
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBoosting the catalytic performance of graphene-supported Pt nanoparticles via decorating with –SnBun: an efficient approach for aqueous hydrogenation of biomass-derived compounds†
Adrián
García-Zaragoza
 a,
Christian
Cerezo-Navarrete
*a,
Pascual
Oña-Burgos
a and
Luis M.
Martínez-Prieto
*ab
a,
Christian
Cerezo-Navarrete
*a,
Pascual
Oña-Burgos
a and
Luis M.
Martínez-Prieto
*ab
aITQ, Instituto de Tecnología Química, Universitat Politècnica de València (UPV), Av. de los Naranjos S/N 46022, Valencia, España. E-mail: chcena@itq.upv.es; luismiguel.martinez@csic.es
bIIQ, Instituto de Investigaciones Químicas (CSIC-Universidad de Sevilla), Departamento de Química Inorgánica, Avda. Americo Vespucio 49, 41092, Sevilla, España
First published on 29th June 2023
Abstract
The pursuit of new catalysts for the aqueous transformation of biomass-derived compounds under mild conditions is an active area of research. In the present work, the selective hydrogenation of 5-hydroxymethylfurfural (HMF) to 2,5-bishydroxymethylfuran (BHMF) was efficiently accomplished in water at 25 °C and 5 bar H2 pressure (after 1 h full conversion and 100% selectivity). For this, a novel nanocatalyst based on graphene-supported Pt NPs decorated with Sn-butyl fragments (–SnBun) has been used. More specifically, Pt NPs supported on reduced graphene oxide (rGO) were functionalized with different equivalents (0.2, 0.5, 0.8 and 1 equiv.) of tributyltin hydride (Bu3SnH) following a surface organometallic chemistry (SOMC) approach. The synthesized catalysts (Pt@rGO/Snx) were fully characterized by state-of-the-art techniques, confirming the presence of Sn-butyl fragments grafted on the platinum surface. The higher the amount of surface –SnBun, the higher the activity of the catalyst, reaching a maximum conversion with Pt@rGO/Sn0.8. Indeed, the latter has proven to be one of the most active catalysts reported to date for the aqueous hydrogenation of HMF to BHMF (estimated TOF = 666.7 h−1). Furthermore, Pt@rGO/Sn0.8 has been demonstrated to be an efficient catalyst for the reduction of other biomass-derived compounds in water, such as furfural, vanillin or levoglucosenone. Here, the catalytic activity is remarkably boosted by Sn-butyl fragments located on the platinum surface, giving a catalyst several times faster than non-functionalized Pt@rGO.
Introduction
From its first conception, the use of metal nanoparticles (MNPs) in catalysis has attracted the attention of the scientific community as they combine the main advantages of heterogeneous (i.e. recyclability and stability) and homogeneous (i.e. high activity) catalysts.1 Due to their small size, high active surface area and particular electronic configuration, MNPs are highly active catalysts.2 However, the large number of surface active sites of different nature makes it difficult to precisely control the selectivity of MNPs in catalysis.3 Therefore, one of the major challenges in catalysis with MNPs is to control their selectivity while maintaining the activity. An interesting strategy to modify the MNP reactivity is through the functionalization of the materials used as supports (i.e. silicas, metal oxides or graphene materials).4 The introduction of new active sites that can work in a collaborative way with MNPs is an efficient way to modulate their catalytic properties. For example, in a recent study it was observed that the inclusion of nitrogen groups into reduced graphene oxide (NH2-rGO) increases the activity and selectivity of graphene-supported MNPs in the hydrogenation of fatty acids to alcohols.5 Another effective way to control the catalytic behaviour of MNPs is the incorporation of a second metal,6 since it modifies the electronic and geometric characteristics of MNPs.7 Among the many examples described in the literature,8 our group has recently studied how the metal composition of bimetallic PtRu alloy-type nanoparticles (NPs) considerably influences their reactivity in hydrogenation reactions.9 A further possible way to modify the electronic and steric properties of MNPs is through surface ligands. As well as in organometallic complexes, surface ligands are able to modulate MNP catalytic properties.10 However, since these coordinating ligands partially block surface active sites, it is necessary to find a compromise between the number of surface ligands and the reactivity of the catalyst. In any case, the functionalization of supported MNPs with organic molecules has been demonstrated to be an excellent tool to control their activity/selectivity.11 For example, Pieters et al. reported a notable enhancement in the selectivity of carbon-supported Ru NPs in H/D isotopic exchange reactions after their functionalization with N-heterocyclic carbene (NHC) ligands.12 Interestingly, surface modifications not only produce an improvement in selectivity, but also are able to increase the MNP stability, as was recently reported on graphene-supported Ru NPs functionalized with pyrene-tagged NHC ligands.13Controlling the formation of well-defined active sites by surface organometallic chemistry (SOMC) is a well-studied technique since the early 80s.14 SOMC is based on generating well-defined surface species by grafting with an organometallic complex the surface of oxides or metals.15 In this way, it is possible to precisely control the density and coordination environment of metal centers and produce heterogeneous catalysts with well-defined catalytically active sites.16 These metal centers can be further modified through subsequent treatments aiming for higher activity, selectivity and robustness. The SOMC methodology has been used in a wide range of catalytic reactions, such as alkane or olefin metathesis,17 epoxidation of alkenes,18 Baeyer–Villiger oxidation,19 dehydrogenation of propane,20 and many others.21 For example, Basset et al. have efficiently employed bimetallic Pt–Sn NPs obtained by SOMC for the selective dehydrogenation of propane.22 The great activity and selectivity of this bimetallic catalyst are mainly due to two promoting effects of tin: (i) electron transfer to the Pt 5d band that generates electron-deficient Sn atoms close enough to electron-rich Pt atoms and makes the surface of the nanoparticles more reactive;23 and (ii) a geometric effect produced by Sn species at the platinum surface that isolates Pt atoms into small ensembles and prevents non-desired side reactions.24
From a sustainable point of view, the production of fine-chemicals from biomass-derived products through catalytic transformations is essential for the future of our planet.25 Among the many platform molecules, 5-hydroxymethylfurfural (HMF) is identified as one of the most important due to its easy obtention by thermal or chemical treatment of biomass.26 HMF is composed of a furan ring and two oxygenated functional groups (–OH and –CHO), and thus the molecule can undergo several catalytic transformations to obtain high-added value products, such as oxidation, hydrogenation, hydrodeoxygenation or decarbonylation.27 Specifically, the selective hydrogenation of HMF to produce 2,5-bishydroxymethylfuran (BHMF) is an attractive transformation due to the many applications that BHMF has as a building block or precursor to the production of other high-value chemicals.28 Although the selective hydrogenation of HMF to BHMF has been recently reported using green solvents,29 the majority of these catalytic transformations are carried out in organic solvents since most catalysts are not compatible with water.30 Therefore, carrying out the selective hydrogenation of HMF in water under mild conditions is always a challenge.
Herein, we present Pt NPs supported on reduced graphene oxide (rGO) functionalized with different equivalents (equiv.) of tributyltin hydride (Bu3SnH). The SOMC approach allowed the selective deposition of Sn complexes on the Pt surface. The synthesized catalysts (Pt@rGO/Snx) were fully characterized by state-of-the-art techniques, revealing the presence of Sn-butyl fragments (–SnBun) on the platinum surface. The catalytic activity of Pt@rGO/Snx has been evaluated in the aqueous hydrogenation of biomass-derived compounds (e.g. HMF) under mild conditions (5 bar H2 and 25 °C), achieving excellent results in terms of activity and selectivity due to the promoting effect of Sn.
Results and discussion
Synthesis, characterization and surface studies
Pt NPs supported on rGO (Pt@rGO) were synthesized and later functionalized with the organometallic tin complex Bu3SnH (Pt@rGO/Snx), following a two-step synthetic route (Scheme 1). First, through organometallic synthesis, Pt NPs with high concentrations of hydrides on their surface were directly generated on rGO (Pt@rGO). For this, the organometallic precursor Pt(NBE)3 (NBE = norbornene) was decomposed in a controlled manner under 3 bar of H2 in a THF dispersion of rGO. In a second step, and with the intention of modifying the catalytic properties of the Pt NPs, Pt@rGO was functionalized with different equivalents of Bu3SnH (Pt@rGO/Snx; x = 0.2, 0.5, 0.8 and 1 equiv.) following an SOMC approach. More specifically, Bu3SnH was added into a THF suspension of Pt@rGO and stirred at room temperature (r.t.) for 24 h (Scheme 1). The Sn complex selectively reacts with the hydride surface-enriched Pt NPs to produce Pt@rGO/Snx. After analyzing the reaction atmosphere resulting from the Pt@rGO/Snx synthesis by gas chromatography (GC), the released butane was clearly detected (see ESI section S2, Fig. S1b†), suggesting that the organotin complex is grafted at the platinum surface as Sn-butyl fragments (–SnBun), as previously observed in similar reported systems.22 This butane comes from the hydrogenolysis of Bu3SnH by hydrogen chemisorbed at the Pt NP surface. The metal contents of Pt@rGO and Pt@rGO/Snx were determined by inductively coupled plasma optic emission spectroscopy (ICP-OES) analysis employing an optimized digestion method (for more details see the Experimental section).31 In all cases, ICP-OES analysis revealed platinum contents close to the theoretical value of 3 wt% (see ESI section S3, Table S2†). On the other hand, tin contents of Pt@rGO/Snx were lower than the theoretical ones, indicating that not all the Sn complex used during the synthesis was finally incorporated into the platinum surface (see ESI section S3, Table S2†). It was observed that as the number of equivalents of Bu3SnH added increases, the Sn % incorporated is lower. For example, for Pt@rGO/Sn0.2 the real number of equiv. of Sn is 0.16 instead of 0.2 (80% of Sn is incorporated), while for Pt@rGO/Sn1 it is 0.33 instead of 1 (33% of Sn is incorporated). This is most likely due to the increased steric hindrance caused by the higher number of surface Sn-butyl fragments in Pt@rGO/Sn1. The lower incorporation of –SnBun was confirmed after analysing the mother liquors resulting from the synthesis of Pt@rGO/Snx by ICP-OES. In all cases, the remaining Sn was found in the mother liquors, proving that the non-incorporated tin fragments were ultimately eliminated by washing with THF during the purification process.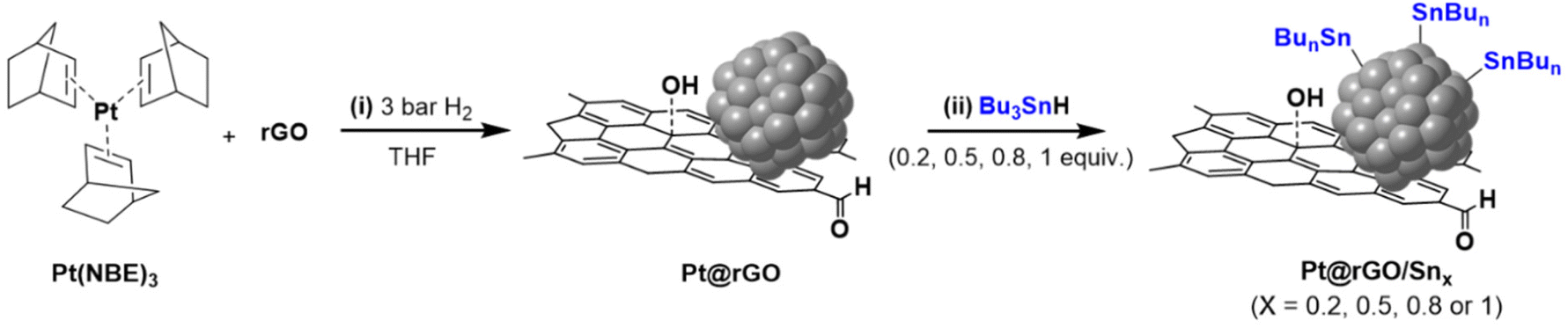 | ||
| Scheme 1 Two-step synthetic route of Pt@rGO/Snx (x = 0.2, 0.5, 0.8 and 1 equiv.) based on the organometallic synthesis of Pt@rGO and the subsequent functionalization with Bu3SnH by SOMC. | ||
The resulting platinum catalysts were characterized by Transmission Electronic Microscopy (TEM) and High-Resolution TEM (HRTEM). The TEM micrographs of unmodified Pt@rGO and functionalized Pt@rGO/Snx revealed the formation of spherical, monodisperse and well-distributed nanoparticles (see Fig. 1 and S2–S4, see ESI section S4†) in all cases. Negligible differences in terms of morphology or distribution size were observed. For example, Pt@rGO NPs present a mean diameter of 2.3 ± 0.7 nm (Fig. 1a), whereas those NPs functionalized with 0.8 equiv. of Bu3SnH exhibit a mean size of 2.1 ± 0.5 nm (Fig. 1b). The HRTEM image of Pt@rGO (Fig. 1c) shows NPs with a characteristic face-centered cubic (fcc) structure of bulk platinum. The same crystalline structure was observed for the tin-functionalized catalyst, Pt@rGO/Sn0.8 (see Fig. 1d). High-angle annular dark-field scanning transmission electron microscopy coupled with energy-dispersive X-ray spectroscopy (HAADF-STEM EDX) of the tin-functionalized systems confirmed the presence of Sn exclusively on the platinum surface. In all cases, the tin was selectively deposited on the Pt NPs avoiding its accumulation on the graphene support (Fig. 1e and Fig. S11, see ESI section S5†). The hydrogenolysis of the organotin complex was preferentially produced by the hydrides chemisorbed at the Pt NP surface, as previously observed.22 Thus, this SOMC approach causes the selective grafting of Sn-butyl fragments onto the Pt surface. Furthermore, by HAADF-STEM EDX the number of Sn equivalents on the platinum surface was also determined. The number of Sn equiv. observed by EDX is close to the values obtained by ICP-OES (see ESI section S3, Table S2†). Such results confirm the good deposition of –SnBun over the platinum surface during the second synthetic step due to the use of the SOMC approach.
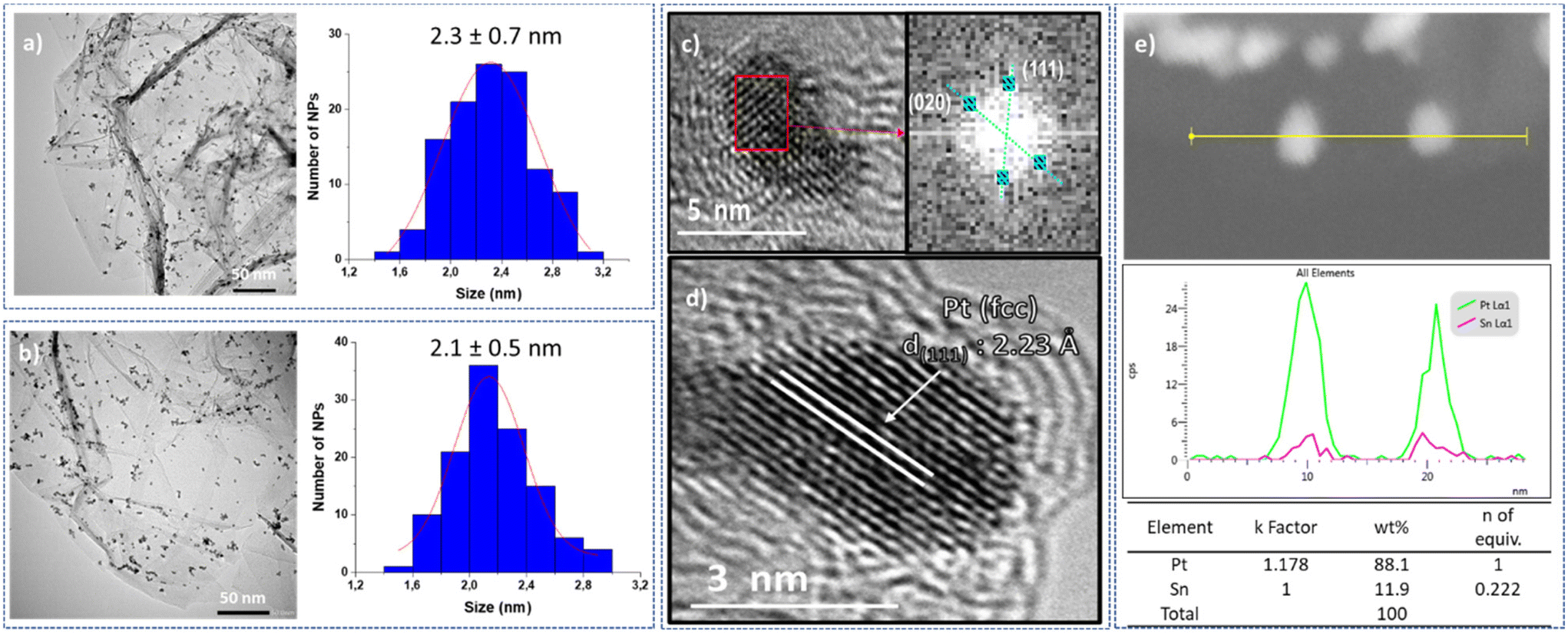 | ||
| Fig. 1 TEM micrographs and size histograms of (a) Pt@rGO and (b) Pt@rGO/Sn0.8. (c) Fourier analysis applied to an HRTEM micrograph of Pt@rGO, which displays reflections to the (111) and (020) atomic planes and correspond to the d-spacing values of 2.23 Å and 1.96 Å, respectively. (d) HRTEM image of Pt@rGO/Sn0.8 showing a lattice fringe spacing of 2.23 Å that corresponds to the Pt (111) crystal plane of metallic Pt. Both HRTEM images reveal the presence of crystalline Pt NPs retaining the fcc structure. (e) HAADF-STEM image and relative composition profile of Pt@rGO/Sn0.8 determined by EDX. | ||
Raman spectroscopy is an established tool to investigate the quality of graphenic materials (defects, exfoliation degree, sp2 character, etc.). Thus, rGO, unmodified Pt@rGO and functionalized Pt@rGO/Sn0.8 were all analyzed by this characterization technique. The Raman spectrum of rGO showed two well-differentiated domains, one located between 1200 and 1700 cm−1 and another one from 2500 to 3250 cm−1 (see ESI section S6, Fig. S14 and S15†). The first domain contains two bands of similar intensities at 1356 and 1594 cm−1, which correspond to D and G bands, respectively. The second domain located at ca. 3000 cm−1 (band 2D) is typically associated with a few layers of graphene (1–2 layers). The ratio between the intensities of the D/G bands (ID/IG = 1.54) is related to a high percentage of defect sites, which are excellent anchoring points for MNPs.32 After the incorporation of Pt NPs on rGO (Pt@rGO), the Raman spectrum did not show any significant differences (see ESI section S6, Fig. S14†). Only a slight decrease of the ratio ID/IG was observed (from 1.54 to 1.48), which is an indication of the decrease of the number of defect sites on the graphene material due to their interaction with the noble metal. Finally, after the surface modification with 0.8 equiv. of the organometallic complex (Pt@rGO/Sn0.8), no significant differences were observed with respect to Pt@rGO.
X-ray Photoelectron Spectroscopy (XPS) has been recently demonstrated to be an appropriate technique to study the coordination mode of surface molecules on metal nanoparticles.33 Thus, the chemical state and coordination of the Sn-butyl fragments at the platinum surface were investigated by XPS. The Sn 3d5/2 signal for Bu3SnH shows a binding energy (BE) of 485.3 eV, which agrees with reported values of similar Sn(IV) alkyl/aryl complexes.34 On the other hand, the Sn 3d5/2 signal of Pt@rGO/Sn0.8 exhibits a higher BE value (485.8 eV), which means that Sn is little more oxidized and indicates that tin-butyl fragments are coordinated to the platinum surface through the Sn atoms (Fig. 2a). The BE of Pt 4f7/2 in the XPS spectrum of Pt@rGO/Sn0.8 is 71.5 eV (Fig. 2c). This signal presents two contributions: (i) a main one located at 71.4 eV is assigned to Pt(0), and (ii) an additional contribution at 72.6 eV, which can be attributed to the partially oxidized Pt surface atoms (Ptδ+). Here the amount of Pt(0) is higher than in the non-functionalized material (Pt@rGO) (see ESI section S8, Fig. S17b†), which confirms that the Pt is little more reduced after functionalization with –SnBun.
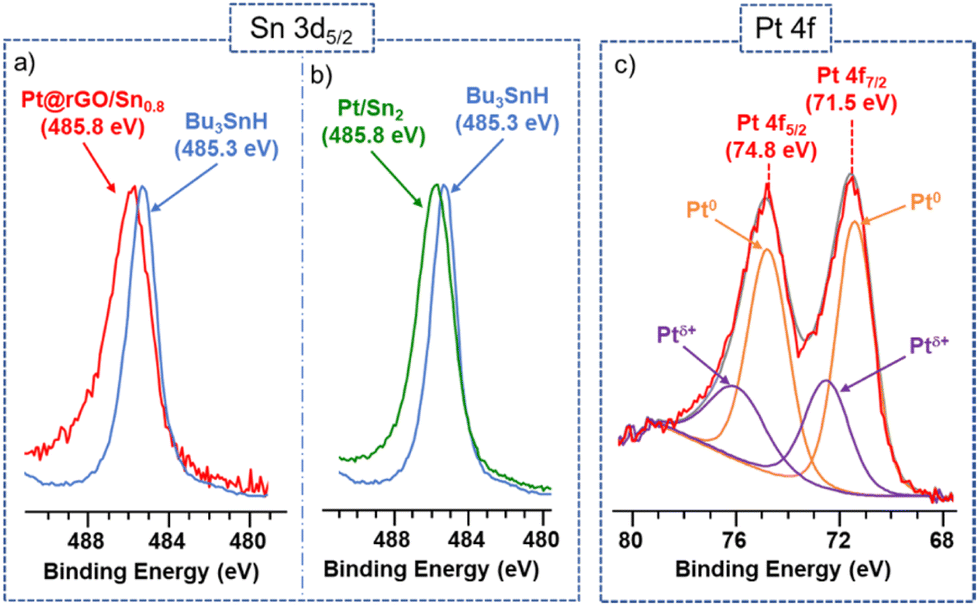 | ||
| Fig. 2 Left: (a and b) X-ray photoelectron spectroscopy (XPS) spectra showing Sn 3d5/2 signals of Pt@rGO/Sn0.8 (red), Pt/Sn2 (green), and Bu3SnH (blue). Right: (c) Pt 4f signals of Pt@rGO/Sn0.8. | ||
Due to the opacity and conductivity of graphene-supported Pt NPs, their surface chemistry could not be investigated by infrared and solid-state MAS-NMR spectroscopic techniques. Thus, in order to better understand the location, chemical nature and coordination of the –SnBun fragments on the metal surface, non-supported Pt NPs directly stabilized with Bu3SnH were prepared as the reference material.
Colloidal Pt NPs ligated by 2 equiv. of Bu3SnH (Pt/Sn2) were synthesized following the same organometallic synthesis of the first step of Scheme 1, but using the organotin complex Bu3SnH as a stabilizer instead of rGO (Scheme 2). Here, the released butane was also identified by analyzing the reaction atmosphere just after the synthesis of Pt/Sn2 (see ESI section 2, Fig. S1a†). This confirms that the Sn-alkyl fragments produced by hydrogenolysis of Bu3SnH at the platinum surface are enough to stabilize these non-supported Pt NPs.
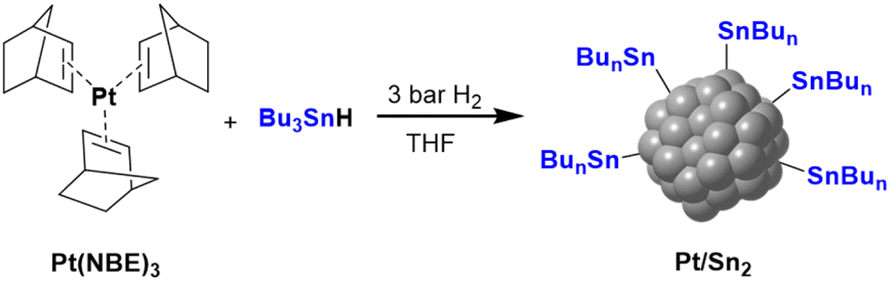 | ||
| Scheme 2 Synthesis of non-supported Pt/Sn2 NPs following the organometallic approach. | ||
The TEM and HRTEM images of Pt/Sn2 showed small, crystalline, and well-distributed Pt NPs with a narrow size distribution (i.e. 1.6 ± 0.5 nm) (see ESI sections S4 and S5, Fig. S5 and S12,† respectively). The crystalline structure and NP size of Pt/Sn2 were confirmed by X-ray powder diffraction (XRD) (see ESI section S9, Fig. S18†). ICP analysis of Pt/Sn2 revealed a platinum content of 49.2 wt% and a tin content of 34.7 wt%, which correspond to the real incorporation of Sn of 1.15 equivalents. The presence of –SnBun fragments at the Pt surface was confirmed by EDX analysis (see ESI section S5, Fig. S13†). Diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) using CO as a probe molecule was performed to investigate the surface chemistry of these colloidal Pt/Sn2 NPs through the location of the surface active sites. It is well-known that CO can coordinate in the bridging (COb) or terminal (COt) mode on Pt NPs.35 Normally, COb bands are located on the faces of MNPs and COt bands are located on their apexes and edges.36 Fig. S16 (see ESI section S7)† shows the DRIFT spectra of Pt/Sn2 before (blue) and after (red) exposure to CO (bubbling CO in a THF dispersion for 5 min). After exposure to CO, the spectrum showed an intense band at 2003 cm−1 corresponding to COt. Interestingly, no COb band was observed, indicating that faces of Pt/Sn2 NPs are not available, likely due to the high tin surface coverage. Thus, the coordination of CO in the terminal mode indicates the presence of surface active sites, mostly located at the edges or apexes of the nanoparticles, even at high surface coverage.
The coordination of Sn-butyl fragments on these colloidal Pt/Sn2 NPs was also investigated by XPS. Also for Pt@rGO/Sn0.8, the Sn 3d5/2 signal of Pt/Sn2 shows a BE of 485.8 eV (Fig. 2b, green). This BE suggests the coordination of the tin-butyl fragments to the platinum surface through the Sn atoms, since it involves the loss of their electron density compared to those of the corresponding organometallic complex Bu3SnH (485.3 eV) (Fig. 2b, blue). The identical BEs for the colloidal and supported Pt NPs (Fig. 2a and b) confirm that in Pt@rGO/Sn0.8 most of the Sn-butyl fragments are coordinated to the platinum surface, in agreement with EDX observations. Fig. S17a (see ESI section S8)† shows the characteristic asymmetric peaks for platinum metal corresponding to the Pt 4f7/2 and Pt 4f5/2 signals of Pt/Sn2. In a similar way to supported Pt NPs (Fig. 2c), the BE of Pt 4f7/2 can be deconvoluted in two components: (i) one at ca. 71.0 eV characteristic of Pt(0), and (ii) another at 72.6 eV attributed to Ptδ+.
Solid state MAS-NMR also confirmed the presence of Sn-butyl fragments on the Pt surface. The 13C CP-MAS spectrum of Pt/Sn2 presents a sharp signal at ca. 13 ppm which belongs to the methyl groups of the surface [–Sn–(CH2)3–CH3]n species. The intense peak at 26 ppm corresponds to the CH2 groups of the alkyl chain and the peak at ca. 127 ppm is attributed to the CH2 groups located next to the Sn atoms (see ESI section S10, Fig. S20†). The latter signal is notably shifted to a lower field compared to that observed in similar systems.22 This could be explained by the proximity of these methylene groups to the platinum surface, which displaces the signal to a higher frequency due to the presence of conduction electrons.37 This result also points that tin-butyl fragments are coordinated to the platinum surface through the Sn atoms, as was previously suggested by XPS (Fig. 2a). To sum up, surface studies on these colloidal organo-tin Pt NPs confirmed the presence of butyl-tin fragments at the Pt surface and the existence of available surface active sites even at high tin surface coverage. We can thus expect some catalytic activity of these non-supported Pt NPs (vide infra, Table 1, entry 3).
| Entry | Catalyst | Substrate | Product | Conversionb,c | Selectivityb |
|---|---|---|---|---|---|
| a Reaction conditions: 0.3 mmol substrate, catalyst (0.1 mol% Pt), 2 mL of H2O, 5 bar H2, 25 °C and 5 h of reaction. b Conversions and selectivities were determined by GC using dodecane as an internal standard, and confirmed by GC-MS. c Metal free reference material (rGO modified with Bu3SnH) showed negligible activity in the hydrogenation of HMF under reaction conditions. d Reaction time: 20 h. e Reaction time: 72 h. | |||||
| 1 | Pt@rGO/Sn0.8 |
|
|
>99% | 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5 = 98:2 :5 = 98:2 |
| 2 | Pt@rGO | 12.3% | 2:5 = 100:0 |
||
| 3 | Pt/Sn2 | 17.9% | 2:5 = 100:0 |
||
| 4 | Pt/Sn2@rGO | 28.8% | 2:5 = 100:0 |
||
| 5 | Pt@rGO/Sn0.8 |
|
|
100% | 8:9 = 99:1 |
| 6 | Pt@rGO | 82.9% | 8:9 = 97:3 |
||
| 7 | Pt@rGO/Sn0.8 |
|
|
>99% | 11:12 = 42:58 |
| 8 | Pt@rGO | 91.2% | 11:12 = 92:8 |
||
| 9 | Pt@rGO/Sn0.8 |
|
|
96.8% | 14:15 = 100:0 |
| 10 | Pt@rGO | 15.5% | 14:15 = 100:0 |
||
| 11d | Pt@rGO/Sn0.8 |
|
|
56.9% | 16 = 100 |
| 12e | Pt@rGO/Sn0.8 | >99% | 16 = 100 | ||
| 13d | Pt@rGO | 13.1% | 16 = 100 | ||
Catalytic studies
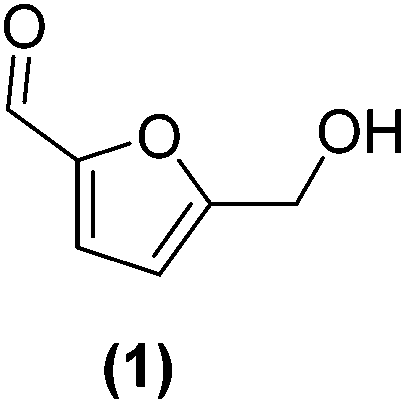
In order to evaluate the catalytic reactivity of rGO-supported Pt NPs decorated with different equiv. of –SnBun, the hydrogenation of 5-hydroxymethylfurfural (HMF, 1) was chosen as a model reaction. HMF is identified as one of the main chemical products derived from biomass,38 being an interesting model molecule since it has two potentially hydrogenable functional groups: (i) the furan ring and (ii) the aldehyde. In addition, the alcohol groups of HMF or its derivatives can be also transformed by hydrodeoxygenation (HDO) processes. This means that HMF can undergo various catalytic transformations to obtain high-value products (Fig. 3a). Among the possible hydrogenated derivatives, 2,5-bishydroxymethylfuran (BHMF, 2) has attracted great interest since it is a potential substitute for petrochemical-based monomers or precursors.28,39 Thus, the selective hydrogenation of HMF to produce BHMF is of special relevance in fine chemistry.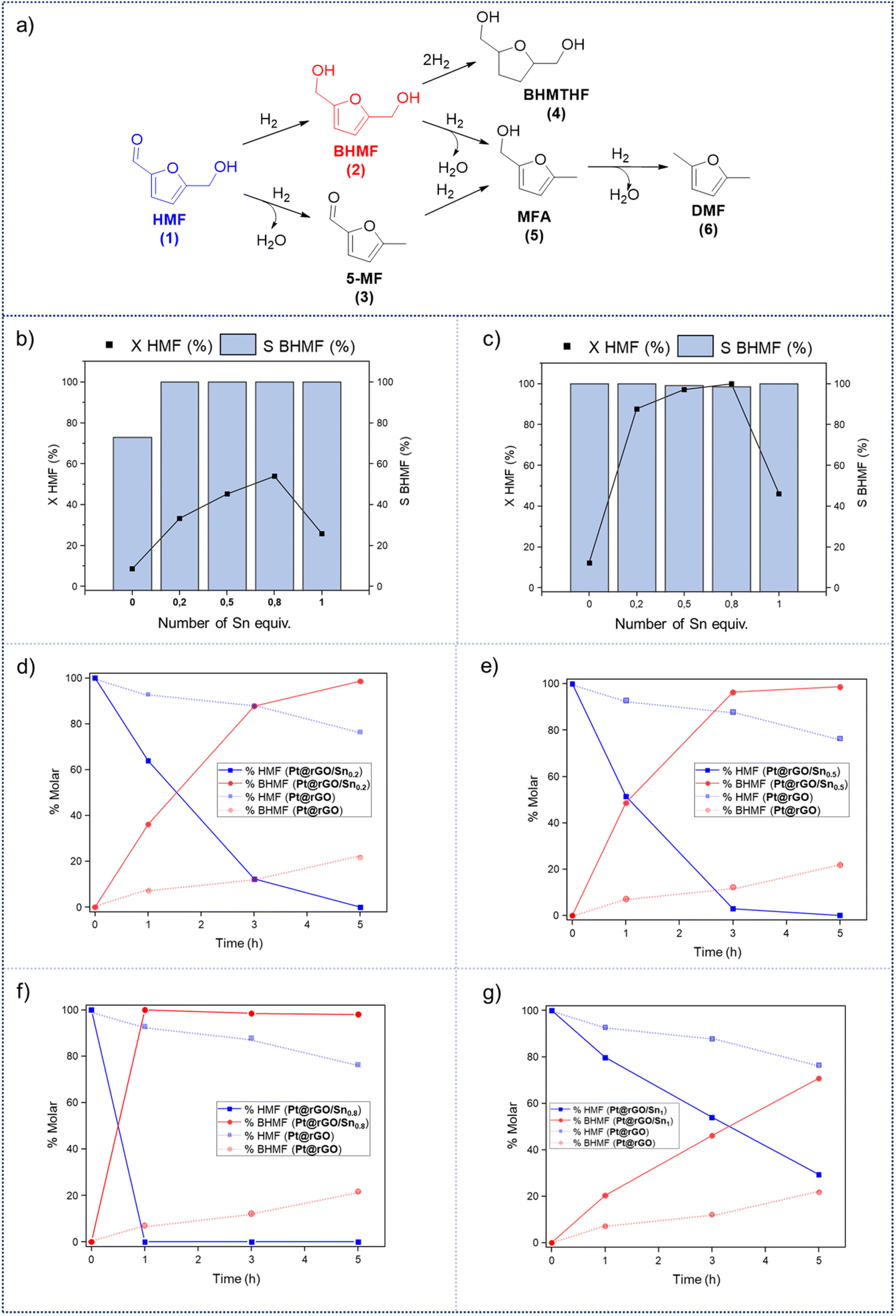 | ||
| Fig. 3 (a) Schematic representation of the hydrogenation/hydrodeoxygenation reaction pathways of HMF into high-value products. Volcano-like behaviour in the HMF hydrogenation activity of Pt@rGO/Snx (x: 0, 0.2, 0.5, 0.8 and 1 equiv.) in (b) THF and (c) H2O after 3 h of reaction [X = conversion (dark blue squares) and S = selectivity (blue bars)]. Hydrogenation of HMF in water using (d) Pt@rGO/Sn0.2, (e) Pt@rGO/Sn0.5, (f) Pt@rGO/Sn0.8 and (g) Pt@rGO/Sn1 as catalysts. Hydrogenation of HMF in water using Pt@rGO is represented in all kinetics as dashed lines. Reaction conditions: 0.2 mmol HMF, 2 mg of Pt@rGO/Snx (0.15 mol% Pt), 2 mL of THF or H2O, 5 bar H2, 25 °C. Conversions and selectivities were determined by GC using dodecane as an internal standard and confirmed by GC-MS. | ||
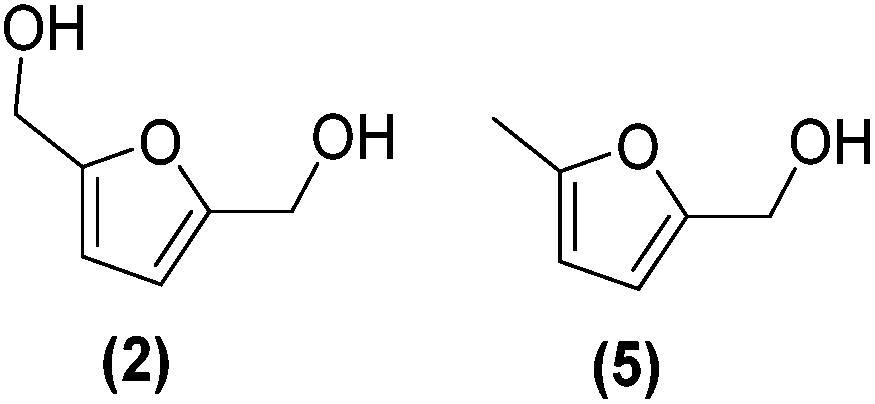
First, the influence of the organo-tin complex on the activity and selectivity of the graphene-supported Pt NPs was investigated in the hydrogenation of HMF in THF. After 3 h of reaction, the non-modified Pt@rGO catalyst showed low conversion (8.5%), producing BHMF (2) as the main product (73% selectivity), but also a considerable amount of the HDO product 2-hydroxymethyl-5-methylfuran (MFA, 5) (Fig. 3b and S19a, see ESI section S10†). On the other hand, after decorating Pt@rGO with 0.2 equivalents of the organotin complex, Pt@rGO/Sn0.2, a higher conversion (33.3%) and an increase in selectivity was observed, obtaining exclusively BHMF (2) (Fig. 3b and S21b, see ESI section S11†). Interestingly, as the number of equiv. increases, the conversion is higher maintaining the selectivity towards BHMF, except for Pt@rGO/Sn1 which showed a lower conversion than all other tin-decorated catalysts (Fig. 3b). This volcano-like behaviour indicates that the addition of –SnBun improves the activity and selectivity of Pt@rGO, reaching a maximum conversion (53.9% after 3 h) with 0.8 equiv. (Pt@rGO/Sn0.8) (Fig. 3b and S21d, see ESI section S11†). However, a higher amount of tin (1 equiv.) is reflected by a notable decrease in the conversion (25.9%), likely due to the high tin surface coverage that blocks most of the surface active sites (Fig. 3b and S21e, see ESI section S11†). Therefore, to maximize the activity and selectivity of Pt@rGO/Snx it is of high importance to decorate with the right amount of –SnBun.
Heterogeneous catalysis in batch reactors normally employs organic solvents. However, most products derived from biomass are more soluble in water,40 which, due to its nontoxicity and environmental compatibility, is considered as the greenest solvent. Thus, the catalytic reactivity of Pt@rGO/Snx was also investigated in the aqueous hydrogenation of HMF. As can be seen in Fig. 3c, similar volcano-like behaviour was observed in water. Nevertheless, the activity and BHMF selectivity of all catalysts studied are much higher than in THF, likely due to the excellent solubility of HMF in water and different operation mechanisms.41 For example, the decoration with a small amount of –SnBun, such as 0.2 equiv., led to a considerable increase in the activity. After 3 h of reaction, Pt@rGO only exhibited an HMF conversion of 12.2%, while the conversion using Pt@rGO/Sn0.2 as a catalyst was 87.7% (Fig. 3d and S22a, b, see ESI section S11†). Furthermore, the most active catalyst, Pt@rGO/Sn0.8, showed full conversion and 100% selectivity towards BHMF after only 1 h of reaction (Fig. 3f and S22d, see ESI section S11†). And again, the functionalization with 1 equiv. reduced the activity of the catalyst due to a high surface coverage, giving a conversion of only 46% (Fig. 3g and S22e, see ESI section S11†). The catalytic activity of Pt@rGO/Sn0.8 in the selective hydrogenation of HMF to BHMF is higher than those of previously reported systems (see ESI section S12, Table S3†). For example, Bell et al. prepared Pt–Sn NPs supported on Al2O3, which were used in the hydrogenation of HMF in ethanol under 14 bar H2 and at 60 °C, exhibiting an estimated TOF of 72 h−1 (see ESI section S12, Table S3† entry 1).29a,42 Shortly after, Kawanami et al. reported a platinum-based catalyst, Pt/MCM-41, which, under mild conditions (35 °C and 8 bar H2), presented almost complete conversion to BHMF.43 Specifically, after 1 h of reaction, Pt/MCM-41 (1 wt%) showed 85% conversion with a BHMF selectivity of 95% (estimated TOF = 566.7 h−1) (see ESI section S12, Table S3† entry 2). On the other hand, Pt@rGO/Sn0.8 prepared in the present work is even faster under milder reaction conditions (5 bar H2 and 25 °C), since it presents an estimated TOF value of 666.7 h−1 (see ESI section S12, Table S3† entry 16), being one of the most active catalysts in the selective hydrogenation of HMF to BHMF in aqueous medium.
Since Pt@rGO/Sn0.8 gave full conversion to the desired product BHMF (2) after only 1 h of reaction time, we decided to increase the substrate/catalyst ratio. More specifically, the catalytic loading was reduced from 0.15 mol% to 0.1 mol% by increasing the amount of initial HMF to 0.3 mmol. After 5 h under these conditions, Pt@rGO exhibited a conversion towards BHMF of only 12.3%, whereas Pt@rGO/Sn0.8 showed complete conversion and almost full selectivity to BHMF, producing only a small amount of MFA (5) (2%) (see Table 1 entries 1 and 2). A kinetic study of HMF hydrogenation using Pt@rGO/Sn0.8 shows that the conversion of HMF practically goes parallel to the formation of BHMF, taking 5 h to reach full conversion (see ESI section S11, Fig. S23b†). On the other hand, non-supported Pt/Sn2 NPs showed moderate activity (17.9% conversion to BHMF after 5 h of reaction; see Table 1 entry 3, and ESI section S11, Fig. S24a†), most likely due to the high degree of aggregation of this colloidal NPs in water (see ESI section S4, Fig. S7†) together with the high tin surface coverage. In fact, after immobilization of Pt/Sn2 on rGO (Pt/Sn2@rGO, see ESI section S4, Fig. S6†) the activity of the NPs increases (28.8% of conversion; Table 1 entry 4 and ESI section S11, Fig. S24b†) due to their higher stability in aqueous medium. These results highlight the importance of the use of rGO as support to enhance the stability/activity of Pt NPs decorated with –SnBun in water.
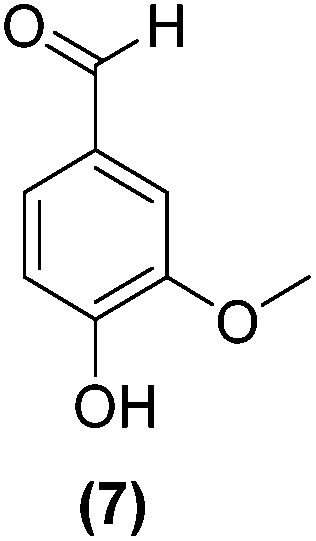
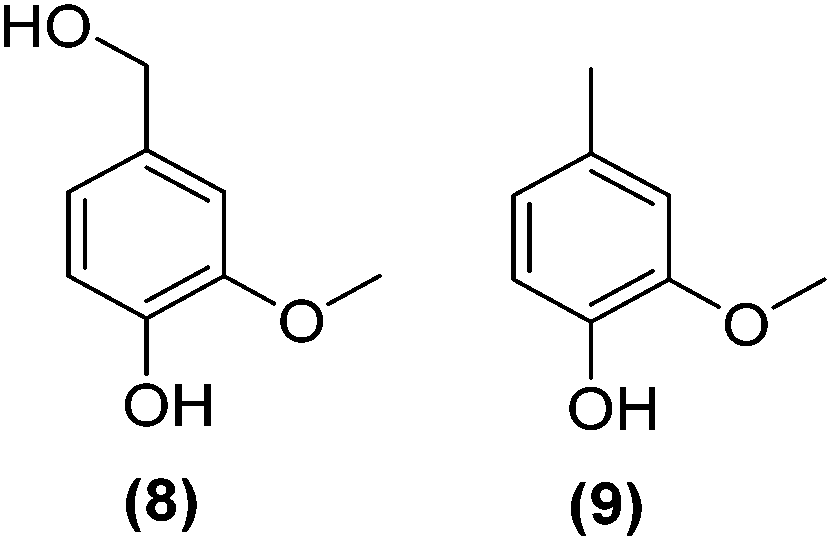
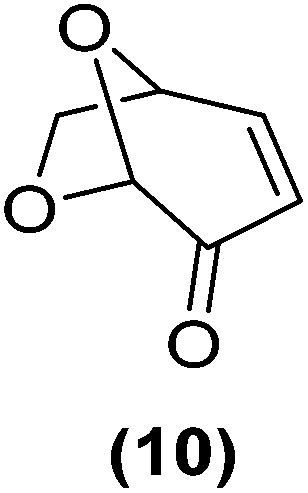
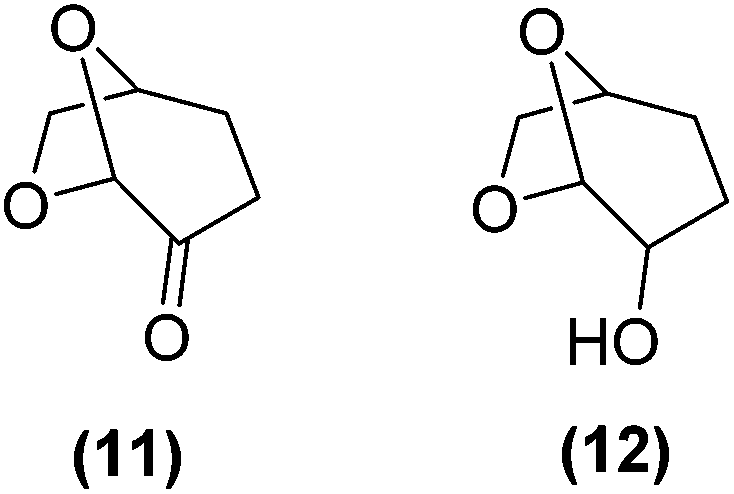
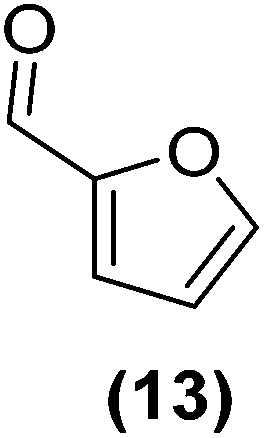
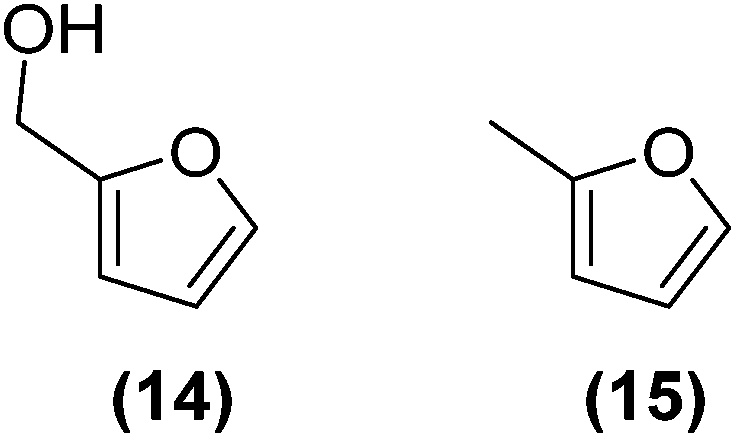
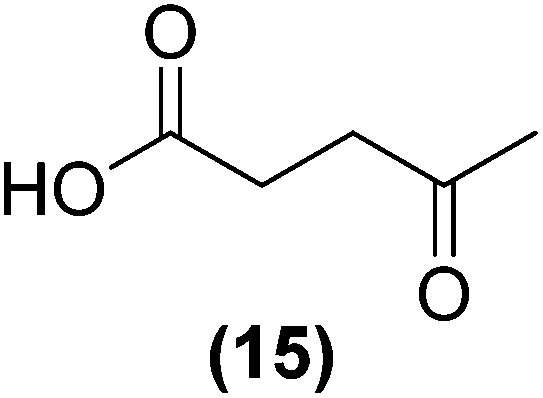
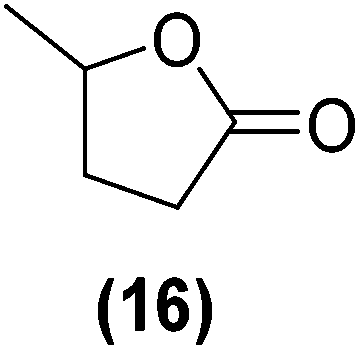
Encouraged by the excellent catalytic activity of Pt@rGO/Sn0.8 in the aqueous hydrogenation of HMF, we also tested it in the hydrogenation of other biomass-derived substrates and compared it with Pt@rGO (Table 1). For example, vanillin (7) was effectively hydrogenated into vanillyl alcohol (8) after 5 h of reaction using Pt@rGO/Sn0.8 as a catalyst (selectivity of 99% at >99% conversion). On the other hand, under the same catalytic conditions (0.1 mol% Pt, H2O, 5 bar H2, 25 °C, 5 h) the monometallic catalyst Pt@rGO presented a conversion of 82.9% and a slightly lower selectivity to the corresponding alcohol (8), due to the formation of a small amount of the HDO product, 2-methoxy-4-methylphenol (9) (see Table 1, entries 5 and 6). In the hydrogenation of levoglucosenone (10) with Pt@rGO/Sn0.8 we observed complete conversion in 5 hours, and a selectivity of 58% towards the fully hydrogenated product, levulinyl alcohol (12) (see Table 1, entry 7). However, at the same reaction time, Pt@rGO showed a slower conversion (91.2%) and 92% selectivity to the product (11), due to its lower reactivity in the hydrogenation of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds compared to the functionalized catalyst (see Table 1, entry 8). This lower catalytic activity was also evidenced by the hydrogenation of furfural (13); while Pt@rGO/Sn0.8 showed a conversion of 96.8% towards the corresponding alcohol (14), Pt@rGO only reached a conversion of 15.5% (see Table 1, entries 9 and 10). Finally, the hydrogenation of levulinic acid (15) into γ-valerolactone was slower with both compared catalysts. After 20 h, Pt@rGO/Sn0.8 showed a conversion of 56.9%, while Pt@rGO showed a conversion of only 13.1% (see Table 1, entries 11 and 13). It was necessary to prolong the reaction time up to 72 h to reach complete conversion (see Table 1, entry 12). In general terms, we can conclude that the presence of –SnBun fragments at the surface of Pt NPs considerably increases their activity in the hydrogenation of polar groups such as aldehydes or ketones (Table 1, entries 1–6 and 9–13). On the other hand, the hydrogenation of non-polar groups such as alkenes, is less affected by the presence of –SnBun as was observed in the hydrogenation of levoglucosenone (10) (Table 1, entries 7 and 8).
O bonds compared to the functionalized catalyst (see Table 1, entry 8). This lower catalytic activity was also evidenced by the hydrogenation of furfural (13); while Pt@rGO/Sn0.8 showed a conversion of 96.8% towards the corresponding alcohol (14), Pt@rGO only reached a conversion of 15.5% (see Table 1, entries 9 and 10). Finally, the hydrogenation of levulinic acid (15) into γ-valerolactone was slower with both compared catalysts. After 20 h, Pt@rGO/Sn0.8 showed a conversion of 56.9%, while Pt@rGO showed a conversion of only 13.1% (see Table 1, entries 11 and 13). It was necessary to prolong the reaction time up to 72 h to reach complete conversion (see Table 1, entry 12). In general terms, we can conclude that the presence of –SnBun fragments at the surface of Pt NPs considerably increases their activity in the hydrogenation of polar groups such as aldehydes or ketones (Table 1, entries 1–6 and 9–13). On the other hand, the hydrogenation of non-polar groups such as alkenes, is less affected by the presence of –SnBun as was observed in the hydrogenation of levoglucosenone (10) (Table 1, entries 7 and 8).
In order to study the stability and heterogeneity of the prepared catalysts, a series of experiments were performed. First, non-modified (Pt@rGO) and tin-functionalized (Pt@rGO/Sn0.8) catalysts were analyzed by TEM and HRTEM after catalysis (i.e. hydrogenation of HMF under standard catalytic conditions). No growth of the Pt NPs was observed in both catalysts, the unmodified Pt@rGO (2.4 ± 0.7 nm), and the functionalized one, Pt@rGO/Sn0.8 (2.3 ± 0.4 nm) (see ESI section S4, Fig. S8b, c and S9b, c,† respectively). Furthermore, by HRTEM we can observe that Pt NPs retain their crystallinity after catalysis (see ESI section S4, Fig. S8a and S9a†). This validates the ability of rGO to efficiently stabilize Pt NPs in the aqueous transformation of biomass-derived compounds. Additionally, the catalyst reusability was evaluated through a multiple addition experiment. Here, the hydrogenation of HMF was performed during nine consecutive additions of a substrate using Pt@rGO/Sn0.8 as a catalyst. To better identify any loss of catalytic activity, HMF was added every hour where the conversion values were around 40%. No evident loss of activity/selectivity was observed, since conversion remains constant with full selectivity towards BHMF (Fig. 4a and Fig. S25, see ESI section S11†). Moreover, TEM analysis of Pt@rGO/Sn0.8 after the multiple addition experiment revealed the presence of small and well-distributed Pt NPs with a similar size to the as-synthesized catalyst (see ESI section S4, Fig. S10†). Finally, to confirm the heterogeneous nature of the catalyst, a filtration experiment was performed. After 1 h under standard catalytic conditions, (i.e. HMF hydrogenation in water), the reaction media was filtered, added into another reactor, and left for four more hours under the same catalytic conditions (5 bar H2 and 25 °C). No change in the catalytic conversion was observed after 2 and 4 h (Fig. 4b). Thus, as can be seen in Fig. 4b, the progress in the reaction after catalyst removal by filtration is marginal (conversion is maintained at ∼43%). ICP analysis after filtration did not show any metal leaching, confirming the heterogeneity and stability of the material during the selective hydrogenation of HMF in aqueous medium.
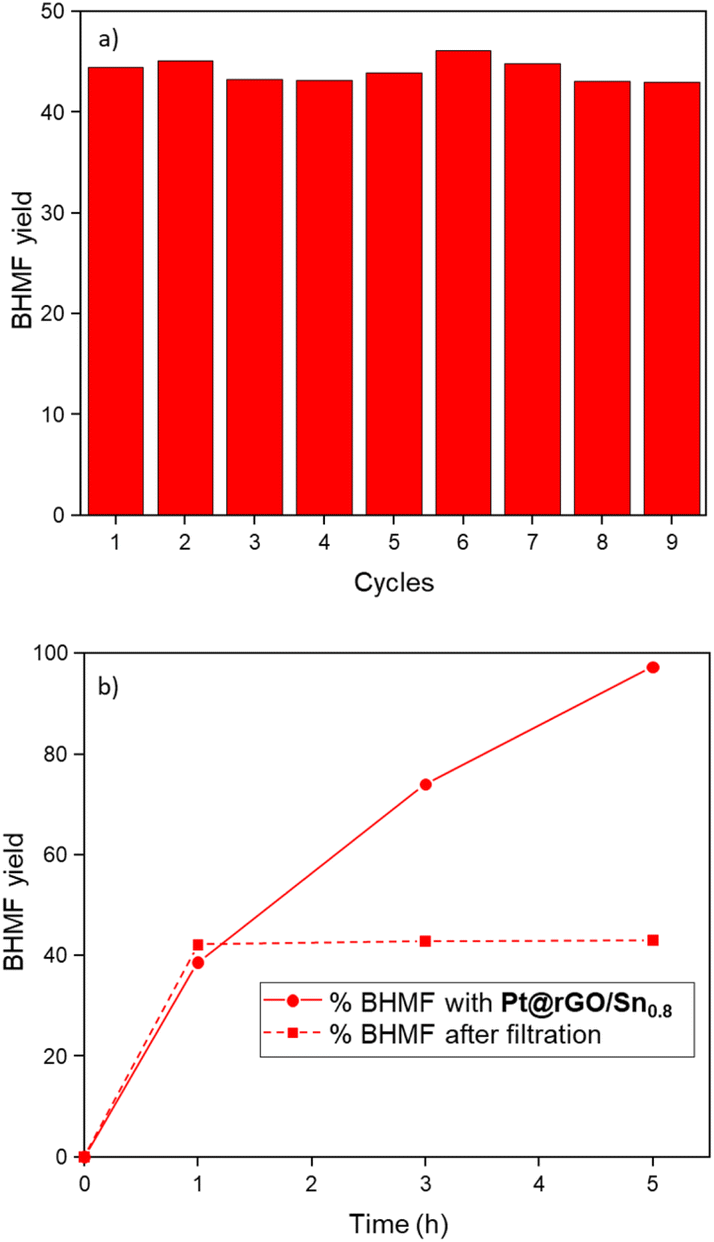 | ||
| Fig. 4 (a) Multiple addition experiment for the hydrogenation of HMF catalyzed by Pt@rGO/Sn0.8. (b) Kinetics of HMF hydrogenation in the presence of Pt@rGO/Sn0.8 and after catalyst removal by filtration. Reaction conditions: 0.3 mmol HMF, 2 mg of Pt@rGO/Sn0.8 (0.1 mol% Pt), 2 mL of H2O, 5 bar H2, 25 °C. Conversions and selectivities were determined by GC using dodecane as an internal standard, and confirmed by GC-MS. | ||
Promoting effect of –SnBun
It is evident that the superior catalytic activity of Sn-functionalized Pt-supported NPs is due to the presence of tin-butyl fragments on the platinum surface. Thus, to study the promoting effect of tin we performed a series of experiments. First, to have an idea about the accessible platinum surface of the unmodified Pt@rGO and functionalized Pt@rGO/Sn0.8 catalysts, CO and H2 chemisorption studies were performed (see ESI section S13, Table S4†). Normally, the presence of surface species blocks the potential adsorption sites,13,44 however, in our case we observed the opposite behaviour. For Pt@rGO, the CO uptake was 6.4 μmol g−1, while for Pt@rGO/Sn0.8, the uptake of CO increased to 9.6 μmol g−1. Thus, the presence of Sn-butyl fragments at the platinum surface increases the amount of CO chemisorbed. This can be related to the capacity of these Sn-decorated catalysts to activate carbonyl groups (vide infra). In a similar way, the H2 chemisorption analysis revealed that for Pt@rGO the H2 uptake was 50.0 μmol g−1, while for Pt@rGO/Sn0.8 the uptake of H2 remained practically constant (50.8 μmol g−1). This demonstrates that even after functionalizing the platinum surface with an organotin complex, the catalyst presents a similar number of accessible active sites. Thus, contrary to what one would expect the presence of tin-butyl fragments at the platinum surface does not decrease the number of surface active sites. Furthermore, thanks to H2 chemisorption it was possible to determine both the particle size and the dispersion of platinum. As can be seen in Table S4 (see ESI section S13),† the sizes observed by H2 chemisorption for the unmodified Pt@rGO and functionalized Pt@rGO/Sn0.8 (1.7 nm in both cases) are in good agreement with the mean size distribution observed by TEM (2.3 ± 0.7 nm for Pt@rGO and 2.1 ± 0.5 nm for Pt@rGO/Sn0.8). On the other hand, the dispersion of both catalysts is around 65–66%.Temperature-programmed reduction (TPR) experiments were performed to compare the reducibility of platinum in both catalysts, Pt@rGO and Pt@rGO/Sn0.8. The TPR profile of Pt@rGO showed a reduction peak at 376.6 °C that shifted to 309.2 °C after the coordination of Sn-butyl fragments to the platinum surface (Pt@rGO/Sn0.8; see ESI section S14, Fig. S26†). This hydrogen consumption corresponds to the reduction of oxidized species of platinum, previously observed by XPS and denoted as Ptδ+.45 It is evident that the presence of –SnBun facilitates Pt reduction, with less temperature being necessary to reduce the catalyst. Likewise, as seen in hydrogen temperature-programmed desorption (H2-TPD) patterns (see ESI section S15, Fig. S27†), Pt@rGO showed two peaks (centered at 305 and 378 °C) attributed to the desorption of two different adsorbed H species,46 while Pt@rGO/Sn0.8 exhibited a broad peak centered at 326 °C, indicating weakened hydrogen binding energy and easier H2 desorption. Therefore, TPR and H2-TPD results reveal that Sn-butyl fragments are modifying the catalysts.
Finally, adsorption of HMF and BHMF on the unmodified and functionalized catalysts was investigated using ultraviolet spectroscopy, as recently reported (see ESI, sections S1 and S16†).47Pt@rGO has lower HMF adsorption capacity than Pt@rGO/Sn0.8. While Pt@rGO only adsorbs 1.16 mg gcat−1 of HMF, Pt@rGO/Sn0.8 adsorbs 8.83 mg gcat−1 (see ESI section S16, Fig. S30†). However, the adsorption trend of the product BHMF is the opposite (see ESI section S16, Fig. S31†). The unmodified Pt@rGO presents a higher adsorption capacity for BHMF than Pt@rGO/Sn0.8 (2.15 mg gcat−1vs. 1.69 mg gcat−1, respectively). Therefore, Pt@rGO/Sn0.8 is capable to adsorb HMF more easily than Pt@rGO, but not BHMF. Comparing the HMF/BHMF adsorption ratios (Fig. 5a), Pt@rGO/Sn0.8 exhibits a much higher one (AHMF/BHMF = 8.83 mg g−1) than Pt@rGO (AHMF/BHMF = 1.55 mg g−1). These results evidence that the coordination of Sn-butyl fragments to the platinum surface facilitates the adsorption of HMF and weakens the adsorption of BHMF, and explain the high selectivity of these catalysts to BHMF. Thus, according to the latter results, the Sn-butyl surface species are probably acting as electron donor “ligands”, which increase the electron density of Pt, and at the same time facilitate the electrophilic activation of the carbonyl groups, as was evidenced by CO chemisorption studies (vide supra).23,48 In fact, XPS analysis showed that after the functionalization of the platinum surface with tin (Fig. 2a), the Sn 3d5/2 signal presents a higher BE (485.9 eV) than Bu3SnH, which means that Sn is more oxidized because it donates its electron density to Pt. Therefore, surface active sites can be described as Pt0–Snδ+, where HMF is easily adsorbed through the carbonyl group via donation of the lone pair of electrons on the oxygen atom, and subsequent hydrogenation to form BHMF by the hydrogen atoms chemisorbed on the Pt surface (Fig. 5b). This, together with the high stability of these supported NPs in water, highlights the high potential of Pt@rGO/Snx catalysts in the aqueous hydrogenation of biomass-derived compounds.
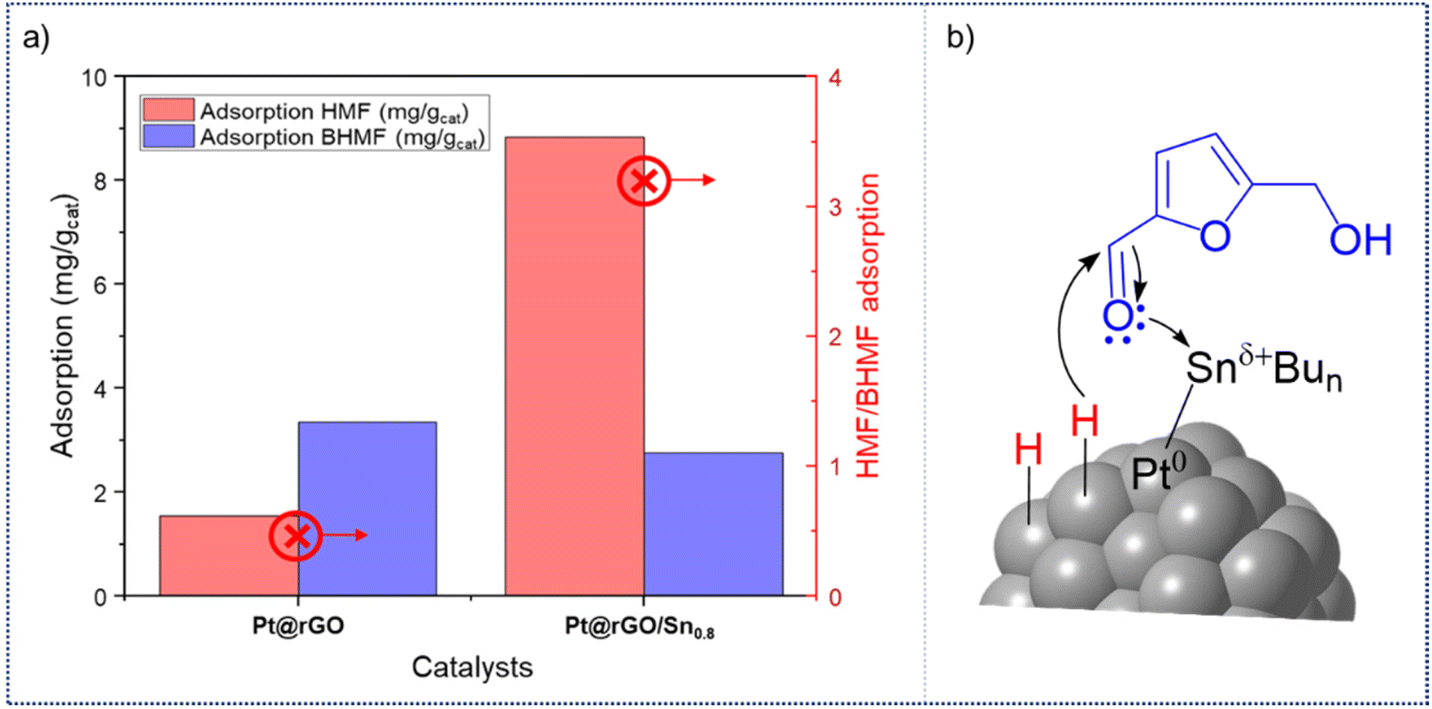 | ||
| Fig. 5 (a) HMF/BHMF adsorption capacities of Pt@rGO and Pt@rGO/Sn0.8. (b) Illustration of the adsorption of HMF on Pt@rGO/Sn0.8. | ||
Conclusions
We have successfully functionalized graphene-supported Pt NPs with different equivalents of –SnBun using Bu3SnH and a SOMC approach (Pt@rGO/Snx; x = 0.2, 0.5, 0.8 and 1 equiv.) for modifying their catalytic properties. The organotin-complex reacts with surface hydrides of the Pt NPs selectively decorating the Pt surface with Sn. Surface characterization studies confirmed the presence of Sn-butyl fragments grafted on the platinum surface. According to the catalysis results, we observed a strong correlation between the number of equivalents of –SnBun and the activity of the rGO-supported Pt NPs using the aqueous hydrogenation of HMF as a model reaction. The greater the amount of surface tin fragments, the higher the yield of BHMF. Interestingly, the maximum conversion was reached with the Pt@rGO/Sn0.8 catalyst, since a higher tin surface coverage (i.e.Pt@rGO/Sn1) led to a significant decrease in the catalytic performance. Indeed, Pt@rGO/Sn0.8 is one of the most active catalysts reported to date (estimated TOF = 666.7 h−1). The reactivity of Pt@rGO/Sn0.8 was also investigated in the aqueous hydrogenation of other biomass-derived compounds (i.e. vanillin, levoglucosenone, furfural or levulinic acid), evidencing the great catalytic activity of these organotin-functionalized Pt NPs. The promoting effect of tin was investigated, demonstrating that Sn-butyl fragments activate the platinum surface and facilitate the HMF adsorption and its subsequent hydrogenation to BHMF. Finally, the stability of this novel catalyst was studied by multiple addition and filtration experiments, demonstrating it to be a stable, reusable, and heterogeneous catalyst. Although the catalytic systems presented herein are based on platinum, a noble metal, the low metal content used during the catalysis (0.1 mol%), together with their excellent activity and the possibility to be reused/recycled, makes them potential candidates for the selective transformation of biomass in aqueous medium under mild reaction conditions.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Instituto de Tecnología Química (ITQ), Instituto de Investigaciones Químicas (IIQ), Consejo Superior de Investigaciones Científicas (CSIC), Universitat Politècnica de València (UPV) and Universidad de Sevilla (US) for the facilities. We also thank the electron microscopy service of the UPV for TEM analysis. The authors also acknowledge Agencia Estatal de Investigación, Ministerio de Ciencia e Innovación (PID2021-126080OA-I00, TED2021-132087A-I00 and RYC2020-030031-I), Junta de Andalucía (ProyExcel_00706, P20_01027 and PYC 20 RE 060 UAL) and the University of Seville (VI PP A.TALENTO; 2022/00000400) for financial support. C. Cerezo-Navarrete is grateful for the Generalitat Valenciana predoctoral fellowship (GVA: ACIF/2019/076).References
- (a) D. Astruc, Nanoparticles and Catalysis, Wiley-VCH, Weinheim, Germany, 2008 Search PubMed; (b) D. Astruc, Introduction: Nanoparticles in Catalysis, Chem. Rev., 2020, 120, 461–463 CrossRef CAS PubMed; (c) P. Serp and K. Philippot, Nanomaterials in Catalysis, Wiley-VCH, Weinheim, Germany, 2013 CrossRef.
- (a) G. Schmid, Nanoparticles: From Theory to Application, Wiley-VCH, Weinheim, Germany, 2006 Search PubMed; (b) L. Liu and A. Corma, Metal Catalysts for Heterogeneous Catalysis: From Single Atoms to Nanoclusters and Nanoparticles, Chem. Rev., 2018, 118, 4981–5079 CrossRef CAS PubMed.
- (a) B. Ni and X. Wang, Face the Edges: Catalytic Active Sites of Nanomaterials, Adv. Sci., 2015, 2, 1500085 CrossRef PubMed; (b) L. Liu, M. López-Haro, C. W. Lopes, C. Li, P. Concepcion, L. Simonelli, J. J. Calvino and A. Corma, Regioselective generation and reactivity control of subnanometric platinum clusters in zeolites for high-temperature catalysis, Nat. Mater., 2019, 18, 866–873 CrossRef CAS PubMed; (c) M. R. Axet and K. Philippot, Catalysis with Colloidal Ruthenium Nanoparticles, Chem. Rev., 2020, 120, 1085–1145 CrossRef CAS PubMed; (d) L. Liu and A. Corma, Confining isolated atoms and clusters in crystalline porous materials for catalysis, Nat. Rev. Mater., 2021, 6, 244–263 CrossRef CAS.
- (a) J. Mazario, M. Parreño Romero, P. Concepción, M. Chávez-Sifontes, R. A. Spanevello, M.-B. Comba, A. G. Suárez and M. E. Domine, Tuning zirconia-supported metal catalysts for selective one-step hydrogenation of levoglucosenone, Green Chem., 2019, 21, 4769–4785 RSC; (b) M. Khawaji and D. Chadwick, Selective oxidation using Au-Pd catalysts: Role of the support in the stabilization of colloidal Au-Pd NPs, Catal. Today, 2020, 348, 203–211 CrossRef CAS; (c) A. Bordet and W. Leitner, Metal Nanoparticles Immobilized on Molecularly Modified Surfaces: Versatile Catalytic Systems for Controlled Hydrogenation and Hydrogenolysis, Acc. Chem. Res., 2021, 54, 2144–2157 CrossRef CAS PubMed; (d) L. Liu, M. Lopez-Haro, J. A. Perez-Omil, M. Boronat, J. J. Calvino and A. Corma, Direct assessment of confinement effect in zeolite-encapsulated subnanometric metal species, Nat. Commun., 2022, 13, 821 CrossRef CAS PubMed.
- L. M. Martínez-Prieto, M. Puche, C. Cerezo-Navarrete and B. Chaudret, Uniform Ru nanoparticles on N-doped graphene for selective hydrogenation of fatty acids to alcohols, J. Catal., 2019, 377, 429–437 CrossRef.
- M. Sankar, N. Dimitratos, P. J. Miedziak, P. P. Wells, C. J. Kiely and G. J. Hutchings, Designing bimetallic catalysts for a green and sustainable future, Chem. Soc. Rev., 2021, 41, 8099–8139 RSC.
- (a) F. Gao and D. W. Goodman, Pd-Au bimetallic catalysts: understanding alloy effects from planar models and (supported) nanoparticles, Chem. Soc. Rev., 2012, 41, 8009–8020 RSC; (b) X. Qi, R. M. Axet, K. Philippot, P. Lecante and P. Serp, Seed-mediated synhtesis of bimetallic ruthenium-platinum nanoparticles efficient in cinnamaldehyde selective hydrogenation, Dalton Trans., 2014, 43, 9283–9295 RSC; (c) N. M. Wilson, P. Priyadarshini, S. Kunz and D. W. Flaherty, Direct synthesis of H2O2 on Pd and AuxPd1 clusters: Understanding effects of alloying Pd with Au, J. Catal., 2018, 357, 163–175 CrossRef.
- (a) K. Föttinger and G. Rupprechter, In Situ Spectroscopy of Complex Surface Reactions on Supported Pd-Zn, Pd-Ga, and Pd(Pt)-Cu Nanoparticles, Acc. Chem. Res., 2014, 47, 3071–3079 CrossRef PubMed; (b) M. Zhou, C. Li and J. Fang, Noble-Metal Based Random Alloy and Intermetallic Nanocrystals: Syntheses and Applications, Chem. Rev., 2021, 121, 736–795 CrossRef CAS PubMed; (c) I. Mustieles, J. M. Asensio and B. Chaudret, Bimetallic Nanoparticles Associating Noble Metals and First-Row Transition Metals in Catalysis, ACS Nano, 2021, 15, 3550–3556 CrossRef PubMed.
- C. Cerezo-Navarrete, Y. Mathieu, M. Puche, C. Morales, P. Concepción, L. M. Martínez-Prieto and A. Corma, Controlling the selectivity of bimetallic platinum-ruthenium nanoparticles supported on N-doped graphene by adjusting their metal composition, Catal. Sci. Technol., 2021, 11, 494–505 RSC.
- (a) L. M. Martínez-Prieto and B. Chaudret, Organometallic Ruthenium Nanoparticles: Synthesis, Surface Chemistry, and Insights into Ligand Coordination, Acc. Chem. Res., 2018, 51, 376–384 CrossRef PubMed; (b) L. M. Martínez-Prieto and P. W. N. M. van Leeuwen, Ligand Effects in Ruthenium Nanoparticle Catalysis. Recent Advances in Nanoparticle Catalysis, Springer, Berlin, Germany, 2020 Search PubMed.
- (a) C. A. Schoenbaum, D. K. Schwartz and J. W. Medlin, Controlling the Surface Environment of Heterogeneous Catalysts Using Self-Assembled Monolayers, Acc. Chem. Res., 2014, 47, 1438–1445 CrossRef CAS PubMed; (b) S. Kunz, Supported, Ligand-Functionalized Nanoparticles: An Attempt to Rationalize the Application and Potential of Ligands in Heterogeneous Catalysis, Top. Catal., 2016, 59, 1671–1685 CrossRef CAS; (c) J. B. Ernst, S. Muratsugu, F. Wang, M. Tada and F. Glorius, Tunable Heterogeneous Catalysis: N-Heterocyclic Carbenes as Ligands for Supported Heterogenenous Ru/K-Al2O3 Catalysts To Tune Reactivity and Selectivity, J. Am. Chem. Soc., 2016, 138, 10718–10721 CrossRef CAS PubMed; (d) P. Tegeder, M. Freitag, K. M. Chepiga, S. Muratsugu, N. Möller, S. Lamping, M. Tada, F. Glorius and B. J. Ravoo, N-Heterocyclic Carbene-Modified Au-Pd Alloy Nanoparticles and, Their Application as Biomimetic and Heterogeneous Catalysts, Chem. – Eur. J., 2018, 24, 18682–18688 CrossRef CAS PubMed; (e) R. Ye, A. V. Zhukhovitskiy, R. V. Kazantsev, S. C. Fakra, B. B. Wickemeyer, F. D. Toste and G. A. Somorjai, Supported Au Nanoparticles with N-Heterocyclic Carbene Ligands as Active and Stable Heterogeneous Catalysts for Lactonization, J. Am. Chem. Soc., 2018, 140, 4144–4149 CrossRef CAS PubMed; (f) D. Ventura-Espinosa, S. Martín, H. García and J. A. Mata, Ligand effects in the stabilization of gold nanoparticles anchored on the surface of graphene: Implications in catalysis, J. Catal., 2021, 394, 113–120 CrossRef CAS.
- A. Palazzolo, T. Naret, M. Daniel-Bertrand, D. Buisson, S. Tricard, P. Lesot, Y. Coppel, B. Chaudret, S. Feuillastre and G. Pieters, Tuning the Reactivity of a Heterogeneous Catalyst using N-Heterocyclic Carbene Ligands for C-H Activation Reactions, Angew. Chem., 2020, 132, 21065–21070 CrossRef.
- A. García-Zaragoza, C. Cerezo-Navarrete, A. Mollar-Cuni, P. Oña-Burgos, J. A. Mata, A. Corma and L. M. Martínez-Prieto, Tailoring graphene-supported Ru nanoparticles by functionalization with pyrene-tagged N-heterocyclic carbenes, Catal. Sci. Technol., 2022, 12, 1257–1270 RSC.
- (a) Y. I. Yermakov, Supported Catalysts Obtained by Interaction of Organometallic Compounds of Transition Elements with Oxide Supports, Catal. Rev., 1976, 13, 77–120 CrossRef; (b) Y. I. Yermakov and B. N. Kuznetsov, Supported metallic catalysts prepared by decomposition of surface organometallic complexes, J. Mol. Catal. A: Chem., 1980, 9, 13–40 CrossRef; (c) A. K. Smith, B. Besson, J. M. Basset, R. Psaro, A. Fusi and R. Ugo, Surface supported metal cluster carbonyls: the surface organometallic chemistry of polymetallic and monometallic osmium carbonyl species formed by depositing various osmium clusters on silica and alumina, J. Organomet. Chem., 1980, 192, C31–C34 CrossRef CAS.
- (a) J. M. Basset and R. Ugo, On the Origins and Development of “Surface Organometallic Chemistry” in Surface Organometallic Chemistry, Wiley-VCH, Weinheim, Germany, 2009 Search PubMed; (b) C. Copéret, Single-Sites and Nanoparticles at Tailored interfaces Prepared via Surface Organometallic Chemistry from Thermolytic Molecular Precursors, Acc. Chem. Res., 2019, 52, 1697–1708 CrossRef PubMed; (c) C. Cóperet, Fuels and energy carriers from single-site catalysts prepared via surface organometallic chemistry, Nat. Energy, 2019, 4, 1018–1024 CrossRef.
- (a) J. D. A. Pelletier and J. M. Basset, Catalysis by Design: Well-Defined Single-Site Heterogeneous Catalysts, Acc. Chem. Res., 2016, 49, 664–677 CrossRef CAS PubMed; (b) M. K. Samantaray, V. D'Elia, E. Pump, L. Falivene, M. Harb, S. O. Chikh, L. Cavallo and J. M. Basset, The Comparison between Single Atom Catalysis and Surface Organometallic Chemistry, Chem. Rev., 2020, 120, 734–813 CrossRef CAS PubMed.
- (a) M. Chabanas, A. Baudouin, C. Copéret and J. M. Basset, A highly Active Well-Definer Rhenium Heterogeneous Catalyst for Olefin Metathesis Prepared via Surface Organometallic Chemistry, J. Am. Chem. Soc., 2001, 123, 2062–2063 CrossRef CAS PubMed; (b) J. M. Basset, C. Copéret, D. Soulivong, M. Taoufik and J. T. Cazat, Metathesis of Alkanes and Related Reactions, Acc. Chem. Res., 2010, 43, 323–334 CrossRef CAS PubMed.
- C. Copéret, M. Chabanas, P. R. Saint-Arroman and J. M. Basset, Homogeneous and Heterogeneous Catalysis: Bridging the Gap through Surface Organometallic Chemistry, Angew. Chem., Int. Ed., 2003, 42, 156–181 CrossRef PubMed.
- A. Corma, M. T. Navarro and M. Renz, Lewis acidic Sn(IV) centers-grafted onto MCM-41 as catalytic sites for the Baeyer-Villiger oxidation with hydrogen peroxide, J. Catal., 2003, 219, 242–246 CrossRef CAS.
- (a) L. M. Martínez-Prieto, J. Marbaix, J. M. Asensio, C. Cerezo-Navarrete, P. F. Fazzini, K. Soulantica, B. Chaudret and A. Corma, Ultrastable Magnetic Nanoparticles Encapsulated in Carbon for Magnetically Induced Catalysis, ACS Appl. Nano Mater., 2020, 3, 7076–7087 CrossRef PubMed; (b) S. Chen, X. Chang, G. Sun, T. Zhang, Y. Xu, Y. Wang, C. Pei and J. Gong, Propane dehydrogenation: catalyst development, new chemistry, and emerging technologies, Chem. Soc. Rev., 2021, 50, 3315–3354 RSC.
- (a) C. Nédez, A. Theolier, F. Lefebvre, A. Choplin, J. M. Basset and J. F. Joly, Surface organometallic chemistry of tin: reactivity of tetraalkyltin complexes and tributyltin hydride toward silica, J. Am. Chem. Soc., 1993, 115, 722–729 CrossRef; (b) Y. Xu, X. Bao and L. Lin, Direct conversion of methane under nonoxidative conditions, J. Catal., 2003, 216, 386–395 CrossRef CAS; (c) F. Rataboul, A. Baudouin, C. Thieuleux, L. Veyre, C. Copéret, J. Thivolle-Cazat, J. M. Basset, A. Lesage and L. Emsley, Molecular Understanding of the Formation of Surface Zirconium Hydrides upon Thermal Treatment under Hydrogen of [(SiO)Zr(CH2tBu)3] by Using Advanced Solid-State NMR Techniques, J. Am. Chem. Soc., 2004, 126, 12541–12550 CrossRef CAS PubMed.
- H. Zhu, D. H. Anjum, Q. Wang, E. Abou-Hamad, L. Emsley, H. Dong, P. Laveille, L. Li, A. K. Samal and J. M. Basset, Sn surface-enriched Pt-Sn bimetallic nanoparticles as a selective and stable catalyst for propane dehydrogenation, J. Catal., 2014, 320, 52–62 CrossRef CAS.
- (a) R. Burch and L. C. Garla, Platinum-tin reforming catalysts: II. Activity and selectivity in hydrocarbon reactions, J. Catal., 1981, 71, 360–372 CrossRef CAS; (b) J. J. H. B. Sattler, J. Ruiz-Martinez, E. Santillan-Jimenez and B. M. Weckhuysen, Catalytic Dehydrogenation of Light Alkanes on Metals and Metal Oxides, Chem. Rev., 2014, 114, 10613–10653 CrossRef CAS PubMed.
- (a) Z. Nawaz, X. Tang, Q. Zhang, D. Wang and W. Fei, SAPO-34 supported Pt-Sn-based novel catalyst for propane dehydrogenation to propylene, Catal. Commun., 2009, 10, 1925–1930 CrossRef CAS; (b) M. Yang, Y. Zhu, X. Zhou, Z. Sui and D. Chen, First-Principles Calculations of Propane Dehydrogenation over PtSn Catalysts, ACS Catal., 2012, 2, 1247–1258 CrossRef CAS.
- (a) G. W. Huber, S. Iborra and A. Corma, Synthesis of Transportation Fuels from Biomass: Chemistry, Catalysts, and Engineering, Chem. Rev., 2006, 106, 4044–4098 CrossRef CAS PubMed; (b) D. M. Alonso, J. Q. Bond and J. A. Dumesic, Catalytic conversion of biomass to biofuels, Green Chem., 2010, 12, 1493–1513 RSC; (c) P. Gazellot, Conversion of biomass to selected chemical products, Chem. Soc. Rev., 2012, 41, 1538–1558 RSC; (d) K. L. Luska, P. Migowski and W. Leitner, Ionic liquid-stabilized nanoparticles as catalysts for the conversion of biomass, Green Chem., 2015, 17, 3195–3206 RSC; (e) J. G. Rosenboom, D. K. Hohl, P. Fleckenstein, G. Stori and M. Morbidelli, Bottle-grade polyethylene furanoate from ring-opening polymerisation of cyclic oligomers, Nat. Commun., 2018, 9, 2701 CrossRef PubMed; (f) H. Cai, C. Li, A. Wang and T. Zhang, Biomass into chemicals: One-pot production of furan-based diols from carbohydrates via tandem reactions, Catal. Today, 2014, 234, 59–65 CrossRef CAS.
- (a) J. C. Serrano-Ruiz, R. Luque and A. Sepúlveda-Escribano, Transformations of biomass-derived platform molecules: from high added-value chemicals to fuels via, aqueous-phase processing, Chem. Soc. Rev., 2011, 40, 5266–5281 RSC; (b) S. Dutta, S. De and B. Saha, A Brief Summary of the Synthesis of Polyester Builidng-Block Chemicals and Biofuels from 5-Hydroxymethylfurfural, ChemPlusChem, 2012, 77, 259–272 CrossRef CAS; (c) J. Slak, B. Pomeroy, A. Kostyniuk, M. Grilc and B. Likozar, A review of bio-refining process intensification in catalytic conversion reactions, separations and purification of hydroxymethylfurfural (H.MF) and furfural, Chem. Eng. J., 2022, 429, 132325 CrossRef CAS.
- (a) Y. Wang, H. Wang, X. Kong and Y. Zhu, Catalytic Conversion of 5-Hydroxymethylfurfural to High-Value Derivatives by Selective Activation of C-O, C=O, and C=C Bonds, ChemSusChem, 2022, 15, e202200421 CAS; (b) A. Messori, A. Fasolini and R. Mazzoni, Advances in Catalytic Routes for the Homogeneous Green conversion of the Bio-Based Platform 5-Hydroxymethylfurfural, ChemSusChem, 2022, 15, e202200228 CrossRef CAS.
- (a) C. Moreau, M. N. Belgacem and A. Gandini, Recent catalytic advances in the chemistry of substituted furans from carbohydrates and in the ensuing polymers, Top. Catal., 2004, 27, 11–30 CrossRef CAS; (b) T. Buntara, S. Noel, P. H. Phua, I. Meliá-Cabrera, J. G. de Vries and H. J. Heeres, Caprolactam from Renewable Resources: Catalytic Conversion of 5-Hydroxymethylfurfural into Caprolactone, Angew. Chem., 2011, 123, 7221–7225 CrossRef; (c) L. Hu, J. Xu, S. Zhou, A. He, X. Tang, L. Lin, J. Xu and Y. Zhao, Catalytic Advances in the Production and Application of Biomass-Derived 2,5-Dihydroxymethylfuran, ACS Catal., 2018, 8, 2959–2980 CrossRef CAS.
- (a) M. Balakrishnan, E. R. Sacia and A. T. Bell, Etherification and reductive etherification of 5-(hydroxymethyl)furfural: 5-(alkoxymethyl)furfurals and 2,5-bis(alkoxymethyl)furans as potential bio-diesel candidates, Green Chem., 2012, 14, 1626–1634 RSC; (b) J. Ohyama, A. Esaki, Y. Yamamoto, S. Arai and A. Satsuma, Selective hydrogenation of 2-hydroxymethyl-5-furfural to 2,5-bis(hydroxymethyl)furan over gold sub-nano clusters, RSC Adv., 2013, 3, 1033–1036 RSC; (c) K. Gupta, R. K. Rai and S. K. Singh, Metal Catalysts for the Efficient Transformation of Biomass-derived HMF and Furfural to Value Added Chemicals, ChemCatChem, 2018, 10, 2326–2349 CrossRef CAS; (d) Q. Chen, T. Li, Y. Zhou, Y. Bi, S. Guo, X. Liu, H. Kang, M. Wang, L. Liu, E. Xing and D. Yang, Selective Hydrogenation of 5-Hydroxymethylfurfural via Zeolite Encapsulation to Avoid Further Hydrodehydroxylation, Ind. Eng. Chem. Res., 2020, 59, 12004–12012 CrossRef CAS.
- (a) R. Alamillo, M. Tucker, M. Chia, Y. Pagán-Torres and J. Dumesic, The selective hydrogenation of biomass-derived 5-hydroxymethylfurfural using heterogeneous catalysts, Green Chem., 2012, 14, 1413–1419 RSC; (b) Q. Cao, W. Liang, J. Guan, L. Wang, Q. Qu, Z. Zhang, X. Wang and X. Mu, Catalytic synthesis of 2,5-bis-methoxymethylfuran: A promising cetane number improver for diesel, Appl. Catal., A, 2014, 481, 49–53 CrossRef CAS; (c) C. Sarkar, R. Paul, S. C. Shit, Q. T. Trinh, P. Koley, B. S. Rao, A. M. Beale, C.-W. Pao, A. Banerjee and J. Mondal, Navigating Copper-Atom-Pair Structural Effect inside a Porous Organic Polymer Cavity for Selective Hydrogenation of Biomass-Derived 5-Hydroxymethylfurfural, ACS Sustainable Chem. Eng., 2021, 9, 2136–2151 CrossRef CAS.
- T. Suoranta, M. Niemelä and P. Perämäki, Comparison of digestion methods for the determination of ruthenium in catalyst materials, Talanta, 2014, 119, 425–429 CrossRef CAS PubMed.
- (a) S. Stankovich, D. A. Dikin, R. D. Piner, K. A. Kohlhaas, A. Kleinhammes, Y. Jia, Y. Wu, S. B. T. Nguyen and R. S. Ruoff, Synthesis of graphene-based nanosheets via chemical reduction of exfoliated graphite oxide, Carbon, 2007, 45, 1558–1565 CrossRef CAS; (b) S. Navalon, A. Dhakshinamoorthy, M. Alvaro and H. Garcia, Metal nanoparticles supported on two-dimensional Graphenes as heterogeneous catalysts, Coord. Chem. Rev., 2016, 312, 99–148 CrossRef CAS.
- (a) F. Bernardi, J. D. Scholten, G. H. Fecher, J. Dupont and J. Morais, Probing the chemical interaction between iridium nanoparticles and ionic liquid by XPS analysis, Chem. Phys. Lett., 2009, 479, 113–116 CrossRef CAS; (b) A. Rehling, K. Schaepe, L. Rakers, B. VonhÅren, P. Tegeder, B. J. Ravoo and F. Glorius, Modular Bidentate Hybrid NHC-Thioether Ligands for the Stabilization of Palladium Nanoparticles in vVarious Solvents, Angew. Chem., Int. Ed., 2016, 55, 5856–5860 CrossRef PubMed; (c) L. M. Martínez-Prieto, E. A. Baquero, G. Pieters, J. C. Flores, E. de Jesffls, C. Nayral, F. Delpech, P. W. N. M. van Leeuwen, G. Lippens and B. Chaudret, Monitoring of nanoparticle reactivity in solution: interaction of L-lysine and Ru nanoparticles probed by chemical shift perturbation parallels regioselective H/D exchange, Chem. Commun., 2017, 53, 5850–5853 RSC.
- See for example: (a) H. Willemen, D. F. Van De Vondel and G. P. Van Der Kelen, An ESCA study of tin compounds, Inorg. Chim. Acta, 1979, 34, 175–180 CrossRef CAS; (b) M. Andersson, J. Blomquist, B. Folkesson, R. Larsson and P. Sundberg, Esca, mössbauer and infrared spectroscopic investigations of a series of tin complexes, J. Electron Spectrosc. Relat. Phenom., 1986, 40, 385–396 CrossRef CAS.
- (a) F. Dassenoy, K. Philippot, T. O. Ely, C. Amiens, P. Lecante, E. Snoeck, A. Mosset, M.-J. Casanove and B. Chaudret, Platinum nanoparticles stabilized by CO and octanethiol, ligands or polymers: FT-IR, NMR, HREM and WAXS studies, New J. Chem., 1998, 22, 703–711 RSC; (b) S. Kinayyigit, P. Lara, P. Lecante, K. Philippot and B. Chaudret, Probing the surface of platinum nanoparticles with 13CO by solid-state NMR and IR spectroscopies, Nanoscale, 2014, 6, 539–546 RSC.
- (a) J. S. Bradley, E. W. Hill, C. Klein, B. Chaudret and A. Duteil, Highly Dispersed Metal Colloids: Spectroscopy and Surface Chemistry in Solution, Stud. Surf. Sci. Catal., 1993, 75, 969–979 CrossRef CAS; (b) F. Novio, K. Philippot and B. Chaudret, Location and Dynamics of CO Co-ordination on Ru Nanoparticles A Solid State NMR Study, Catal. Lett., 2010, 140, 1–7 CrossRef CAS.
- (a) P. K. Babu, H. S. Kim, E. Oldfield and A. Wieckowski, Electronic Alterations Caused by Ruthenium in Pt-Ru Alloy Nanoparticles as Revealed by Electrochemical NMR, J. Phys. Chem. B, 2003, 107, 7595–7600 CrossRef CAS; (b) L. M. Martinez-Prieto, I. Cano, A. Marquez, E. A. Baquero, S. Tricard, L. Cusinato, I. del Rosal, R. Poteau, Y. Coppel, K. Philippot, B. Chaudret, J. Campora and P. W. N. M. van Leeuwen, Zwitterionic amidinates as effective ligands for platinum nanoparticle hydrogenation catalysts, Chem. Sci., 2017, 8, 2931–2941 RSC; (c) L. M. Martínez-Prieto, L. Rakers, A. M. López-Vinasco, I. Cano, Y. Coppel, K. Philippot, F. Glorius, B. Chaudret and P. W. N. M. van Leeuwen, Soluble Platinum Nanoparticles Ligated by Long-Chain N-Heterocyclic Carbenes as Catalysts, Chem. – Eur. J., 2017, 23, 12779–12786 CrossRef PubMed.
- R. J. van Putten, J. C. van der Waal, E. de Jong, C. B. Rasrendra, H. J. Heeres and J. G. de Vries, Hydroxymethylfurfural, A Versatile Platform Chemical Made from Renewable Resources, Chem. Rev., 2013, 113, 1499–1597 CrossRef CAS PubMed.
- (a) C. Zeng, H. Seino, J. Ren, K. Hatanaka and N. Yoshie, Bio-Based Furan Polymers with Self-Heating Ability, Macromolecules, 2013, 46, 1794–1802 CrossRef CAS; (b) T. Wang, J. Wei, H. Liu, Y. Feng, X. Tang, X. Zeng, Y. Sun, T. Lei and L. Lin, Synthesis of renewable monomer 2,5-bishydroxymethylfuran from highly concentrated 5-hydroxymethylfurfural in deep eutectic solvents, J. Ind. Eng. Chem., 2020, 81, 93–98 CrossRef CAS.
- (a) T. Kitanosono, K. Masuda, P. Xu and S. Kobayashi, Catalytic Organinc Reactions in Water toward Sustainable Society, Chem. Rev., 2018, 118, 679–746 CrossRef CAS PubMed.
- (a) M. Cardona-Farreny, P. Lecante, J. Esvan, C. Dinoi, I. del Rosal, R. Poteau, K. Philippot and M. R. Axet, Bimetallic RuNi nanoparticles as catalysts for upgrading biomass: metal dilution and solvent effects on selectivity shifts, Green Chem., 2021, 23, 8480–8500 RSC; (b) C. Cerezo-Navarrete, I. M. Marin, H. García-Miquel, A. Corma, B. Chaudret and L. M. Martínez-Prieto, Magnetically Induced Catalytic Reduction of Biomass-Derived Oxygenated Compounds in Water, ACS Catal., 2022, 12, 8462–8475 CrossRef CAS.
- Turn over frequencies (TOFs) were calculated considering the number of moles of HMF consumed and the number of moles of metal as previously reported: A. P. Umpierre, E. de Jesús and J. Dupont, Turnover Numbers and Soluble Metal Nanoparticles, ChemCatChem, 2011, 3, 1413–1418 CrossRef CAS.
- M. Chatterjee, T. Ishizaka and H. Kawanami, Selective hydrogenation of 5-hydroxymethylfurfural to 2,5-bis-(hydroxymethyl)furan using Pt/MCM-41 in an aqueous medium: a simple approach, Green Chem., 2014, 16, 4734–4739 RSC.
- (a) H. Verbeek and W. M. H. Sachtler, The study of the alloys of platinum and tin chemisorption, J. Catal., 1976, 42, 257–267 CrossRef CAS; (b) F. Humblot, F. Lepeltier, J. P. Candy, J. Corker, O. Clause, F. Bayard and J. M. Basset, Surface Organometallic Chemistry on Metals: Formation of a Stable Sn(n,-C4H9) Fragment as a Precursor of Surface Alloy Obtained by Stepwise Hydrogenolysis of Sn(n-C4H9)4 on a Platinum Particle Supported on Silica, J. Am. Chem. Soc., 1998, 120, 137–146 CrossRef CAS; (c) F. Humblot, J. P. Candy, F. Le Peltier, B. Didillon and J. M. Basset, Surface Organometallic Chemistry on Metals: Selective Dehydrogenation of Isobutane into Isobutene on Bimetallic Catalysts Prepared by Reaction of Tera n-Butyltin on Silica-Supported Platinum Catalyst, J. Catal., 1998, 179, 459–468 CrossRef CAS; (d) E. M. Crabb, R. Marshall and D. Thompsett, Carbon Monoxide Electro-oxidation Properties of Carbon-Supported PtSn Catalyst Prepared Using Surface Organometallic Chemistry, J. Electrochem. Soc., 2000, 147, 4440–4447 CrossRef CAS; (e) J. Singh, R. C. Nelson, B. C. Vicente, S. L. Scott and J. A. van Bokhoven, Electronic structure of alumina-supported monometallic Pt and bimetallic PtSn catalysts under hydrogen and carbon monoxide environment, Phys. Chem. Chem. Phys., 2010, 12, 5668–5677 RSC.
- G. Aguilar-Ríos, M. Valenzuela, P. Salas, H. Armendáriz, P. Bosch, G. del Toro, R. Silva, V. Bertín, S. Castillo, A. Ramírez-Solís and I. Schifter, Hydrogen interactions and catalytic properties of platinum-tin supported on zinc aluminate, Appl. Catal., A, 1995, 127, 65–75 CrossRef.
- (a) M. A. P. da Silva and M. Schmal, Reduction of NO by CO on Pt-MoO3/γ-Al2O3 catalysts, Catal. Today, 2003, 85, 31–37 CrossRef; (b) O. A. Bariås, A. Holmen and E. A. Blekkan, Propane Dehydrogenation over Supported Pt and Pt-Sn Catalysts: Catalyst Preparation, Characterization, and Activity Measurements, J. Catal., 1996, 158, 1–12 CrossRef.
- Y. Zhang, A. Rezayan, K. Wang, J. Wang, C. C. Xu and R. Nie, On-Demand, Highly Tunable, and Selective 5-Hydroxymethylfurfural Hydrogenation to Furan Diols Enabled by Ni and Ni3Ga Alloy Catalysts, ACS Catal., 2023, 13, 803–814 CrossRef CAS.
- (a) P. Claus, Selective hydrogenation of α,β-unsaturated aldehydes and other C=O and C=C bonds containing compounds, Top. Catal., 1998, 5, 51–62 CrossRef CAS; (b) P. V. Samant, M. F. R. Pereira and J. L. Figueiredo, Mesoporous carbon supported Pt and Pt-Sn catalysts for hydrogenation of cinnamaldehyde, Catal. Today, 2005, 102–103, 183–188 CrossRef CAS; (c) P. Mäki-Arvela, J. Hájek, T. Salmi and D. Y. Murzin, Chemoselective hydrogenation of carbonyl compounds over heterogeneous catalysts, Appl. Catal., A, 2005, 292, 1–49 CrossRef; (d) J. P. Stassi, P. D. Zgolicz, S. R. de Miguel and O. A. Scelza, Formation of different promoted metallic phases in PtFe and PtSn catalysts supported on carbonaceous materials used for selective hydrogenation, J. Catal., 2013, 306, 11–29 CrossRef CAS; (e) T. B. L. W. Marinelli and V. Ponec, A Study on the Selectivity in Acrolein Hydrogenation on Platinum Catalysts: A Model for Hydrogenation of α,β-Unsatured Aldehydes, J. Catal., 1995, 156, 51–59 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3nr02083e |
| This journal is © The Royal Society of Chemistry 2023 |