DOI:
10.1039/D3NR01625K
(Paper)
Nanoscale, 2023,
15, 10749-10754
Histidine–DNA nanoarchitecture as laccase mimetic DNAzymes†
Received
8th April 2023
, Accepted 10th June 2023
First published on 16th June 2023
Abstract
Herein, we report on the construction of Cu–histidine (His)–DNA hybrids as laccase-mimetic DNAzymes. Cu–His–DNAzymes showed remarkable activity in a colorimetric oxidation reaction between 2,4-dichlorophenol and 4-aminoantipyrine. Our results provide new insights for the systematic construction of tailor-made active sites for biomimetics.
Introduction
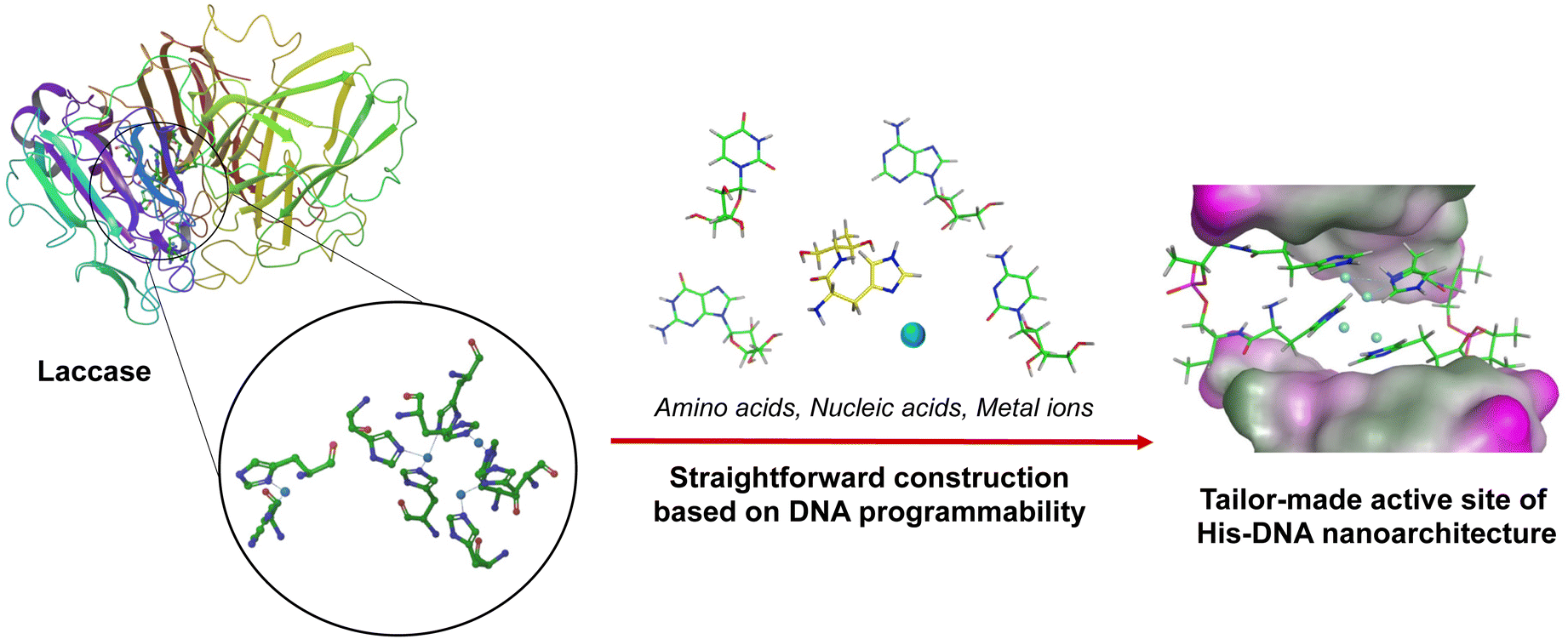
Nucleic acids have received immense attention as versatile biomaterials that can play several roles across the various sciences.1 They offer many advantages, including automated chemical synthesis, thermal stability, programmability, and creation of a diverse range of tertiary structures constructed from hydrogen bonding between the four bases (i.e., A, T/U, G, C). By leveraging these intrinsic features, DNA nanotechnology has made momentous advances in chemical biology, materials science, medicinal chemistry, and pharmacology.2 In addition, recent studies have demonstrated the previously unnoticed function of DNA as a bioscaffold for catalysis.3 These papers have reported the versatility of DNA-based hybrid catalysts for various chemical reactions, such as Diels–Alder reactions, Friedel–Crafts alkylation, and Michael addition.4 In addition, the chemical modification of native nucleic acids with various functional moieties has facilitated further improvements to their capability. These functional moieties can be sequence-specifically implemented on nucleic acid strands due to advances in phosphoramidite chemistry and solid-phase DNA synthesis.5 These pioneering works on DNA/RNA modification with ligands, fluorophores, and the precursors of biorthogonal reactions have created an opportunity for modulating molecular machinery using synthetic oligonucleotides.6 Amino acid residues are very promising building blocks for modifying DNA oligonucleotides because they are abundant natural materials that display diverse functional characteristics.7 By hybridizing amino acid residues with oligonucleotides, the utility and effectiveness of nucleic acids can be reinforced. For instance, the conjugation of cationic polypeptides to nucleic acid-based therapeutic drugs led to charge-neutralization and thus improved cellular uptake efficiency.8 Nucleopeptides have also been widely used as a unit for fabricating supramolecular nanocarriers, fibers, and hydrogels through alternative hydrogen bond formation between amino acid side chains and nucleotides.9 Due to the broad potential of biomedical and industrial applications, a handy and straightforward approach is required to synthesize amino acids-conjugated oligonucleotides. Our group has been involved in the development of modified oligonucleotides and has established a systematic strategy to incorporate amino acid residues into oligonucleotides via a D-threoninol linker.10 We have named these amino acid-conjugated DNA as amino acid–nucleic acid hybrids (ANHs). In a previous study we synthesized histidine-conjugated DNA oligonucleotides and demonstrated their capability as metal reservoirs. Histidine-containing DNA (His–DNA) was successfully used as an asymmetric catalyst for Diels–Alder reactions. His–DNA–hemin complexes showed remarkable catalytic performance in 2,2′-azino-bis(3-ethylbenzothiazoline-6-sulfonic acid) oxidation. We have further exploited ANHs to construct an advanced aptamer library as a post-SELEX modification approach. We have generated amino acid-functionalized thrombin-binding aptamers and successfully demonstrated their utility with enhancement of thrombin inhibition activity and nuclease resistance.11 Encouraged by the remarkable performance of ANHs, we have moved forward to the goal of exploiting ANHs that can mimic enzymes by the construction of active sites on DNA strands with key amino acids. Laccase is a class of phenol oxidases widely applied in environmental remediation by catalyzing the oxidation of toxic phenols and amines.12 Because laccase reactions use molecular oxygen as the electron acceptor to release water without the production of hydrogen peroxide, the use of laccase is attracting attention in terms of “green” catalysis. This has led to efforts toward the development of laccase-mimetic materials.16
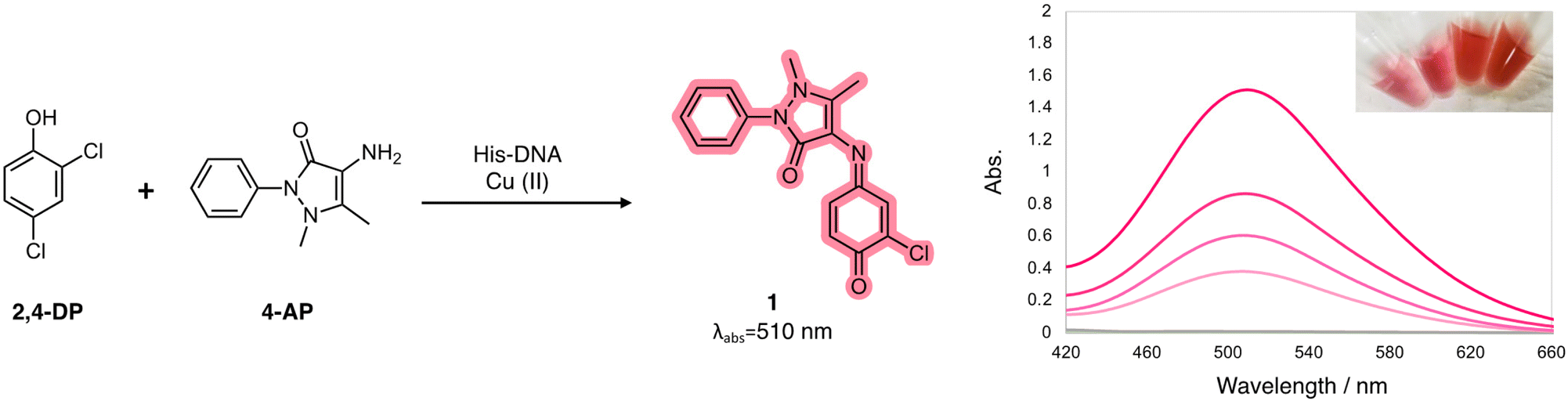
For instance, enzyme immobilization and directed evolution have been explored to overcome vulnerable aspects of natural enzymes, and metal–organic framework-based nanozymes were reported to expand applications of laccase.13,17 In the active site of laccase, four copper ions are coordinated with multiple histidine residues. We postulated a rational design for a laccase-mimetic apoenzyme by incorporating histidine residue on the various DNA structures. It would be activated as the holoenzyme for catalysis via the coordination of copper ions (Fig. 1). Here we report on the construction of advanced Cu–histidine (His)–DNA hybrids as laccase-mimetic DNAzymes and their catalytic performance in a standard colorimetric oxidation reaction between 2,4-dichlorophenol and 4-aminoantipyrine (Scheme 1).
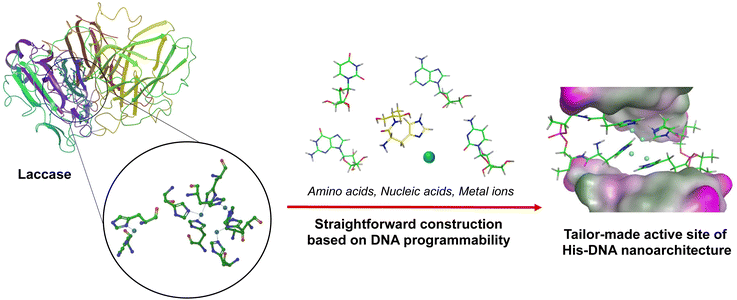 |
| | Fig. 1 A laccase-mimetic Cu–His–DNAzyme containing multiple histidine residues in the active sites. | |
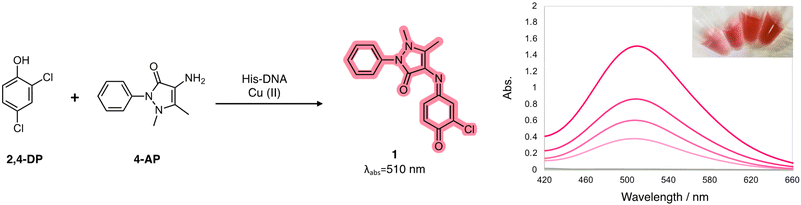 |
| | Scheme 1 Colorimetric oxidation reaction between phenolic compound 1 and aromatic amine 2 catalyzed by laccase-mimetic Cu–His–DNAzymes. | |
Results and discussion
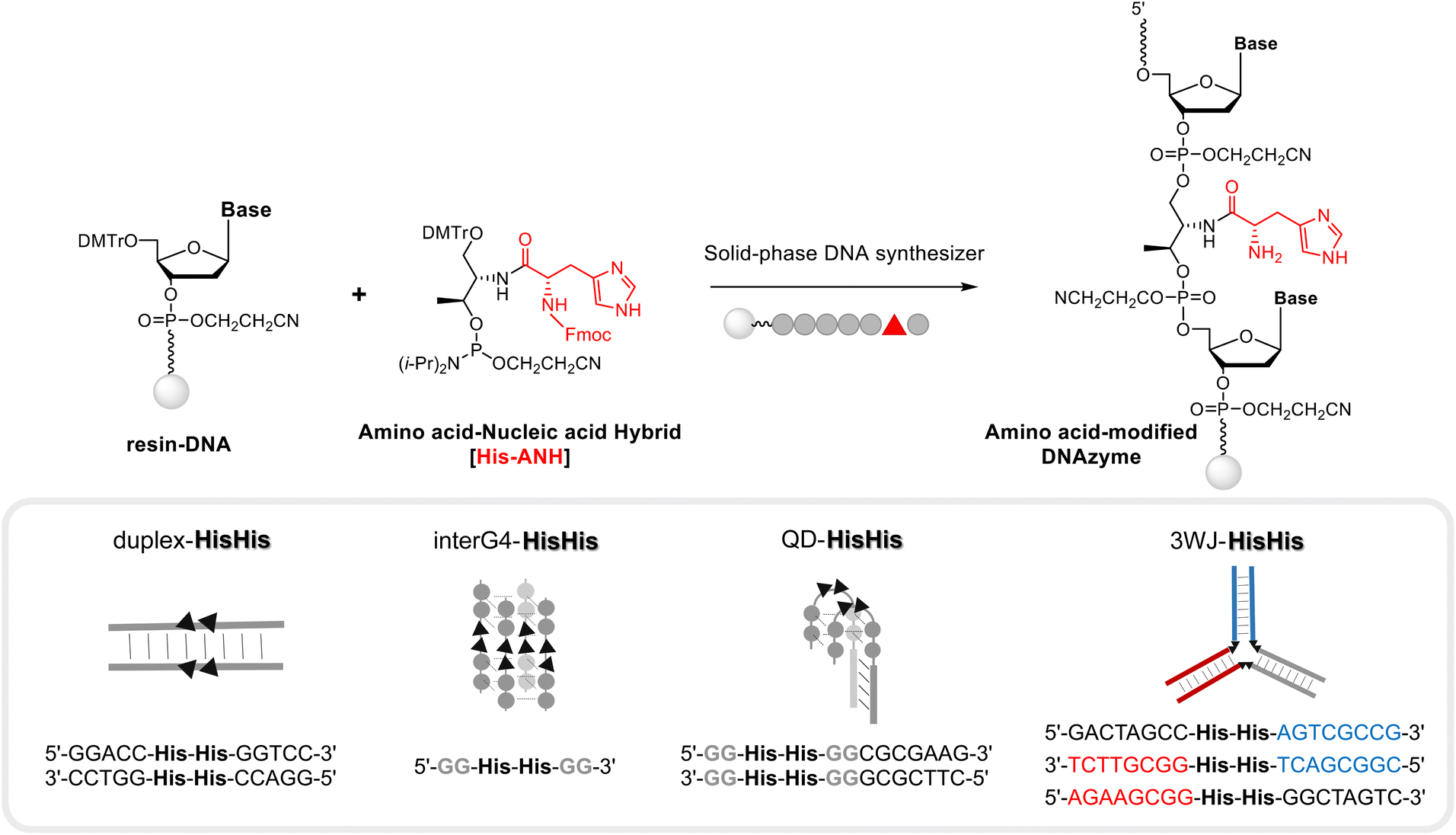
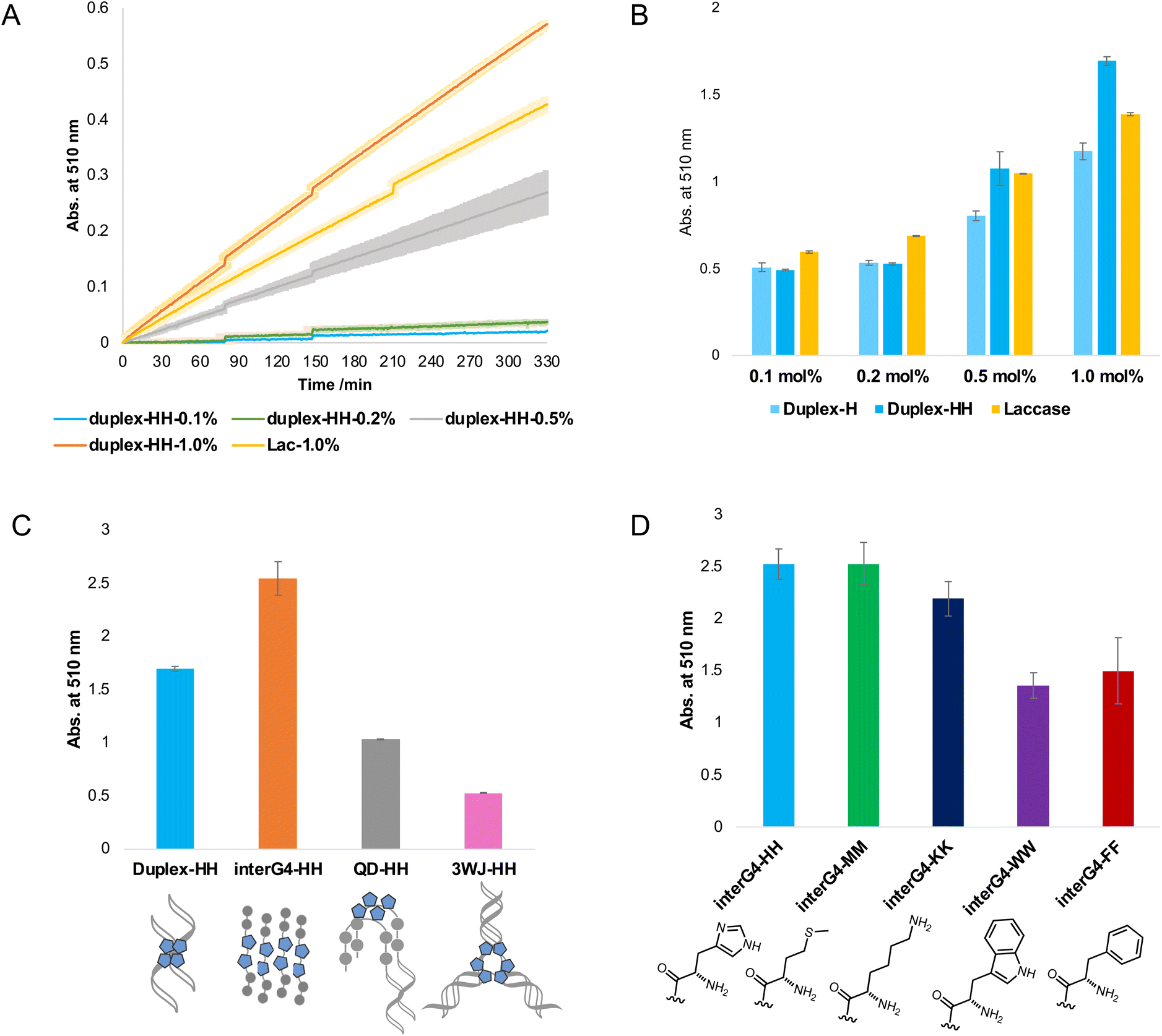
A series of DNA metalloenzymes functionalized with His residues were designed based on the copper-coordinated histidine-rich catalytic domain of laccase (PDB ID: 2X88). A threoninol-tethered histidine residue was synthesized as reported previously.10,11 To construct an active site capable of coordinating multiple copper ions, His-containing phosphoramidites were sequence-specifically installed on oligonucleotides through automated solid-phase DNA synthesis (Scheme 2). His-modified DNAs with palindromic sequences (5′-GGACC-His-His-GGTCC-3′) were obtained in high yield (overall synthetic yields ∼70%) after the consecutive incorporation of His modules. Together with canonical B-DNA, diverse tertiary DNA structures including a G-quadruplex, a quadruplex–duplex hybrid, and three-way junction (3WJ) DNAs were designed as scaffolds for His functionalization (Table S1†). All oligomers were purified through reverse HPLC and then identified using MALDI-TOF-MS spectroscopy. Circular dichroism (CD) analysis provides structural information on DNA, and we confirmed that the His-functionalized oligomers possessed the desired conformations (Fig. S1†). Besides canonical B-helical structures of duplex–HH and 3WJ–HH, interG4–HH was characterized as an intermolecular parallel G-quadruplex in potassium ion solution. We also observed the antiparallel G-quadruplex of QD–HH in the presence of potassium. To investigate the thermal stability of the novel His–DNA hybrids, we measured melting temperatures (Tm) in the absence and presence of Cu(II) ions (Fig. S2†). For both duplex–HH and interG4–HH, we observed significant increases in Tm values in the presence of Cu(II) ions (ΔTm = 6.8 °C and 2.5 °C, respectively). For QD–HH, a remarkable thermal stabilization effect (ΔTm = 14.6 °C) was observed on the addition of Cu(II) ions. Considering the structural characterization through spectroscopy, computational modelling was done to gain further insight into the structural motifs of the His–DNA nanostructures. The plausible structures of His–DNA hybrids were simulated with reported PDB structures and CHARM force-field parameters. As shown in Fig. S6–S9,† the energetically minimized structures suggest well-constructed microenvironments for metal–histidine coordination in the designed active sites. Next, we proceeded to validate the catalytic ability of His–DNA metalloenzymes as multicopper oxidases. The oxidation of 2,4-dichlorophenol (2,4-DP) and 4-aminoantipyrine (4-AP) was chosen as the model reaction (Scheme 1). A His–DNA-based catalysis system was thus constructed by mixing His–DNAs and various equivalents of Cu(NO3)2 in 20 mM MOPS buffer (pH 7.0) with 100 mM NaCl. Substrates 2,4-DP and 4-AP were then added to the His–DNA solution such that the final catalyst loading was 0.1 mol% for duplex DNA and 0.1–1.0 mol% for the catalytic metal. Because product 1 absorbs visible light (510 nm, magenta, Fig. S5†), the reaction progress was quantified through absorption spectroscopy (Fig. 2A and B). The rapid increase of the absorption intensity at 510 nm indicated that His-functionalized DNAzymes were able to catalyze the reaction. The reaction plateaued after 12 h (Fig. S3B†). We then explored other transition metals to investigate whether they were able to catalyze the phenol oxidation reaction (Fig. S4†). However, none of them resulted in an absorption band at 510 nm. The use of DNA in the absence of Cu(II) did not afford an absorption band of target product 1 (data not shown). This suggests that Cu(II) ions is necessary for the present reaction. We examined the reactivity of duplex–HH DNA with various Cu(II) ion concentrations (Fig. 2B). We found that the reactivity of Cu–His–DNAzymes increased significantly as the concentration of Cu(II) ions increased and exhibited comparable catalytic performance to that of laccase even under low Cu(II) ion (0.1 mol%) concentrations. We settled on 1.0 mol% Cu(II) ions and 0.1 mol%-modified oligomers as optimized conditions for further investigation. In a previous study, we observed that the catalytic performance of DNA-based hybrid catalysts varied depending on the tertiary structures of DNA.14 For instance, a duplex-based DNA hybrid catalyst showed very low reactivity in the asymmetric Michael addition reaction, whereas a G-quadruplex DNA hybrid catalyst resulted in excellent yield and stereoselectivity. Accordingly, we conducted the laccase-mediated oxidation reaction with various Cu–His–DNAzymes and found that DNA structures significantly affected the reaction progress (Fig. 2C). InterG4–HH DNA constructed with a four-stranded parallel quadruplex bearing 8 His modules for the catalytic pocket significantly promoted the oxidation reaction as reported by previous studies based on G-quadruplex-mediated catalytic systems.18 In addition, kinetic parameters including Vmax, kcat and Km could be calculated by Michaelis–Menten equation (Table S2†). Based on the analysis of kinetic parameters, it was observed that Cu–His–DNAzymes exhibited significant catalytic efficiency (kcat/Km) compared to laccase under the given reaction conditions. A better binding affinity (Km) was observed with interG4–HH DNA and duplex–HH DNA afforded a higher turnover number (kcat). A three-way junction DNA (3WJ–HH) showed relatively lower catalytic activities that other catalysts. These results indicate that the microenvironment of active site generated by DNA tertiary structures could determine the capability of ANH-based DNAzymes. Subsequently, we asked whether the incorporation of other amino acid residues could mediate the oxidation reaction as laccase mimics. Thus, we introduced four different ANH modules (i.e., Lys, Met, Trp, and a Phe), into the concise intermolecular G-quadruplex-forming DNA sequences (Scheme S1†). DNA was found to be functionalized with aromatic amino acids (i.e., Trp and Phe), which gave lower yields of the chromogenic product. In contrast, DNA functionalized with amino acids that enabled metal ion coordination (i.e., Met- and Lys-containing intermolecular G-quadruplexes) displayed similar catalytic ability to His–DNAs. The results for Met-modified DNA are particularly noteworthy in that methionine participates in Cu(II) coordination in the catalytic pocket and provides catalytic performance as a novel laccase surrogate. This suggests that ANH-based DNAzymes can exhibit native enzyme-like behaviour by the combination of suitable amino acid residues in oligonucleotides. Further, the catalytic performances of the present modular DNAzymes can be further improved through the optimization of DNA structures and amino acid residues as demonstrated above.
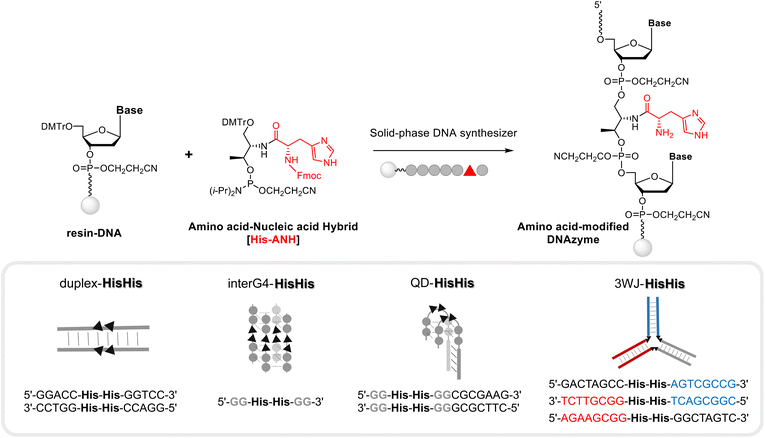 |
| | Scheme 2 Modular strategy for constructing a library of amino acid-modified DNAzymes via solid-phase DNA synthesis (for the synthesis details and identification of various DNAzymes, please see the ESI†). | |
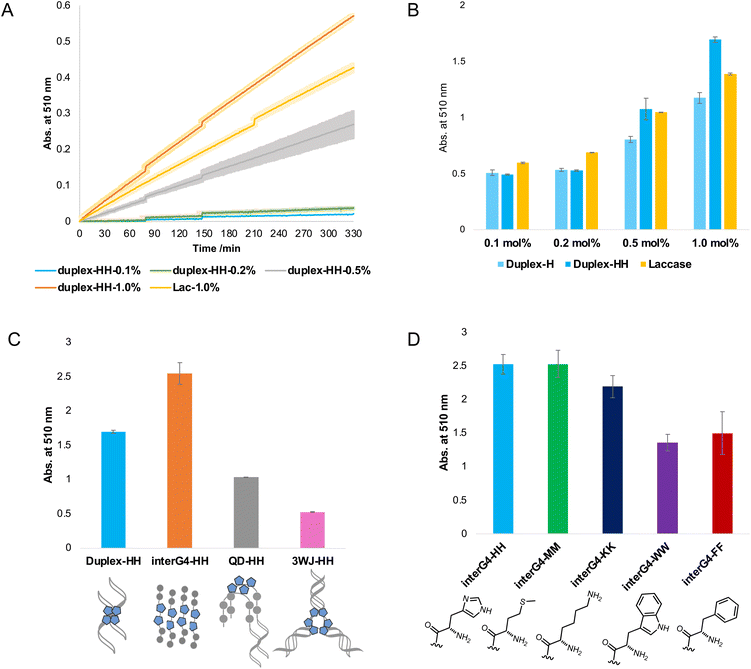 |
| | Fig. 2 Catalytic performance of laccase-mimetic Cu–His–DNAzymes for oxidation reactions. (A) Absorption spectra of the reaction progress (5.5 h). (B) Activity comparison under various copper ion concentration after the reaction for 12 h. (C) Activity comparison catalyzed by His–DNA with various secondary structures. (D) Activity comparison with inter-G4-containing various ANH modules. All reactions were performed in triplicate. | |
Conclusions
In conclusion, we developed a laccase mimic using a DNA scaffold with minimal amino acid componentry. The Cu–His–DNAzyme activity was modulated their catalytic performance with various design features such as DNA tertiary structures and DNA/amino acid sequences. Our results demonstrate the potential of rationally designed DNAzymes as an alternative strategy to protein-based enzymes and inorganic nanozymes:15 (1) DNAzymes are biodegradable, biohybrid catalysts that are harmless to the environment; (2) they are highly versatile because their spatial structure and the capability as metal deposit can be modulated; (3) the molar mass of our DNAzyme is ca. 7.3 kDa, spanning 20 nm, which is remarkably compact compared with the native protein (Trametes Versicolor laccase, 22 kDa) and Cu-GMP nanozyme (particle size, about 1500 nm).13f By employing a straightforward technology, tailor-made active sites could be systematically designed and constructed with ANH modules. We hope the present study will trigger new insights in a variety of research fields and inspire the further application of an ANH modular assembly for biomimetics.
Author contributions
Soyoung Park (S. P.) and Ji Hye Yum (J. H. Y.) conceptualized and designed research. J. H. Y. carried out the synthesis and characterization of the compounds and analyzed the data. Tomotaka Kumagai (T. K.) and Daisuke Hori (D. H) validated the data. S. P. and J. H. Y. wrote original draft. S. P., D. H. and T. K. edited the manuscript. S. P. and Hiroshi Sugiyama (H. S.) supervised the project and reviewed the manuscript. S. P. and H. S. acquired the research funding and provided resources.
Conflicts of interest
The authors declare no competing financial interest.
Acknowledgements
We would like to thank Karin Nishimura (Graduate School of Engineering, Kyoto University) for technical assistance in obtaining the mass spectra of synthesized compounds. S. P. appreciate the financial support by Grant-in-Aid for Transformative Research Area (A) [23H04076; “Material Symbiosis”]. This work was also supported by AMED under 22ama121025j0001 (Platform Project for Supporting Drug Discovery and Life Science Research (BINDS)) to S. P. and JSPS 20H05936 and 21H04705 to H. S.
References
-
(a) N. C. Seeman, Nature, 2003, 421, 427 CrossRef PubMed;
(b) S. M. Douglas, H. Dietz, T. Liedl, B. Högberg, F. Graf and W. M. Shih, Nature, 2009, 459, 414 CrossRef CAS PubMed.
-
(a) M. Zahid, B. Kim, R. Hussain, R. Amin and S. H. Park, Nanoscale Res. Lett., 2013, 8, 1 CrossRef PubMed;
(b) Y. Ke, C. Castro and J. H. Choi, Annu. Rev. Biomed. Eng., 2018, 20, 375 CrossRef CAS PubMed;
(c) J. L. Mergny and D. Sen, Chem. Rev., 2019, 119, 6290 CrossRef CAS PubMed.
-
(a) S. Park and H. Sugiyama, Angew. Chem., Int. Ed., 2010, 49, 3870 CrossRef CAS PubMed;
(b) A. Rioz-Martínez and G. Roelfes, Curr. Opin. Chem. Biol., 2015, 25, 80 CrossRef PubMed;
(c) S. Park and H. Sugiyama, Molecules, 2012, 17, 12792 CrossRef CAS PubMed;
(d) S. K. Silverman, Trends Biochem. Sci., 2016, 41, 595 CrossRef CAS PubMed;
(e) N. Duchemin, I. Heath-Apostolopoulos, M. Smietana and S. Arseniyadis, Org. Biomol. Chem., 2017, 15, 7072 RSC;
(f) J. H. Yum, S. Park and H. Sugiyama, Org. Biomol. Chem., 2019, 17, 9547 RSC;
(g) J. H. Yum, H. Sugiyama and S. Park, Chem. Rec, 2022, e202100333 CAS.
-
(a) S. Park, K. Ikehata, R. Watabe, Y. Hidaka, A. Rajendran and H. Sugiyama, Chem. Commun., 2012, 48, 10398 RSC;
(b) S. Park, L. Zheng, S. Kumakiri, S. Sakashita, H. Otomo, K. Ikehata and H. Sugiyama, ACS Catal., 2014, 4, 4070 CrossRef CAS;
(c) S. Park, I. Okamura, S. Sakashita, J. H. Yum, A. Chiranjit, L. Gao and H. Sugiyama, ACS Catal., 2015, 5, 4708 CrossRef CAS;
(d) J. H. Yum, S. Park, R. Hiraga, I. Okamura, S. Notsu and H. Sugiyama, Org. Biomol. Chem., 2019, 17, 2548 RSC.
- W. Bannwarth, Helv. Chim. Acta, 1988, 71, 1517 CrossRef CAS.
-
(a) N. S. Oltra and G. Roelfes, Chem. Commun., 2008, 6039 RSC;
(b) W. Xu, K. M. Chan and E. T. Kool, Nat. Chem., 2017, 9, 1043 CrossRef CAS PubMed;
(c) A. H. El-Sagheer and T. Brown, Chem. Soc. Rev., 2010, 39, 1388 RSC.
-
(a) M. Sarikaya, C. Tamerler, A. K.-Y. Jen, K. Schulten and F. Baneyx, Nat. Mater., 2003, 2, 577 CrossRef CAS PubMed;
(b) M. Sato and T. J. Webster, Expert Rev. Med. Devices, 2004, 1, 105 CrossRef CAS PubMed.
- P. Geotti-Bianchini, J. Beyrath, O. Chaloin, F. Formaggio and A. Bianco, Org. Biomol. Chem., 2008, 6, 3661 RSC.
-
(a) K. Baek, A. D. Noblett, P. Ren and L. J. Suggs, ACS Appl. Bio Mater., 2019, 2, 2812 CrossRef CAS PubMed;
(b) S. Fleming and R. V. Ulijn, Chem. Soc. Rev., 2014, 43, 8150 RSC;
(c) F. Gelain, A. Horii and S. Zhang, Macromol. Biosci., 2007, 7, 544 CrossRef CAS PubMed.
- S. Park, H. Matsui, K. Fukumoto, J. H. Yum and H. Sugiyama, RSC Adv., 2020, 10, 9717 RSC.
- J. H. Yum, T. Ishizuka, K. Fukumoto, D. Hori, H.-L. Bao, Y. Xu, H. Sugiyama and S. Park, ACS Biomater. Sci. Eng., 2021, 7, 1338 CrossRef CAS PubMed.
-
(a) P. Giardina, V. Faraco, C. Pezzella, A. Piscitelli, S. Vanhulle and G. Sannia, Cell. Mol. Life Sci., 2010, 67, 369 CrossRef CAS PubMed;
(b) J.-R. Jeon, P. Baldrian, K. Murugesan and Y.-S. Chang, Microb. Biotechnol., 2012, 5, 318 CrossRef PubMed;
(c) C. Galli, C. Madzak, R. Vadalà, C. Jolivalt and P. Gentili, ChemBioChem, 2013, 14, 2500 CrossRef CAS PubMed;
(d) B. Ghosh, R. Saha, D. Bhattacharya and M. Mukhopadhyay, Bioresour. Technol. Rep., 2019, 6, 268 CrossRef;
(e) A. D. Moreno, D. Ibarra, M. E. Eugenio and E. Tomas-Pejo, J. Chem. Technol. Biotechnol., 2020, 95, 481 CrossRef CAS.
-
(a) S. K. S. Patel, H. Choi and J.-K. Lee, ACS Sustainable Chem. Eng., 2019, 7, 13633 CrossRef CAS;
(b) L. Zhang, W. Tang, T. Ma, L. Zhou, C. Hui, X. Wang, P. Wang, C. Zhang and C. Chen, RSC Adv., 2019, 9, 38935 RSC;
(c) L. Zhang, H. Cui, Z. Zou, T. M. Garakani, C. Novoa-Henriquez, B. Jooyeh and U. Schwaneberg, Angew. Chem., Int. Ed., 2019, 58, 4562 CrossRef CAS PubMed;
(d) M. A. Molina, J. Díez-Jaén, M. Sánchez-Sánchez and R. M. Blanco, Catal. Today, 2022, 390–391, 265 CrossRef CAS;
(e) S. Patra, S. Sene, C. Mousty, C. Serre, A. Chaussé, L. Legrand and N. Steunou, ACS Appl. Mater. Interfaces, 2016, 8, 20012 CrossRef CAS PubMed;
(f) H. Liang, F. Lin, Z. Zhang, B. Liu, S. Jiang, Q. Yuan and J. Liu, ACS Appl. Mater. Interfaces, 2017, 9, 1352 CrossRef CAS PubMed.
- J. H. Yum, H. Sugiyama and S. Park, Org. Biomol. Chem., 2020, 18, 6812 RSC.
-
(a) H. Liang, F. Lin, Z. Zhang, B. Liu, S. Jiang, Q. Yuan and J. Liu, ACS Appl. Mater. Interfaces, 2017, 9, 1352 CrossRef CAS PubMed;
(b) J. Wu, X. Wang, Q. Wang, Z. Lou, S. Li, Y. Zhu, L. Qin and H. Wei, Chem. Soc. Rev., 2018, 48, 1004 RSC;
(c) D. Jiang, D. Ni, Z. T. Rosenkrans, P. Huang, X. Yan and W. Cai, Chem. Soc. Rev., 2019, 48, 3683 RSC;
(d) Y. Huang, J. Ren and X. Qu, Chem. Rev., 2019, 119, 4357–4412 CrossRef CAS PubMed;
(e) X. Xu, J. Wang, R. Huang, W. Qi, R. Su and Z. He, Catal. Sci. Technol., 2021, 11, 3402 RSC.
-
(a) J. Yang, W. Li, T. B. Ng, X. Deng, J. Lin and X. Ye, Front. Microbiol., 2017, 8, 832 CrossRef PubMed;
(b) T. Brugnari, D. M. Braga, C. S. A. Santos, B. H. C. Torres, T. A. Modkovski, C. W. I. Haminiuk and G. M. Maciel, Bioresour. Bioprocess, 2021, 8, 131 CrossRef;
(c) I. Bassanini, E. E. Ferrandi, S. Riva and D. Monti, Catalysts, 2021, 11, 26 CrossRef CAS;
(d) L. Lei, X. Yang, Y. Song, H. Huang and Y. Li, New J. Chem., 2022, 46, 3541 RSC.
-
(a) J. Wang, R. Huang, W. Qia, R. Sua, B. P. Binks and Z. He, Appl. Catal., B, 2019, 254, 452 CrossRef CAS;
(b) S. Shams, W. Ahmad, A. H. Memon, Y. Wei, Q. Yuan and H. Liang, RSC Adv., 2019, 9, 40845 RSC;
(c) A. Koyappayil, H. T. Kim and M.-H. Lee, J. Hazard. Mater., 2021, 412, 125211 CrossRef CAS PubMed;
(d) R. Su and Z. He, Catal. Sci. Technol., 2021, 11, 3402 RSC;
(e) L. Huang, Y. Tang, J. Wang, X. Niu, J. Zhou and Y. Wu, Sens. Actuators, B, 2023, 391, 134052 CrossRef CAS.
-
(a) E. Golub, H. B. Albada, W.-C. Liao, Y. Biniuri and I. Willner, J. Am. Chem. Soc., 2016, 138, 164 CrossRef CAS PubMed;
(b) H. B. Albada, J. W. Vries, Q. Liu, E. Golub, N. Klement, A. Herrmann and I. Willner, Chem. Commun., 2016, 52, 5561 RSC;
(c) H. B. Albada, E. Golub and I. Willner, Chem. Sci., 2016, 7, 3092 RSC;
(d) J. Chen, Y. Zhang, M. Cheng, Y. Guo, J. Šponer, D. Monchaud, J.-L. Mergny, H. Ju and J. Zhou, ACS Catal., 2018, 8, 11352 CrossRef CAS;
(e) P. M. Punt and G. H. Clever, Chem. Sci., 2019, 10, 2513 RSC;
(f) P. M. Punt and G. H. Clever, Chem. – Eur. J., 2019, 25, 13987 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *ab and
Soyoung
Park
*ab and
Soyoung
Park