M. Ballauff and Y. Lu, Nano-Micro Lett., 2019, 11, 83 CrossRef CAS PubMed.
- X. Ye, X. Shi, H. Zhong, T. Wang, J. Duo, B. Jin and F. Jin, Green Chem., 2022, 24, 813–822 RSC.
- Y. Zhao, T. Zhao, Y. Cao, J. Sun, Q. Zhou, H. Chen, S. Guo, Y. Wang, Y. Zhen, X.-J. Liang and S. Zhang, ACS Nano, 2021, 15, 6517–6529 CrossRef CAS PubMed.
- S. Wunder, Y. Lu, M. Albrecht and M. Ballauff, ACS Catal., 2011, 1, 908–916 CrossRef CAS.
- Y. Chen, X. Ge, Y. Cao, C. Yao, J. Zhang, G. Qian, X. Zhou and X. Duan, Ind. Eng. Chem. Res., 2022, 61, 6427–6435 CrossRef CAS.
- Y. R. Mejía and N. K. Reddy Bogireddy, RSC Adv., 2022, 12, 18661–18675 RSC.
- J. Sun, J. Zhang, M. Zhang, M. Antonietti, X. Fu and X. Wang, Nat. Commun., 2012, 3, 1139 CrossRef.
- X. Pan, R. M. Sarhan, Z. Kochovski, G. Chen, A. Taubert, S. Mei and Y. Lu, Nanoscale, 2022, 14, 6888–6901 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3nr02607h |
| ‡ These two authors contributed equally to this manuscript. |
| This journal is © The Royal Society of Chemistry 2023 |