
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Large-scale synthesis of 2D-silica (SiOx) nanosheets using graphene oxide (GO) as a template material†
Björn K.
Birdsong
a,
Billy W.
Hoogendoorn
a,
Fritjof
Nilsson
 ad,
Richard L.
Andersson
a,
Antonio J.
Capezza
a,
Mikael S.
Hedenqvist
a,
Stefano
Farris
b,
Antonio
Guerrero
c and
Richard T.
Olsson
*a
ad,
Richard L.
Andersson
a,
Antonio J.
Capezza
a,
Mikael S.
Hedenqvist
a,
Stefano
Farris
b,
Antonio
Guerrero
c and
Richard T.
Olsson
*a
aDepartment of Fibre and Polymer Technology, KTH Royal Institute of Technology, Teknikringen 58, 11428, Stockholm, Sweden. E-mail: rols@kth.se
bDeFENS – Department of Food, Environmental and Nutritional Sciences Food Packaging Laboratory, Via Celoria 2, 20133, Milan, Italy
cDepartment of Chemical Engineering, Escuela Politécnica Superior, Universidad de Sevilla, 41011, Sevilla, Spain
dMid Sweden University, 85170 Sundsvall, Sweden
First published on 26th July 2023
Abstract
Graphene oxide (GO) was used in this study as a template to successfully synthesize silicon oxide (SiOx) based 2D-nanomaterials, adapting the same morphological features as the GO sheets. By performing a controlled condensation reaction using low concentrations of GO (<0.5 wt%), the study shows how to obtain 2D-nanoflakes, consisting of GO-flakes coated with a silica precursor that were ca. 500 nm in lateral diameter and ca. 1.5 nm in thickness. XPS revealed that the silanes had linked covalently with the GO sheets at the expense of the oxygen groups present on the GO surface. The GO template was shown to be fully removable through thermal treatment without affecting the nanoflake morphology of the pure SiOx-material, providing a methodology for large-scale preparation of SiOx-based 2D nanosheets with nearly identical dimensions as the GO template. The formation of SiOx sheets using a GO template was investigated for two different silane precursors, (3-aminopropyl) triethoxysilane (APTES) and tetraethyl orthosilicate (TEOS), showing that both precursors were capable of accurately templating the graphene oxide template. Molecular modeling revealed that the choice of silane affected the number of layers coated on the GO sheets. Furthermore, rheological measurements showed that the relative viscosity was significantly affected by the specific surface area of the synthesized particles. The protocol used showed the ability to synthesize these types of nanoparticles using a common aqueous alcohol solvent, and yield larger amounts (∼1 g) of SiOx-sheets than what has been previously reported.
Introduction
Silica has been extensively used as a functional polymer filler material in gels/lubricants/plastics and composites due to its mechanical strength, thermal stability, low toxicity, and possibilities for further functionalization.1–4 The Stöber reaction, developed in the 1960s, remains one of the most common aqueous methods of producing spherical silica nanoparticles (50–2000 nm diameter).5,6 The morphology of the particles stems from the reaction mechanism involved in the formation of the particles, where an initial nucleation process is followed by a growth stage governed by condensation onto the surface of the growing particles.7,8 The specific condensation, in turn, is related to the chemical structure of the precursor, the concentration of the precursor, the reaction temperature, the water-to-alcohol ratio, pH, and the presence of catalysts, all of which can be considered as tools for controlling the condensation reactions.9–11 It is challenging to form particles with more complex shapes, such as rods and sheets, because the condensation reactions must be steered toward promoting anisotropic growth.12 One method of providing this growth restriction is to use an already existing template material as the initial nucleation growth site for the in situ condensation, thus allowing for replicating the template morphology while simultaneously limiting the growth in at least one dimension.13–17Templating reactions have been reported as successfully strategies to obtain flake-shaped 2D-nanomaterials when preparing transition metals (e.g., gold and platinum) and metal oxides, such as TiO2, ZnO, and Fe2O3,18,19 where graphene oxide (GO) was used as the template materials. Tian et al.20 also obtained ultrathin 2D-metal oxide nanoplatelets (ZnO, Al2O3, Mn2O3, CuO) in the space between the stacked GO sheets, which provided confinement of the metal oxide phase during its formation due to the strong affinity between the carboxyl functional GO-sheets.20 Interestingly, graphene oxide was used to synthesize transition metal dichalcogenides (TMDs) templates, where the graphene formed when the molybdenum disulfide (MoS2) atomic TMDs-layer was reduced to Mo as the temperature reached 750 °C.21 Furthermore, various thin metal films have been synthesized on monolayer MoS2-sheets due to their favorable lattice parameters for single metal ion deposition, i.e., Al, Ag, Cu, Mn, Mo, Ni, Pd, Ru, Re, and Zn.22 However, although many confirmed successfully templating reactions have been reported, several challenges exist before these materials can be manufactured on larger scales for industrial implementation. Firstly, the template material must have uniform and well-defined surface properties and sizes that match the required dimensions for the replicates, i.e., the novel materials formed. Furthermore, methods to remove the template without affecting the synthesized material are essential for a templating reaction to be a viable process.20 Finally, to make these novel 2D-material structures of industrial interest, the reactions and synthesis procedures must show economic viability and the possibility of upscaling to industrial levels.
In this study, a method to produce 2D silica oxide (SiOx) materials with a lateral width of ca. 500 nm and a thickness of ca. 1.5 nm was developed. The synthesized 2D materials were formed via adsorption and condensation directly on the outer surfaces of the graphene oxide (GO) templates. The minimal amount of GO required during the solution-based synthesis allowed for record-large scale (∼1 g) synthesis of SiOx material. A newly developed and highly effective high-temperature treatment at 580 °C ensured a full removal of the GO template without having a detrimental impact on the formed SiOx nanosheets. The replication factor of the GO's lateral-sized dimensions was high and matched the prepared SiOx-material with high accuracy, 500 ± 200 nm. The bulk suspension showed, from rheological measurements, a shear thinning behavior for both the SiOx nanosheets and GO (synonymous with sheet particle morphologies), and demonstrated a high specific surface area often exhibited by 2D nanoparticles. Due to their unique size, reproducible morphology, and large surface area, the synthesized particles make good candidates as future barrier materials, carriers of catalysts, and in nano-electronics. The synthesized material also replicated the finest structures at the nanoscale and demonstrated significant durability to the thermal treatment conditions, allowing for the template's robust thermal removal. Amino-functional (3-aminopropyl) triethoxysilane (APTES) or tetraethyl orthosilicate (TEOS) were used as SiOx precursor materials in this study, firstly reporting GO as a material for templating 2D SiOx sheets.
Experimental section
Materials
Graphene oxide (GO) was purchased from Angstron materials (Dayton, USA) as an aqueous suspension with a solid content of 0.5 wt%.23 The GO-flakes had a reported thickness of 1–1.2 nm and a maximum lateral width of 554 nm.23 3-Aminopropyl triethoxysilane (APTES, ≥98%), tetraethyl orthosilicate (TEOS, ≥99.0%), fumed silica particles (22 wt%, pH = 10, Evonik Industries), ethanol (≥99.5%), 2-propanol (≥99.7%), and ammonia (NH3 (aq), 28–30%) were purchased from Sigma-Aldrich and used as obtained. MilliQ water (MQw), (18.2 MΩ, pH = 7.0), was used as the aqueous medium.Preparation of SiOx-coated GO
GO was functionalized by adding 18.53 ml of the as-received aqueous GO suspension to 250 ml HDPE Nalgene bottles. 2-Propanol (iPROH, see Table 1 and Fig. 1) was added to the GO suspension before sonicating the mixture for 10 min at room temperature, using a Branson 2510 sonication bath (V = 2.8 L, ultrasonic peak output = 100 W, f = 42 kHz). The iPROH was added to control the hydrolization of the SiOx precursors. 0.1 ml of NH3 (aq) as catalyst was added to the mixture under vigorous stirring before adding the SiOx precursor (see Table 1). The bottles were sealed and left to react under ambient conditions for 40 h with constant stirring. The reactions were terminated by removing any unreacted SiOx precursor using centrifugation at 8000 rpm for 3 × 10 min and exchanging the transparent supernatant with ethanol between each cycle for removal of unreacted SiOx precursors. The final product was diluted and dispersed in 100 ml using MQw. | ||
| Fig. 1 Illustration of reaction scheme. (a) GO dispersion, (b) silanization of GO dispersion into APTES-coated GO (AGO) and TEOS-coated GO (TGO), (c) freezing methods investigated, (d) freeze-drying of AGO and TGO, (e) heat treatment of AGO and TGO (586 °C 2 hours). | ||
| Sample name | 2-propanol (ml) | APTES (ml) | TEOS (ml) |
|---|---|---|---|
| See Table S1† for more details. | |||
| APTES-coated GO | 71.37 | 10 | — |
| TEOS-coated GO | 71.37 | — | 10 |
Post-treatment of SiOx-coated GO
The refinement of the coated GO material, see Fig. 1a and b for a simplified reaction scheme, was done by performing two different freezing methods as illustrated in Fig. 1. In method one, referred to as fast freezing (see Fig. 1c), 30 μl droplets of the obtained suspension were dropped onto a copper surface that was half immersed in liquid nitrogen. This freezing resulted in fully frozen droplets in t < 1 s. In the second freezing method, referred to as slow freezing (see Fig. 1c), 30 μl droplets were placed on an identical copper surface before placing the surface in a freezer (T = −21 °C) for 1.5–2 min, allowing fully frozen droplet to form at t ≅ 100 s. For both cases, the fully frozen droplets were freeze-dried for 24 h using a Scanvac coolsafe 55-4 vacuum drier at 0.37 mbar. As a comparison, the same freezing methods were performed on the untreated GO material. The pure SiOx sheets were obtained from the freeze-dried material after heat treatment (T = 586 °C, 2 h, 1 atm), using a high-temperature oven (T + M M9-1200), resulting in a white powder consisting of pure SiOx.Characterization
Results and discussion
Effect of GO template on silane particle morphology
Fig. 2a–c shows the results associated with the graphene oxide (GO) templating reaction providing pure silica sheets of almost identical size as the GO template. The inserted size distributions show that the sheets were of lateral dimensions 500 ± 184 nm after removal of the GO material by heat-treatment at 586 ± 5 °C for 2 h (see Fig. 2c and Table 2). The optimal heat-treatment temperature and time (2 h) was established as effective for removing the GO templateafter several heating experiments ranging from 550 to 620 °C, further validated by thermogravimetric measurements (Fig. S1†) and the energy dispersive X-ray data (XPS) shown in Fig. 2e. The formation of the silica sheets and their preservation of the GO morphology (Fig. 2a) relied on having the GO present as a suspended and never dried template at the earliest stages of the condensation reaction, as well as using an optimized quantity of silica precursor, for the formation of the condensed SiOx sheets. While both the silica precursors (aminopropyl-triethoxy and tetra-ethoxy functional silanes, APTES and TEOS, respectively) repeatedly resulted in on average ca. 1.5 nm thick particles (Fig. 2c), the APTES-coated GO was found to form 1–2 layer coatings, while TEOS-coated GO formed 1–3 coating layers (Table 2). The difference in coating layers and in the specific surface areas, where APTES-derived SiOx resulted in an SSA value of 247.6 m2 g−1 compared to TEOS-derived SiOx resulting in a SSA of 86.6 m2 g−1, suggested that multi-layer coatings had resulted with an overall reduction in SSA for the TEOS-derived SiOx compared to the GO template (Table 2). It appeared that the deposition of the silica precursor to the GO template particles also was more uneven in the case of TEOS due to the less homogenous condensation behavior of the silica precursor, forming small protrusions from the surface of the GO (Fig. 2b and c). After the coating reaction and heat-treatment, it was evident that the smooth and uniform surface seen in the ca. 1 nm thick GO sheets (Fig. 2a), had been replaced by a slightly rougher SiOx surface (Fig. 2c). However, the material retained the overall sheet morphology of the GO template. Furthermore, the ability of the GO sheets to roll back slightly at the edges (Fig. 2c and 5c) was lost in the replicated SiOx material, suggesting that the particles are stiffer in the plane of the SiOx sheets compared to the GO template. The slightly reduced lateral dimensions of the SiOx sheets 500 ± 184 nm may indicate that extensively curved edges of GO sheets are more challenging to replicate in the SiOx materials. Fig. S2† shows the result of precipitating the above SiOx materials without the GO template, demonstrating that no sheet morphology could be observed. Fig. 2d depicts commercial spherical SiOx nanoparticles sized ca. 20 nm, known as fumed silica (FS),24 which were characterized as a reference material for comparing the rheological effects shown in Fig. 2f.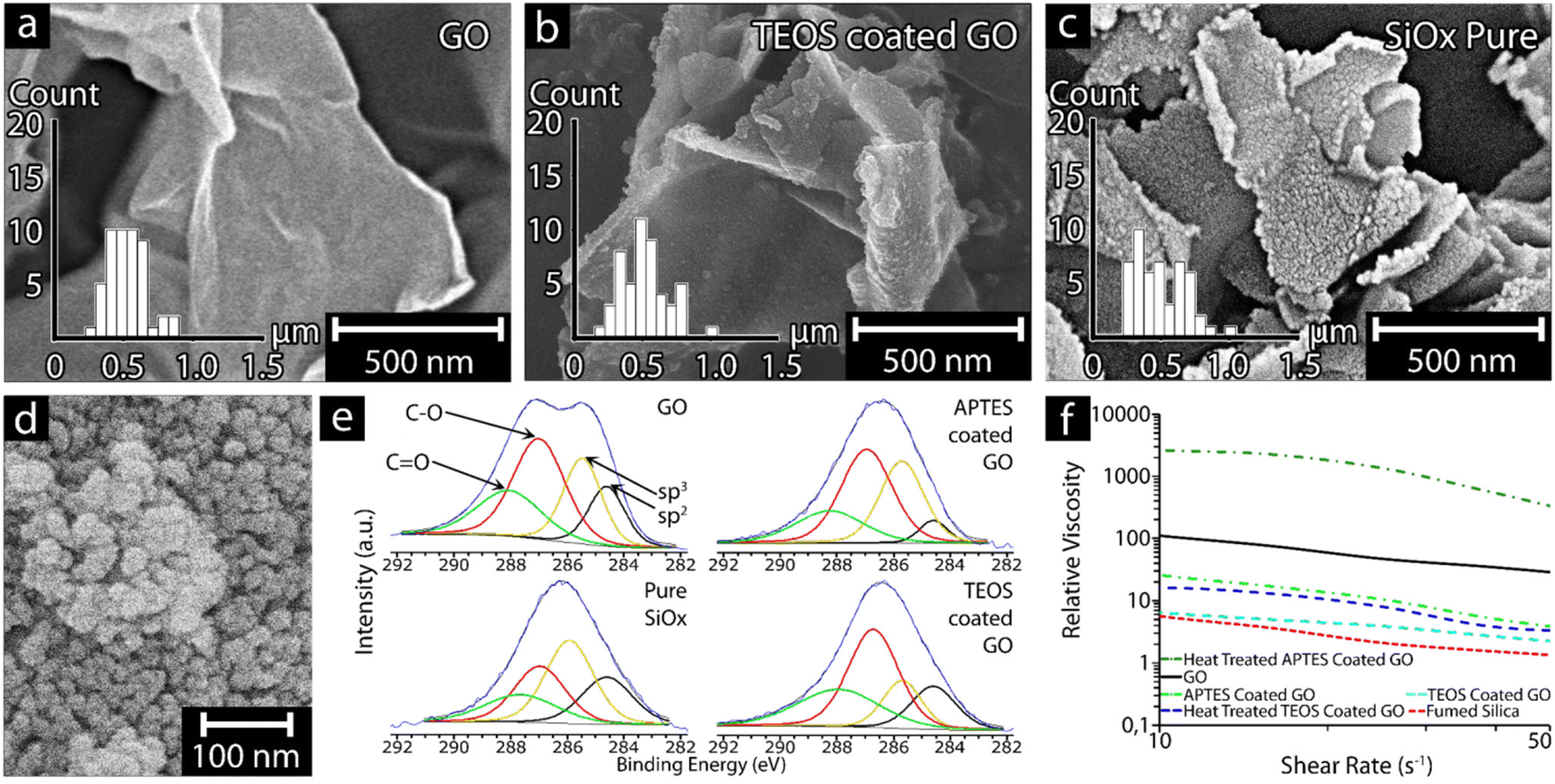 | ||
| Fig. 2 Micrographs of freeze-dried GO sheets (a) and GO sheets functionalized with tetra-ethoxy functional silane (TEOS) before (b) and after thermal removal of the GO template (c). The histograms depict the lateral size distributions using the longest axis for each measurement. (d) shows a micrograph of fumed silica particles (FS) as obtained from the manufacturer. (e) shows XPS spectra of the material depicted in (a–c); also, the APTES-modified GO is included for comparison and validation of the coating reaction and consolidation of silica precursor on the GO sheets. (f) rheology plots showing relative viscosity vs. shear rate for the materials depicted in (a–d). | ||
| Average particle thicknessa (nm) | SSA (m2 g−1) | Lateral dimensionsa (nm) | Number of SiOx precursor coating layers | Simulated monolayer coating thickness (nm) | ||||
|---|---|---|---|---|---|---|---|---|
| Pre-heat treatment | Post-heat treatment | Pre-heat treatment | Post-heat treatment | Pre-heat treatment | Post-heat treatment | |||
| a Average particle thickness was determined from 9 measurements using AFM, while the lateral dimensions were determined from 50 measurements using ImageJ from SEM images. AFM images are included as Fig. S3.† b Thickness of a single GO layer, including OH groups on both sides. The SSA for GO was determined to be 140 m2 g−1. The superscript letters mean significant differences with a confident level of 95% (p < 0.05). | ||||||||
| GO | 0.9 ± 0.1a | — | 140.0 | — | 533 ± 134A | — | — | 0.65b |
| GO + TEOS | 1.5 ± 1.0b | 1.6 ± 0.9b | — | 86.6 | 516 ± 168A | 500 ± 184A | 1–3 | 0.55 |
| GO + APTES | 1.4 ± 0.8b | 1.5 ± 0.8b | — | 247.6 | 585 ± 306A | 471 ± 185A | 1–2 | 0.8 |
Fig. 2e shows the deconvoluted high-resolution C 1s XPS spectra of GO, SiOx precursor-coated GO, and heat-treated SiOx-coated GO, revealing a completely different pattern for pristine GO and functionalized GO before and after heat treatment. More specifically, the saddle-like pattern of GO, which is associated with graphene after severe oxidation,25,26 implies the simultaneous presence of sp2 (284.6 eV) and sp3 (285.4 eV) C atoms, typical of the graphitic and hybridized components. The two other peaks are associated with C–O bonds from hydroxyls or epoxides (287 eV) and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds from carbonyls (288 eV), which agrees with data presented by Vryonis et al.27 The condensation reaction with APTES resulted in an increase of the sp3-peak (relative to the sp2 peak), which is consistent with a reduction in the non-graphitic, non-conductive property of the silica-based functionalization on the GO template. This increase was at the expense of hydroxyl and epoxide groups of GO, as indicated by the simultaneous decrease of the peak at 287 eV. When TEOS was used as a precursor, the resulting deconvolution spectrum was unexpectedly similar to the pristine GO (with the exception of less intense sp2 and sp3 peaks), suggesting that TEOS was less efficient at reacting with the oxygen functional groups on the surface of the GO. Finally, the deconvolution spectrum for the heat-treated SiOx-coated GO (pure SiOx) showed that the non-graphitic component was the dominant material. However, there was still graphitic material present, as well as oxygen-bearing carbon groups, which can plausibly be due to insufficient heat treatment for complete removal of the carbon phase from the pure SiOxsheets.
O bonds from carbonyls (288 eV), which agrees with data presented by Vryonis et al.27 The condensation reaction with APTES resulted in an increase of the sp3-peak (relative to the sp2 peak), which is consistent with a reduction in the non-graphitic, non-conductive property of the silica-based functionalization on the GO template. This increase was at the expense of hydroxyl and epoxide groups of GO, as indicated by the simultaneous decrease of the peak at 287 eV. When TEOS was used as a precursor, the resulting deconvolution spectrum was unexpectedly similar to the pristine GO (with the exception of less intense sp2 and sp3 peaks), suggesting that TEOS was less efficient at reacting with the oxygen functional groups on the surface of the GO. Finally, the deconvolution spectrum for the heat-treated SiOx-coated GO (pure SiOx) showed that the non-graphitic component was the dominant material. However, there was still graphitic material present, as well as oxygen-bearing carbon groups, which can plausibly be due to insufficient heat treatment for complete removal of the carbon phase from the pure SiOxsheets.
Si 2p XPS was performed to investigate potential silicon oxide differences due to the condensation of the SiOx precursor, where a clear difference between the APTES and TEOS-coated GO was observed in the spectra (Fig. S6†). The APTES-coated GO showed two peaks, at 101.9 eV attributed to Si–C and at 103 eV attributed to Si–OH. The Si–C peak was only observed in the APTES-coated GO, being a characteristic peak for the aminosilane.28–31 In comparison the TEOS-coated GO showed three peaks at 102.9, 104.2, and 105.7 eV, which where attributed to Si–OH, SiO2, and Si–O–Si, respectively.29,32,33 After heat treatment, no new bonds where observed for the TEOS-coated GO, instead a shift in intensity occurred as SiO2 increased markedly in comparison to Si–OH and Si–O–Si, from the conversion of Si–OH and Si–O–Si bonds into SiO2.34 In contrary, APTES-coated GO showed a different shift in its spectrum after the heat treatment, where a shift of ca. 2 eV occurred for the entire spectra. Furthermore, the resolved bonds found prior to the heat treatment had been completely replaced by the same bonds (104.1 and 105.5 eV) found in the heat-treated TEOS-coated GO (Fig. S6d†) corresponding to SiO2 and Si–O–Si respectively. The results suggests that the heat treatment converted the APTES-coated GO into a similar materials as the heat-treated TEOS-coated GO. The reason for the difference in the spectra prior to heat treatment (Fig. S6a and b†) is attributed to the more effective condensation of TEOS in comparison to APTES, resulting in a more crosslinked SiOx network, while APTES is a more compromised cross-linked network, on the surface of the GO as seen by the absence of Si–O–Si and SiO2 bonds (Fig. S6a†).35 A possible explination as to why the Si–OH groups remains in the heat-treated TEOS-coated GO (Fig. S6d†) and not in the heat-treated APTES-coated GO (Fig. S6c†) is that TEOS forms a more randomly condensed network on the GO (Table 2), preventing some of the OH functional groups from converting in to SiO2. This explination is supported by the carbon (C 1s) peak seen in Fig. S7†, which changed significantly for the APTES materials (b and c) while remaining relatively unchanged for the TEOS materials (d and e), indicating trapped material within the heat-treated TEOS-coated GO materials.
Fig. 2f shows the shear thinning response of the 0.5 wt% GO and fumed silica (FS) particles suspended in MQw (black and red, respectively). A significant difference in relative viscosity can be observed within the measured shear rates between 10 and 50 s−1 for the two systems. The 2D GO sheets exhibited a relative viscosity of 110 at 10 s−1, which is ca. 20 times higher than the spherical FS particles at the same shear rate. In comparison, the 0.5 wt% APTES-coated GO and TEOS-coated GO particles suspended in ethanol (light green and light blue, respectively) show similar relative viscosity patterns, being in between the values of the 2D GO sheets and the spherical FS particles. Both APTES and TEOS-coated GO exhibited a lower relative viscosity than the GO but higher than the FS particles. The APTES-coated GO showed the highest relative viscosity of the two SiOx precursors throughout the entire shear range, with a value of ca. 26 (at 10 s−1) compared to TEOS-coated GO (ca. 6 at the same shear rate). However, the APTES-coated GO is shown to have a significant shear thinning over the shear rate range, decreasing by a value of ca. 23, compared to the seemingly more stable TEOS-coated GO, which only had a relative viscosity reduction of ca. 5.
Heat-treating the coated GO particles; (APTES-coated GO (dark green), and TEOS-coated GO (dark blue), respectively) had the same effect regardless of the precursor,where the measured relative viscosity increased. However, the degree of the increase in relative viscosity varied between the heat-treated APTES-coated and TEOS-coated GO, where heat-treated APTES-coated GO showed an increase by ca. 100 times, compared to the mild increase for heat-treated TEOS-coated GO (only 2 times). The increase for the heat-treated TEOS-coated GO resulted in a comparable relative viscosity as the APTES-coated GO particles, thus approaching the rheological behavior of the GO sheets (see Fig. 2f). The heat-treated APTES-coated GO particles exhibited the highest recorded relative viscosity of all the particles tested, even surpassing the GO particles. In addition, the specific surface area (SSA) of the heat-treated SiOx-coated particles using APTES was ca. 3 times higher compared to heat-trated TEOS-coated GO, confirming the differences between using APTES/TEOS as precursors. Comparing the relative viscosity of the APTES and TEOS SiOx particles against the GO particles, the trend was that particles with larger SSA exhibited a higher relative viscosity.36 Furthermore, particles with a higher aspect ratio have a higher relative viscosity, which can be seen when comparing the heat-treated APTES-coated GO particles with an aspect ratio of ca. 174 compared to the spherical FS particles with an aspects ratio of 1. The effect possibly stems from the spherical morphology of the FS particles.37,38 However; additional interactions can significantly affect the rheology of the particle suspensions, including particle network formation associated with hydrogen bonding and π-stacking.39,40 In the case of the APTES-coated GO, the amino groups present on the surface may also have increased the interaction volume of the particles by forming layers of adsorbed water molecules increasing the interacting volume of the particles.41
The rheological behavior of the nanoparticle suspensions showed that a shear thinning effect was always observed, regardless of surface functional groups or morphology (see Fig. 2f). However, comparing the decrease in relative viscosity of the particles, it was found that FS, GO, TEOS-coated GO, and heat-treated TEOS-coated GO decreased by ca. 70–80%, while the APTES-coated GO and heat-treated APTES-coated GO showed a slightly higher decrease of ca. 90%, indicating that the APTES-modified particles were less shear stable in comparison to the TEOS particles. The the heat-treated APTES-coated GO particles showed a significantly higher relative viscosity than all other particles. It is suggested that the higher relative viscosity of the heat-treated APTES-coated GO was due to the highly uniform thin particles obtained and the highest SSA of all the tested particles (see Table 2). The results suggest that the SSA of particles has a large impact on the viscosity of the material and that using APTES resulted in the highest relative viscosity.
High-temperature removal of the GO-template
Fig. 3 shows the infrared spectra of freeze-dried samples of the virgin GO-material (Fig. 3a), the silane-coated GO with (APTES-coated GO, Fig. 3a) and without amino functionalities (TEOS-coated GO, Fig. 3b). The pure SiOx sheets obtained after thermal treatment at 586 °C for 2 h(replicated from the GO-template), are also displayed in Fig. 3a and b for their respective SiOx precursor material (APTES or TEOS). For the GO-reference sheets, the FTIR measurements showed three major peaks in the range 1800–1000 cm−1 (Fig. 3a). The peaks corresponded to the functional carbonyl-, conjugated carbon–carbon, and carbon–oxygen groups (bond stretching) at 1710–1720, 1618 and 1050–1150 cm−1, respectively,42,43 although at relatively low intensity compared to the large shoulder present in the range 3000–3500 cm−1, which was associated with the adsorbed water and the hydroxyl groups (O–H bond stretching).44 After high-temperature thermal treatment at 586 °C, the formed pure SiOx-sheets had in common the formation of a more uniform strong peak at ca. 1060 cm−1, which is synonymous to a more crosslinked SiOx-network (Fig. 3a and b).45–47 The more crosslinked SiOx-network is represented by the merging of the two smaller peaks at 1188 cm−1 and 950 cm−1, corresponding to the asymmetric stretching of Si–O–Si and the stretching of Si–O (silanol) groups, respectively.48 In a less constrained network, these two peaks are typically accompanied by an additional peak at 920 cm−1, which was more prominent in the case of the amino-functional SiOx-network. Finally, the peak observed at approximately 1640 cm−1 in the pure SiOx (Fig. 3a) corresponded to the amide groups, which explains its absence in the TEOS-based materials.49 Overall, it was evident that the carbon contained in the material was removed during the high-temperature treatment, something which was also seen in the mass loss curves shown in Fig. S1.† The mass losses observed initially corresponded to the removal of physically absorbed water (T ≤ 150 °C),50 and at the higher temperatures of 300–350 °C, the removal of oxygen-containing functional groups such as epoxy and carboxylic groups.51 Finally, the combustion of the carbon framework (GO template) was apparent, occurring at 500–600 °C,51,52 forming the pure SiOx material (Fig. 3a and b). Fig. S1† further shows the specific oxidation patterns associated with the GO template and the removal of oxygen, hydroxyl, and epoxy groups associated with the surface of GO (compare XPS data in Fig. 2e).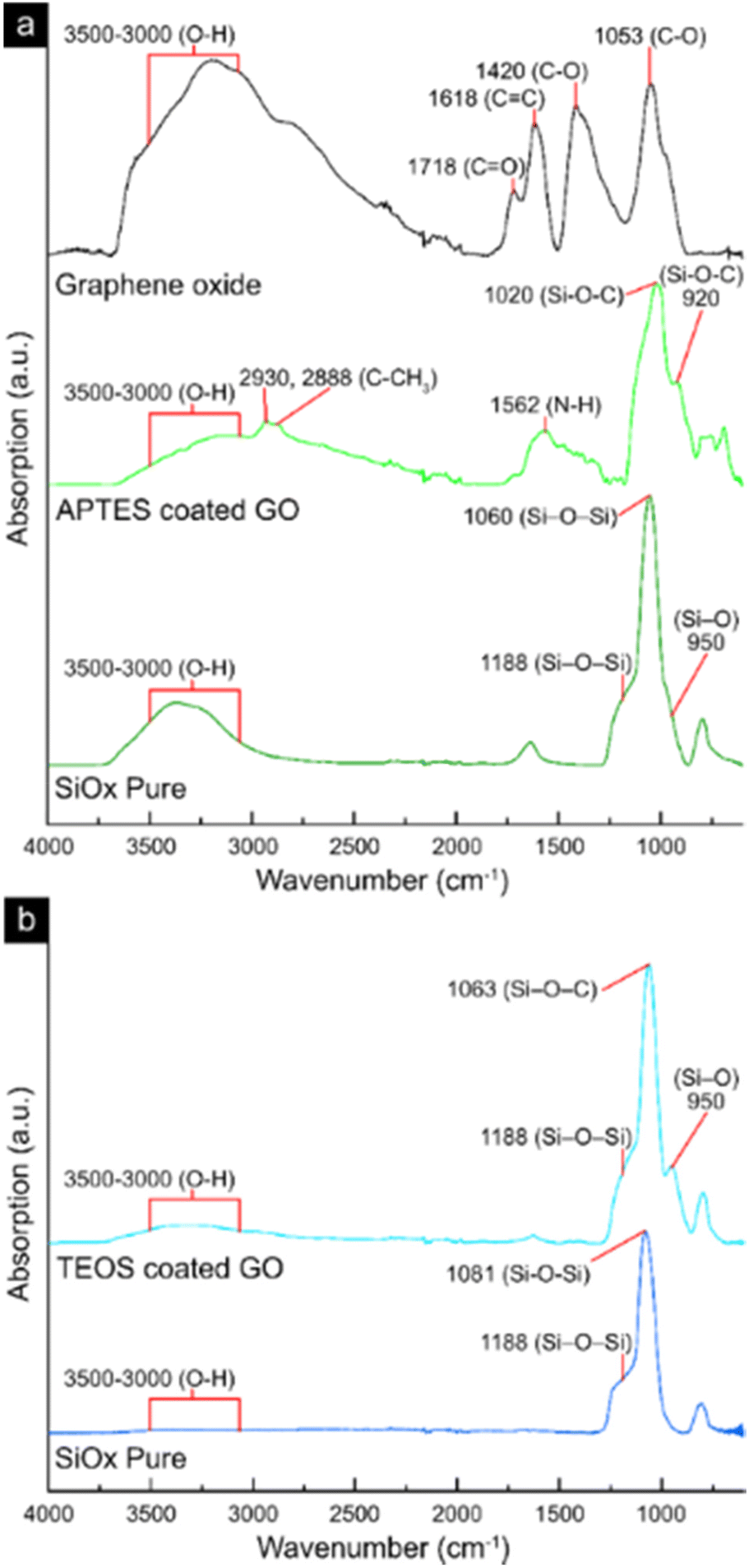 | ||
| Fig. 3 FTIR-spectra of freeze-dried samples of GO, APTES-coated GO, pure SiOx obtained from APTES-coated GO (a), TEOS-coated GO, and pure SiOx obtained from TEOS-coated GO (b). | ||
Fig. 4 shows the advantage of carrying out the GO-templating reactions in the suspended state, using only alcohol and water as the reaction medium. Although the reactions required extensive time to ensure completion (40 h), the alcohol allowed for a slow and gradual deposition and condensation of the SiOx-precursor (APTES and TEOS, see Table 2), which emphasized itself as uniformly shaped and extremely thin silica sheets (Fig. 4). Accordingly, the controlled large-scale preparation of the hybrid SiOx/GO-material was not limited by the size of the reaction, as long as excessively rapid condensation of the precursor material to the template could be avoided, which would occur with excessive amounts of water present in the reactions (Fig. 4). Fig. 4a demonstrate the pure SiOx-sheets responsible for the rheological behavior shown in Fig. 2f, as spread-out freeze-dried material on the surface of the SEM-sample holder. The sheets can be observed as randomly oriented clusters with low sheet-to-sheet adherence; see Fig. 4b (inset). The record large-scale GO template synthesis is also demonstrated from the inset photographs (Fig. 4c–e), where the GO, the SiOx coated GO, and the pure SiOx-sheets are shown as powders. The preparation of the powders required controlled freeze-drying conditions to avoid extensive sheet aggregation; this was the case for all nanosheet materials in this study. Fig. 5 demonstrates the effect of the freezing rate before carrying out the freeze drying procedure on the aqueious suspension containing the pure GO-sheets. These sheets contained a more limited amount of hydroxyl groups on their surface compared to the glassy hybrid SiOx/GO sheets obtained (see XPS, Fig. 2e).
 | ||
| Fig. 4 Micrograph depicting the homogenous 2D TEOS SiOx material after heat treatment (a and b). Photo of materials at various stages freeze-dried GO (c), freeze-dried silanized GO (d), and heat-treated silanized GO (e). The images were taken after the freeze drying, demonstrating solely the effect of the freezing rate. | ||
 | ||
| Fig. 5 Effect of suspension freezing rate on GO using a freeze time of t < 1 s (a–c), and t ≅ 100 s (d–f); insets do not represent the shown magnified area. | ||
Effect of suspension freeze rate on GO Morphology
Fig. 5 depicts GO that was frozen at a fast-freezing time (Fig. 5a–c, t < 1 s) and a slow freezing rate (Fig. 5d–f, t ≅ 100 s). The fast freezing resulted in a highly uniform and porous-like material, which, on a macroscopic scale, could be reproduced as large pieces of foam, see Fig. 5a. The material shown in Fig. 5a consists of sheet-like structures, further highlighted in Fig. 5b and c, appearing as a 3-dimensional web of bent, twisted and rolled up GO sheets, similar to what had previously been described by Guex et al. (2017).26 The individual GO sheets measured 530 ± 130 nm in lateral width (inset Fig. 2a), which was 96.4% of the documented size reported by the manufacturer (500–550 nm).23 Additionally, no apparent anisotropy could be observed within the network of associated GO sheets (Fig. 5b), i.e., when the rapid freezing had been carried out. The relevance of the rapid freezing, in the context of replicating the 2D sheet morphology of the GO into the SiOx sheets, is visible from Fig. 5d–f. Fig. 5d shows individual GO sheets that always were associated into large “super-sheets” when slowly freezing the GO suspension. Fig. 5f highlights that the super-sheets consisted of individual GO nanosheets assembled in a parallel stacked manner, as opposed to the foams from the fast freezing consisting of individual GO sheets. The material orientation was further visible as the super-sheets had no perpendicular direction, instead adopting a more layered formation in relation to the neighboring super-sheets (Fig. 5d and e). Further magnification shows the GO super-sheets having smooth surfaces with a decreased presence of features such as bending, splitting, or looping compared to the individual sheets (comp. Fig. 5e and b). The formation of the large super-sheets led to a significant size increase, going from 0.5 μm using the fast freezing method, to more than 100 μm in lateral dimensions using the slow freezing method, which stemmed from merging of individual sheets due to of the slower freezing rate.53,54 The size difference of the GO sheets could be attributed to the formation of ice crystals, which were steered towards forming larger ice crystals during the slower freezing process, compared to when the freezing rate was fast.54,55 It is suggested that the slower freezing rate allowed the individual GO sheets to be moved along with the ice crystal boundaries, forming the larger compressed GO super-sheets.53 This super-sheet formation with multiple individual sheets compressed into larger sheets play an important role in forming π–π interactions, leading to a more stable material.40 Overall, the micrographs reveal that the choice of freezing method during the treatment of the GO sheets, as well as the SiOx coated sheets, has a major impact on the 3D structure of the formed foam materials, as well as the morphology of the individual sheets. However, the morphology-phenomena was likely also affected by the condensation reaction itself and the choice of SiOx precursor when preparing the SiOx materials.Effect of silane structure on coating morphology
Fig. 6 shows the SiOx-coated GO before and after the heat treatment using APTES (Fig. 6a and b) and TEOS (Fig. 6c and d) as the silane precursor. While the dimensions of the APTES-coated GO and the TEOS-coated GO have been shown to have similar lateral dimensions both before and after heat treatment (see Table 2), the micrographs reveal differences in the sheet appearances resulting from the choice of SiOx precursor. While the APTES-coated GO sheets appear smooth and flexible, able to deform into conformations that form twisted, bent, and rolled-up structures, the slightly thicker (see Table 2) TEOS-coated GO appears as rigid sheets, seemingly unable to deform to the same degree. These differences are preserved after the heat treatment, indicating that the morphological traits of the coated GO were transferred to the pure SiOx nanosheets. Accordingly, it is suggested that the structure of the individual sheets was attributed to the coating thickness, which in turn, resulted from the varying configurations and stereochemistry of the SiOx precursors. | ||
| Fig. 6 Micrographs showing individual nanosheet structure for the APTES-coated GO, pre and post-heat treatment (a and b), compared to TEOS-coated GO, pre and post-heat-treatment (c and d), insets do not represent the shown magnified area. All micrographs were taken on samples frozen using a freezing time of t < 1 s. Models used for theoretical monolayer calculations for GO, APTES-coated GO, and TEOS-coated GO (e–g). Model of double layer coating obtained using TEOS (h). | ||
Fig. 6e–h shows the theoretical models of monolayer and bilayer coatings on the surface of the GO nanosheet template made from atomistic simulation models. The GO depicted in Fig. 6e shows the OH functional groups present on the surface; these functional groups are attributed to the location where the SiOx precursors would condensate and form the initial nucleating condensation structure. The models seen in Fig. 6f and g demonstrate a slight difference in the theoretical coating thickness, even when monolayer coatings are achieved. Stemming from the bulky carbon chain functional group in APTES. Although the theoretical monolayer coating thickness is larger for APTES, the bulky carbon chain limits further condensation, thus limiting the number of coating layers. This limitation may explain the virtually no difference in the measured thickness as seen in Table 2. Using Eqn (1), it is possible to calculate the number of coating layers present on the GO template:
 | (1) |
The results suggest that, regardless of using APTES or TEOS, the resulting coatings where monolayers. However, after heat treatment, the particle thickness remained relatively unchanged, possibly due to multiple SiOx sheets packing together (simulation model seen in Fig. 6h). Furthermore, the AFM measurements shows a variance high enough that multiple coating layers may occur, with two layers forming for APTES and up to three layers being recorded when TEOS was used (Table 2). The cause of this difference is due to the available reaction sites on the SiOx precursors, where APTES contains one less reactive site compared to the four OH groups present in TEOS, thus restricting the growth of APTES away from the surface of the GO. The difference in thickness between the theoretical monolayer thickness and the measured monolayer thickness is also likely attributed to the possible formation of defects during the condensation of the SiOx precursors, either due to the reaction conditions or defects in the GO template. It is also possible that the nature of the condensation reaction for the TEOS precursor, to a greater extent, was affected by competing reactions, shown as small grain structures on the derived sheets, see Fig. S8.† Overall, it is argued that the difference in coating thickness is caused by the variation in functionalities present in the SiOx precursors as the reason for the difference in the behavior of the final SiOx nanosheets. The theoretical maximum yield was estimated using the amount of APTES and TEOS described in Table 1. The yield was calculated by dividing the moles of silane that can theoretically react with the GO (estimating one oxygen per seven carbon atoms) with the total amount of SiOx produced if all silane reacted with GO. Thus, taking 10 ml APTES and TEOS results in a theoretical maximum yield of 2.17 to 2.07%, respectively. This demonstrates that the choice of SiOx precursor will have an impact on the morphology and behavior of the SiOx nanoparticle sheets. The suggested silica precursors open up for further exploration of 2D silica oxide materials and provide an alternative synthesis method for smooth mono and bi-layer nanoparticles such as physical exfoliation.56,57
Conclusion
Graphene oxide (GO) nanosheets were used as a template for the preparation of silicon oxide (SiOx) 2D nanoparticles, where the morphology of the GO sheets was successfully replicated to the SiOx-nanoparticles with near identical sizes and thicknesses as the template. The optimized condensation reactions allowed for coherent and uniformly coated GO sheets with a total thickness of ca. 1.5 nm for two different silane precursors, i.e., (3-aminopropyl) triethoxysilane (APTES) and tetraethyl orthosilicate (TEOS). However, the two SiOx precursors displayed different condensation characteristics when used at the same concentration. Computational modeling of the theoretical SiOx precursor monolayers and AFM measurements revealed that using APTES resulted in 1–2 layer coatings while TEOS resulted in 1–3 layers coatings. The removal of the organic carbon template (GO) and isolation of the SiOx sheets was possible by a controlled thermal treatment at 586 ± 5 °C, where carbon was thermally removed (as verified by XPS data), while the formed SiOx sheets remained intact. Furthermore, the nanoparticles showed an increase in the specific surface area up to ca. 77%, compared to the original GO template after the heat treatment due to the realization of smooth, thin, uniform and complete GO template coverage, which upon thermal high-temperature carbon template removal opened up for additional sheet exposure. Rheological measurements showed a shear thinning behavior for both coated and heat-treated SiOx particles and demonstrated that high specific surface area, often exhibited by 2D nanoparticles, markedly affected the relative viscosity of the particle suspension. To accurately determine the true dimensions of the virgin GO template, the coated GO sheets, and pure SiOx nanoparticles, the study included the development of a simple freezing protocol to minimize artifacts during the isolation from the reaction medium. The preservation of the virgin GO morphology relied on minimizing any sheet associations from occurring in the GO suspension prior to the templating reaction and drying. It was found that rapid freezing conditions were essential to reduce particle aggregation due to ice crystal formation during the freeze-drying process. Lastly, the degree of bending of the SiOx-sheets observed via SEM suggests that the choice of SiOx precursor affects the stiffness of the resulting SiOx material. This novel type of solution-based templating reaction reveals how scalable methods of developing SiOx-based nanoparticles are possible, where the morphology of the template solely limits the morphology. Such types of SiOx 2D nanoparticles can be envisioned in numerous applications, such as electronics, semiconductors, energy storage, and catalysis materials.Author contributions
B. K. B. carried out all laboratory experiments included in this study. B. K. B. also performed all characterizations involving microscopy, AFM, FTIR, TGA, and rheology. B. K. B., B. W. H., and R. T. O. wrote the manuscript. S. F. conducted XPS measurements and co-wrote the XPS interpretation. F. N. contributed with theoretical simulations and calculations and co-wrote the simulation interpretation. A. J. C., M. H., and A. G. reviewed rheological data and interpretations and provided input on improvements. A. J. C. also contributed to discussing and formulating some of the freezing discussions. B. W. H. critically reviewed the manuscript and participated in conversations about obtained results and implications. R. L. A. reviewed the manuscript and, with R. T. O., recognized the concept of using graphene oxide as a templating material to synthesize silicon oxide 2D nanosheets.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to acknowledge Anna Smed for her assistance during the investigation of freezing techniques and the financial contributions from the Swedish Research Council (VR). Bo Rydins Stiftelsen (Grant F 30/19) is acknowledged for the financial support to A. J. C.References
- Developments in the Formulation and Reinforcement of Concrete, ed. S. Mindess, Woodhead Publishing, 2nd edn, Elsevier Science, 2019, vol. iv Search PubMed.
- L. Wang, Z. Wang, H. Yang and G. Yang, The study of thermal stability of the SiO2 powders with high specific surface area, Mater. Chem. Phys., 1999, 57, 260–263, DOI:10.1016/S0254-0584(98)00226-0.
- Y.-X. Yang, et al., Evaluation of the toxicity of food additive silica nanoparticles on gastrointestinal cells, J. Appl. Toxicol., 2014, 34, 424–435, DOI:10.1002/jat.2962.
- C. A. Schlecht and J. A. Maurer, Functionalization of glass substrates: mechanistic insights into the surface reaction of trialkoxysilanes, RSC Adv., 2011, 1, 1446, 10.1039/c1ra00421b.
- W. Stöber, A. Fink and E. Bohn, Controlled growth of monodisperse silica spheres in the micron size range, J. Colloid Interface Sci., 1968, 26, 62–69, DOI:10.1016/0021-9797(68)90272-5.
- G. Kickelbick, in The Sol–Gel Handbook, 2015, pp. 225–244 Search PubMed.
- T. Matsoukas and E. Gulari, Dynamics of growth of silica particles from ammonia-catalyzed hydrolysis of tetra-ethyl-orthosilicate, J. Colloid Interface Sci., 1988, 124, 252–261, DOI:10.1016/0021-9797(88)90346-3.
- Y. Han, et al., Unraveling the Growth Mechanism of Silica Particles in the Stöber Method: In Situ Seeded Growth Model, Langmuir, 2017, 33, 5879–5890, DOI:10.1021/acs.langmuir.7b01140.
- N. Plumeré, A. Ruff, B. Speiser, V. Feldmann and H. A. Mayer, Stöber silica particles as basis for redox modifications: Particle shape, size, polydispersity, and porosity, J. Colloid Interface Sci., 2012, 368, 208–219, DOI:10.1016/j.jcis.2011.10.070.
- M. A. Bourebrab, et al., Influence of the initial chemical conditions on the rational design of silica particles, J. Sol-Gel Sci. Technol., 2018, 88, 430–441, DOI:10.1007/s10971-018-4821-9.
- A. Issa and A. Luyt, Kinetics of Alkoxysilanes and Organoalkoxysilanes Polymerization: A Review, Polymer, 2019, 11, 537, DOI:10.3390/polym11030537.
- I. Castano, et al., Chemical Control over Nucleation and Anisotropic Growth of Two-Dimensional Covalent Organic Frameworks, ACS Cent. Sci., 2019, 5, 1892–1899, DOI:10.1021/acscentsci.9b00944.
- B. Singh and V. Polshettiwar, Solution-phase synthesis of two-dimensional silica nanosheets using soft templates and their applications in CO2 capture, Nanoscale, 2019, 11, 5365–5376 RSC.
- Y. Xue, et al., A simple and controllable graphene-templated approach to synthesise 2D silica-based nanomaterials using water-in-oil microemulsions, Chem. Commun., 2016, 52, 575–578, 10.1039/C5CC06941F.
- Z. Lu, et al., Synthesis of Ultrathin Silicon Nanosheets by Using Graphene Oxide as Template, Chem. Mater., 2011, 23, 5293–5295, DOI:10.1021/cm202891p.
- L. Kan, B. Zheng and C. Gao, Graphene-templated approach to ultrathin silica nanosheets, Chin. Sci. Bull., 2012, 57, 3026–3029, DOI:10.1007/s11434-012-5252-6.
- D. Liu, et al., Cellulose nanofibril core–shell silica coatings and their conversion into thermally stable nanotube aerogels, J. Mater. Chem. A, 2015, 3, 15745–15754, 10.1039/c5ta03646a.
- V. S. Marangoni, L. D. Germano, C. C. C. Silva, E. A. De Souza and C. M. Maroneze, Engineering two-dimensional gold nanostructures using graphene oxide nanosheets as a template, Nanoscale, 2018, 10, 13315–13319, 10.1039/c8nr02855a.
- Y. Saito, et al., Filling the Gaps between Graphene Oxide: A General Strategy toward Nanolayered Oxides, Adv. Funct. Mater., 2015, 25, 5683–5690, DOI:10.1002/adfm.201501358.
- Z. Tian, C. Wei and J. Sun, Recent advances in the template-confined synthesis of two-dimensional materials for aqueous energy storage devices, Nanoscale Adv., 2020, 2, 2220–2233, 10.1039/d0na00257g.
- J. Wu, L. Ma, A. Samanta, M. Liu, B. Li, Y. Yang, J. Yuan, J. Zhang, Y. Gong, J. Lou, R. Vajitai, B. Yakobson, A. K. Singh, C. S. Tiwary and P. M. Ajayan, Adv. Mater. Interfaces, 2017, 4, 1600866, DOI:10.1002/admi.201600866.
- A. C. Domask, K. A. Cooley, B. Kabius, M. Abraham and S. E. Mohney, Room Temperature van der Waals Epitaxy of Metal Thin Films on Molybdenum Disulfide, Cryst. Growth Des., 2018, 18, 3494–3501, DOI:10.1021/acs.cgd.8b00257.
- N. Werk, Graphene Oxide Aqueous Dispersion, 2022. https://www.nanowerk.com/nanocatalog/Graphene/N002-PS/details/1739 Search PubMed.
- O. W. Flörke, et al., Silica, in Ullmann's Encyclopedia of Industrial Chemistry, 2008. DOI:10.1002/14356007.a23_583.pub3.
- A. Ganguly, S. Sharma, P. Papakonstantinou and J. Hamilton, Probing the Thermal Deoxygenation of Graphene Oxide Using High-Resolution In Situ X-ray-Based Spectroscopies, J. Phys. Chem. C, 2011, 115, 17009–17019, DOI:10.1021/jp203741y.
- L. G. Guex, et al., Experimental review: chemical reduction of graphene oxide (GO) to reduced graphene oxide (rGO) by aqueous chemistry, Nanoscale, 2017, 9, 9562–9571, 10.1039/c7nr02943h.
- O. Vryonis, T. Andritsch, A. S. Vaughan and P. L. Lewin, An alternative synthesis route to graphene oxide: influence of surface chemistry on charge transport in epoxy-based composites, J. Mater. Sci., 2019, 54, 8302–8318, DOI:10.1007/s10853-019-03477-w.
- S. Ray, D. Mishra, A. M. Strydom and P. Papakonstantinou, Magnetic behavioural change of silane exposed graphene nanoflakes, J. Appl. Phys., 2015, 118, 115302, DOI:10.1063/1.4930932.
- S. Ishak, S. Mandal, H.-S. Lee and J. K. Singh, pH-controlled synthesis of sustainable lauric acid/SiO2 phase change material for scalable thermal energy storage, Sci. Rep., 2021, 11, 15012, DOI:10.1038/s41598-021-94571-0.
- W. A. Talavera-Pech, et al., Effects of different amounts of APTES on physicochemical and structural properties of amino-functionalized MCM-41-MSNs, J. Sol-Gel Sci. Technol., 2016, 80, 697–708, DOI:10.1007/s10971-016-4163-4.
- G. Jakša, B. Štefane and J. Kovač, XPS and AFM characterization of aminosilanes with different numbers of bonding sites on a silicon wafer, Surf. Interface Anal., 2013, 45, 1709–1713, DOI:10.1002/sia.5311.
- Y. He, Z. Shan, T. Tan, Z. Chen and Y. Zhang, Ternary Sulfur/Polyacrylonitrile/SiO2 Composite Cathodes for High-Performance Sulfur/Lithium Ion Full Batteries, Polymer, 2018, 10, 930, DOI:10.3390/polym10080930.
- C. D. Wagner, A. V. Naumkin, A. Kraut-Vass, J. W. Allison, C. J. Powell and J. R. Rumble Jr., NIST Standard Reference Database, 2003, 20 Search PubMed https://www.xpsfitting.com/search/label/Silicon .
- G. Zheng, et al., Controlling Surface Oxides in Si/C Nanocomposite Anodes for High–Performance Li–Ion Batteries, Adv. Energy Mater., 2018, 8, 1801718, DOI:10.1002/aenm.201801718.
- T. Noisser, G. Reichenauer and N. Hüsing, In Situ Modification of the Silica Backbone leading to Highly Porous Monolithic Hybrid Organic–Inorganic Materials via Ambient Pressure Drying, ACS Appl. Mater. Interfaces, 2014, 6, 1025–1029, DOI:10.1021/am404005g.
- D. P. Bentz, C. F. Ferraris, M. A. Galler, A. S. Hansen and J. M. Guynn, Influence of particle size distributions on yield stress and viscosity of cement–fly ash pastes, Cem. Concr. Res., 2012, 42, 404–409, DOI:10.1016/j.cemconres.2011.11.006.
- S. Mueller, E. W. Llewellin and H. M. Mader, The effect of particle shape on suspension viscosity and implications for magmatic flows, Geophys. Res. Lett., 2011, 38, L13316, DOI:10.1029/2011gl047167.
- Y. Xie, Z. Yang and D. Barreto, The Influence of Particles’ Aspect Ratio on the Shear Behaviour of Granular Materials, 2017, vol. 188 Search PubMed.
- W. Tesfai, P. Singh, Y. Shatilla, M. Z. Iqbal and A. A. Abdala, Rheology and microstructure of dilute graphene oxide suspension, J. Nanopart. Res., 2013, 15, 1989, DOI:10.1007/s11051-013-1989-3.
- H. Bai, C. Li, X. Wang and G. Shi, On the Gelation of Graphene Oxide, J. Phys. Chem. C, 2011, 115, 5545–5551, DOI:10.1021/jp1120299.
- S. Zhang and X. Han, Effect of different surface modified nanoparticles on viscosity of nanofluids, Adv. Mech. Eng., 2018, 10, 168781401876201, DOI:10.1177/1687814018762011.
- Z. Bojarska, M. Mazurkiewicz-Pawlicka, S. Gierlotka and Ł. Makowski, Production and Properties of Molybdenum Disulfide/Graphene Oxide Hybrid Nanostructures for Catalytic Applications, Nanomaterials, 2020, 10, 1865, DOI:10.3390/nano10091865.
- R. A. Rochman, S. Wahyuningsih, A. H. Ramelan and Q. A. Hanif, Preparation of nitrogen and sulphur Co-doped reduced graphene oxide (rGO-NS) using N and S heteroatom of thiourea, IOP Conf. Ser.: Mater. Sci. Eng., 2019, 509, 012119, DOI:10.1088/1757-899x/509/1/012119.
- B. D. Ossonon and D. Bélanger, Synthesis and characterization of sulfophenyl-functionalized reduced graphene oxide sheets, RSC Adv., 2017, 7, 27224–27234, 10.1039/c6ra28311j.
- A. D. Ross and K. K. Gleason, Effects of condensation reactions on the structural, mechanical, and electrical properties of plasma-deposited organosilicon thin films from octamethylcyclotetrasiloxane, J. Appl. Phys., 2005, 97, 113707, DOI:10.1063/1.1923163.
- Y. Lin, T. Y. Tsui and J. J. Vlassak, Octamethylcyclotetrasiloxane-Based, Low-Permittivity Organosilicate Coatings, J. Electrochem. Soc., 2006, 153, F144, DOI:10.1149/1.2202120.
- C. Rau and W. Kulisch, Mechanisms of plasma polymerization of various silico-organic monomers, Thin Solid Films, 1994, 249, 28–37, DOI:10.1016/0040-6090(94)90081-7.
- S. Musić, N. Filipović-Vinceković and L. Sekovanić, Precipitation of amorphous SiO2 particles and their properties, Braz. J. Chem. Eng., 2011, 28, 89–94, DOI:10.1590/s0104-66322011000100011.
- M. Vakili, et al., Ultrasound-Assisted Preparation of Chitosan/Nano-Activated Carbon Composite Beads Aminated with (3-Aminopropyl)Triethoxysilane for Adsorption of Acetaminophen from Aqueous Solutions, Polymer, 2019, 11, 1701, DOI:10.3390/polym11101701.
- Y. Sehlleier, A. Abdali, S. Schnurre, H. Wiggers and C. Schulz, Surface functionalization of microwave-plasma synthesized silica nanoparticles for enhancing stability of dispersions, J. Nanopart. Res., 2014, 16, 2557, DOI:10.1007/s11051-014-2557-1.
- F. Farivar, P. Lay Yap, R. U. Karunagaran and D. Losic, Thermogravimetric Analysis (TGA) of Graphene Materials: Effect of Particle Size of Graphene, Graphene Oxide and Graphite on Thermal Parameters, C, 2021, 7, 41, DOI:10.3390/c7020041.
- P. L. Yap, S. Kabiri, D. N. H. Tran and D. Losic, Multifunctional Binding Chemistry on Modified Graphene Composite for Selective and Highly Efficient Adsorption of Mercury, ACS Appl. Mater. Interfaces, 2019, 11, 6350–6362, DOI:10.1021/acsami.8b17131.
- P. Munier, K. Gordeyeva, L. Bergström and A. B. Fall, Directional Freezing of Nanocellulose Dispersions Aligns the Rod-Like Particles and Produces Low-Density and Robust Particle Networks, Biomacromolecules, 2016, 17, 1875–1881, DOI:10.1021/acs.biomac.6b00304.
- S. Deville, E. Saiz and A. P. Tomsia, Ice-templated porous alumina structures, Acta Mater., 2007, 55, 1965–1974, DOI:10.1016/j.actamat.2006.11.003.
- B. Wicklein, et al., Thermally insulating and fire-retardant lightweight anisotropic foams based on nanocellulose and graphene oxide, Nat. Nanotechnol., 2015, 10, 277–283, DOI:10.1038/nnano.2014.248.
- C. Büchner, et al., A Large-Area Transferable Wide Band Gap 2D Silicon Dioxide Layer, ACS Nano, 2016, 10, 7982–7989, DOI:10.1021/acsnano.6b03929.
- C. Büchner, et al., Bending Rigidity of 2D Silica, Phys. Rev. Lett., 2018, 120, 226101, DOI:10.1103/physrevlett.120.226101.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3nr01048a |
| This journal is © The Royal Society of Chemistry 2023 |