
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Enhanced catalytic performance derived from coordination-driven structural switching between homometallic complexes and heterometallic polymeric materials†
Gracjan
Kurpik
 ab,
Anna
Walczak
ab,
Grzegorz
Markiewicz
ab,
Jack
Harrowfield
c and
Artur R.
Stefankiewicz
*ab
ab,
Anna
Walczak
ab,
Grzegorz
Markiewicz
ab,
Jack
Harrowfield
c and
Artur R.
Stefankiewicz
*ab
aCenter for Advanced Technology, Adam Mickiewicz University in Poznań, Uniwersytetu Poznańskiego 10, 61-614 Poznań, Poland. E-mail: ars@amu.edu.pl
bFaculty of Chemistry, Adam Mickiewicz University in Poznań, Uniwersytetu Poznańskiego 8, 61-614 Poznań, Poland
cInstitut de Science et d'Ingénierie Supramoléculaires, Université de Strasbourg, 8 allée Gaspard Monge, 67083 Strasbourg, France
First published on 8th May 2023
Abstract
A bifunctional ligand 4,4-dimethyl-1-(pyridin-4-yl)pentane-1,3-dione (HL) able to provide two distinct coordination sites, i.e. anionic β-diketonate (after deprotonation) and neutral pyridine, has been used in the synthesis of Ag(I), Pd(II) and Pt(II) complexes that then have been applied as metalloligands for the construction of new heterometallic polymeric materials. The ambidentate nature of L− enables switching between different modes of coordination within mononuclear complexes or their conversion into polymeric species in a fully controllable way. The coordination-driven processes can be triggered by various stimuli, i.e. a metal salt addition or acid–base equilibria, and presents an efficient strategy for the generation of metallosupramolecular materials. As a consequence of self-assembly, new multimetallic coordination aggregates have been synthesized and characterized in depth in solution (1H NMR, ESI-MS) as well as in the solid state (XPS, SEM-EDS, FTIR, pXRD, TGA). Furthermore, the Pd-based assemblies have been found to be efficient catalyst precursors in the Heck cross-coupling reaction, demonstrating a direct impact of compositional and morphological differences on their catalytic activity.
Introduction
Metal–ligand interactions are the primary driving force in the generation of simple coordination compounds, as well of metallosupramolecular architectures with a high degree of complexity and a precisely defined structure.1 Taking into account the character of a coordinate bond, i.e. its high strength but commonly dynamic nature, coordination-driven self-assembly processes are characterized by high controllability at the molecular level with respect to directionality, reversibility and post-synthetic switchability.2 The supramolecular transformations induced by different external physicochemical stimuli, e.g. changes in concentration,3,4 acid–base equilibria,5,6 stoichiometry,7,8 cation/anion exchange,9,10 or radiation,11,12 lead to entirely new systems of diverse chemical composition, structure, topology and properties. Thus, externally driven interconversion processes and other structural modifications play an increasingly important role in the design and construction of functional materials that constitute a competitive approach to classical synthesis.2,13Ambidentate pyridyl-β-diketonate ligands derived from pyridyl-1,3-diketones are an interesting class of organic species commonly used as building blocks in coordination and metallosupramolecular chemistry.14–16 Such units are capable of binding metal-ion centers in more than one way through different donor-atom combinations. The distinct nature of coordination sites, i.e. anionic β-diketonate and neutral pyridine, makes them good donors for a wide range of metal cations, both hard and soft acids according to HSAB theory.17,18 In most cases, they act as O,O-chelates or as simple N-donors through the pyridine ring, but other coordination modes can be achieved.19,20 Due to this structural variety they have found a number of applications in the construction of sophisticated coordination assemblies, including complex compounds,21–24 macrocycles,14 multimetallic polymers,20,25,26 metal–organic frameworks (MOFs)27,28 and metallocages.29–31
Although a number of reports on metallosupramolecular assemblies based on pyridyl-β-diketonate-derived ligands are available in the literature, we focused on an unexplored aspect, namely the switchable nature of species with such units as well as conducting studies on the application potential of new heterometallic aggregates. In this work the ditopic molecule 4,4-dimethyl-1-(pyridin-4-yl)pentane-1,3-dione (HL) was employed to synthesize a series of Ag(I), Pd(II) and Pt(II) coordination compounds which were further converted into more complex metallosupramolecular assemblies. Particular efforts have been undertaken to ensure an efficient strategy for multistage supramolecular transformations between the resultant systems triggered by external stimuli. The present work describes coordination-driven structural switching between a variety of mononuclear complexes based on the ligand HL/L− and Ag(I), Pd(II) and Pt(II) cations. As a consequence of metallosupramolecular self-assembly, new heterometallic polymeric materials have been obtained and their physicochemical characterization is described ahead. Additionally, the diversity in their functionality has been illustrated by their use as catalyst precursors in the Heck cross-coupling reaction.
Results and discussion
Synthesis and characterization of complexes
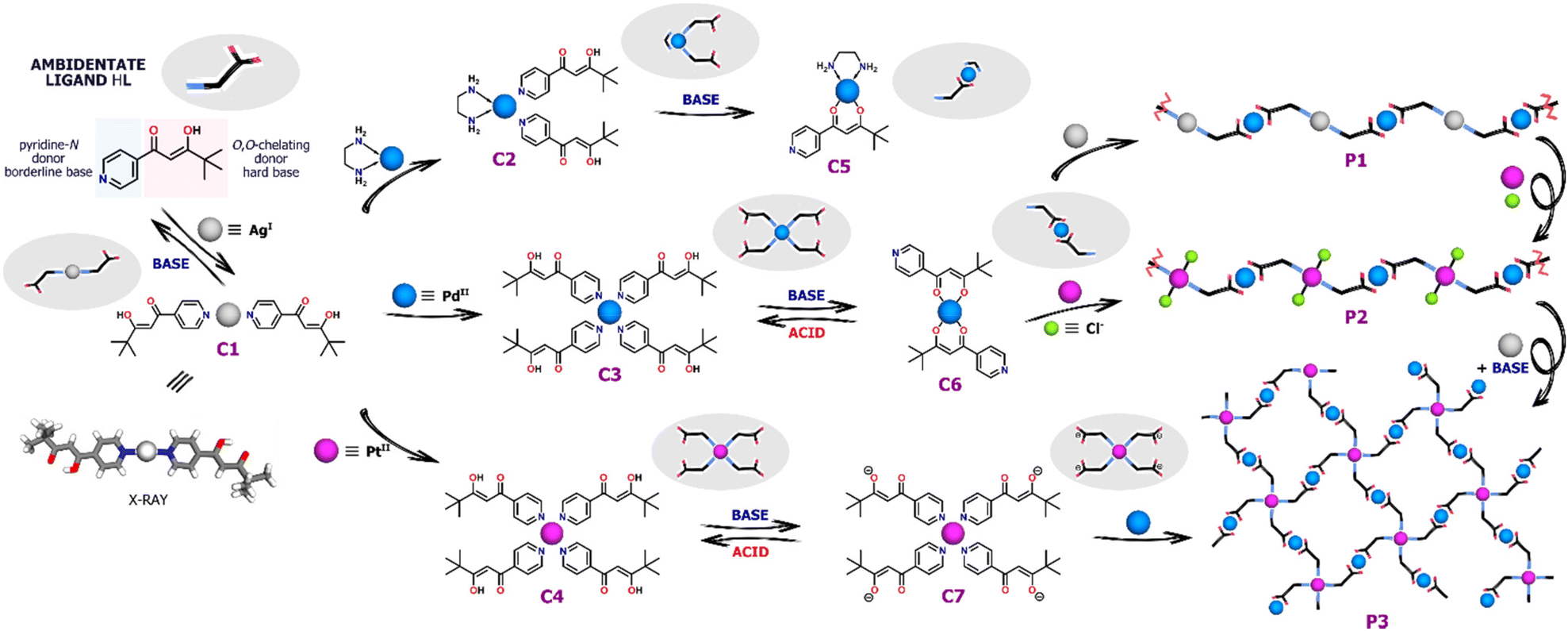
As shown in Fig. 1, the pyridyl-β-diketone ligand HL acts as the starting material and main building block for all coordination species presented. Its ambidentate nature was used to design, obtain and convert a series of complexes with Ag(I), Pd(II) and Pt(II) cations. All the coordination compounds presented herein can be directly synthesized by complexation reactions between HL and appropriate metal salts. Pd(II) and Pt(II) complexes, i.e. [Pd(HL)4](NO3)2 (C3), [Pt(HL)4](NO3)2 (C4) and [PdL2] (C6) were synthesized according to literature procedures.22,23 The compound [Ag(HL)2]NO3 (C1) was synthesized by the reaction of HL (2 equiv.) with AgNO3 (1 equiv.) carried out in DCM/EtOH (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v) at room temperature for 18 h. The generation and purity of the desired complex were confirmed in solution via1H NMR spectroscopy and ESI-MS spectrometry (Fig. S1 and S2†). Moreover, slow evaporation of the methanol solution provided crystals, suitable for single-crystal X-ray diffraction measurements. The complex C1 crystallizes in the monoclinic space group P21/n and has an almost ideal linear geometry of Ag(I) coordination where the N–Ag–N angle is equal to 170.53°. It is worth emphasizing that C1 occurs as a dimer in the crystal (Fig. S13†), where two molecules are linked by a weak metal–metal interaction Ag⋯Ag (3.08 Å),32,33 but this was not confirmed in solution. The β-diketone units remained in their protonated enol form and were not involved in the formation of coordinate bonds. The crystal and structure refinement data are given in the ESI, Table S1.†
:1, v/v) at room temperature for 18 h. The generation and purity of the desired complex were confirmed in solution via1H NMR spectroscopy and ESI-MS spectrometry (Fig. S1 and S2†). Moreover, slow evaporation of the methanol solution provided crystals, suitable for single-crystal X-ray diffraction measurements. The complex C1 crystallizes in the monoclinic space group P21/n and has an almost ideal linear geometry of Ag(I) coordination where the N–Ag–N angle is equal to 170.53°. It is worth emphasizing that C1 occurs as a dimer in the crystal (Fig. S13†), where two molecules are linked by a weak metal–metal interaction Ag⋯Ag (3.08 Å),32,33 but this was not confirmed in solution. The β-diketone units remained in their protonated enol form and were not involved in the formation of coordinate bonds. The crystal and structure refinement data are given in the ESI, Table S1.†
 | ||
| Fig. 1 General scheme showing the supramolecular transformations within coordination-driven assemblies based on Ag(I), Pd(II) and Pt(II) ions and the pyridyl-β-diketonate ligand HL. | ||
Two structurally distinct Pd(II) complexes of the ligand HL and ethylenediamine (en) co-ligand were observed in solution via1H NMR spectroscopy but neither could be isolated in pure form. The reaction of [Pd(en)(NO3)2] with 2 equiv. of HL in CD3CN resulted in the formation of [Pd(en)(HL)2](NO3)2 (C2) whereas the deprotonation of HL (1 equiv.) with Et3N prior to the addition of metal salt gave [Pd(en)L]NO3 (C5). Hence, depending on the reaction conditions applied one or two molecules of the ligand HL occupy two available positions in Pd(II) sphere and coordinate by O,O-chelate or N-donors, respectively. 1H NMR spectroscopy as well as ESI-MS spectrometry allowed unequivocal determination of the structures and molecularity of the new compounds generated (Fig. S3–S6†).
Structural switching
In addition to their direct syntheses, the complexes can be converted into other species by addition of either appropriate metal salts or acid/base, as shown by 1H NMR spectroscopy (Fig. 2 and see ESI, Fig. S14–S19†). As previously mentioned, the ligand HL reacted with AgNO3 to give the disubstituted coordination compound C1 although it decomposes under the influence of base (Fig. S14†). After addition of 0.5 equiv. of Et3N only upfield shifted signals from the deprotonated ligand L− were observed. Therefore, the instability of C1 under basic conditions excluded its further coordination through O,O-chelation requiring prior deprotonation.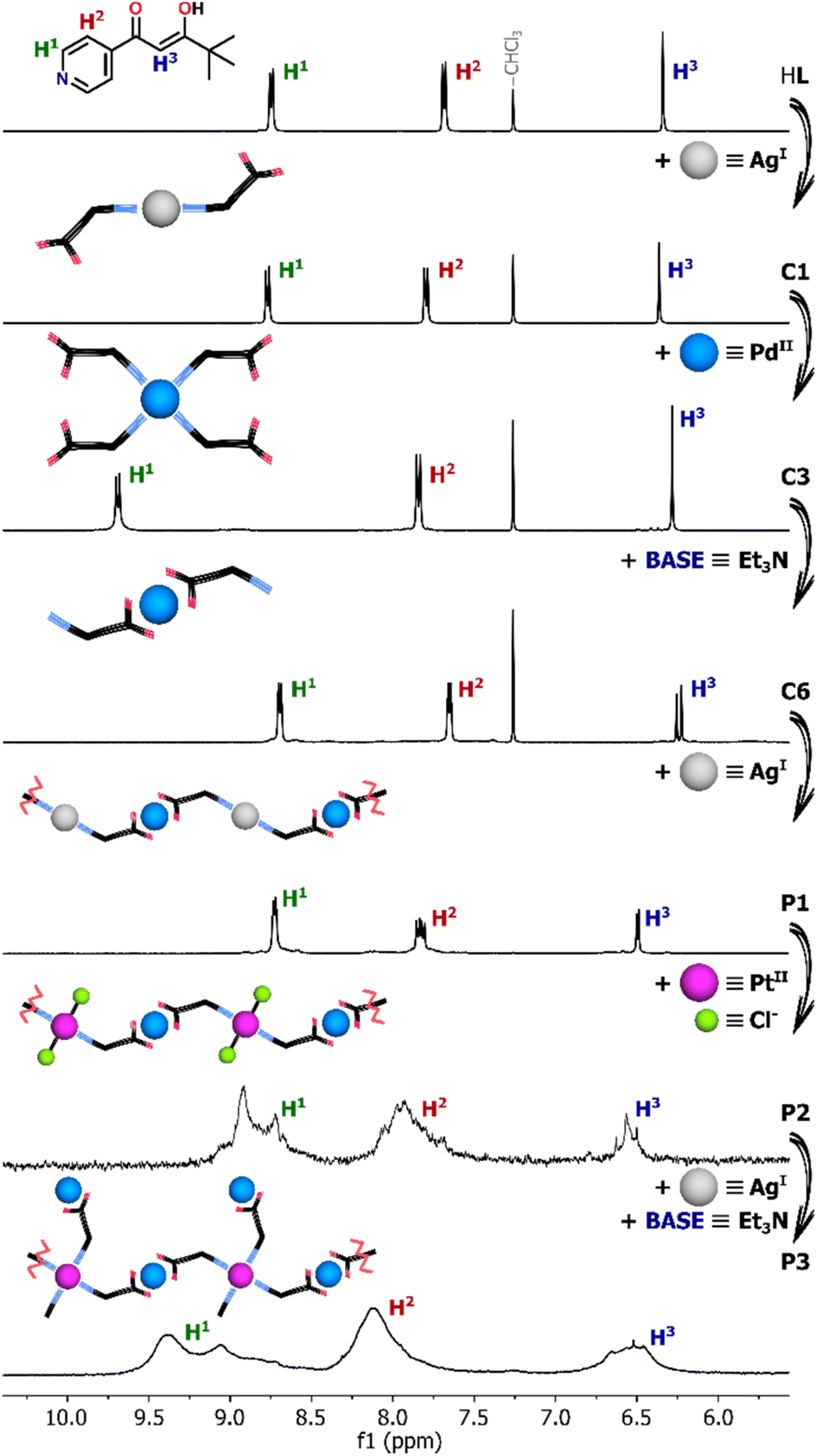 | ||
| Fig. 2 Parts of 1H NMR spectra (600 MHz, CDCl3 or DMSO-d6 for P1–P3) showing the coordination-driven transformations triggered by the addition of metal salt or base. | ||
Nevertheless, the Ag(I) complex C1 can be completely transformed into Pd(II) or Pt(II) species upon addition of the appropriate metal salt. As a result of Pd(NO3)2 addition, gradual disappearance of the C1 signals was observed, along with the emergence of new intense signals for the Pd(II) compound, which was found to be the four-coordinate complex C3 (Fig. S15†). As shown by 1H NMR titration experiments, the change in the coordination number from 2 to 4 was clearly illustrated by the significantly downshifted signal (Δδ = ∼0.8 ppm) from the protons near pyridine-N atoms (H1). Similar behavior was observed when C1 was titrated with [Pd(en)(NO3)2]. Two molecules of the ligand HL derived from the degradation of the Ag(I) complex completed the coordination sphere of Pd(II) leading to the formation of the heteroleptic compound C2 (Fig. S16†).
In contrast, no additional signals or shifts were initially detected during the transformation of C1 triggered by Pt(II) ions. The Ag(I) complex appeared to be stable as Pt(NO3)2 was added, not undergoing any obvious reaction. The generation of the desired complex C4 was noticed only after several days due to the greater kinetic inertness of Pt(II) ion in comparison to Pd(II), for which no kinetic hysteresis was observed during the experiment (Fig. S17 and S18†).
The Pd(II) and Pt(II) coordination compounds generated by these metal ion exchange reactions of C1 were subjected to the addition of base (Et3N). Pd(II) complexes C3 and C6 can be easily interconverted between N- and O,O-donor forms with 100% efficiency by deprotonation/protonation of the enolic centre.22 Addition of base to the cationic N-bound complex C3 leads to linkage rearrangement and formation of neutral O,O-chelated counterparts, a reaction which can be reversed by addition of methanesulfonic acid (MSA). Similarly, titration of the heteroleptic species C2 with Et3N caused a change in the coordination mode via release of one N-bound ligand molecule and the formation of monocharged complex C5 (Fig. S19†). No further transformation into the homoleptic complex C6 coordinated by two β-diketonate units occurred despite the addition of a large excess of Et3N (10 equiv.), consistent with the high stability of the Pd(en) unit. In contrast to its Pd(II) analogue, the Pt(II) complex C4 does not undergo any conversion under basic conditions even after several days. The switching between cationic and neutral species is completely retarded by the kinetic inertness of Pt(II). Addition of a stoichiometric amount of Et3N results only in deprotonation of the uncoordinated enol units and formation of anions [PtL4]2− (C7), while the N-bound complex structure remains essentially intact.23
Synthesis of heterometallic polymers
Due to the presence of accessible coordination sites capable of reacting with metal cations, the complexes C6 and C7 (the deprotonated form of C4) can act as metalloligands. As a pyridine-N donor, Pd(II) complex C6 was expected to be a good ligand for Ag(I) and for this reason C6 was reacted with AgOTf (or AgNO3) in anticipation of the formation of a new multimetallic aggregate [PdAgxL2]x+n (P1), where x ≈ 1. In this structure, the O,O-chelating and pyridine-N donors were expected to be involved in coordination with Pd(II) and Ag(I), respectively, with Ag(I) being two-coordinate, as in complex C1. During a 1H NMR titration of C6 with AgOTf, a progressive decrease in signal intensity was observed as a result of the precipitation of a new product (Fig. S20–S21†) along with minor downfield chemical shifts that could be attributed to the coordination of Ag(I) cations by pyridine-N. Full disappearance of 1H NMR signals from the Pd(II) metalloligand C6 and complete product precipitation (P1) were observed after the addition of 1 equiv. of Ag(I), corresponding to the expected reaction stoichiometry.Next, C6 was reacted with an equimolar amount of PtCl2 in the expectation that retention of the chlorido ligands would favor formation of Pt(II) centers involving just two pyridine-N donors.34,35 The reaction was performed in MeCN at 80 °C for 24 h, giving an orange precipitate, consistent with the formation of a coordination polymer P2. In order to verify the composition of the aggregate, we synthesized an analogue of the complex C6 – [Pd(bpm)2],36 which lacks pyridyl donors. In the control experiment, Pt(II) salt was added to the solution of [Pd(bpm)2] under the reaction conditions, but no clear changes were observed in the 1H NMR spectra (Fig. S23†). Thus, the Pd/Pt exchange could be unambiguously excluded in the structure of the monomer C6, confirming the preservation of O,O-coordination for Pd(II) ions. The isolated material P2 was highly insoluble in most common solvents and only in DMSO-d6 it was possible to record 1H NMR spectrum which showed severely broadened signals, indicative of the generation of the complex metalloaggregates in this solvent (Fig. S9†).
The capacity to deprotonate the enol units of C4 without causing complex decomposition or isomerization prompted us to react C7 with Pd(NO3)2 under basic conditions. The formation of a new species (P3) was observed and again broad and highly overlapped set of signals for all of the aromatic and methine protons were observed in the 1H NMR spectrum (Fig. S11†). This broadening is characteristic of and consistent with the presence of complex metallosupramolecular assemblies. The spectrum contained no signal from the enol group of a diketone moiety (≈16 ppm), indicating complete coordination between this group and Pd(II), possibly as a result of the creation of a complex branched oligomeric structure. The control experiment carried out for a simple Pt(II) complex with pyridine ligands also excluded the exchange of inert Pt(II) ions with Pd(II). As indicated by the 1H NMR spectra (Fig. S24†), its structure was retained in the reaction environment, which showed that only deprotonated O,O-chelates could be involved in the Pd(II) coordination.
The coordination-driven switching strategies developed for the mononuclear complexes C1–C7 were successfully adapted to the transformations of the coordination polymers P1–P3 as well. In order to exchange Ag(I) cations with Pt(II), PtCl2 was added to the solution of P1 in DMSO-d6, which resulted in the precipitation of AgCl and a clear color change. The transformation, followed by 1H NMR spectroscopy, indicated the formation of a new coordination complex, as illustrated by the significant broadening of downshifted signals in a spectrum consistent with that of P2. Furthermore, the polymer P2 was converted into P3 in a two-step process by adding AgNO3 and, subsequently, Et3N. First, a counterion exchange from Cl− to NO3− was carried out by removing chlorido co-ligands, followed by rearrangement of the structure in a basic environment.34 The labile NO3− anions in the coordination sphere of Pt(II) can be readily replaced under basic conditions, leading to the generation of a two-dimensional lattice with Pt(II) ions bound by four pyridine-N donors, which was demonstrated by strong downfield shifts in the 1H NMR spectrum after this conversion.
Characterization of heterometallic materials
As expected for coordination polymers, their solubility in most solvents was very limited, which effectively hampered their full characterization in solution. Although the 1H NMR spectra for P1–P3 described above are not readily interpreted, their profiles are consistent with the formation of larger aggregates. High-resolution ESI-MS spectrometry also confirmed the successful generation of the desired heteronuclear complexes. The mass spectra recorded for P1–P3 demonstrated the presence of the expected fragmentary constituents consisting of chains of successive monomers connected by additional metal ions. All of the identified peaks were in good agreement with their calculated theoretical distribution, allowing the composition and heterometallic nature of the aggregates to be established (Fig. S8, S10 and S12†).The metal content of the polymeric materials P1–P3 was determined via ICP-MS analysis, which enabled confirmation of the presence of the two different metals expected, i.e. Ag and Pd for P1; Pd and Pt for P2, P3 (Table S2†). The percentage contribution of individual metal ions corresponds well to the values calculated for each polymer, with minor deviations perhaps attributable to the lack of any means of ensuring sample purity. XPS spectroscopy confirmed the identity of the different metal mixtures in the three heterometallic polymers (Fig. S25–S27†). The XPS spectra of P1–P3 showed signals indicating the presence of Ag(I), Pd(II), and Pt(II) ions, as well as elements from the organic unit: C, O, N.
To further investigate the complex formation, ATR-FTIR analysis was performed in order to confirm the involvement of specific chemical groups in coordination (Fig. S28–S34†). The absorption bands in the structure of the ligand HL have been assigned by the comparison of its spectrum with the spectra of the reference compounds containing the indicative fragments, i.e. pinacolone, acetylacetone, pyridine and methyl isonicotinate (Fig. S35†). It resulted in the identification of characteristic band at 1611 cm−1 related to the stretching vibration of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond of β-diketonate unit (νCO) and four bands in the region 1585–1427 cm−1 from CN and CC bond vibrations of the heterocyclic ring (νpy). With regard to the coordination assemblies, their recorded IR spectra were consistent with the assumed bonding modes.37 For the mononuclear compounds, the shifts of the absorption bands to lower wavenumbers corresponding to β-diketonate (C6) or pyridine ring (C2, C4) demonstrate the existence of the metal coordination bonding and confirm the complexation reaction of the ligand HL with the appropriate metal ions, whereas the other bands are observed almost in the same position (Fig. S36†). FTIR spectra show that the generation of the heterometallic polymers P1–P3 results in the clear red shifts of the bands characteristic of both νCO and νpy vibrations of the ligand functional groups (Fig. 3). Furthermore, in the spectra of C2, C4 and P1, we observed the appearance of an additional band in the region of 1250–1300 cm−1, originating from the stretching vibrations of the O–NO2 bond due to the presence of NO3− counterions in the complex structures.
O bond of β-diketonate unit (νCO) and four bands in the region 1585–1427 cm−1 from CN and CC bond vibrations of the heterocyclic ring (νpy). With regard to the coordination assemblies, their recorded IR spectra were consistent with the assumed bonding modes.37 For the mononuclear compounds, the shifts of the absorption bands to lower wavenumbers corresponding to β-diketonate (C6) or pyridine ring (C2, C4) demonstrate the existence of the metal coordination bonding and confirm the complexation reaction of the ligand HL with the appropriate metal ions, whereas the other bands are observed almost in the same position (Fig. S36†). FTIR spectra show that the generation of the heterometallic polymers P1–P3 results in the clear red shifts of the bands characteristic of both νCO and νpy vibrations of the ligand functional groups (Fig. 3). Furthermore, in the spectra of C2, C4 and P1, we observed the appearance of an additional band in the region of 1250–1300 cm−1, originating from the stretching vibrations of the O–NO2 bond due to the presence of NO3− counterions in the complex structures.
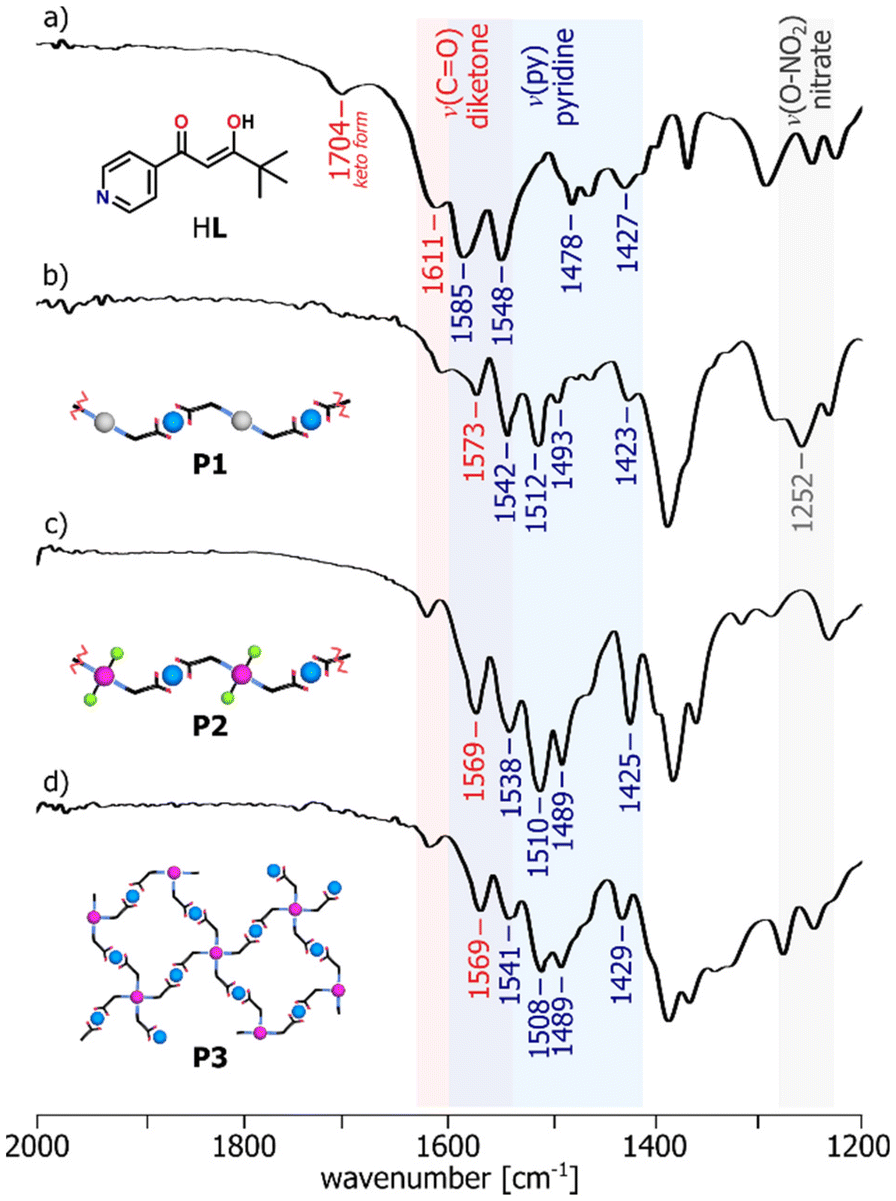 | ||
| Fig. 3 ATR-FTIR spectra in the 2000–1200 cm−1 region of: the ligand HL (a) and the polymers P1–P3 (b–d), showing the involvement of the functional groups in metal binding. | ||
The bimetallic materials were also characterized via scanning electron microscopy (SEM) to establish their morphology (Fig. S43, S45 and S47†). The images were recorded with an accelerating voltage of 10 kV and magnification up to ×30000 using an LFD in solid state. The morphologies of the coordination aggregates P1–P3 indicate the existence of irregular polymer networks depending on their one- or two-dimensional structure. The SEM images of P1–P3 illustrate the formation of quasi-spherical particles of various sizes in the nanometer range. As shown in Fig. 4, all the species show a tendency to agglomerate into larger clusters and the one-dimensional polymers P1–P2 intertwine with each other, creating rounded, disordered shapes. For comparison, SEM images were also recorded for the metalloligands C1, C4 and C6 (Fig. S49–S51†). In contrast to P1–P3, the mononuclear complexes appear as single microcrystals with regular shapes. The SEM results are consistent with the data obtained via powder X-ray diffraction (pXRD) studies. The pXRD patterns with a set of sharp and intense reflections indicate the crystallinity of the samples C1, C4 and C6 (Fig. S37–S39†). In contrast, the diffractograms recorded for P1–P3 established their amorphous character (Fig. S40–S42†), which, however, can be advantageous with regard to their use as (pre)catalysts, facilitating dispersion in the reaction medium.
 | ||
| Fig. 4 SEM images and EDS elemental mapping of the sample composition for (a) P1; (b) P2 and (c) P3. The same magnification indicated in the graphic was used for all EDS images. | ||
EDS mapping provided the topographic distribution of individual elements in the samples (Fig. 4 and S44, S46, S48†). Comparison of the false-color EDS mapping images demonstrated the uniform and common location of the metal pairs in each solid, consistent with their identity as heterometallic coordination polymers.
Thermogravimetric analysis (TGA) was conducted to study the thermal stability of the compounds P1–P3 and compare their properties with the metalloligands C4 and C6 used as building blocks in their synthesis. The experiments were performed in the temperature range 30–600 °C under N2 with a heating rate of 10 °C min−1. The TGA curves of the polymeric materials P1–P3 have similar profiles and show higher stability than the mononuclear analogues C4 and C6 (Fig. S52–S57†). In the DTG curves (Fig. S58†), the main peaks which indicate the rate of decomposition are always shifted towards higher temperatures when P1–P3 are compared to the corresponding mononuclear units, attributed to the retardation of volatilization within the polymers. The heterometallic aggregates show no weight loss of up to ∼250 °C, whereas C4 and C6 begin to decompose at 160 and 210 °C, respectively. The stepwise decomposition via gradual removal of organic components from the structure of compounds P2–P3 leads to the formation of the mixture of appropriate metal oxides – PdO and PtO2 as the residue (calcd. 44.76% and found 47.01% for P2; calcd 38.53% and found 35.21% for P3). The polymer P1 turned out to be the most stable, as illustrated by the very gentle course of the TGA curve. In the tested temperature range, it underwent thermal degradation only partially, because the weight loss was equal to 24.44% whereas the mixture of thermal decomposition products, i.e. PdO and metallic silver in the final residue should constitute 29.84% of the sample mass.
Catalytic studies
The ability to combine in close and ordered proximity different catalytically active metal ions in the polymers presently described renders them of particular interest for evaluation of the influence of one metal on the activity of another. The remarkable utility of Pd complexes as catalysts for cross-coupling reactions led us to choose familiar reactions of this kind for an initial assessment of the catalytic properties of the polymers and their precursors. The reaction screening revealed a high potential of the polymeric species P1–P3 to be applied as precatalysts in various Pd-catalyzed C–C coupling reactions, such as the Heck, Suzuki–Miyaura and Sonogashira reactions. Considering the most promising results in the former process, we decided to assess the influence of the compositional differences between homometallic complexes and heterometallic materials on their catalytic activity in this reaction. Thus, the Heck cross-coupling between iodobenzene and styrene catalyzed by P2 was chosen as a model reaction for the development of reaction conditions. All the optimization procedures are summarized in the ESI, Table S3.† The percentage of Pd in all the structures was determined via ICP-MS analysis and the results were used to calculate the catalyst loading. Different variations in terms of solvents and bases were tested using a 0.1 mol% Pd. Of all the combinations, the pair of Et3N and DMSO gave the expected (E)-stilbene in the highest GC yield after 18 h. Further experiments revealed that the catalyst loading could be reduced to relatively low concentration – 0.05 mol%. This concentration was sufficient to guarantee an almost quantitative conversion along with excellent selectivity achievable after 6 h, whereas the use of 0.01 mol% significantly extended the reaction time to 18 h. Taking economic and environmental considerations into account, subsequent catalytic reactions were carried out in DMSO at 100 °C for 6 h using Et3N (2 equiv.) as a base and 0.05 mol% Pd. Importantly, the reactions were performed without the need to exclude air or water. Furthermore, the metalloaggregates P1–P3 showed high stability under ambient conditions exceeding several months of storage without any decomposition or loss of catalytic activity, which is beneficial in terms of their utility as catalyst precursors.A series of catalytic tests was performed to evaluate and compare the performance of other coordination assemblies containing Pd(II) ions in the Heck cross-coupling reaction. For the selected set of aryl iodides and olefins (Fig. 5a), the complex C6, a building block of all the coordination polymers, had a lower efficiency under the same total Pd loading (GC yields in the range of 60–68%). Roughly the same results (55–70%) were obtained using a mixture of [Pd(bpm)2] and PtCl2, i.e. the constituents of the polymer P2, lacking the pyridine units and thus unable to unite under the catalytic reaction conditions. In both cases, the lower GC yields are possibly due to the absence of neighboring metal ions compared to the heterometallic aggregates. A number of catalytic centers in a single molecule significantly increases catalytic activity through a higher local concentration of Pd which is demonstrated in considerably enhanced effectiveness in the reactions catalyzed by P1–P3. This positive effect, defined in the literature for dendrimers,38–41 coordination polymers,42,43 and recently for polynuclear complexes,44 was also observed in this case and provides an explanation of the apparent differences between mononuclear units and P1–P3. The results of the comparative tests for P1–P3 demonstrated high efficiency of all the species, slightly variable as indicated by the different reaction yields. The lower catalytic activity of P3 might be explained due to its morphology since the catalytic sites in the structure of the two-dimensional polymer P3 might be less accessible compared to its linear counterparts P1–P2. Nevertheless, the extension of the reaction time to 12 h allowed attainment of yields comparable to those with the precatalyst P2. The local concentration enhancement could possibly be weaker in the case of reactions catalyzed by P1. The presence of labile Ag(I) ions as linkers between Pd(II) units might promote polymer fragmentation under reaction conditions that limits the influence of the nuclearity effect. On the other hand, very inert Pt(II) ions are able to retain the polymer structure of P2 more effectively. Thus, a number of differences between the mononuclear complexes and coordination polymers P1–P3, i.e. varied nuclearity, composition, dimensionality and morphology, had a significant impact on the course of catalytic reactions and, consequently, on the obtained reaction yields.
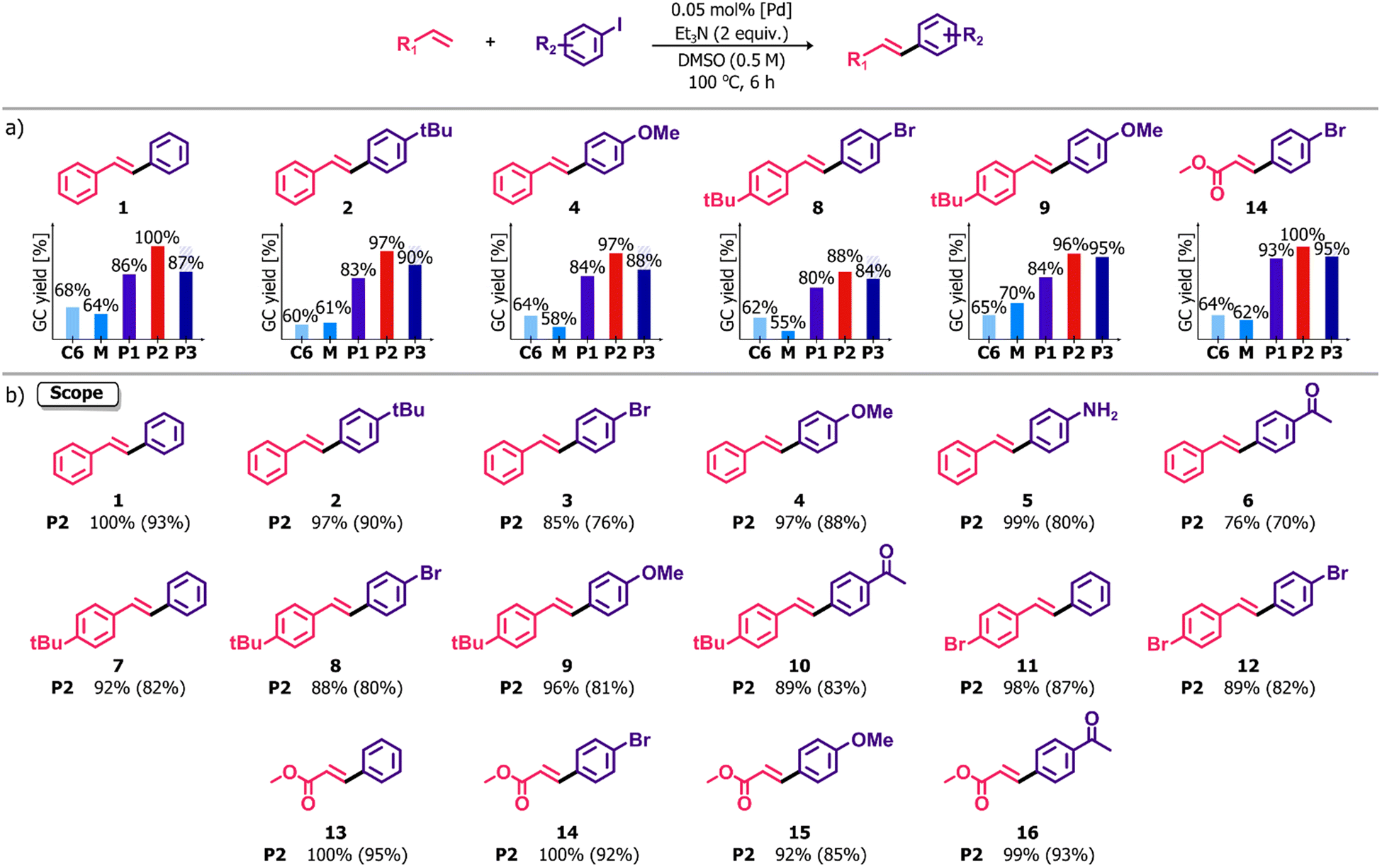 | ||
| Fig. 5 (a) Comparative tests showing different catalytic activity of the complex C6, a mixture of [Pd(bpm)2] and PtCl2 (M), and the coordination polymers P1–P3 in the Heck cross-coupling reaction. (b) Scope of the Heck cross-coupling catalyzed by P2. Reaction conditions: aryl iodide (0.5 mmol, 1 equiv.), olefin (0.5 mmol, 1 equiv.), Et3N (1.0 mmol, 2 equiv.) and the Pd(II) complex (0.05 mol% Pd) were stirred in DMSO (1 mL) at 100 °C for 6 h. Yields determined by GC-MS measurements of aryl iodide decay. The yields in parentheses are those of the isolated compounds. | ||
With these optimized conditions in hand, we undertook studies of the scope and limitations of this system in the Heck cross-coupling reaction between a series of structurally diversified substrates. The investigations were carried out with the use of P2 as a catalyst precursor, identified to be the most effective among the investigated. As shown in Fig. 5b, the high efficiency of P2 was revealed regardless of the presence of electron donating (–OMe, –NH2, –tBu) or electron withdrawing (–COMe, –Br) functional groups in the reactant molecules. In all cases, the stilbene derivatives were formed in high-to-excellent isolated yields, ranging from 70% to 93%. Moreover, the reaction of methyl acrylate with a set of aryl iodides enabled the synthesis of coupling products in excellent yields (85–95%). It is noteworthy that the reaction system exhibited great chemoselectivity towards aryl iodides because no cross-coupling involving bromoarene moieties, as either olefin or haloarene coupling partners, was observed. In all cases, the selectivity in the generation of reaction products with the (E)-configuration of double bond was very high and completely independent of the nature of substrates since (Z)-isomers never exceeded 10% as determined via GC-MS distribution of both isomers.
Conclusions
In summary, the ambidentate nature of the ligand HL and its conjugate base L− allows the generation of distinct complex compounds with modifiable structure and tunable properties. The availability of free coordination sites enables their utilization as metalloligands in further complexation reactions. The work also defines an effective strategy of coordination-driven structural switching processes for a specific group of coordination systems based on pyridyl-β-diketone ligands. As a consequence, we have successfully synthesized and analyzed new polymeric materials based on precious metals. The structural features, morphology and stability of these coordination assemblies were determined via NMR, ESI-MS ICP-MS, XPS, FTIR, SEM-EDS and TG analyses, which unambiguously confirmed their heterometallic character. Furthermore, the coordination polymers reported in this paper exhibit not only structural variety but, more importantly, diverse functionality, as reflected in their catalytic activity. The polymers have been found to be efficient and versatile precatalysts in the Heck cross-coupling reaction within a scope of structurally distinct substrates.Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was funded by the National Science Centre in Poland (grant SONATA BIS 2018/30/E/ST5/00032 – ARS).References
- E. C. Constable and C. E. Housecroft, Chem. Soc. Rev., 2013, 42, 1429–1439 RSC.
- W. Wang, Y.-X. Wang and H.-B. Yang, Chem. Soc. Rev., 2016, 45, 2656–2693 RSC.
- K. Uehara, K. Kasai and N. Mizuno, Inorg. Chem., 2010, 49, 2008–2015 CrossRef CAS PubMed.
- S. Tashiro, M. Tominaga, T. Kusukawa, M. Kawano, S. Sakamoto, K. Yamaguchi and M. Fujita, Angew. Chem., Int. Ed., 2003, 42, 3267–3270 CrossRef CAS PubMed.
- J. Beswick, V. Blanco, G. De Bo, D. A. Leigh, U. Lewandowska, B. Lewandowski and K. Mishiro, Chem. Sci., 2015, 6, 140–143 RSC.
- V. Blanco, A. Carlone, K. D. Hänni, D. A. Leigh and B. Lewandowski, Angew. Chem., Int. Ed., 2012, 51, 5166–5169 CrossRef CAS PubMed.
- Q.-F. Sun, S. Sato and M. Fujita, Nat. Chem., 2012, 4, 330–333 CrossRef CAS PubMed.
- S. Hiraoka, T. Yi, M. Shiro and M. Shionoya, J. Am. Chem. Soc., 2002, 124, 14510–14511 CrossRef CAS PubMed.
- O. Jurček, P. Bonakdarzadeh, E. Kalenius, J. M. Linnanto, M. Groessl, R. Knochenmuss, J. A. Ihalainen and K. Rissanen, Angew. Chem., Int. Ed., 2015, 54, 15462–15467 CrossRef PubMed.
- H. T. Chifotides and K. R. Dunbar, Acc. Chem. Res., 2013, 46, 894–906 CrossRef CAS PubMed.
- S. Chen, L.-J. Chen, H.-B. Yang, H. Tian and W. Zhu, J. Am. Chem. Soc., 2012, 134, 13596–13599 CrossRef CAS PubMed.
- M. Han, R. Michel, B. He, Y.-S. Chen, D. Stalke, M. John and G. H. Clever, Angew. Chem., Int. Ed., 2013, 52, 1319–1323 CrossRef CAS PubMed.
- V. Blanco, D. A. Leigh and V. Marcos, Chem. Soc. Rev., 2015, 44, 5341–5370 RSC.
- A. D. Burrows, M. F. Mahon, C. L. Renouf, C. Richardson, A. J. Warren and J. E. Warren, Dalton Trans., 2012, 41, 4153–4163 RSC.
- C. J. McMonagle, P. Comar, G. S. Nichol, D. R. Allan, J. González, J. A. Barreda-Argüeso, F. Rodríguez, R. Valiente, G. F. Turner, E. K. Brechin and S. A. Moggach, Chem. Sci., 2020, 11, 8793–8799 RSC.
- G.-L. Wang, Y.-J. Lin, H. Berke and G.-X. Jin, Inorg. Chem., 2010, 49, 2193–2201 CrossRef CAS PubMed.
- R. G. Pearson, J. Chem. Educ., 1968, 45, 581–587 CrossRef CAS.
- T.-L. Ho, Chem. Rev., 1975, 75(1), 1–20 CrossRef CAS.
- M. J. Mayoral, P. Cornago, R. M. Claramunt and M. Cano, New J. Chem., 2011, 35, 1020–1030 RSC.
- A. Walczak, G. Kurpik and A. R. Stefankiewicz, Int. J. Mol. Sci., 2020, 21(17), 6171 CrossRef CAS PubMed.
- S.-L. Lee, F.-L. Hu, X.-J. Shang, Y.-X. Shi, A. L. Tan, J. Mizera, J. K. Clegg, W.-H. Zhang, D. J. Young and J.-P. Lang, New J. Chem., 2017, 41, 14457–14465 RSC.
- A. Walczak and A. R. Stefankiewicz, Inorg. Chem., 2018, 57, 471–477 CrossRef CAS PubMed.
- A. Walczak, H. Stachowiak, G. Kurpik, J. Kaźmierczak, G. Hreczycho and A. R. Stefankiewicz, J. Catal., 2019, 373, 139–146 CrossRef CAS.
- R. A. A. Abdine, G. Kurpik, A. Walczak, S. A. A. Aeash, A. R. Stefankiewicz, F. Monnier and M. Taillefer, J. Catal., 2019, 376, 119–122 CrossRef CAS.
- M. Dudek, J. K. Clegg, C. R. K. Glasson, N. Kelly, K. Gloe, K. Gloe, A. Kelling, H.-J. Buschmann, K. A. Jolliffe, L. F. Lindoy and G. V. Meehan, Cryst. Growth Des., 2011, 11, 1697–1704 CrossRef CAS.
- P. C. Andrews, G. B. Deacon, R. Frank, B. H. Fraser, P. C. Junk, J. G. MacLellan, M. Massi, B. Moubaraki, K. S. Murray and M. Silberstein, Eur. J. Inorg. Chem., 2009, 2009, 744–751 CrossRef.
- G.-G. Hou, Y. Liu, Q.-K. Liu, J.-P. Ma and Y.-B. Dong, Chem. Commun., 2011, 47, 10731–10733 RSC.
- B. Chen, F. R. Fronczek and A. W. Maverick, Inorg. Chem., 2004, 43, 8209–8211 CrossRef CAS PubMed.
- S. Sanz, H. M. O'Connor, V. Martí-Centelles, P. Comar, M. B. Pitak, S. J. Coles, G. Lorusso, E. Palacios, M. Evangelisti, A. Baldansuren, N. F. Chilton, H. Weihe, E. J. L. McInnes, P. J. Lusby, S. Piligkos and E. K. Brechin, Chem. Sci., 2017, 8, 5526–5535 RSC.
- H.-B. Wu and Q.-M. Wang, Angew. Chem., Int. Ed., 2009, 48, 7343–7345 CrossRef CAS PubMed.
- S. Sanz, H. M. O'Connor, P. Comar, A. Baldansuren, M. B. Pitak, S. J. Coles, H. Weihe, N. F. Chilton, E. J. L. McInnes, P. J. Lusby, S. Piligkos and E. K. Brechin, Inorg. Chem., 2018, 57, 3500–3506 CrossRef CAS PubMed.
- H.-L. Zhu, X.-M. Zhang, X.-Y. Liu, X.-J. Wang, G.-F. Liu, A. Usman and H.-K. Fun, Inorg. Chem. Commun., 2003, 6, 1113–1116 CrossRef CAS.
- E. M. Njogu, B. Omondi and V. O. Nyamori, J. Coord. Chem., 2015, 68, 3389–3431 CrossRef CAS.
- G. Kurpik, A. Walczak, M. Gołdyn, J. Harrowfield and A. R. Stefankiewicz, Inorg. Chem., 2022, 61, 14019–14029 CrossRef CAS PubMed.
- N. A. Lewis, S. Pakhomova, P. A. Marzilli and L. G. Marzilli, Inorg. Chem., 2017, 56, 9781–9793 CrossRef CAS PubMed.
- M. Kołodziejski, A. Walczak, Z. Hnatejko, J. Harrowfield and A. R. Stefankiewicz, Polyhedron, 2017, 137, 270–277 CrossRef.
- K. Nakamoto, Infrared Spectra of Inorganic and Coordination Compounds, Wiley-Interscience, 1970 Search PubMed.
- B. Helms and J. M. J. Fréchet, Adv. Synth. Catal., 2006, 348, 1125–1148 CrossRef CAS.
- R. S. Bagul and N. Jayaraman, J. Organomet. Chem., 2012, 701, 27–35 CrossRef CAS.
- E. Delort, T. Darbre and J.-L. Reymond, J. Am. Chem. Soc., 2004, 126, 15642–15643 CrossRef CAS PubMed.
- D. Wang and D. Astruc, Coord. Chem. Rev., 2013, 257, 2317–2334 CrossRef CAS.
- M. Kołodziejski, A. J. Brock, G. Kurpik, A. Walczak, F. Li, J. K. Clegg and A. R. Stefankiewicz, Inorg. Chem., 2021, 60, 9673–9679 CrossRef PubMed.
- S. Kitagawa, R. Kitaura and S.-I. Noro, Angew. Chem., Int. Ed., 2004, 43, 2334–2375 CrossRef CAS PubMed.
- G. Kurpik, A. Walczak, M. Gilski, J. Harrowfield and A. R. Stefankiewicz, J. Catal., 2022, 411, 193–199 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2244267. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3nr01298k |
| This journal is © The Royal Society of Chemistry 2023 |