
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
In situ observation of the on-surface thermal dehydrogenation of n-octane on Pt(111)†
Daniel
Arribas
 a,
Víctor
Villalobos-Vilda
a,
Ezequiel
Tosi
ab,
Paolo
Lacovig
b,
Alessandro
Baraldi
bc,
Luca
Bignardi
b,
Silvano
Lizzit
b,
José Ignacio
Martínez
a,
Pedro Luis
de Andres
ad,
Alejandro
Gutiérrez
ef,
José Ángel
Martín-Gago
a and
Pablo
Merino
*a
a,
Víctor
Villalobos-Vilda
a,
Ezequiel
Tosi
ab,
Paolo
Lacovig
b,
Alessandro
Baraldi
bc,
Luca
Bignardi
b,
Silvano
Lizzit
b,
José Ignacio
Martínez
a,
Pedro Luis
de Andres
ad,
Alejandro
Gutiérrez
ef,
José Ángel
Martín-Gago
a and
Pablo
Merino
*a
aInstituto de Ciencia de Materiales de Madrid, c/Sor Juana Inés de la Cruz, 3, Spain. E-mail: pablo.merino@csic.es
bElettra-Sincrotrone Trieste S.C.p.A., S.S. 14 km 163.5, Trieste, Italy
cPhysics Department, University of Trieste, Via Valerio 2, 34127 Trieste, Italy
dOn leave of absence at nanoteeche@surfaces Laboratory, Swiss Federal Laboratories for Materials Science and Technology, 8600-Dübendorf, Switzerland
eApplied Physics Department, Universidad Autónoma de Madrid, Calle Francisco Tomás y Valiente, 7, 28049 Madrid, Spain
fInstituto Nicolás Cabrera, Cantoblanco, Calle Francisco Tomás y Valiente, 7, 28049 Madrid, Spain
First published on 4th July 2023
Abstract
The catalytic dehydrogenation of alkanes constitutes a key step for the industrial conversion of these inert sp3-bonded carbon chains into other valuable unsaturated chemicals. To this end, platinum-based materials are among the most widely used catalysts. In this work, we characterize the thermal dehydrogenation of n-octane (n-C8H18) on Pt(111) under ultra-high vacuum using synchrotron-radiation X-ray photoelectron spectroscopy, temperature-programmed desorption and scanning tunneling microscopy, combined with ab initio calculations. At low activation temperatures, two different dehydrogenation stages are observed. At 330 K, n-C8H18 effectively undergoes a 100% regioselective single C–H bond cleavage at one methyl end. At 600 K, the chemisorbed molecules undergo a double dehydrogenation, yielding double bonds in their carbon skeletons. Diffusion of the dehydrogenated species leads to the formation of carbon molecular clusters, which represents the first step towards poisoning of the catalyst. Our results reveal the chemical mechanisms behind the first stages of alkane dehydrogenation on a platinum model surface at the atomic scale, paving the way for designing more efficient dehydrogenation catalysts.
1 Introduction
Alkanes, saturated hydrocarbons with the chemical formula CnH2n+2, find their main applications in energy production and as solvents and building blocks for more complex molecules in the chemical industry.1–3 Although the global energy sector is expected to experience gradual decarbonization in response to climate change, the size of the global market for hydrocarbon-based products (including alpha olefins, plastic precursors, aliphatic and aromatic solvents, and detergents) is expected to grow annually between 4 and 6 percent in the coming years.4,5 Alkanes are ideal for providing carbon skeletons to which other functional groups are added to produce more complex hydrocarbons, but such modifications first require to overcome large energy barriers to activate the inherently inert C(sp3)–H bonds. For this reason, their catalytic dehydrogenation provides an interesting strategy to increase the economic and chemical values of the otherwise unreactive alkanes.6,7 Among the most widely used catalysts are those based on platinum, due to its high selectivity towards C–H bond cleavage, usually in the form of alumina- or zeolite-supported platinum (nano)crystallites.5,8,9 However, the required temperatures and the continuous input of carbon-based reactants lead to catalyst poisoning by accumulation of carbonaceous species on the reactive sites, after which an expensive regeneration process is required to restore the original catalytic properties of platinum.Efforts have been made to investigate the on-surface alkane dehydrogenation processes on transition metals using theoretical calculations10–13 and techniques such as temperature-programmed desorption, infrared spectroscopy, or X-ray photoelectron spectroscopy.14–19 Such studies have made it possible to determine statistical and thermodynamic parameters of the dehydrogenation reactions on these substrates (reactivity, selectivity, or activation energy, among others). Most of them are based on theoretical simulations to study dehydrogenation since the experimental data at the atomic scale are relatively scarce in the literature. Moreover, the focus is generally placed on the dehydrogenation products of lighter alkanes (less than six carbon atoms), which require high temperatures for dehydrogenation, favoring the poisoning of the catalyst. Alternatively, for long unbranched alkanes (n-C44H90 and n-C32H66), scanning probe microscopy studies have been carried out on Au(110),20–22 Pt(110)20 and Cu(111),23 in which linear polymerization, C–C bond cracking and cascade dehydrogenation were respectively reported.
In this work, we investigate the dehydrogenation products from n-octane (n-C8H18) deposited on Pt(111) under ultra-high vacuum (UHV) conditions from an atomic-scale perspective. We take advantage of the high intensity and energy resolution of synchrotron radiation for the in situ characterization of the on-surface reactions by X-ray photoelectron spectroscopy (XPS). Temperature-programmed desorption (TPD) experiments enable us to follow the release of hydrogen from the system. Scanning tunneling microscopy (STM) images allow the morphological characterization of dehydrogenated products with submolecular resolution. The complementary information provided by the experimental data combined with ab initio calculations allows us to determine in detail the mechanism of dehydrogenation reactions and the corresponding energy barriers. Our results show that n-octane undergoes two initial stages of dehydrogenation on the Pt(111) surface. In the first one, a terminal C(sp3)–H bond is cleaved and a C–Pt bond is formed to stabilize the radical. In the second one, a double dehydrogenation occurs at the third and fourth C atoms, which, together with the diffusion and aggregation of adsorbates, results in the formation of carbon clusters that represents the first step towards catalyst poisoning.
2 Experimental and computational details
2.1 Experimental details
Our experiments were performed under UHV conditions with a base pressure of <2 × 10−10 mbar. Pt(111) crystals were cleaned by cycles of Ar+ sputtering at 1.5 kV followed by two temperature flashes at 1100 K. We made the first flash under an O2 atmosphere to remove carbon contamination and the second one under ultra-high vacuum conditions or a hydrogen atmosphere to remove residual chemisorbed oxygen. The surface was considered clean once the typical surface core level shift was observed in the Pt-4f7/2 spectrum24 or after direct inspection by STM. n-Octane molecules (Sigma-Aldrich, purity >99%) were introduced, without further purification, from an external quartz evaporator through a leak valve by direct evaporation at room temperature until the desired coverage was reached or until the surface became saturated (typically after 100 seconds at a pressure between 0.4 and 4 × 10−8 mbar), according to the C-1s total area in XPS spectra. The sample temperature was controlled in the range between 80 K and 1100 K using a liquid nitrogen cryostat combined with an annealing stage. The temperature was measured by means of a thermocouple welded to the back of the sample and a pyrometer (emissivity: ε = 0.1).We performed our synchrotron radiation XPS and TPD experiments at the SuperESCA beamline of the Elettra Synchrotron facility (Italy). High-resolution XPS spectra were acquired at normal emission with the sample held at 77 K. The spectra shown in this work correspond to the normalized sum of 30 sequential scans. The photon energy was set to 400 eV to maximize high surface sensitivity and photoionization cross-sections for both Pt-4f7/2 and C-1s core levels. We observed no beam-induced damage to the samples during spectra acquisition. For the analysis of the spectra, we subtracted a Shirley background and fitted each individual contribution with a Gaussian–Lorentzian sum that approximates a Voigt function. The free parameters for each peak were the binding energy, the area, the full width at half maximum (FWHM, typically about 400 meV), and the Gaussian–Lorentzian ratio. No further restrictions were imposed to the parameters, apart from requiring consistent values for all peaks in all spectra. For clarity, our XPS fits do not include vibrational profiles for each peak; rather we consider the vibrations later in the analysis of the intensities and for the assignment of each peak.
For TPD experiments (heating rate of 2 K s−1), the mass spectrometer head, equipped with a quartz glass cylinder with a diameter similar to that of the sample, was approached to the single crystal so that the background signal was minimized. The measurable masses ranged from 1 to 100 atomic mass units. We acquired several TPD spectra to ensure reproducibility. A custom population dynamics model was employed to fit the experimental TPD results. Alkane dehydrogenation and rehydrogenation processes and hydrogen recombination to yield molecular H2 were described with rate constants by following the Arrhenius equation. The pre-exponential factors and the activation energies were used as free parameters and H2 was assumed to desorb immediately after its formation.25 Simulated TPD spectra were convoluted with a Gaussian envelope (σ = 15 K) to account for the observed experimental broadening.
STM measurements were performed in an in-house UHV system after n-octane deposition on the Pt(111) substrate. n-Octane coverages were high enough to saturate the surface, typically after a dosage of 1–5 Langmuir. To reveal the different chemical species that appear on the surface upon thermal dehydrogenation, we carried out measurements after briefly annealing the samples to the corresponding temperatures and allowing them to cool to room temperature. Images were acquired in constant current mode with a chemically etched tungsten tip conditioned under UHV by standard procedures and post-processed using the WSxM software.26
2.2 Computational methods
We conducted density functional theory calculations to simulate the different processes at the atomic level. To this aim, we set up a periodic model for the alkane adsorbed on a 7 × 4 three-layered slab cut of Pt(111). The bottom layer was constrained to remain at bulk positions, while the other two layers were allowed to relax and adjust to the adsorption of the molecule. The calculations were performed with the code CASTEP,27 using a plane-wave basis and soft pseudopotentials with Koelling–Hamon relativistic corrections.28 The exchange and correlation energy was computed within the Perdew–Burke–Ernzerhof generalized-gradient approximation.29 Thresholds for converged geometry configurations are 10−6 eV for total energies, 10−2 eV Å−1 for residual forces, and 10−3 Å for the maximum displacement of atoms in the last three iterations. Energy barriers between stationary points were obtained by applying a quadratic complete Linear Synchronous Transit algorithm.30FIREBALL was employed to simulate the theoretical STM images based on the established interfacial geometries along the different dehydrogenation stages.31 FIREBALL uses a local-orbital formulation to achieve self-consistency on the orbital occupation numbers, calculated using the orthonormal Löwdin orbitals. In our calculations, we used a basis set of optimized single and double numerical atomic orbitals: sp3d5 for C and Pt and ss* for H. Tunneling currents for the STM images were computed using the Keldysh–Green function formalism, together with the first-principles tight-binding Hamiltonian obtained from the FIREBALL code, as explained in full detail elsewhere.32,33 Our STM theoretical simulation approach includes a detailed description of the electronic properties of both the W-tip and the sample simultaneously. All computed theoretical STM images were obtained under constant-current scanning conditions by moving the W-tip perpendicularly to the sample in each STM scanning step to search for a pre-selected fixed value of the tunnel current, mimicking the experimental procedure.
3 Results and discussion
In the first step, we characterized the on-surface thermal dehydrogenation of n-octane on Pt(111) by TPD and high-resolution XPS experiments. Fig. 1a shows the TPD evolution of molecular hydrogen (mass-to-charge ratio, m/z = 2) for a saturated layer of n-octane deposited at 77 K on Pt(111), measured over a wide temperature range (100–800 K). The curve displays two main hydrogen desorption features: an intense peak at 325 K, preceded by a small step-like feature at 220 K and followed by a shoulder at 390 K, and a less intense and broader peak at 600 K with a shoulder at 700 K. In the temperature range between 250 K and 270 K, partial desorption of n-octane occurs, as evidenced by the presence of alkane fragments in the mass spectrometer (m/z = 14, 15, 27, 28, 39, and 42, Fig. S5†) and by a decrease in the total intensity of the C-1s signal in XPS. On the other hand, molecular hydrogen desorption starts at temperatures as low as 220 K. Considering that the diffusion energy barrier of atomic hydrogen is much lower than that of recombination (around 0.058 eV for diffusion compared to 0.87 eV for desorption34), it can be assumed that all m/z = 2 contributions to the TPD spectra come from n-octane dehydrogenation followed by almost immediate recombination and desorption. | ||
| Fig. 1 (a) Temperature programmed desorption of molecular hydrogen (m/z = 2) obtained at a temperature ramp of 2 K s−1 after n-octane deposition at 77 K. The red line corresponds to a simulated TPD spectrum (see the text). Insets show the proposed structural models for the physisorbed and increasingly dehydrogenated states of n-octane on Pt(111), according to DFT calculations. (b)–(d) Deconvoluted high-resolution C-1s XPS spectra obtained at 77 K after flash-annealing to 290, 375 and 675 K, respectively. All spectra are normalized to their maximum for clarity, being their relative intensities 1, 0.89, and 0.70, respectively. | ||
According to the experimental data, three different chemical stages can be established in their corresponding temperature ranges, representing different degrees of dehydrogenation of the molecules. The temperature limits for each range were defined as the temperatures of the main maxima in the TPD spectrum (stage 1: 100 K–330 K; stage 2: 330 K–600 K; stage 3: 600 K–900 K, depicted in blue, yellow and green, respectively in Fig. 1a). We fit the TPD spectrum with an Arrhenius-based dynamic rate model (Fig. 1a red line). In this model, dehydrogenation reactions are governed by first-order dynamic equations in which the constants follow the Arrhenius equation, 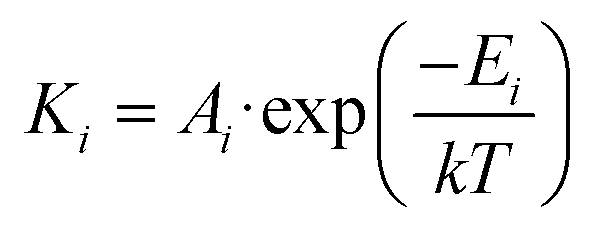 , where k corresponds to Boltzmann's constant, Ai corresponds to the pre-exponential factors, and Ei to the activation energies, of each reaction.14,25 Dehydrogenation populates the surface with atomic hydrogen, whose recombination and desorption yielding gas phase molecular hydrogen proceed as a second-order reaction. In our model, we assume that diffusion of atomic hydrogen is faster than recombination over the measured temperature range (for further details, see the ESI, section 1†). The good agreement of the model with the TPD experimental data demonstrates that the observed chemical changes can be correlated with well-defined activation energies that quantitatively compare with barriers obtained from DFT calculations of the most probable energy path (see the last paragraph of this section and Fig. 5).
, where k corresponds to Boltzmann's constant, Ai corresponds to the pre-exponential factors, and Ei to the activation energies, of each reaction.14,25 Dehydrogenation populates the surface with atomic hydrogen, whose recombination and desorption yielding gas phase molecular hydrogen proceed as a second-order reaction. In our model, we assume that diffusion of atomic hydrogen is faster than recombination over the measured temperature range (for further details, see the ESI, section 1†). The good agreement of the model with the TPD experimental data demonstrates that the observed chemical changes can be correlated with well-defined activation energies that quantitatively compare with barriers obtained from DFT calculations of the most probable energy path (see the last paragraph of this section and Fig. 5).
The chemical evolution with the temperature and the proposed division into three temperature ranges is also supported by temperature-resolved XPS (Fig. S3†). Fig. 1b–d show high-resolution XPS spectra obtained within each of the three temperature regions. Within each stage, the line shape of the XPS C-1s peak remains almost unchanged, while there are clear differences in the line shape between the different stages. Additionally, we introduce a detailed analysis of the XPS spectra by deconvolution into basic components. The stark differences evidence strong chemical changes in the molecules. In the next sections, we show that the combination of XPS, TPD, STM and DFT results allows us to determine the atomic-scale configuration of the three stages and assign them to (i) physisorption of intact n-octane molecules in the range of 100 K–330 K, (ii) dehydrogenation of a methyl termination in the range of 330 K–600 K, and (iii) fast dehydrogenation of two adjacent carbons, formation of a double bond and molecular aggregation, in the range of 600 K–900 K.
3.1 Physisorption of n-octane on Pt(111)
Fig. 2a and b present STM images of n-octane after deposition on Pt(111) at room temperature, characteristic of the first stage (100–330 K) identified in the TPD experiments. The images show straight, 1.15 ± 0.15 nm long features with an apparent height of h = 0.90 ± 0.10 Å, following the [1![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 0] high symmetry directions (see schematics in Fig. 2c and Fig. S9†). The measured dimensions and the morphology suggest that the observed features correspond to single, intact, non-dehydrogenated n-octane molecules adsorbed on the surface (Fig. 2d and e). No apparent morphological modifications (i.e., cis–trans isomerization or on-surface fragmentation) were observed. The surface density amounts to approximately 0.5 molecules per nm2. We notice that tip scanning can induce, in some cases, small displacement of the molecules around their adsorption positions but no tip- or thermal-induced diffusion was detected. The absence of self-assembled molecular islands suggests a repulsive lateral interaction between adsorbates.
0] high symmetry directions (see schematics in Fig. 2c and Fig. S9†). The measured dimensions and the morphology suggest that the observed features correspond to single, intact, non-dehydrogenated n-octane molecules adsorbed on the surface (Fig. 2d and e). No apparent morphological modifications (i.e., cis–trans isomerization or on-surface fragmentation) were observed. The surface density amounts to approximately 0.5 molecules per nm2. We notice that tip scanning can induce, in some cases, small displacement of the molecules around their adsorption positions but no tip- or thermal-induced diffusion was detected. The absence of self-assembled molecular islands suggests a repulsive lateral interaction between adsorbates.
 | ||
| Fig. 2 (a) and (b) STM images obtained at room temperature (Vbias = 0.82 V and I = 0.43 nA) after n-octane deposition at that temperature. Panel (b) corresponds to the dashed square area in panel (a). (c) Schematic representation of n-octane distribution on the Pt(111) surface. Different colors denote molecules following the three different crystallographic directions. (d)–(f) Schematic illustration, simulated and experimental STM images of a single physisorbed molecule. Simulations were conducted under the Keldysh–Green formalism. (g) Ball-and-stick model of the optimized physisorbed n-octane on Pt(111). | ||
To further understand the adsorption geometry in this temperature range, we performed DFT simulations of the minimum energy configuration of an n-octane molecule on a Pt(111) surface. The calculations showed that the molecules physisorb horizontally following the high-symmetry directions in an all-trans configuration with the plane defined by the carbon zig-zag skeleton tilted 14° with respect to the platinum surface. Such a result slightly differs from the common assumption that n-alkane molecules lie flat on the (111) surface, with the zig-zag skeleton parallel to the surface plane,35,36 and showcases the need for advanced calculations for interpreting the atomistic structures of small hydrocarbons on surfaces. The DFT-based STM simulations are in good agreement with the experimental observations (Fig. 2f). In this tilted physisorbed configuration, one of the two H atoms of each methylene group lies closer to the surface while the other protrudes to the vacuum side. In our DFT simulations, we observed neither significant bond deformation nor torsion in agreement with previously reported infrared (IR) experiments.35,37,38
High-resolution XPS confirms this physisorption pattern. Spectra taken at 290 K (Fig. 1b) can be deconvoluted into four different contributions at 283.10 eV, 283.50 eV, 283.95 eV and 284.35 eV, which can be interpreted in terms of a weak interaction of n-octane with Pt(111). n-Octane has two types of non-equivalent carbon atoms: two carbon atoms at the methyl-ends (–CH3) and six internal carbon atoms (–CH2–).39 In addition, the XPS spectra of hydrocarbons show a characteristic vibrational profile due to the simultaneous excitation of quantum vibrational states of the core-excited state (i.e. the final state).40–46 This vibrational excitation produces a loss of kinetic energy of the photoelectrons, giving rise to satellite peaks at higher binding energies. The main excited vibrational mode is the C–H stretching mode (2950 cm−1 ≈ 0.36 eV). Therefore, each C-1s carbon peak is expected to have at least one vibrational replica at higher energies (around +0.36 eV). Taking this into account, we can assign the highest binding energy peak (284.30 eV, blue dashed curve in Fig. 1b) to a vibrational replica of the most intense peak at 283.95 eV (blue curve), the latter assigned to the most abundant internal –CH2– carbon atoms. Similarly, up to 25% of the intensity at this energy (283.95 eV) is caused by the vibrational replica of the peak at 283.50 eV (ref. 40) (green curve), which is assigned to the methyl carbon atoms at the end of the chains (see section 2 of the ESI† for further details). Finally, we assigned the peak at 283.10 eV (and a small part of that at 283.50 eV due to vibrations) to the remnants of a denser monolayer phase, detectable at lower temperatures, that desorbs mainly between 240 and 280 K (see Fig. S3†).
3.2 First dehydrogenation stage
After annealing n-octane at 400 K for 100 seconds, the morphology of the adsorbates changes, as can be seen in the constant current STM images shown in Fig. 3a and b – characteristic of the second stage (330–600 K) identified in the TPD experiments. According to these images, the molecules display a bright protrusion (h = 1.55 ± 0.05 Å) at one of their ends and a tail that retains the aspect and height of physisorbed n-octane. A slight increase of the length (L = 1.35 ± 0.08 nm) was also observed with respect to the physisorbed state due to the presence of the protrusion. In this state, molecules are stable and no diffusion or displacement can be induced by manipulation with the tip. Similar results were obtained for deposition at 520 K (Fig. S10†). A comparison between experimental and simulated STM images allows us to identify the new products as singly dehydrogenated n-octane molecules forming a chemical bond (i.e., chemisorbed) with the platinum surface. Dehydrogenation occurs at one of the methyl ends of the molecules, yielding a Pt-bonded n-octyl group. This C–Pt strong covalent bond prevents the thermally-induced desorption of the products. Although in DFT calculations, the carbon skeleton remains aligned with the high symmetry direction, simulated STM images show the molecules as elongated features with their axes tilted by 21° ± 1° with respect to the main crystallographic directions. According to our DFT results (Fig. 3g), a deformation of the carbon skeleton towards the surface takes place at one end of the molecule as a result of the C–Pt bond formation, which introduces an asymmetry in simulated STM images (Fig. 3e). However, the non-dehydrogenated end of the molecule stays in a tilted-on configuration at a distance very similar to that of the physisorbed case.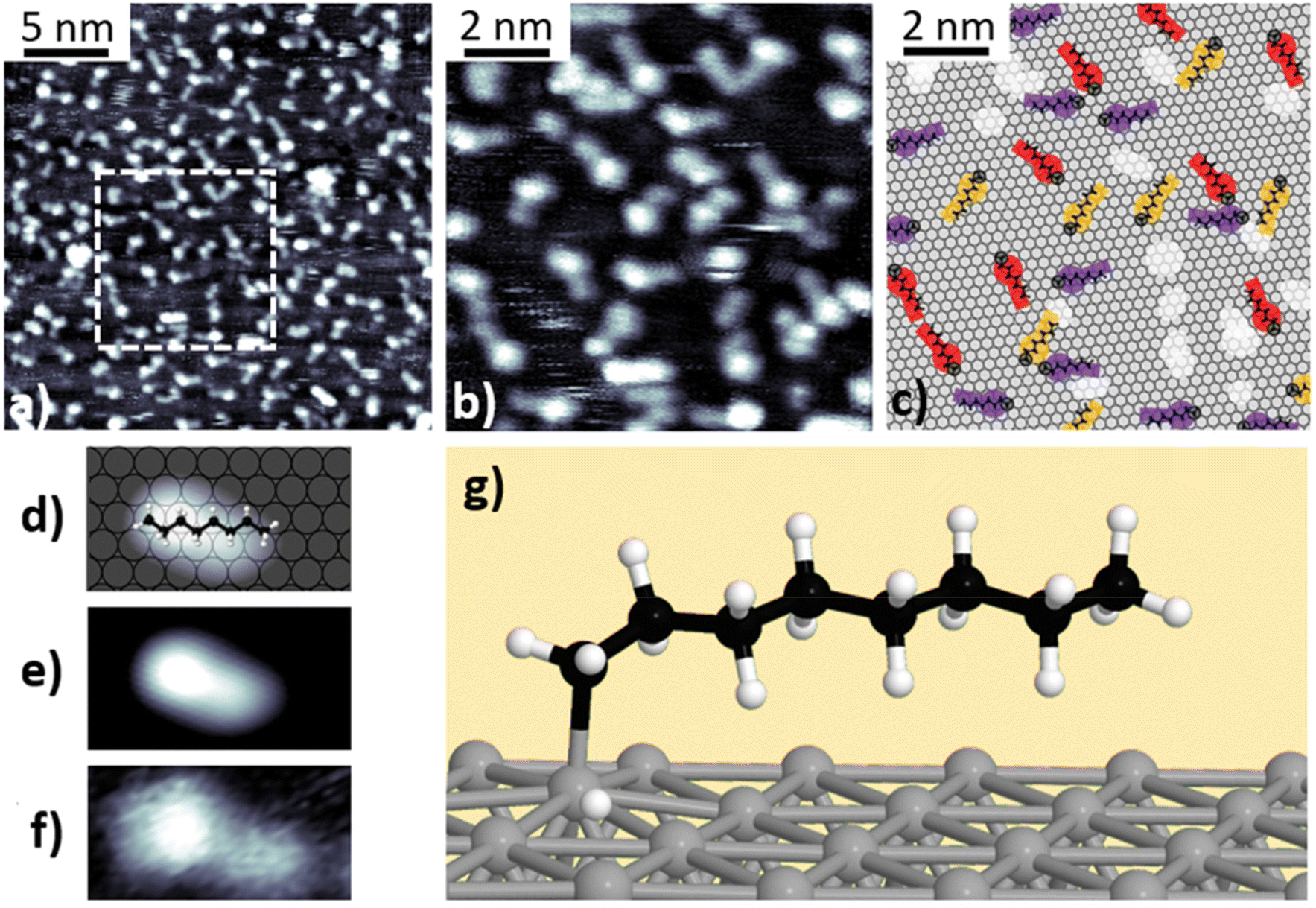 | ||
| Fig. 3 (a) and (b) STM images obtained at room temperature (Vbias = 0.75 V and 0.83 V and I = 0.43 nA) after n-octane deposition at 400 K. Panel (b) corresponds to the dashed square area in (a). (c) Proposed schematic model for image (b). Pt atoms bonding to the molecules are marked with a dark circle. Colors indicate molecules following different crystallographic directions. (d), (e) and (f) respectively. Schematic illustration, simulated and experimental STM images of a single chemisorbed molecule. Simulations were conducted under the Keldysh–Green formalism. (g) Ball-and-stick model of the optimized singly dehydrogenated chemisorbed n-octane on Pt(111). | ||
Fig. 1c shows the high-resolution C-1s XPS spectrum of a dehydrogenated sample after flash-annealing at 375 K. In addition to the previously mentioned contributions, a new narrow component appears at 283.7 eV (grey curve in Fig. 1c). The fitted FWHM of the new component is 0.3 eV, which is narrower than the rest of the components (around 0.45 eV). We assign this component to the C–Pt bond at the methyl end of the chemisorbed molecules.47 Compared to the C-1s spectrum of physisorbed molecules, the intensity of the component associated with the methyl end (283.5 eV, green peak) is halved, leaving the ratio between the C–Pt bond and the methyl components at approximately 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. In addition, a small shift of 0.1 eV towards lower binding energies was observed upon chemisorption. The peaks associated with the internal carbon atoms (283.95 and 284.35 eV, solid and dashed blue curves in Fig. 1c) underwent minimal intensity changes as expected for the unaltered methylene-based backbone. The contribution of the component associated with the low-temperature layer remnants (283.10 eV) becomes negligible because desorption of this weakly bonded species is more efficient at higher temperatures. The almost complete disappearance of this component explains the 10% decrease in the global intensity of the C-1s XPS signal with respect to the sample annealed at 290 K.
:1. In addition, a small shift of 0.1 eV towards lower binding energies was observed upon chemisorption. The peaks associated with the internal carbon atoms (283.95 and 284.35 eV, solid and dashed blue curves in Fig. 1c) underwent minimal intensity changes as expected for the unaltered methylene-based backbone. The contribution of the component associated with the low-temperature layer remnants (283.10 eV) becomes negligible because desorption of this weakly bonded species is more efficient at higher temperatures. The almost complete disappearance of this component explains the 10% decrease in the global intensity of the C-1s XPS signal with respect to the sample annealed at 290 K.
3.3 Second dehydrogenation stage
Annealing above 600 K results in a second dehydrogenation process, corresponding to the third stage (600–800 K) identified in the TPD experiments. In Fig. 4, we present the results for a submonolayer n-C8H18/Pt(111) sample after thermal treatment at 675 K for 10 seconds. In the STM images shown in Fig. 4a and b, the surface appears covered by a collection of lobed structures of different shapes and dimensions, with an apparent height between 1.8 Å and 3.2 Å. In some cases, the dimensions of these structures exceed those of a single n-octane molecule, and their shapes are far from the rod-like appearance of intact molecules. This suggests that they are composed of molecular aggregates (as illustrated in Fig. 4c), indicating that on-surface diffusion and intermolecular interactions increase at temperatures above 520 K (see Fig. S10†). Since alkanes are saturated molecules, C–C bond formation between them is inefficient under normal conditions. Therefore, we propose that the appearance of intramolecular unsaturated C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds makes the interaction possible between molecules and the subsequent aggregation of some of them.
C bonds makes the interaction possible between molecules and the subsequent aggregation of some of them.
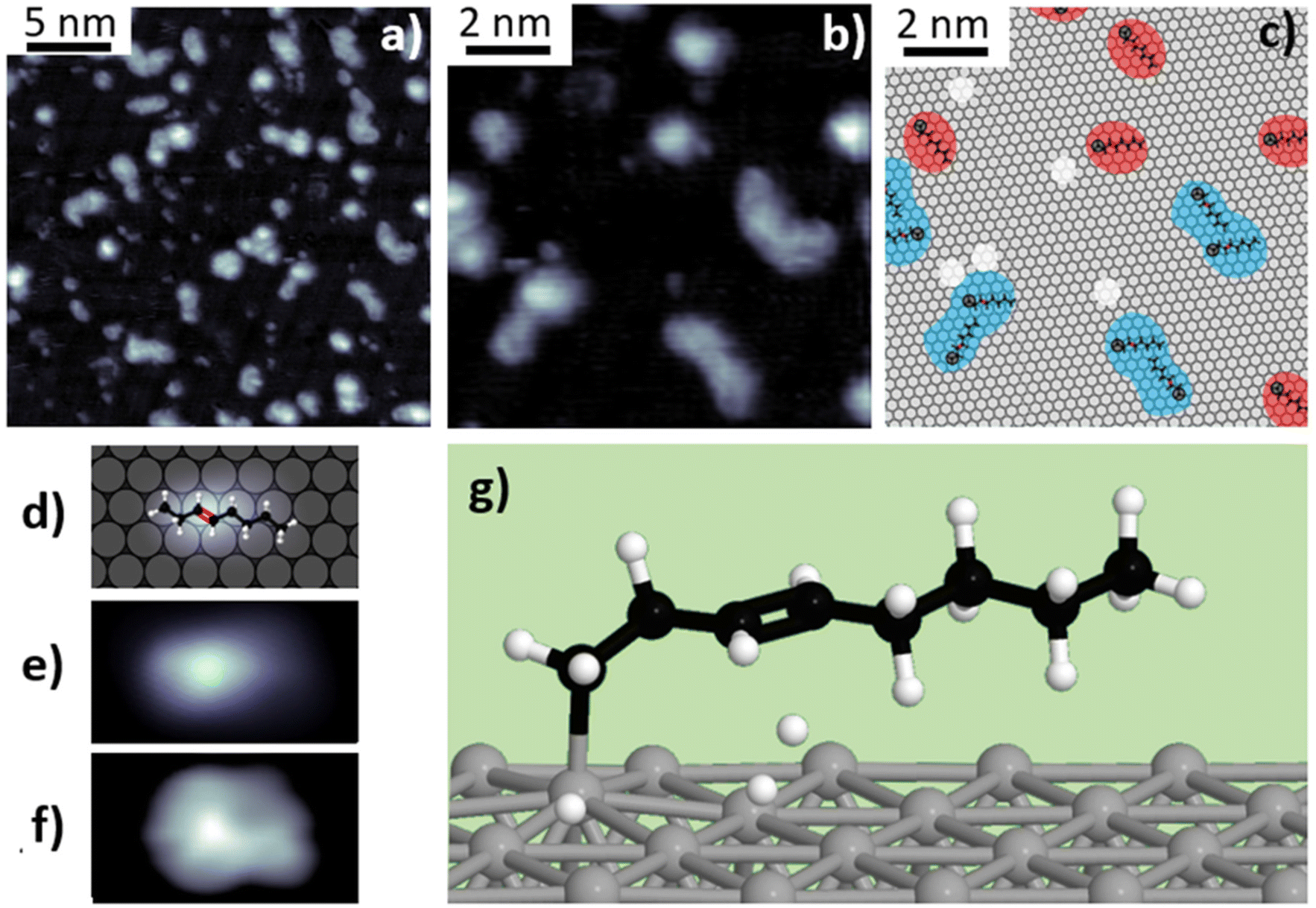 | ||
| Fig. 4 (a) and (b) STM images obtained at room temperature (Vbias = 0.52 V, I = 0.63 nA) after n-octane deposition at 675 K. (c) Proposed schematic model representing features in panel (b). Each color indicates different types of structures. DFT models of the triply dehydrogenated molecules are superimposed. Features formed by a molecule are displayed in red and those formed by two or more are marked in blue. (d)–(f) Schematic illustration, simulated and experimental STM images of a triply dehydrogenated molecule. Simulations were conducted under the Keldysh–Green formalism. (g) Ball-and-stick model of the optimized triply dehydrogenated chemisorbed n-octane on Pt(111). | ||
To determine the nature of the structures observed in the STM images, we performed an analysis of the high-resolution XPS data. The C-1s XPS spectrum obtained after thermal treatment at 675 K (Fig. 1d) reveals the changes in the relative intensities of the different components. The contributions assigned to the methylene groups (–CH2–), at 283.95 eV and 284.35 eV (solid and dashed blue peaks in Fig. 1d), decreased by 47%, while the components at 283.4 eV and 283.7 eV (light green and grey curves) increased by 53% and 25%, respectively. We assigned the latter component to the C–Pt bonds, whose increase implies a larger interaction between carbon atoms and platinum, hindering adsorbate desorption even at temperatures higher than 675 K (see Fig. S3 and S5†). The increase of this component suggests that further dehydrogenation is taking place, which would result in the formation of more C–Pt bonds and eventually lead to possible structural changes of n-octane, such as fragmentation or branching of the molecule. Regarding the component at 283.4 eV, it has been reported that the XPS contribution corresponding to ethylene di-σ-bonded to the surface appears at this particular energy,48 so this component probably arises from σ-related interaction between molecules containing CC and the Pt surface. DFT simulations show that a double dehydrogenation at the third and fourth carbon atoms is energetically favored, explaining the decrease in the intensity of the XPS peak assigned to –CH2– carbon atoms. A total intensity reduction of 20% was observed in the overall XPS C-1s signal with respect to the sample treated at 375 K, most likely due to the direct desorption of some chemisorbed molecules.
3.4 Dehydrogenation energy reaction path
To fully understand the processes involved in the catalytic dehydrogenation of n-octane, we have calculated the most probable reaction path for an energetically favored sequential dehydrogenation of this molecule on Pt(111), as shown in Fig. 5. The energy of the physisorbed state is taken as the reference (Eref), and the effective energy path (Fig. 5c) is defined as ΔE = EC − Eref − n·EH.23,49 In this equation, EC corresponds to the calculated energy for each configuration along the reaction path, n is the number of hydrogen atoms cleaved from the molecule in previous steps, and EH is the adsorption energy of a single hydrogen atom. EH is estimated to be half the energy of the recombinative desorption of two hydrogen atoms (EH = −0.46 ± 0.09 eV,25,50–52 further details in the ESI, section 6†).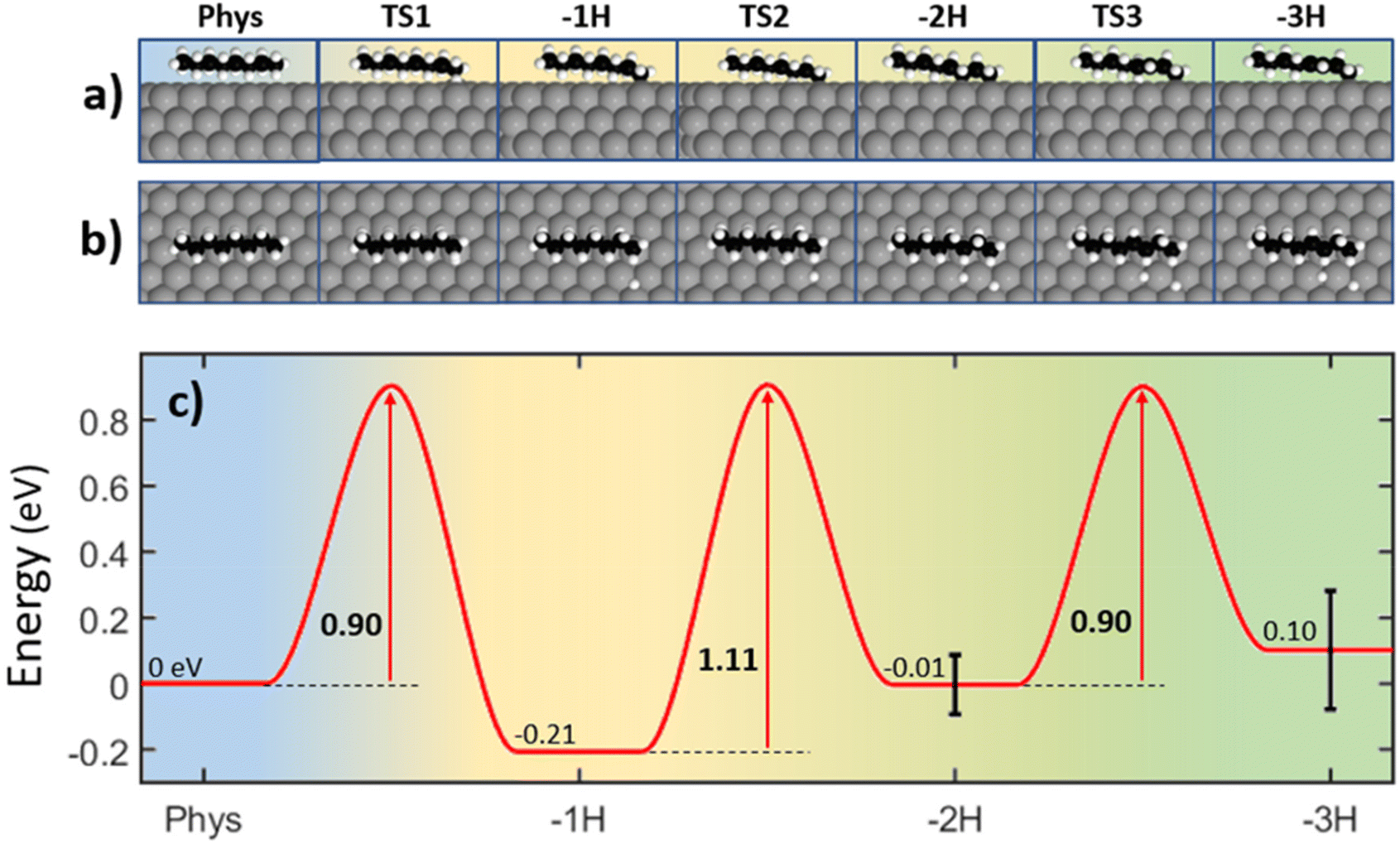 | ||
| Fig. 5 (a) and (b) Lateral and top views of the different dehydrogenation stages of n-octane on Pt(111). (c) Energy profile for the sequential dehydrogenation reactions of n-octane as obtained in our calculations. In each step, the adsorption energy of hydrogen atoms produced in previous stages (EH = −0.46 ± 0.09 eV per H atom) has been subtracted. | ||
The first dehydrogenation step involves the exothermic activation of a methyl-end C–H bond with an energy barrier of 0.90 eV. The second dehydrogenation occurs at the third carbon atom in an endothermic reaction presenting a barrier of 1.11 eV followed by an endothermic C–H bond cleavage at the fourth carbon atom yielding a double bond between the third and fourth carbon atoms, with an energy barrier of 0.90 eV. The energy barriers of the effective dehydrogenation processes (Fig. 5c) are in good agreement with the activation energies extracted from the measured TPD spectrum (Fig. 1a). The activation energy for the main peak in TPD appearing at 320 K is 0.85 eV and the one associated to the shoulder appearing at 380 K is 1.06 eV (see Table S1†), compared with that of the calculated first transition state (TS1) of 0.9 eV. We attribute the main peak to molecules adsorbed on the terraces, while the shoulder would correspond to molecules adsorbed on defective surface regions (e.g., steps). At temperatures above 550 K, dehydrogenation continues. The second peak in the TPD spectrum at 600 K requires an activation energy of 1.74 eV and the corresponding shoulder of 2.02 eV. These values are consistent with simultaneous dehydrogenation at the third and fourth carbon atoms leaving two adsorbed hydrogen atoms, for which the activation energy can be estimated as the addition of the barriers calculated for the second and third dehydrogenation. The total intensity of the TPD contributions at 600 K and 700 K is just a third of that of the first peak, implying that not all the molecules doubly dehydrogenate following this path. Therefore, isomerization, cyclization, and aggregation processes without hydrogen desorption or desorption of hydrocarbon fragments may also contribute to the morphological changes observed by STM (Fig. 4a and b). We propose that the formation of unsaturated bonds, such as the double bonds formed in the second dehydrogenation stage above 600 K, is instrumental in cluster formation and further adsorbate aggregation, representing a preliminary step for the catalytic poisoning of the surface and coke formation.
4 Conclusion
We have studied the thermal dehydrogenation of n-octane on Pt(111) using nanoscopic, spectroscopic and theoretical methods that complement themselves. STM experiments combined with simulated STM images allow us to identify the chemical structure of the on-surface dehydrogenated products. The results are supported by XPS and TPD spectra. At near room temperature, physisorbed n-octane forms a disordered phase in which molecules align along the main crystallographic directions. Above 300 K, it dehydrogenates at one methyl end, yielding an n-octyl group bonded to a platinum atom with high efficiency and regioselectivity. Above 550 K, subsequent dehydrogenation and diffusion of the adsorbates lead to unsaturated species containing CC bonds that tend to cluster. We have modeled the TPD spectrum using population dynamics equations, which is in good agreement with the experimental activation energies and the energy barriers calculated by DFT. Our investigations of surface-catalyzed alkane dehydrogenation provide new insights into the atomistic mechanisms of such industrially relevant reactions.
Author contributions
D. Arribas – investigation, data acquisition, formal analysis, preparation and editing of the manuscript. V. Villalobos-Vilda, E. Tosi, P. Lacovig, A. Baraldi, L. Bignardi, and S. Lizzit – data acquisition at Elettra Synchrotron. J. I. Martínez and P. de Andres – theoretical calculations. A. Gutierrez, J. A. Martín-Gago, and P. Merino – conceptualization, supervision, project administration, and funding acquisition. All authors have discussed and approved the manuscript.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
The authors acknowledge financial support from projects PID2021-12509OA-I00, PID2020-113142RB-C21, TED2021-129999B-C31, TED2021-129416A-I00, RYC2020-029800-I and EUR2021-122006 funded by MCIN/AEI/10.13039/501100011033. D. Arribas acknowledges the Spanish Ministry of Universities, FPU-2019 predoctoral grant (FPU19/04556).References
- B. V. Vora, Development of dehydrogenation catalysts and processes, Top. Catal., 2012, 55, 1297–1308 CrossRef CAS.
- A. Deneyer, et al., Alkane production from biomass: Chemo-, bio- and integrated catalytic approaches, Curr. Opin. Chem. Biol., 2015, 29, 40–48 CrossRef CAS PubMed.
- I. Amghizar, L. A. Vandewalle, K. M. Van Geem and G. B. Marin, New Trends in Olefin Production, Engineering, 2017, 3, 171–178 CrossRef CAS.
- Global Market Insights ( https://www.gminsights.com/industry-analysis/) Last consulted: 13/01/2023 Report IDs: GMI1664, GMI2131, GMI2182, GMI1794, GMI4892, GMI2049, GMI2216, GMI1516, and GMI761, G. Global Market Insights reports.
- J. J. H. B. Sattler, J. Ruiz-Martinez, E. Santillan-Jimenez and B. M. Weckhuysen, Catalytic dehydrogenation of light alkanes on metals and metal oxides, Chem. Rev., 2014, 114, 10613–10653 CrossRef CAS PubMed.
- K. Griesbaum, et al., Hydrocarbons, Ullmann's Encycl. Ind. Chem., 2012, 18, 133–189 Search PubMed.
- D. Sanfilippo and P. N. Rylander, Hydrogenation and Dehydrogenation, Ullmann's Encycl. Ind. Chem., 2012, 18, 451–471 Search PubMed.
- Z. Nawaz, Light alkane dehydrogenation to light olefin technologies: A comprehensive review, Rev. Chem. Eng., 2015, 31, 413–436 CAS.
- Y. Zhang, W. Yao, H. Fang, A. Hu and Z. Huang, Catalytic alkane dehydrogenations, Sci. Bull., 2015, 60, 1316–1331 CrossRef CAS.
- Z. J. Zhao, C. C. Chiu and J. Gong, Molecular understandings on the activation of light hydrocarbons over heterogeneous catalysts, Chem. Sci., 2015, 6, 4403–4425 RSC.
- G. Sun, J. T. Fuller, A. N. Alexandrova and P. Sautet, Global Activity Search Uncovers Reaction Induced Concomitant Catalyst Restructuring for Alkane Dissociation on Model Pt Catalysts, ACS Catal., 2021, 11, 1877–1885 CrossRef CAS.
- X. Ding, et al., Adsorption and dehydrogenation of C2-C6n-alkanes over a Pt catalyst: A theoretical study on the size effects of alkane molecules and Pt substrates, Phys. Chem. Chem. Phys., 2020, 22, 21835–21843 RSC.
- P. Raybaud, et al., From γ-alumina to supported platinum nanoclusters in reforming conditions: 10 years of DFT modeling and beyond, J. Catal., 2013, 308, 328–340 CrossRef CAS.
- S. L. Tait, Z. Dohnálek, C. T. Campbell and B. D. Kay, N-alkanes on Pt(111) and on C (0001) Pt (111): Chain length dependence of kinetic desorption parameters, J. Chem. Phys., 2006, 125, 234308 CrossRef PubMed.
- J. S. Jarvis, et al., Inhibiting the Dealkylation of Basic Arenes during n-Alkane Direct Aromatization Reactions and Understanding the C6Ring Closure Mechanism, ACS Catal., 2020, 10, 8428–8443 CrossRef CAS.
- W. L. Manner, G. S. Girolami and R. G. Nuzzo, The C-H⋯interaction and reactivity differences of n-octane on the (1 × 1) and (5 × 20) Surfaces of Pt(100), Langmuir, 1998, 14, 1716–1724 CrossRef CAS.
- J. F. Weaver, M. A. Krzyzowski and R. J. Madix, Direct dissociative chemisorption of alkanes on Pt(111): Influence of molecular complexity, J. Chem. Phys., 2000, 112, 396–407 CrossRef CAS.
- P. Ye and A. J. Gellman, Transition state for alkyl group hydrogenation on Pt(111), J. Am. Chem. Soc., 2008, 130, 8518–8526 CrossRef CAS PubMed.
- C. R. Arumainayagam, G. R. Schoofs, M. C. McMaster and R. J. Madix, Dynamics of molecular adsorption of ethane with Pt(111): A supersonic molecular beam study, J. Phys. Chem., 1991, 95, 1041–1047 CrossRef CAS.
- Z. Cai, et al., Linear alkane C-C bond chemistry mediated by metal surfaces, ChemPhysChem, 2015, 16, 1356–1360 CrossRef CAS PubMed.
- Z. Cai, et al., Linear Alkane Polymerization on Au-Covered Ag(110) Surfaces, J. Phys. Chem. C, 2018, 122, 24209–24214 CrossRef CAS.
- D. Zhong, et al., Linear alkane polymerization on a gold surface, Science, 2011, 334, 213–216 CrossRef CAS PubMed.
- X. Li, et al., Direct transformation of n-alkane into all-trans conjugated polyene via cascade dehydrogenation, Natl. Sci. Rev., 2021, 8(10), nwab093 CrossRef PubMed.
- A. Baraldi, Structure and chemical reactivity of transition metal surfaces as probed by synchrotron radiation core level photoelectron spectroscopy, J. Phys.: Condens. Matter, 2008, 20, 093001 CrossRef.
- B. Poelsema, K. Lenz and G. Comsa, The dissociative adsorption of hydrogen on Pt(111): Actuation and acceleration by atomic defects, J. Chem. Phys., 2011, 134(7), 074703 CrossRef PubMed.
- I. Horcas, et al., WSXM: A software for scanning probe microscopy and a tool for nanotechnology, Rev. Sci. Instrum., 2007, 78, 013705 CrossRef CAS PubMed.
- S. J. Clark, et al., First principles methods using CASTEP, Z. Kristallogr., 2005, 220, 567–570 CAS.
- D. D. Koelling and B. N. Harmon, A technique for relativistic spin-polarised calculations, J. Phys. C: Solid State Phys., 1977, 10, 3107–3114 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized gradient approximation made simple, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- T. A. Halgren and W. N. Lipscomb, The synchronous-transit method for determining reaction pathways and locating molecular transition states, Chem. Phys. Lett., 1977, 49, 225–232 CrossRef CAS.
- J. P. Lewis, et al., Advances and applications in the FIREBALL ab initio tight-binding molecular-dynamics formalism, Phys. Status Solidi B, 2011, 248, 1989–2007 CrossRef CAS.
- J. M. Blanco, F. Flores and R. Pérez, STM-theory: Image potential, chemistry and surface relaxation, Prog. Surf. Sci., 2006, 81, 403–443 CrossRef CAS.
- J. M. Blanco, et al., First-principles simulations of STM images: From tunneling to the contact regime, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 70, 1–9 CrossRef.
- D. Borodin, et al., Quantum effects in thermal reaction rates at metal surfaces, Science, 2022, 377, 394–398 CrossRef CAS PubMed.
- O. Endo, M. Nakamura, K. Amemiya and H. Ozaki, Thermal dehydrogenation of n-alkane on Au(111) and Pt(111) surface, Surf. Sci., 2019, 681, 32–37 CrossRef CAS.
- H. M. Zhang, Z. X. Xie, B. W. Mao and X. Xu, Self-Assembly of Normal Alkanes on the Au (111) Surfaces, Chem. – Eur. J., 2004, 10, 1415–1422 CrossRef CAS PubMed.
- A. R. Bishop, G. S. Girolami and R. G. Nuzzo, Structural Models and Thermal Desorption Energetics for Multilayer Assemblies of the n-Alkanes on Pt(111), J. Phys. Chem. B, 2000, 104, 754–763 CrossRef CAS.
- M. Yang and G. A. Somorjai, Adsorption and reactions of C6 hydrocarbons at high pressures on Pt(111) single-crystal surfaces studied by sum frequency generation vibrational spectroscopy: Mechanisms of isomerization and dehydrocyclization of n-hexane, J. Am. Chem. Soc., 2004, 126, 7698–7708 CrossRef CAS PubMed.
- K. Weiss, et al., XPS and XAS investigation of condensed and adsorbed n-octane on a Cu(1 1 0) surface, J. Electron Spectrosc. Relat. Phenom., 2003, 128, 179–191 CrossRef CAS.
- L. J. Sæthre, et al., High-resolution C 1s photoelectron spectra of methane, ethene, propene, and 2-methylpropene, Phys. Rev. A: At., Mol., Opt. Phys., 1997, 55, 2748–2756 CrossRef.
- L. J. Sæthre, T. D. Thomas and S. Svensson, Markovnikov addition to alkenes. A different view from core-electron spectroscopy and theory, J. Chem. Soc., Perkin Trans. 2, 1997, 749–755, 10.1039/a606562g.
- T. D. Thomas, L. J. Saethre, S. L. Sorensen and S. Svensson, Vibrational structure in the carbon 1s ionization of hydrocarbons: Calculation using electronic structure theory and the equivalent-cores approximation, J. Chem. Phys., 1998, 109, 1041–1051 CrossRef CAS.
- T. Karlsen, et al., Toward the spectrum of free polyethylene: Linear alkanes studied by carbon 1s photoelectron spectroscopy and theory, J. Am. Chem. Soc., 2002, 124, 7866–7873 CrossRef CAS PubMed.
- V. Myrseth, J. D. Bozek, E. Kukk, L. J. Sathre and T. D. Thomas, Adiabatic and vertical carbon 1s ionization energies in representative small molecules, J. Electron Spectrosc. Relat. Phenom., 2002, 122, 57–63 CrossRef CAS.
- Z. Zou, et al., In Situ Observation of C–C Coupling and Step Poisoning during the Growth of Hydrocarbon Chains on Ni(111), Angew. Chem., Int. Ed., 2023, 62, e2022132 Search PubMed.
- H. Tetlow, et al., Ethylene decomposition on Ir(111): Initial path to graphene formation, Phys. Chem. Chem. Phys., 2016, 18, 27897–27909 RSC.
- A. Pinardi, Tailoring on-surface chemistry of (hetero) aromatics on transition metal surfaces, Thesis, 2013 Search PubMed.
- N. Freyer, G. Pirug and H. P. Bonzel, C(1s) spectroscopy of hydrocarbons adsorbed on Pt(111), Surf. Sci. Lett., 1983, 126, A118 CrossRef.
- J. Björk, S. Stafström and F. Hanke, Zipping up: Cooperativity drives the synthesis of graphene nanoribbons, J. Am. Chem. Soc., 2011, 133, 14884–14887 CrossRef PubMed.
- C. Yu, et al., H2 thermal desorption spectra on Pt(111): A density functional theory and kinetic Monte Carlo simulation study, Catalysts, 2018, 8, 26–28 CrossRef.
- S. Saerens, et al., The Positive Role of Hydrogen on the Dehydrogenation of Propane on Pt(111), ACS Catal., 2017, 7, 7495–7508 CrossRef CAS.
- Z. Lian, et al., Revealing the Janus Character of the Coke Precursor in the Propane Direct Dehydrogenation on Pt Catalysts from a kMC Simulation, ACS Catal., 2018, 8, 4694–4704 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3nr02564k |
| This journal is © The Royal Society of Chemistry 2023 |