
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Nanoscale pores introduced into paper via mesoporous silica coatings using sol–gel chemistry†
J. J.
Mikolei
a,
D.
Richter
a,
R.
Pardehkhorram
a,
C.
Helbrecht
b,
S.
Schabel
b,
T.
Meckel
 c,
M.
Biesalski
c,
M.
Ceolin
d and
A.
Andrieu-Brunsen
*a
c,
M.
Biesalski
c,
M.
Ceolin
d and
A.
Andrieu-Brunsen
*a
aErnst-Berl Institut für Technische und Makromolekulare Chemie; Macromolecular Chemistry – Smart Membranes; Technische Universität Darmstadt, Alarich-Weiss-Strasse 8, D-64287 Darmstadt, Germany. E-mail: annette.andrieu-brunsen@tu-darmstadt.de
bPaper Technology and Mechanical Process Engineering; Technische Universität Darmstadt, Alexanderstraße 8, 64283 Darmstadt, Germany
cErnst-Berl Institut für Technische und Makromolekulare Chemie; Macromolecular and Paper Chemistry; Technische Universität Darmstadt, Alarich-Weiss-Strasse 8, D-64287 Darmstadt, Germany
dInstituto de Investigaciones Fisicoquímicas Teóricas y Aplicadas; Universidad Nacional de La Plata and CONICET, Diag. 113 y 64 (1900), La Plata, Argentina
First published on 2nd May 2023
Abstract
Mesopores, with diameters between 2 and 50 nm, not only increase the specific surface area, but also generate hierarchically porous materials with specific properties such as capillary fluid transport, ion specific pore accessibility, or size exclusion. Paper is a strongly hierarchical, porous material with specific properties, such as capillary force-driven fluid transport. However, paper fibers change their morphology during the initial step of wood disintegration. This results in changes of the porous fiber structure. In particular paper fibers loose their mesopores during the final drying step in the fabrication process. Here, we investigate silica mesopore formation in paper by sol–gel chemistry and evaporation induced self-assembly to specifically introduce and rationally design mesopore formation and distribution in cotton linter and eucalyptus sulfate paper sheets. We demonstrate the importance of synchronizing the solvent evaporation rate and capillary fluid velocity to ensure mesopore formation as well as the influence of the fiber type and sol–gel solution composition. The combination of argon and krypton sorption, SAXS, TEM and CLSM provides systematic analysis of the porous structure and the silica distribution along the cellulose paper fiber length and cross-section. These results provide a deeper understanding of mesopore formation in paper and how the latter is influenced by paper fluidic properties.
Introduction
Nanoporous and hierarchically porous materials offer a large platform for several applications in the field of transport, sensing, and separation processes.1 Upon nanopore functionalization, e.g. using stimulus responsive-polymers, complex transport, sensing and separation allowing charge specific ion concentration or exclusion have been demonstrated.2 Materials with a hierarchical porous structure, such as paper, inherit material properties specifically based on this structural hierarchy. Inexpensive, portable, and easy to use paper-based devices have been implemented in the field of point-of-care diagnostics, sensor technology, or lab-on-a-chip applications benefiting from capillary driven fluid transport and paper functionalization.3 However, the intrinsic porous structure of the fibres is affected by drying and rewetting processes, in particular during wood disintegration which is the initial step in pulp fibre preparation. Lovikka et al. showed that the specific surface area of dried pulp decreased by about more than 95% compared to that of wet pulp by nitrogen adsorption measurements.4 The decrease of the specific surface area is ascribed to the collapse of the pores and especially affects the size range of mesopores. S. Cichosz et al. explained the collapse of the pores as a combination of capillary forces and the surface tension of water.5,6 The reduction of specific surface area is ascribed to the collapse of the pores due to dehydration while drying. This process is known as hornification and it is described in the work of Mo et al.7 When rewetting the cellulose fibres, fibre swelling occurs which leads to an increase in the specific surface area.4,8 Nevertheless, rewetting does not restore the initial specific surface area and thus the fibre porosity as well as the specific surface area of the fibres remain below the initial value detected before the drying procedure.4,7 To preserve the fibre porosity, drying techniques such as freezing or critical point drying can be used as shown in the work of Lovikka et al. and Mo et al.4,8 However, during the paper production, fibres are exposed to different temperatures, which inevitably leads to fibre drying and thus to pore collapse. Because of the structural changes due to the collapse of the mesopores of paper fibres, direct pore functionalization for introducing sensing, gating, and separation properties in ions and small molecules in paper is not trivial. Therefore, composite materials containing metal–organic frameworks (MOFs)9 or silica nanoparticles10 can provide a stable porous environment which is already able or which can be functionalized in a further step to sense, separate or gate ions or molecules. Abdelhamid et al. and Wang et al. showed the possibility of heavy metal ion sensing and removal through the introduction of ZIF-8 or Zn-BTC into lab engineered papers.11,12 The sensing function of the paper MOF composite material is mainly based on the coordination of the metal in the MOF. Besides heavy metals like Pb(II),12 As(V)13 or Cr(II),14 paper MOF composite materials can also be used for removing organic compounds from water like toluene15 and benzotriazole,16 and even the removal of the drug aspirin17 is possible. MOF paper composite materials can also be used as humidity sensors as shown by Rauf et al.18 integrating a MIL-96 (AL) MOF film on commercially available cotton textiles taking advantage of its high affinity for water which allows the detection of humidity with a detection limit of 0.71% RH. Wang et al. showed that it is possible to detect small biomolecules like micro RNAs with paper-based sensors through the integration of gold nanoparticles and copper containing MOFs.19 Another strategy to insert stable nanoscale pores into paper is to introduce mesoporous silica nanoparticles like SBA-15. Moreira et al. demonstrated that amine functionalized SBA-15 particles with immobilized horseradish peroxidase can be used for the detection of ascorbic acid in paper.10 Another example of paper modified with SBA-15 as an enzyme immunoassay is presented in the work of Scala-Benuzzi et al.20 Again, amino functionalized SBA-15 particles were brought into paper and the anti-ethinylestradiol antibody ethinylestradiol was immobilized in SBA-15. Based on an enzymatic reaction, the synthetically produced estrogen ethinylestradiol which is used in hormone replacement therapies was detectable and the quantity determinable. Ghosh et al. showed that ultra-fast detection of Cr(VI) ions is possible with an amino functionalized silica nanoparticle paper composite material, which makes this material an easy to handle sensing device.21 Also, the amount of histidine and cysteine in human urine is detectable with paper SBA-15 composite materials as shown in the publication of Razavi et al.22 All these studies added porous particles to a paper substrate but did not directly introduce nanoscale pores into the cellulose paper fibre itself.Ceramic coatings are suitable to template the fibre and on the other hand for fibre coating. During templating, the ceramic penetrates the complete fibre whereby the structure of the fibre is transferred to the ceramic material, resulting in a negative of the fibre itself.23,24 The templating of properties into ceramic was used by Persson et al. to investigate the cell-wall structure of fibres.25 However, with coating the wetting properties of the fibre can be modified.24 In our previous work, we could demonstrate that sol–gel chemistry and evaporation induced self-assembly (EISA) are well suited to influence the cellulose paper wettability as well as the fluid transport velocity with dense and porous ceramic coatings. Due to dense silica coatings, the wettability of the cellulose paper can be tuned from highly hydrophilic to water exclusion.24,26 With the introduction of well-defined pores into paper, the hydrophilic character remains and the fluid transport through the paper can be accelerated compared to unmodified cellulose papers.27 Interestingly, the hydrophilic character of the cellulose paper–mesoporous ceramic hybrid material can be switched into hydrophobic by the functionalization of the pores with a stimulus responsive polymer which allows the control of the accessibility of the pores.26 This finding demonstrates the importance of nanopores in papers and thus their targeted introduction. Besides the control and design of the wettability and fluid transport in cellulose paper, these coatings can be asymmetrically distributed along the paper length or cross-section generating Janus paper allowing even side selective and directed oil–water separation.24,26,27
Here we present the systematic investigation and tuning of silica mesopore formation in cotton linter and eucalyptus sulphate paper sheets by sol–gel chemistry and EISA. Mesopore formation during dip-coating based on the EISA process only occurs if the critical micellar concentration (CMC) of the pore forming template is reached upon solvent evaporation. Using krypton and argon 87 K adsorption, transmission electron microscopy (TEM), and small angle X-ray scattering (SAXS) measurements, new insights into the mesopore formation during the EISA process were gained. The interplay of the capillary fluid transport velocity and solvent evaporation rate together with micelle forming template concentration and CMC was determined to be crucial factors in tuning mesopore formation in paper. Correlating 87 K argon adsorption and confocal laser scanning microscopy (CLSM), the silica distribution along the cellulose paper fibre length and cross-section as well as its templating properties were investigated showing mesoporous silica formation on the outer fibre surface as well as inside the fibre lumen.
Results and discussion
Interplay of solvent evaporation, critical micellar concentration, and capillary transport velocity for mesopore formation in paper
To understand the influence of paper on mesopore formation during the functionalization by sol–gel chemistry and the dip-coating based EISA-process, laboratory-engineered cotton linter and eucalyptus sulphate paper sheets were functionalized with mesoporous silica. Therefore, sol–gel solutions containing the precursor tetraethoxysilane (TEOS) and three different concentrations of a mesopore forming template Pluronic® F127 (TEOS ethanol ratio 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20; 1:40 and 1:80) were used. After dip-coating, the silica coated paper samples undergo a thermal post-treatment and the mesopore template is chemically extracted afterwards (Fig. 1, Fig. S1† and Experimental details). The obtained hybrid silica–paper material is analysed with respect to mesopore formation. Interestingly, cotton linter and eucalyptus sulphate paper show a template concentration and cellulose fibre type dependent formation of mesopores as deduced from argon adsorption isotherms and SAXS measurements (Fig. 2a, b, 3a and b). Mesopore formation requires reaching the CMC of the mesopore forming template Pluronic® F 127 upon solvent evaporation from the applied cotton linter paper sheets. The CMC seems to be reached suitably fast only when using the sol–gel solution containing a TEOS:ethanol ratio of 1:20 and thus a high template concentration. When using sol–gel solutions with lower template concentrations with TEOS:ethanol ratios of 1:40 or 1:80 the CMC is not reached sufficiently fast during the coating process, and thus no mesopore formation occurs. In contrast to cotton linter paper sheets, mesopores are generated within the silica coating of eucalyptus sulphate paper sheets when using a sol–gel solution with an intermediate (TEOS:EtOH = 1:40) template concentration (Fig. 3b). This probably indicates a faster solvent evaporation due to faster capillary transport and thus micelle as well as mesopore formation occurs at lower template concentration in eucalyptus sulphate paper sheets as compared to cotton linter paper sheets.
:20; 1:40 and 1:80) were used. After dip-coating, the silica coated paper samples undergo a thermal post-treatment and the mesopore template is chemically extracted afterwards (Fig. 1, Fig. S1† and Experimental details). The obtained hybrid silica–paper material is analysed with respect to mesopore formation. Interestingly, cotton linter and eucalyptus sulphate paper show a template concentration and cellulose fibre type dependent formation of mesopores as deduced from argon adsorption isotherms and SAXS measurements (Fig. 2a, b, 3a and b). Mesopore formation requires reaching the CMC of the mesopore forming template Pluronic® F 127 upon solvent evaporation from the applied cotton linter paper sheets. The CMC seems to be reached suitably fast only when using the sol–gel solution containing a TEOS:ethanol ratio of 1:20 and thus a high template concentration. When using sol–gel solutions with lower template concentrations with TEOS:ethanol ratios of 1:40 or 1:80 the CMC is not reached sufficiently fast during the coating process, and thus no mesopore formation occurs. In contrast to cotton linter paper sheets, mesopores are generated within the silica coating of eucalyptus sulphate paper sheets when using a sol–gel solution with an intermediate (TEOS:EtOH = 1:40) template concentration (Fig. 3b). This probably indicates a faster solvent evaporation due to faster capillary transport and thus micelle as well as mesopore formation occurs at lower template concentration in eucalyptus sulphate paper sheets as compared to cotton linter paper sheets.
 | ||
| Fig. 1 Schematic illustration of the interplay between evaporation rate, critical micellar concentration, capillary transport and withdrawal speed during the dip-coating procedure based on the EISA process and the necessary conditions for pore formation on paper. | ||
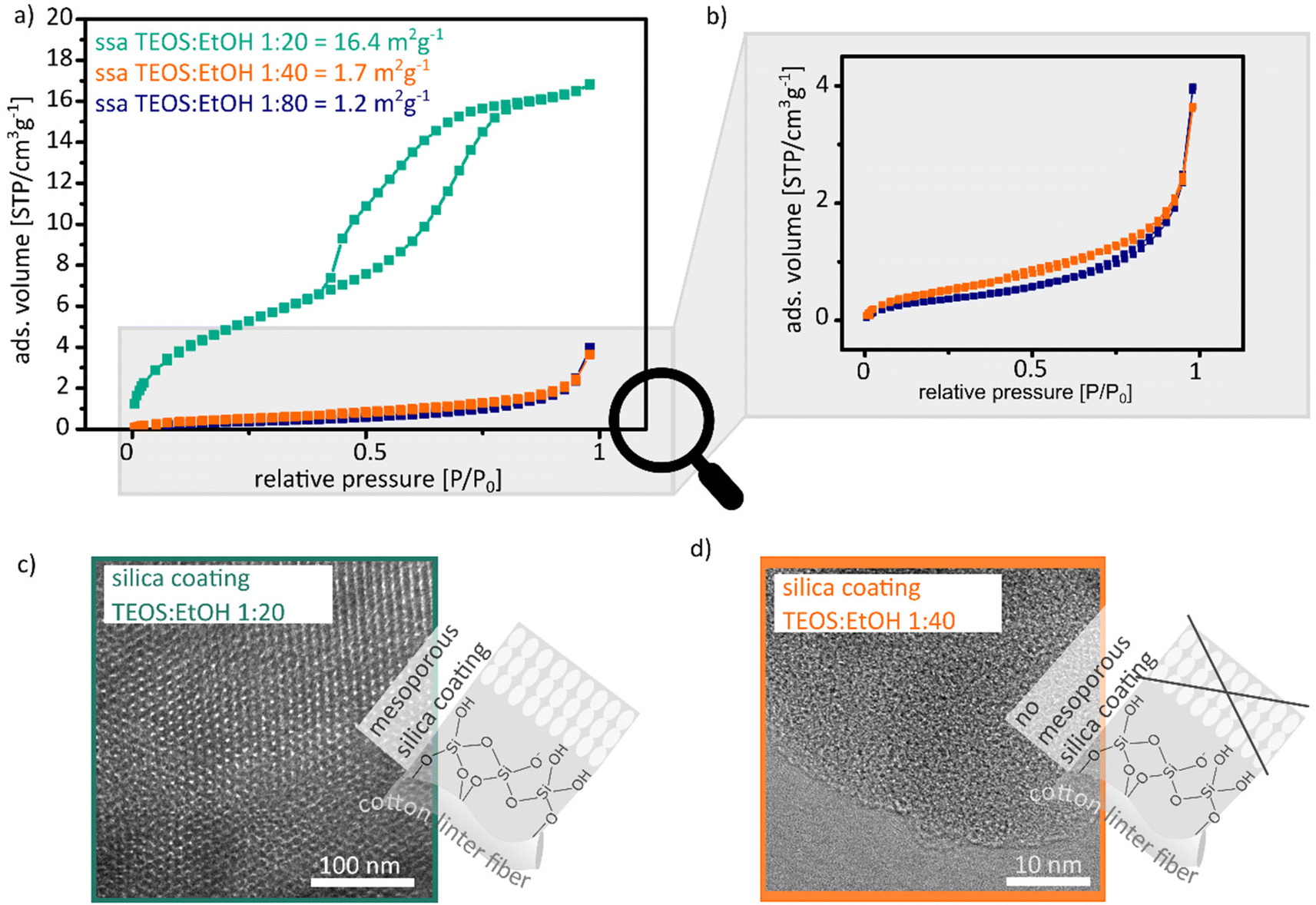 | ||
| Fig. 2 (a) Argon adsorption isotherms measured at 87 K between 0 and 1 relative pressure of silica cotton linter hybrid paper sheets, which were coated with sol–gel solution containing 1:20 (dark cyan), 1:40 (orange) and 1:80 (blue) TEOS:EtOH ratios after degassing for 12 h at 80 °C. (c) and (d) TEM images of the silica coating with the 1:20 (c) and the 1:40 (d) TEOS:EtOH ratio after the thermal removal of the paper at 500 °C for 2 h. | ||
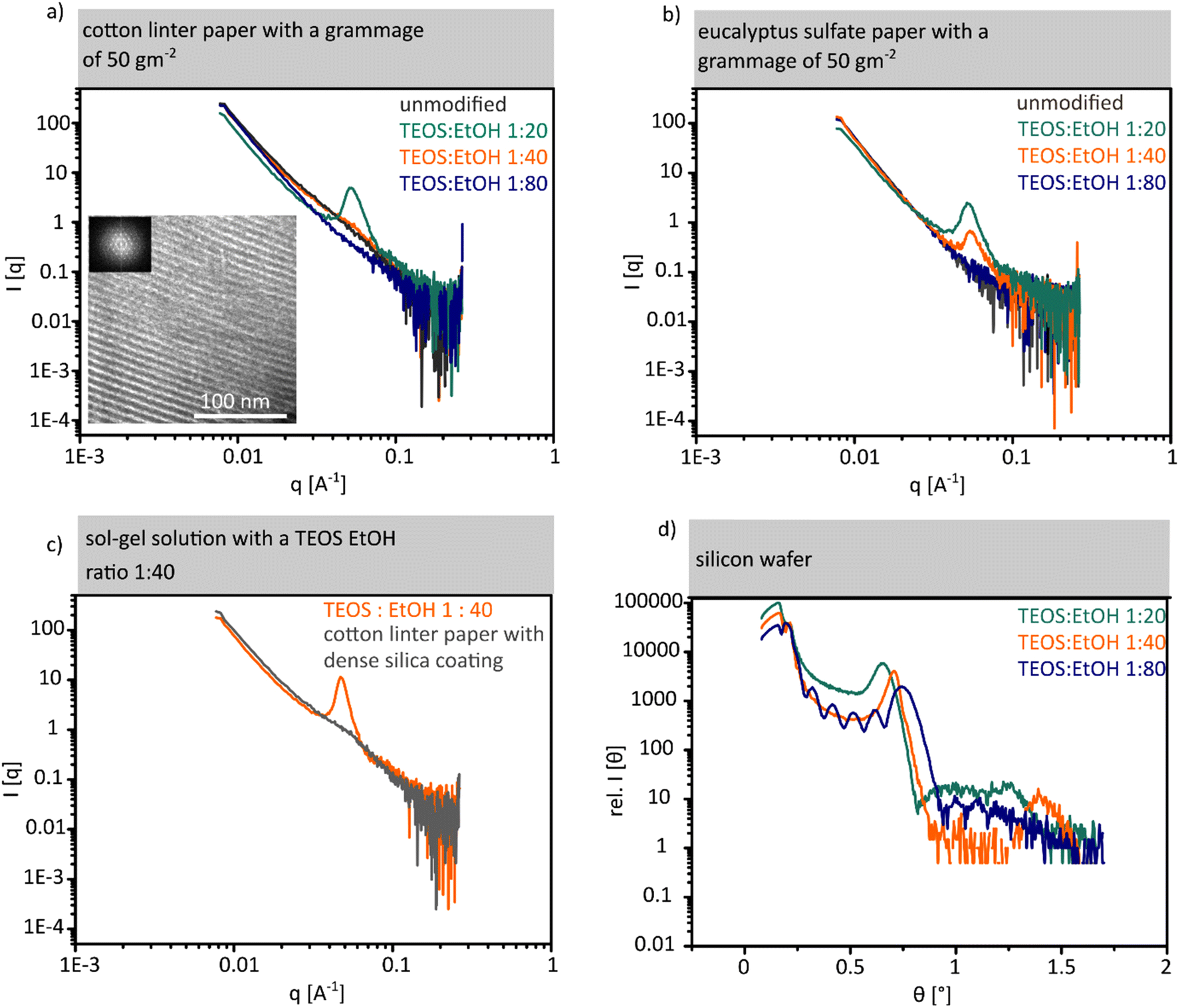 | ||
| Fig. 3 (a) and (b) SAXS results of unmodified cotton linter and eucalyptus sulphate paper with high (cyan), medium (orange) and low (blue) concentration of the template in the sol–gel solutions. TEM image and the corresponding FFT of the mesoporous silica coating after the removal of the fibre (c) SAXS results for dense silica followed by a mesoporous silica coating with medium concentration sol–gel solution coated cotton linter paper. (d) Results of the XRR experiments on silicon wafer coated with high, medium and low concentration sol–gel solutions. | ||
Analysing the argon adsorption isotherms measured at 87 K, cotton linter paper sheets coated with a TEOS:EtOH ratio of 1:20 show a IV(a) shaped isotherm with a H2(b) hysteresis loop (Fig. 2a dark cyan). This type of isotherm indicates mesopore formation in the paper sheet as well as the presence of pore blocking during the desorption process.28,29 The specific surface area increases from 1 m2 g−1 (11-point krypton BET) for unmodified cotton linter paper sheets to 16.4 m2 g−1 (11-point BET of the argon measurement) after mesoporous silica coating which is a 13× relative increase. Based on the isotherm as well as the hysteresis loop type, fibre swelling can be excluded. In gas adsorption, fibre swelling is indicated by a type II isotherm with a H3 hysteresis loop.4,8,30 Upon increasing the TEOS:EtOH ratio from 1:20 to 1:40 and 1:80 and thus decreasing template concentration, an argon adsorption isotherm of type II with a minimal occurrence of a H3 hysteresis loop is obtained, which indicates nonporous and microporous silica coating (Fig. 2a and b orange and blue isotherm).28,29 Thus, a specific surface area of 1.7 m2 g−1 for the 1:40 ratio and a specific surface area of 1.2 m2 g−1 for the 1:80 ratio are obtained, which indicate the absence of mesopore formation in accordance with the shape of the adsorption isotherms. No increase in the specific surface area as compared to unmodified cotton linter papers (specific surface area of 1 m2 g−1) is observed for these samples showing a type II isotherm. The observations from argon adsorption are consistent with TEM images (Fig. 2c and d). Mesopores with a diameter of 6 nm are observed in the TEM images from cotton linter paper with a 1:20 ratio coating (Fig. 2c). In comparison, the TEM images of cotton linter papers with a 1:40 coating show only micropores with a pore diameter <1 nm (Fig. 2d). Table 1 summarizes isotherm as well as hysteresis loop type, specific surface area and mesopore size for the cotton linter paper hybrid materials coated with 1:20, 1:40 and 1:80 TEOS:EtOH ratio containing sol–gel solutions.
:20, 1:40 and 1:80 TEOS:EtOH ratio containing sol–gel solutions
| Unmodified paper | High template con. | Intermediate template con. | Low template conc. | |
|---|---|---|---|---|
| TEOS:EtOH (1:20) |
TEOS:EtOH (1:40) |
TEOS:EtOH (1:80) |
||
| Isotherm type | Krypton 11 point BET | IV (a) | II | II |
| Hysteresis loop type | — | H2 (b) | H3 | H3 |
| 11 point MBET [m2 g−1] | 1; R = 0.9995 | 16.4; R = 0.9999 | 1.7; R = 0.9999 | 1.2; R = 0.9995 |
| Pore size [nm] | — | 6 | <1 | <1 |
Along with SAXS measurements of unmodified cotton linter papers (Fig. 3a grey SAXS measurement), cotton linter paper coated using sol–gel solutions with high (Fig. 3a, cyan SAXS measurement), intermediate (Fig. 3a, orange SAXS measurement), and low (Fig. 3a, blue SAXS measurement) template concentrations are consistent with argon adsorption and TEM measurements. The SAXS measurement of the paper sheet coated with a high template concentration shows a broad Bragg peak at 0.51 nm−1. This peak is caused by scattering centres in the mesopore size range which are arranged in a regular distance of 12.3 nm in accordance with the interpore distance obtained from TEM images (Fig. 3a TEM image). In silica coated cotton linter paper samples using a sol–gel solution with an intermediate as well as a low template concentration (Fig. 3 orange and blue SAXS measurements) and in unmodified cotton linter paper sheets, no Bragg peaks are observed (Fig. 3a grey SAXS measurement) indicating the absence of ordered mesopores in accordance with gas sorption results (Fig. 2a and b).
For eucalyptus sulphate paper coated with both sol–gel solutions with high (Fig. 3b cyan SAXS measurement) and intermediate (Fig. 3b orange SAXS measurement) template concentrations, a Bragg peak at 0.51 nm−1 (Fig. 3b) is observed indicating mesopore formation under both conditions in contrast to cotton linter paper sheets. Therefore, the Bragg peak of the eucalyptus sulphate papers coated with sol–gel solution with a high template concentration shows a higher intensity as compared to the eucalyptus paper sheet coated with intermediate template concentration of TEOS:EtOH = 1:40. The sol–gel solutions do not only differ in their template concentration but also in the precursor concentration. With a higher TEOS precursor concentration, a larger amount of silica coating is deposited and with this a thicker coating layer seems to be formed. Based on TGA measurements a silica coating amount of 6 wt% on eucalyptus sulphate paper was deposited (SI eqn (1)†) when using a sol–gel solution with a TEOS:ethanol ratio of 1:20 (high template concentration). With a TEOS:ethanol ratio of 1:40 (intermediate template concentration) 3.5 wt% silica is deposited onto the paper sheet. A lower amount of silica coating contains a smaller quantity of scattering centres in the silica coating which results in the lower intensity of the Bragg signal as compared to that due to a higher silica amount containing more scattering centres. Table 2 summarizes the conditions under which mesopore formation occurs on cotton linter paper, eucalyptus sulphate paper and planar silicon wafer. Assuming that a higher capillary fluid transport velocity relative to the evaporation rate is necessary for reaching the CMC and thus for mesopore formation in paper, a sufficient reduction in capillary fluid imbibition speed should result again in mesopore formation even under conditions where this did not happen with faster capillary fluid transport. One possibility to reduce the fluid transport velocity of paper is to deposit a dense silica coating. SAXS measurements of dense silica coated cotton linter paper do not show a Bragg signal in the range of mesopores (Fig. 3c). Using the sol–gel solution (TEOS:EtOH = 1:40) with an intermediate concentration of the template did not result in mesopore formation in unmodified cotton linter paper sheets. A Bragg signal is observed at 0.51 nm−1 for dense silica coated cotton linter papers demonstrating that on slowing down the capillary fluid transport in cotton linter paper sheet with the use of a dense silica coating, mesopore formation becomes again possible. Furthermore, mesopore formation is observed for all three applied sol–gel solution compositions showing a highly ordered arrangement when coating them onto a planar silicon wafer. The porosity and the pore arrangement of the thin mesoporous silica films on silicon wafers were investigated by X-ray reflectometry (XRR). Fig. 3d shows XRR measurements for all three silica films on the wafer. A critical angle below 0.22° (the value expected for bulk SiO2) is observed, which clearly indicates that the film has a lower density arising from the mesoporous structure. Moreover, the observation of diffraction peaks indicates that the mesoporous structure displays a high degree of crystallinity (also confirmed by GISAXS experiments). Film density, interplanar distances and film thickness are presented in the ESI (Fig. S4†). These results clearly indicate that mesopore formation in paper sheets using dip-coating and EISA strongly depends on the TEOS:EtOH ratio and thus on the template concentration as well as the fibre type which is related to the interplay of the evaporation rate reaching the CMC and the solution capillary transport velocity. The CMC has to be reached relatively fast as otherwise the paper fibre structure seems to prevent micelle and thus mesopore formation. This is attributed to a non-synchronized capillary solution transport into paper, solvent evaporation, and critical micellar concentration and not reaching the CMC in a suitable timescale before the mesopore forming template is probably trapped into the cellulose paper fibres not being able to arrange into micelles anymore. Therefore, for papers with a high capillary transport rate, higher evaporation and a high template concentration are required in order to reach the critical micellar concentration rapidly, and thus allow mesopore formation and arrangement. Consequently, mesopore formation on cotton linter and eucalyptus sulphate papers is related to different capillary flow velocities (for cotton linter: 2.7 ± 0.2 cm s−1 and eucalyptus sulphate: 2 ± 0.16 cm s−1) in the corresponding paper sheets which is also caused by differences in the fibre origin (Fig. S3 and S10†). The capillary flow velocity affects the available time for solvent evaporation and thus for reaching the CMC. This accordingly also influences micelle formation which results in mesopores after calcination. In cotton linter paper sheets, the fluid transport is faster than in eucalyptus sulphate paper despite having identical grammage and refining degree (Fig. S3†). Consequently, the capillary transport of the sol–gel solution is faster in cotton linter paper while the evaporation rate is constant. Therefore, the evaporation rate determines the time to reach the CMC which then induces micelle arrangement required for mesopore formation upon calcination. Thus, faster capillary transport seems to allow uptake and transport of the sol–gel solution into the cellulose paper fibres before reaching the CMC which apparently prevents sufficiently rapid micelle formation and arrangement and thus prevents mesopore formation.
:20, 1:40 and 1:80 TEOS:EtOH ratio containing sol–gel solutions
| TEOS:EtOH ratio |
Cotton linter paper | Eucalyptus sulfate paper | Silicon wafer |
|---|---|---|---|
| High capillary transport velocity = 2.7 ± 0.2 cm−1 | Low capillary transport velocity = 2 ± 0.16 cm−1 | No capillary transport velocity = 0 cm−1 | |
| 1:20 |
Yes | Yes | Yes |
| High template conc. | |||
| 1:40 |
No | Yes | Yes |
| Intermediate template conc. | |||
| 1:80 |
No | No | Yes |
| Low template conc. |
Localization of the silica coating as well as pore formation on and in cotton linter fibres
For understanding mesopore formation and mesoporous silica coating properties such as the specific surface area, pore size and pore size distribution and the local distribution of the silica coating in the paper sheet, both the silica-paper hybrid material and the mesoporous silica coating after cellulose paper fibre calcination were analysed by adsorption and CLSM imaging. Due to the low amount of mesoporous silica coating (up to 8 wt%) the specific surface area of the silica-hybrid material increases from 1 m2 g−1 for unmodified paper to 16.4 m2 g−1and mesopores with a pore diameter of 5.7 nm are inserted into the paper sheet (Fig. 4a and c). Therefore, the mesoporous silica coating amount of up to 8 wt% does not change the structural paper composition on the micrometre scale (Fig. 4b). There are two options to obtain the specific surface area of the mesoporous silica coating without the paper material. First the sample amount from the gas adsorption measurement can be reduced to the coating amount based on the silica residue obtained by TGA measurements or second by the thermal removal and the gas adsorption measurement of the remaining silica coating (information regarding the surface area estimation can be taken from the ESI†). | ||
| Fig. 4 (a), (d) and (h) argon 87 K adsorption isotherms for the silica-paper hybrid material (cyan), the mesoporous silica coating after the thermal cellulose removal (light grey) and for the dense silica coating after the thermal cellulose removal (black). The paper silica hybrid material as well as the silica residue after the thermal removal of the cellulose were degassed at 80 °C for 12 h. (c), (f) and (j) Pore size and pore size distribution of the silica-paper hybrid material, the mesoporous and dense silica coating after the thermal cellulose removal. (b), (e) and (i) SEM images of the silica-paper hybrid material (b), the mesoporous (e) and dense (i) silica coating after the thermal cellulose removal. (g) and (k) TEM images of the mesoporous (g) and dense (k) silica coating after the thermal removal of the cellulose at 500 °C of 2 h. | ||
Upon thermally removing cellulose fibres at 500 °C for 2 h only the silica remains. This remaining silica shows fibre-like structures with cracks indicating that the coating bursts upon fibre calcination (Fig. 4e). The isotherm of the remaining silica coating has a type IV(a) shape and a H2(a) hysteresis loop (Fig. 4d).28,29 Compared to the silica paper hybrid material, the argon adsorption isotherm for the mesoporous silica after cellulose removal has a steeper increase of adsorbed volume in the relative pressure range from 0.25 to 0.5. At a relative pressure higher than 0.5, gas uptake does not increase significantly further and a constant volume is reached. The small but steady increase of the adsorbed volume in the hysteresis loop indicates a change of the pore size and pore size distribution in the silica coating which can be caused by the thermal removal of the cellulose. Based on the BET method, a specific surface area of 305 m2 g−1 is determined for the remaining “paper-free” mesoporous silica coating obtained by a thermal treatment up to 500 °C. Compared to the silica-paper hybrid material, this is an increase of 19 times. This is a 6 times higher specific surface area than the specific surface area after the sample amount adjustment based on the TGA results. The specific surface area considering only the weight of the mesoporous silica coating was calculated to a specific surface area of 206 m2 g−1, which is within the range of the specific surface area of never dried pulp. Furthermore, after the thermal paper calcination the remaining silica contains micropores with a pore diameter of 2 nm and mesopores with a pore diameter of 3.7 nm and 5.7 nm, with a broad peak for the mesopores showing a maximum at 3.7 nm and a shoulder at 5.7 nm (Fig. 4f). Fig. 4g shows the corresponding TEM image of the mesoporous silica coating after the thermal removal of the cellulose fibres. In the TEM image spherical shaped pores with a diameter of 6 nm are observed. The pores have a hexagonal arrangement in the silica coating. The pore size distribution for the silica coating after the paper removal by calcination shows that during the thermal treatment the micropores and the mesopores with a pore diameter of 2 nm and 5.7 nm remain but also pores with a diameter of 3.7 nm are detected. The differences between the specific surface area of the mesoporous silica coating before and after paper calcination are attributed to the imprinting or templating of the cellulose paper fibre structure into the silica coating. During the coating process the sol–gel solution is distributed in the entire fibre. After the thermal removal of the cellulose paper fibres, the imprinted structures in the silica coating are accessible for the gas during the gas adsorption measurement which leads to a larger specific surface area for the “paper-free” mesoporous silica after the thermal removal of the cellulose as compared to the mathematically adjusted value.
The imprinting properties of the fibre are also indicated by the specific surface area of 584 m2 g−1 from cotton linter papers coated with dense silica after the thermal removal of the cellulose (Fig. 4h and i SEM image of the remaining dense silica coating). This large increase in the specific surface area as compared to 1 m2 g−1 for cotton linter paper with a dense silica coating (Fig. S6a†) is caused by micropores with a pore size of 1.7 nm (Fig. 4j and k TEM image) and is attributed to the imprinting of the fibre into the dense silica coating. Table 3 summarises the isotherm as well as hysteresis loop type, specific surface area, pore size and pore size distribution of mesoporous or dense silica coated cotton linter paper as hybrid material or after the thermal removal of the cellulose fibre.
| Mesoporous silica paper hybrid material | Mesoporous silica coating of the hybrid material | Mesoporous silica coating after thermal paper removal | Dense silica paper hybrid material | Dense silica coating after thermal paper removal | |
|---|---|---|---|---|---|
| Isotherm type | IV (a) | IV (a) | IV (a) | Kr 77 K 11 point BET | II |
| Hysteresis loop type | H2 (b) | H2 (b) | H2 (a) | — | H2 (a) |
| 11 point MBET [m2 g−1] | 16.4; R = 0.9999 | 206; R = 0.9999 | 305; R = 0.9988 | 1; R = 0.9995 | 584; R = 0.9997 |
| Pore size [nm] | 2; 5.7 | 2; 5.7 | 2; 3.7 | — | 1.7 |
| Pore size distribution [nm] | 4.9–6.9 | 4.9–6.9 | 3.1–5.8 | 1.4–2.8 | |
| Bragg Peak | Yes | Yes | No | No |
The distribution of the silica coating within the cellulose paper fibres was analysed by fluorescence microscopy imaging (CLSM). To visualize the outer and inner cellulose paper fibre surfaces, the cotton linter fibre was stained with calcofluor white (CFW) before coating with silica. The silica coating was stained with rhodamine B (RhoB) which was added to the dense silica forming sol–gel solution as already demonstrated in previous studies.24,27,31 Single cotton linter fibres were coated with the fluorophore containing sol–gel solution via dip-coating and underwent a thermal post-treatment before they were characterized by CLSM recording the z-stacks (Fig. S9†) to analyse the silica distribution along the fibre length (Fig. 5a and b) and at fibre cross-sections (Fig. 5a and c). On the molecular level, cotton linter fibres largely consist of cellulose polymers. During cell wall synthesis, several cellulose polymer chains bundle together to form microfibrils which again arrange to the next larger structural unit, the macrofibrils.32 Later, the cell wall producing cell dries out and leaves an empty lumen surrounded by this cell wall, which is then harvested as cotton or cotton linters (Fig. 5a). Due to this hierarchical arrangement of cellulose into micro- and macrofibrils, micro-, meso- as well as macropores are present in paper cellulose fibre. After thermal treatment, mesopores in the cellulose paper fibre largely collapse and only micro- and macropores remain.4,5 Based on the resolution limitations of the CLSM, individual micro- and macrofibrils cannot be resolved but the fibre outer and inner surfaces as well as the fibre lumen can be distinguished from one another (Fig. 5a–c). Fig. 5b shows a lateral (XY) confocal scan of a dense silica coated single fibre. In the figure, the CFW stained fibre is shown in cyan and the rhodamine B dyed dense silica coating in magenta. A very thin and homogeneous rhodamine B containing silica layer is located at the outer surface of the fibre. Also, a rhodamine B signal can be detected at the inner surface of the cellulose paper fibre and in the fibre lumen, as shown in an axial (XZ) confocal scan of the fibre's cross-section (Fig. 5c). For the fibre shown in Fig. 5b, the fibre lumen is filled with the dense silica coating. The silica distribution along the fibre cross-section was analysed from the line profile of the intensity values of the pixels for rhodamine B dye along the fibre cross-section. High grey values for rhodamine B and with this for the silica were obtained at the outer and inner surface of the fibre. Based on the pixel intensity and signal width, most of the silica coating is located in the fibre lumen (Fig. 5d). All of the 25 investigated single cotton linter fibres which were coated with silica by dip coating have silica on the outer fibre surface. In more than 50% of the fibres, silica is also present on the inner fibre surface and in more than 40% the silica coating fills the fibre lumen (Fig. 5e).
 | ||
| Fig. 5 (a) Structure of a cotton linter fibre.32 (b) CLSM image of a confocal cross section of the fibre length. The fibre walls are stained with CFW and is shown in the CLSM image in cyan blue. The silica coating is marked with rhodamine B and is shown in magenta. (c) CLSM image of a dense silica coated fibre cross section with the same colour code. (d) CFW and rhodamine B intensity profiles along the fibre cross section. (e) Relative frequency of the silica at the outer and inner surfaces of the fibre and as well as in the fibre lumen. | ||
The CSLM results demonstrate that the sol–gel solution reaches the cotton linter fibre along the entire cross-section. As a result, silica is not only present on the outer cellulose paper fibre surface but also on the inner wall and in the fibre lumen. These observations indicate that mesopore formation takes place outside as well as in the fibre lumen of cellulose paper fibre.
Experimental section
Reagents
All chemicals and solvents were purchased from Merck and used as received.Paper fabrication
Lab-engineered papers were produced with cotton linter fibres which were provided by the Eifeltor Mühle factory and eucalyptus sulphate fibres. The cotton linter pulp and the eucalyptus sulphate pulp were refined in a Voith LR 40 laboratory refiner (SEL 0.7 J m−1, set 3–1.6–60) with an effective specific energy of 100 kW h t−1. From these pulps, lab-engineered paper sheets with a grammage of 50 g−2 ± 3% were fabricated using a conventional Rapid-Koethen (FRANK-PTI GmbH, Birkenau, Germany) hand sheet maker according to DIN 54358 and ISO 5269/2 in the absence of additives and fillers. Fiber and paper characteristics can be taken from Tables S2 and S3 in the ESI.†Mesoporous and dense silica coatings
Mesoporous silica coatings were synthesized via sol–gel chemistry based on the precursor tetraethoxysilane (TEOS) and the template Pluronic® F127 according to our previous studies.24,26 Three different coating solutions with the respective reagent ratios shown in Table 4 were prepared and stirred at room temperature for 24 h. For the dense silica coating, the sol–gel solution with the TEOS:ethanol ratio of 1:20 and without Pluronic® mesopore template was used.
| High template concentration | Medium template concentration | Low template concentration | ||||
|---|---|---|---|---|---|---|
| Molar ratio | Solution composition | Molar ratio | Solution composition | Molar ratio | Solution composition | |
| TEOS | 1 | 13.18 mL | 1 | 6.59 mL | 1 | 3.30 mL |
| EtOH | 20 | 67.56 mL | 40 | 67.56 mL | 80 | 67.56 mL |
| Pluronic® F127 | 0.05 | 3.64 g | 0.05 | 1.82 g | 0.05 | 0.91 g |
| Water | 5 | 5.21 mL | 5 | 2.60 mL | 5 | 1.30 mL |
| conc. HCl | 0.01 | 0.05 mL | 0.01 | 0.03 mL | 0.01 | 0.01 mL |
Dry cotton linter (water content: 0.053 wt%; fluid flow velocity: 2.7 ± 0.2 cm−1) and eucalyptus sulphate (water content: 0.063 wt%; fluid flow velocity: 2 ± 0.16 cm−1) paper strips (acclimatized at 50 ± 2% RH and 23 ± 1 °C) with a dimension of 2 × 6 cm were dip-coated using different sol–gel solutions under a relative humidity of 50 (±5) % and a temperature of 25 (±1) °C.26,33 30 ml of sol–gel solution was used and 9 cm2 of the paper strips was covered with the sol–gel solution for 5 s before the paper strips were withdrawn with a speed of 2 mm s−1. Subsequently, the paper sheets coated with the template containing sol–gel solutions were aged for 1 h under the same conditions as the dip-coating was performed before they underwent thermal post-treatment. Papers with a dense silica coating directly undergo the thermal post-treatment without the aging step. For the thermal post-treatment, the paper sheets with the mesoporous as well as with the dense silica coating were placed in an oven (Carbolite Gero GmbH, Neuenhausen, Germany).
The thermal treatment was carried out starting with a temperature increase from 25 °C to 60 °C in 10 min. After reaching 60 °C, the temperature was maintained constant for 1 h, followed by a second temperature increase from 60 °C to 130 °C in 10 min. The final temperature of 130 °C was maintained for 2 h before cooling to ambient temperature by waiting. For removing the cellulose paper fibres, a subsequent temperature increase to 500 °C with a rate of 1 °C min−1 was performed. The temperature of 500 °C was maintained for 2 h before cooling to room temperature. After the thermal treatment up to the final temperature of 130 °C, the template in mesoporous silica coated cotton linter paper samples was chemically extracted by placing the samples in 125 mL of acidic ethanol (0.01 M HCl) for three days. After the chemical extraction, the samples were dried under ambient conditions at 25 °C for 24 h. The change in the sample weight during the post-treatment steps is shown in Fig. S1b, c† and Fig. 6.
 | ||
| Fig. 6 TGA measurement of unmodified cotton linter paper. The different mass loss areas are connected to structural changes of the paper. | ||
Thermogravimetric analysis (TGA)
TGA analysis was performed on a TGA 1 instrument (Mettler Toledo, Gießen, Germany) and the evaluation of the obtained curves was performed with the corresponding program Star1 (Mettler Toledo, Gießen, Germany). The mesoporous silica coated paper sample is placed in a 100 μL Al crucible and it is heated from 25 °C to 600 °C at a rate of 10 °C min−1 under constant air flow of 30 mL min−1. The maximum temperature of 600 °C was maintained for 10 min.Scanning electron microscopy (SEM)
SEM images were obtained using an ECO 10 (Zeiss, Aalen, Germany) scanning microscope operating at an acceleration voltage of 10 kV in a high vacuum mode. Before performing SEM imaging, a 10 nm thick platinum(80)/palladium(20) (ESG Edelmetall-Service Rheinstetten; charge number IN0145660) layer was applied with a Cressington 208HR sputter coater (TESCAN GmbH, Dortmund, Germany).Argon and krypton adsorption
Surface area determination of unmodified paper samples and dense silica powder were performed using krypton gas sorption at 77 K in a relative pressure range of 0.05 to 0.3 using an Autosorb iQ (Anton Paar, Ostfildern-Scharnhausen, Germany). With argon adsorption at 87 K, full isotherms in the relative pressure range of 0 to 1 were measured. Based on the argon isotherms the specific surface area, pore size and the pore size distribution of mesoporous silica coated paper samples as well as of the coating material after combustion of the fibre were determined. Before each measurement, the samples were degassed at 80 °C for 12 h under high vacuum. The measured adsorption isotherms were evaluated based on the Brunauer–Emmett–Teller (BET; 11 points between 0.05 and 0.3 P/P0) model and the NLDFT kernels using the program ASiQwin (Anton Paar, Ostfildern-Scharnhausen, Germany).Transmission electron microscopy (TEM)
TEM images were recorded on a JEM 2100F microscope (JOEL, Freisingen, Germany) with a maximum resolution of 2.2 Å at an acceleration voltage of 200 kV. TEM samples were prepared by drop-casting ∼5 μL of the silica film dispersion in ethanol (sonicated for five minutes beforehand) on carbon-coated copper grids (3.05 mm, 200 mesh size) followed by air-drying overnight.Small angle X-ray scattering (SAXS) and grazing-incidence small-angle X-ray scattering (GISAXS)
SAXS experiments were performed in a XEUSS 1.0 SAXS setup (XENOCS, Grenoble, France). Monochromatic X-rays (λ = 0.15419 nm) were produced with a GENIX 3D micro-focus tube. The incoming X-ray beam was collimated to have a size at the sample position of 0.5 × 0.5 mm2. Scattered photons were detected using a PILATUS 100 K detector placed at D = 2500 mm sample to detector distance (calibrated using Silver Behenate as standard). XRR and GISAXS experiments were performed under similar conditions except for the beam size that was maintained at 0.15 × 0.15 mm2 at the sample position.Confocal laser scanning microscopy (CLSM)
For the localization of the silica coating along the fibre length and the fibre cross-section, cotton linter fibres were stained with calcofluor white (CFW) while stirring in a 2.5 × 10−7 M ethanolic CFW solution for 1 h. The CFW stained fibres were dried under air and fixed on a 3D-printed perforated grid (Fig. S7†). The labelling of the silica coating was achieved by dip coating the fibres into a pre-condensed dense silica sol–gel solution containing 20 μM rhodamine B. The dip coating of the single fibres was carried out under the same conditions as described above under mesoporous and dense silica coatings for the paper modification. Confocal microscopic imaging was performed using a TCS SP8 microscope (Leica, Wetzlar, Germany) equipped with a 63× water objective and HyD detector. Images of the fibres were then recorded as three-dimensional z-stacks detecting the fluorescence of CFW (410–483 nm) and rhodamine B (557–707 nm) under excitation with a 405 nm and 552 nm laser, respectively. Data processing of the obtained images was conducted with the software Fiji,34 including two steps of background subtraction and application of a median filter. Line profiles of the corrected grey value data within an appropriate confocal section were finally evaluated to identify structural characteristics of the fibres and to analyse and correlate the position of the silica coating with the latter (Fig. S9†).Conclusions
The combination of insights from argon adsorption, SAXS, TEM and CLSM measurements for cotton linter and eucalyptus sulphate paper sheets coated by sol–gel chemistry and EISA under varying TEOS precursor, and thus mesopore-forming template concentrations, reveals that the solvent evaporation rate, the critical micellar concentration, and the capillary transport velocity are the determining factors for mesopore formation in paper. Therefore, the paper properties and especially the cellulose paper fibre type, the fibrillation degree, the fibre content as well as the pulping and bleaching process are crucial as they determine the capillary fluid flow velocity in paper. The CLSM results furthermore showed the silica distribution along the cellulose paper fibre length and the cellulose paper fibre cross-section, demonstrating the presence of silica both at the outer surface and inside the fibre lumen. Therefore, the processes occurring within the paper cellulose fibre wall at small length scales remain an interesting open question for future studies. Based on this fundamental understanding of mesopore formation and placement in paper sheets, we aim to broaden the application potential of paper in separation, sensing, and transport or even generating hierarchically porous silica paper sheets after thermal removal of the cellulose paper fibres.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors kindly acknowledge the financial support by the German Research Foundation (DFG) in the project AN1301/8 and the PAK-962 as well as the German Academic Exchange Service (DAAD) for supporting the research stay at the Instituto de Investigaciones Físicoquimicas Teóricas y Aplicadas (INIFTA-UNLP/CONICET) in La Plata, Argentina where the SAXS experiments were performed. MC is a staff member of CONICET Argentina. In addition, the authors thank Prof. E. Dörsam and Carl Fridolin Weber (IDD, Technische Universität Darmstadt, Germany) for supporting with the determination of the paper surface roughness. Finally, the authors thank Prof. O. Azzaroni for the opportunity to work in his research group at INIFTA in La Plata, Argentina.References
- C. Sanchez, P. Belleville, M. Popall and L. Nicole, Chem. Soc. Rev., 2011, 2, 696 RSC.
- (a) G. Pérez-Mitta, M. E. Toimil-Molares, C. Trautmann, W. A. Marmisollé and O. Azzaroni, Adv. Mater., 2019, 37, 1901483 CrossRef PubMed; (b) R. Pardehkhorram and A. Andrieu-Brunsen, Chem. Commun., 2022, 34, 5188 RSC.
- E. Noviana, T. Ozer, C. S. Carrell, J. S. Link, C. McMahon, I. Jang and C. S. Henry, Chem. Rev., 2021, 121, 11835 CrossRef CAS PubMed.
- V. A. Lovikka, P. Khanjani, S. Väisänen, T. Vuorinen and T. C. Maloney, Microporous Mesoporous Mater., 2016, 326 CrossRef CAS.
- S. Cichosz and A. Masek, Polym. Degrad. Stab., 2019, 33 CrossRef CAS.
- S. Park, R. Venditti, H. Jameel and J. Pawlak, Carbohydr. Polym., 2006, 1, 97 CrossRef.
- W. Mo, K. Chen, X. Yang, F. Kong, J. Liu and B. Li, Carbohydr. Polym., 2022, 119434 CrossRef CAS PubMed.
- W. Mo, F. Kong, K. Chen and B. Li, Wood Sci. Technol., 2022, 3, 867 CrossRef.
- H. N. Abdelhamid and A. P. Mathew, Coord. Chem. Rev., 2022, 214263 CrossRef CAS.
- C. M. Moreira, M. L. Scala-Benuzzi, E. A. Takara, S. V. Pereira, M. Regiart, G. J. Soler-Illia, J. Raba and G. A. Messina, Talanta, 2019, 186 CrossRef CAS PubMed.
- H. N. Abdelhamid, D. Georgouvelas, U. Edlund and A. P. Mathew, Chem. Eng. J., 2022, 136614 CrossRef CAS.
- N. Wang, X.-K. Ouyang, L.-Y. Yang and A. M. Omer, ACS Sustainable Chem. Eng., 2017, 11, 10447 CrossRef.
- M. Schelling, M. Kim, E. Otal, M. Aguirre and J. P. Hinestroza, Cellulose, 2020, 11, 6399 CrossRef.
- S. Bo, W. Ren, C. Lei, Y. Xie, Y. Cai, S. Wang, J. Gao, Q. Ni and J. Yao, J. Solid State Chem., 2018, 135 CrossRef CAS.
- X. Cui, X. Sun, L. Liu, Q. Huang, H. Yang, C. Chen, S. Nie, Z. Zhao and Z. Zhao, Chem. Eng. J., 2019, 898 CrossRef CAS.
- H. Zhu, X. Yang, E. D. Cranston and S. Zhu, Adv. Mater., 2016, 35, 7652 CrossRef PubMed.
- J. Cui, X. Xu, L. Yang, C. Chen, J. Qian, X. Chen and D. Sun, Chem. Eng. J., 2020, 125174 CrossRef CAS.
- S. Rauf, M. T. Vijjapu, M. A. Andrés, I. Gascón, O. Roubeau, M. Eddaoudi and K. N. Salama, ACS Appl. Mater. Interfaces, 2020, 26, 29999 Search PubMed.
- H. Wang, Y. Jian, Q. Kong, H. Liu, F. Lan, L. Liang, S. Ge and J. Yu, Sens. Actuators, B, 2018, 561 Search PubMed.
- M. L. Scala-Benuzzi, E. A. Takara, M. Alderete, G. J. A. A. Soler-Illia, R. J. Schneider, J. Raba and G. A. Messina, Microchem. J., 2018, 287 CrossRef CAS.
- R. Ghosh, S. Gopalakrishnan, T. Renganathan and S. Pushpavanam, Sci. Rep., 2022, 1, 5673 CrossRef PubMed.
- F. Razavi and H. Khajehsharifi, Chem. Pap., 2021, 7, 3401 CrossRef.
- (a) A. Ott, J. Peter, L. Wiehl, V. Potapkin, U. I. Kramm, H.-J. Kleebe, R. Riedel and E. Ionescu, Int. J. Appl. Ceram. Technol., 2022, 2, 838 CrossRef; (b) K. E. Shopsowitz, J. A. Kelly, W. Y. Hamad and M. J. MacLachlan, Adv. Funct. Mater., 2014, 3, 327 CrossRef.
- M. Nau, N. Herzog, J. Schmidt, T. Meckel, A. Andrieu-Brunsen and M. Biesalski, Adv. Mater. Interfaces, 2019, 18, 1900892 CrossRef.
- P. V. Persson, J. Hafrén, A. Fogden, G. Daniel and T. Iversen, Biomacromolecules, 2004, 3, 1097 CrossRef PubMed.
- C. Dubois, N. Herzog, C. Rüttiger, A. Geißler, E. Grange, U. Kunz, H.-J. Kleebe, M. Biesalski, T. Meckel, T. Gutmann, M. Gallei and A. Andrieu-Brunsen, Langmuir, 2017, 1, 332 CrossRef PubMed.
- J. J. Mikolei, L. Neuenfeld, S. Paech, M. Langhans, M. Biesalski, T. Meckel and A. Andrieu-Brunsen, Adv. Mater. Interfaces, 2022, 19, 2200064 CrossRef.
- M. Thommes, K. Kaneko, A. V. Neimark, J. P. Olivier, F. Rodríguez-Reinoso, J. Rouquerol and K. S. W. Sing, Pure Appl. Chem., 2015, 87, 1051 CrossRef CAS.
- C. Schlumberger and M. Thommes, Adv. Mater. Interfaces, 2021, 4, 2002181 CrossRef.
- M. Kimura, Z.-D. Qi, H. Fukuzumi, S. Kuga and A. Isogai, Cellulose, 2014, 5, 3193 CrossRef.
- T. Fujii, A. Ishii and M. Anpo, J. Photochem. Photobiol., A, 1990, 2, 231 CrossRef.
- (a) H. Zhu, Z. Jia, Y. Chen, N. Weadock, J. Wan, O. Vaaland, X. Han, T. Li and L. Hu, Nano Lett., 2013, 7, 3093 CrossRef PubMed; (b) S. Zhu, S. K. Biswas, Z. Qiu, Y. Yue, Q. Fu, F. Jiang and J. Han, Prog. Mater. Sci., 2023, 101025 CrossRef CAS; (c) T. Li, C. Chen, A. H. Brozena, J. Y. Zhu, L. Xu, C. Driemeier, J. Dai, O. J. Rojas, A. Isogai, L. Wågberg and L. Hu, Nature, 2021, 7844, 47 CrossRef PubMed.
- (a) D. Grosso, A. R. Balkenende, P. A. Albouy, A. Ayral, H. Amenitsch and F. Babonneau, Chem. Mater., 2001, 5, 1848 CrossRef; (b) C. Sanchez, C. Boissière, D. Grosso, C. Laberty and L. Nicole, Chem. Mater., 2008, 3, 682 CrossRef; (c) G. J. Soler-Illia and P. Innocenzi, Chemistry, 2006, 17, 4478 CrossRef PubMed; (d) C. J. Brinker, Y. Lu, A. Sellinger and H. Fan, Adv. Mater., 1999, 11, 579 CrossRef CAS.
- J. Schindelin, I. Arganda-Carreras, E. Frise, V. Kaynig, M. Longair, T. Pietzsch, S. Preibisch, C. Rueden, S. Saalfeld, B. Schmid, J.-Y. Tinevez, D. J. White, V. Hartenstein, K. Eliceiri, P. Tomancak and A. Cardona, Nat. Methods, 2012, 7, 676 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3nr01247f |
| This journal is © The Royal Society of Chemistry 2023 |