
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Results of an interlaboratory comparison for characterization of Pt nanoparticles using single-particle ICP-TOFMS†
Lyndsey
Hendriks
*a,
Robert
Brünjes
 b,
Sara
Taskula
b,
Jovana
Kocic‡
c,
Bodo
Hattendorf
c,
Garret
Bland§
d,
Gregory
Lowry
d,
Eduardo
Bolea-Fernandez¶
e,
Frank
Vanhaecke
e,
Jingjing
Wang
f,
Mohammed
Baalousha
f,
Marcus
von der Au
g,
Björn
Meermann
g,
Timothy Ronald
Holbrook||
h,
Stephan
Wagner**
h,
Stasia
Harycki
i,
Alexander
Gundlach-Graham
i and
Frank
von der Kammer
*b
b,
Sara
Taskula
b,
Jovana
Kocic‡
c,
Bodo
Hattendorf
c,
Garret
Bland§
d,
Gregory
Lowry
d,
Eduardo
Bolea-Fernandez¶
e,
Frank
Vanhaecke
e,
Jingjing
Wang
f,
Mohammed
Baalousha
f,
Marcus
von der Au
g,
Björn
Meermann
g,
Timothy Ronald
Holbrook||
h,
Stephan
Wagner**
h,
Stasia
Harycki
i,
Alexander
Gundlach-Graham
i and
Frank
von der Kammer
*b
aTOFWERK AG, Thun, Switzerland. E-mail: lyndsey.hendriks@tofwerk.com
bDepartment of Environmental Geosciences, University of Vienna, Vienna, Austria. E-mail: frank.kammer@univie.ac.at
cSwiss Federal Institute of Technology (ETH), Zurich, Switzerland
dCarnegie Mellon University (CMU), Pittsburgh, USA
eGhent University, Department of Chemistry, Atomic & Mass Spectrometry – A&MS research group, Ghent, Belgium
fUniversity of South Carolina (USC), Columbia, USA
gFederal Institute for Materials Research and Testing (BAM) – Division 1.1 – Inorganic Trace Analysis, Berlin, Germany
hHelmholtz-Centre for Environmental Research (UFZ), Leipzig-Halle, Germany
iIowa State University (ISU), Ames, USA
First published on 16th June 2023
Abstract
This study describes an interlaboratory comparison (ILC) among nine (9) laboratories to evaluate and validate the standard operation procedure (SOP) for single-particle (sp) ICP-TOFMS developed within the context of the Horizon 2020 project ACEnano. The ILC was based on the characterization of two different Pt nanoparticle (NP) suspensions in terms of particle mass, particle number concentration, and isotopic composition. The two Pt NP suspensions were measured using icpTOF instruments (TOFWERK AG, Switzerland). Two Pt NP samples were characterized and mass equivalent spherical sizes (MESSs) of 40.4 ± 7 nm and 58.8 ± 8 nm were obtained, respectively. MESSs showed <16% relative standard deviation (RSD) among all participating labs and <4% RSD after exclusion of the two outliers. A good agreement was achieved between the different participating laboratories regarding particle mass, but the particle number concentration results were more scattered, with <53% RSD among all laboratories, which is consistent with results from previous ILC studies conducted using ICP-MS instrumentation equipped with a sequential mass spectrometer. Additionally, the capabilities of sp-ICP-TOFMS to determine masses on a particle basis are discussed with respect to the potential for particle density determination. Finally, because quasi-simultaneous multi-isotope and multi-element determinations are a strength of ICP-TOFMS instrumentation, the precision and trueness of isotope ratio determinations were assessed. The average of 1000 measured particles yielded a precision of below ±1% for intensity ratios of the most abundant Pt isotopes, i.e.194Pt and 195Pt, while the accuracy of isotope ratios with the lower abundant isotopes was limited by counting statistics.
1. Introduction
Nanotechnology has become one of the most significant and innovative fields of material science. Thanks to their unique physical and chemical properties, nanoparticles (NPs) are used extensively in various industrial activities and consumer products.1,2 The prospect of unforeseen and undesirable biological effects that may result from the enhanced reactivity and special properties of nanomaterials (NMs) has triggered increased attention from regulators, the public, and the scientific community. Consequently, dedicated methods and analytical tools are required to adequately characterize the physicochemical properties of NMs, their aggregates, and transformation products to support the development of nanotechnology, as well as to assess their potential toxicological effects. Different international research programs aim to understand the impacts of NMs and develop analytical tools and standard operation procedures (SOPs) for NM-risk assessment.Inductively coupled plasma mass spectrometry (ICP-MS) is a well-established analytical technique for multi-element analysis with high sensitivity and precision, and its potential for single-particle analysis was initially outlined in 2003 by Degueldre et al.3 Subsequently, a rapid development of the technique, with improved hardware and dedicated software, took place, leading to a continuously growing number of publications in the field.4–6
Today, single-particle ICP-MS (sp-ICP-MS) is a widely accepted technique within the scientific community and is routinely applied for the determination of particle size and number concentration of metallic and metal–oxide NPs in various sample types.7 The methodology has already been validated through several interlaboratory comparisons (ILC) using ICP-MS instrumentation equipped with a quadrupole-based (QMS) or sector-field (SFMS) mass spectrometer.8–10 However, in spite of their successful use in many routine applications, the information attainable by sequential scanning-based ICP-MS instrumentation still remains limited for NP analysis. The ion cloud produced by a nanoparticle gives rise to a short transient event with a duration of approximately 200–1000 μs;11,12 and despite the development of sp-ICP-QMS and -SFMS with dwell time <1 ms for monitoring of these fast transient signals, sequential mass analysers can only be used to monitor signals from one or a maximum of two nuclides with different mass-to-charge (m/z) ratios.13 Even in these multi-m/z approaches, a short time delay is required for ion clearance after m/z-switching, and so a fraction of the signal is unavoidably lost, which results in only partial detection of two or more nuclides per particle transient event. Subsequently, the observation of only an unknown fraction of a particle event per nuclide hampers accurate quantification.
Although this fast-switching dual-element method is a non-negligible step in the direction of multi-element sp-ICP-MS, the measurement of only one or two nuclide types per NP event is not sufficient for many applications, ranging from NPs analysis in the environment, to nanotoxicology studies, to biological and medical applications, to NP characterization in material sciences, to forensics. As an example, the determination of the intrinsic elemental “fingerprint” of NPs allows one to answer the question of whether the NPs detected are of natural or anthropogenic origin.14–16 Comparatively, for nanotoxicology studies, in which cells are exposed to NPs and their uptake is investigated, multi-element information is required to shed some light onto possible assimilation mechanisms.17 For forensics and geochronology questions, the elemental and isotopic signatures contained in the individual NPs could provide key information regarding their origin and (trans-)formation.18 The common denominator in all previous applications relies on fast and simultaneous multi-element detection. While multi-element detection is possible with single-collector instruments, a long analysis time is required to repeatedly determine all nuclides of interest and address a sufficient number of NP events for reliable statistics. Furthermore, this approach provides an averaged multi-element composition for ensembles only, and not a multi-element composition on an individual particle basis.19 Multi-collector (MC) ICP-MS instruments with Nier–Johnson geometry and an array of ion counting detectors allow for simultaneous multi-element data collection but only within a limited m/z range. The number of analytes that are recordable in the specific mass range is limited to 9–15 isotopes depending on the number of collectors on the instrument.20 Wider m/z ranges can principally be acquired using Mattauch–Herzog type MC-ICPMS21 but our experience with the only commercial instrument currently available indicates that the signal/noise of the Faraday-strip-based detection system prevents the acquisition of low intensity, short signals from individual NPs.
Consequently, in order to gain a more comprehensive picture, not only is simultaneous and full multi-element detection required, but this should take place on an individual particle basis.
A fundamental solution to surmount the shortcomings of scanning-type mass analysers is the use of (quasi-)simultaneous full-spectrum mass analysers, such as a time-of-flight mass spectrometer22,23 (TOFMS). With its high-speed mass spectral acquisition and simultaneous monitoring of all nuclides, ICP-TOFMS is a game-changing technology in terms of the use of ICP-MS in multi-element single-particle analysis.4,24,25 While the lead in sp-ICP-MS taken by QMS and SFMS instruments can be explained by the initial limited sensitivity of ICP-TOFMS instruments, current designs have improved significantly and now provide sensitivities comparable to QMS instruments.23,26 The same fundamental principles described elsewhere4 apply to single-particle experiments with ICP-TOFMS, in which a dilute suspension of NPs is introduced into the plasma to generate individual ion clouds. The difference here, as opposed to sequential mass analysers, is that the full composition of each ion cloud is measured, providing multi-elemental composition information for every individual particle. Thanks to the fast and simultaneous detection capabilities of TOFMS instruments, the full elemental mass range (m/z 6 to m/z 238) can be recorded at time scales appropriate for single-particle analysis. This feature is particularly attractive for screening purposes of NPs mixtures, as a priori knowledge of the particle composition is not required and the complete elemental fingerprints of the different NPs types can be revealed. However, it should be noted that although sp-ICP-TOFMS holds great promise, it also brings about specific challenges, such as those regarding thresholding, data evaluation, and accurate multi-element NP detection. The underlying principle of multi-element particle analysis is that, if multiple mass-channels present coincident signals, these are assumed to stem from the same particle. However, although unlikely, these signals may originate from independent particles, which are reaching the plasma at the same time. Consequently, the particles will not be viewed as independent particles but instead as a multi-element particle. The probability of such events can be determined by coincidence analysis and limited using adequate dilution.25
The capabilities of sp-ICP-TOFMS have already been demonstrated through numerous different case studies, including multi-element NP characterization,26,27 distinguishing between naturally occurring (NNPs) and engineered nanoparticles (ENPs),16,28,29 analysis of surface water30–32 and wastewater samples,33 soil samples,34–36 gunshot residues,37,38 microplastics,39,40 as well as more exotic samples such as polar ice dust41 and space-station dust particles.42 While all these works highlighted the benefits of TOFMS for multi-element NP analysis, the sp-ICP-TOFMS methodology has not yet been validated. Hence, compared to previous ILCs, the ILC presented here aimed to assess the performance of sp-ICP-TOFMS for the characterization of Pt NPs, in terms of mass, size, and particle number concentration (PNC). An additional aim was to assess the precision and trueness of isotope ratio determination on a single-particle basis.
2. Materials and methods
2.1. Study concept
The objectives of the ILC were to evaluate the accuracy and precision of the measurements of particle composition, mass, size, and PNC using the dedicated particle workflow in TOFpilot43 with icpTOF instruments (TOFWERK AG, Switzerland). As an additional point of comparison, the precision and trueness of isotope ratio determination was targeted. All participating labs were asked to follow the SOP for sp-ICP-TOFMS, which was developed within the framework of the Horizon 2020 project ACEnano and can be found in the ESI.† In short, the participants were asked to measure the mass and size distribution, particle number concentration (PNC) and isotope ratios for two Pt NPs samples.2.2. Test materials
Dilute suspensions of commercially available monodisperse nominal 50 nm and 70 nm Pt NPs were analysed in this ILC (see Table 1 for manufacturer specifications). From the TEM images of the manufacturer, the Pt NPs do not appear to consist of solid metal but rather seem to be porous aggregates of smaller Pt NPs.44 For the determination of the transport efficiency (TE), suspensions of 100 nm Au NPs were also provided. All NP samples were purchased from NanoComposix (San Diego, US), divided into multiple aliquots, and supplied to the participating laboratories.| Nominal 50 nm Pt | Nominal 70 nm Pt | Nominal 100 nm Au | |
|---|---|---|---|
| Diameter ± STD (TEM) | 46 ± 4 nm | 70 ± 4 nm | 97 ± 11 nm |
| Mass concentration | ∼0.05 mg mL−1 | ∼0.05 mg mL−1 | ∼0.05 mg mL−1 |
| Particle concentration (calculated) | 4.6 × 1010 particles per mL | 1.3 × 1010 particles per mL | 5.7 × 109 particles per mL |
| Particle surface | Bare (citrate) | Bare (citrate) | Bare (citrate) |
| Solvent | 2 mM sodium citrate | 4 mM sodium citrate | 2 mM sodium citrate |
2.3. Participants
A total of nine independent laboratories operating sp-ICP-TOFMS participated in this ILC. Among the participating laboratories, six are situated in Europe and three in North America. The analysis was performed using different models of the icpTOF instruments (TOFWERK AG, Switzerland); including two icpTOF S2, three icpTOF R and four icpTOF 2R units. In short, due to the difference in length of the flight tube, the icpTOF S2 offers the highest sensitivity as opposed to the icpTOF 2R which offers the highest mass resolution.25,452.4. Measurement procedure & particle analysis workflow
The SOP was sent to all participants. The participants were requested to optimize their instrument to the best of their capability and choose the dilution factor for the analysis according to the sample introduction setup used (see Table S1 in the ESI†). No requirements for the duration of the measurement were set, but a minimum of 1000 particles was to be recorded for reliable statistics.The NP workflow in the TOFpilot software, i.e. Single-Particle Analysis, includes experiment setup and subsequent data processing modules. Quantification is based on the method developed by Pace et al.46 using liquid standards for sensitivity calibration and the particle size method for the determination of the TE. The TOFpilot user interface facilitates the setup of a complete sequence, including blanks, ionic standards, NP standards, and NP samples, as well as data acquisition parameters such as integration time, measurement time, and dilution factor. At the end of the sequence, the data is automatically processed, and the experiment output is summarized in a pdf report, which includes detailed graphical results of the calibration curves and the relevant histograms (integrated signal intensity and mass distributions). The processed data is all saved in the form of csv-files for further post-processing. The SOP for this particular workflow is provided in the ESI.† A TOFpilot version of 2.8.8 or higher was recommended for data evaluation.
The major parameters to be reported were particle mass, Pt-isotope ratios, PNC, and particle size. While the first three parameters are determined from the raw data through data treatment in TOFpilot, the size and standard deviation were calculated individually by the participants. The mass can be converted into an equivalent spherical size assuming the density of the material is known (see eqn (1)):
 | (1) |
It should be noted here that based on the visual information provided by the TEM images, the Pt NPs under investigation are not solid spheres but aggregates of smaller Pt NPs. Thus, smaller sizes (respectively lighter masses) and higher PNC can be expected as opposed to the values stated by the manufacturer. In this regard a particle size determined by sp-ICP-MS should rather be considered as mass equivalent spherical size (MESS) than its microscopic appearance.
2.5. Statistical evaluation
The statistical analysis of the reported results was carried out according to the ACEnano deliverable D5.1 Guidelines for ILCs for validation of harmonised SOPs.47For the individual laboratory results, the individual average masses and standard deviations are reported and the overall mass average and SD of all nine laboratories have been taken as the apparent true values to calculate the individual laboratory z-scores according to eqn (2):
 | (2) |
Since no suitable standard reference material for a multi-isotope NP was available at the time, the correctness of the particle size and particle number concentration determination could not be assessed by comparison to the true value. Hence, the average of all participating laboratories was considered as the apparent true value, as long as the reported results were not scattered too much or did not show strong systematic deviations. Deviations from manufacturer values are also discussed.
For the isotope ratio precision, despite the lack of suitable reference material, the natural abundances of the different Pt isotopes can be used to calculate the true isotope ratios, with their abundance ratios to 195Pt spanning from 0.0004 to 0.9736. The relative abundances according to IUPAC and the IRMM-010 certified reference material are: 190Pt = 0.01289%, 192Pt = 0.7938%, 194Pt = 32.81%, 195Pt = 33.79%, 196Pt = 25.29% and 198Pt = 7.308%.48 Due to the low abundance of 190Pt, the measurement of its relative abundance was omitted and only the isotope ratios of 192Pt, 194Pt, 196Pt and 198Pt relative to 195Pt were requested from the participating laboratories in this study.
3. Results and discussion
All nine laboratories provided results, and none of them reported issues with the software or the provided SOP.The results regarding particle mass and size, PNC, and isotope ratios are discussed separately in the following sections. The linearity of the method was not investigated specifically here, but has been addressed elsewhere and covers six orders of magnitude for dissolved ionic standards,23 and >4 orders of magnitude at the single-particle level, which translates to ∼1.5 orders of magnitude in terms of NP diameter.49 Additional parameters such as repeatability and robustness were not requested from the participants and therefore will not be discussed here.
3.1. Particle mass and size
The average masses of the nominal 50 nm (manufacturer-determined particle diameter (TEM): 46 nm, corresponding to 1.09 fg) and 70 nm Pt NPs (manufacturer-determined particle diameter (TEM) 70 nm, corresponding to 3.85 fg) were determined to be 0.75 ± 0.25 fg and 2.42 ± 0.84 fg, respectively, which converts into 40.4 ± 7 nm and 58.8 ± 8 nm, respectively, when the density of solid bulk Pt is used in eqn (1). The participants were requested to report average mass and standard deviation, as well as the corresponding spherical size equivalent with the corresponding standard deviations. All reported values are presented in Fig. 1. The z-scores, which provide the number of standard deviations (SDs) that a single result deviates from the average, are depicted in Fig. 2. All results reported were within +2 and −2.5 SDs, seven out of nine within ±0.5 SD. For both NP samples under investigation, seven laboratories reported sizes close to the average diameters of 40.4 nm and 58.8 nm, while two laboratories reported deviating values. Lab no.2 reported for both Pt NPs suspensions larger sizes (55.1 nm and 71.6 nm), while smaller sizes were reported by Lab no.8 (26.1 nm and 41.3 nm). Since per lab, the sizes were either systematically biased high or low for both NPs samples, something might have gone wrong with the calibration and/or determination of the TE. Interestingly, although similar average size diameters were obtained, very different size SDs were reported. The magnitude of the SDs was not found to be linked to instrument type, nor to shipping distance, but because the corresponding sizes were calculated by the operators, an operator bias might be possible (Fig. 1b). Indeed, when comparing to the SDs of the masses which are determined by the software during data processing, such large variations are not present (Fig. 1a). Consequently, when converting from mass to size, a similar pattern in terms of standard deviations should be observed. Thus the large variation in SD for the size of the NPs can be explained by incorrect error propagation. The size SDs were re-calculated based on the mass data (Fig. 1c) and all the odd SD variations disappeared except for one dataset (lab no. 2). This high SD observed for both the mass and size data can potentially be attributed to agglomeration effects and suggests an insufficient dilution of the sample or a delay between dilution and measurement. Although the analyzed test samples were monodisperse samples, and thus no large size differences were expected, it should be noted that sp-ICP-TOFMS may also provide additional information regarding the size dispersity of the samples as was demonstrated here with the observation of potential agglomerates. | ||
| Fig. 1 (a) Results for the particle mass calculated by TOFpilot and reported by the participants for the nominal 50 nm Pt NPs and the nominal 70 nm Pt NPs. (b) Results for the mass equivalent spherical size reported by the participants after conversion to corresponding spherical sizes for the nominal 50 nm Pt NPs and nominal 70 nm Pt NPs. The two dotted lines represent the calculated average values for the nominal 50 nm and 70 nm Pt NPs, for mass and size respectively, while the shaded beige and blue rectangle represents the size range specified by the manufacturer, i.e. 46 ± 4 nm and 70 ± 4 nm respectively. Some artificially large standard deviations were reported due to incorrect error propagation. (c) Results for the particle diameter after re-calculation of the size SDs based on the SDs of the mass data by applying conventional error propagation rules. | ||
 | ||
| Fig. 2 Z-Score per lab for (a) the nominal 50 nm Pt NPs and (b) the nominal 70 nm Pt NPs, for which the ILC average of 40.4 and 58.8 nm were set as the expected values respectively. All reported results were comprised within +2 and −2.5 SDs of the average value. If the outliers (lab no.2 and no.8) were excluded, all remaining results lie within ±0.5 SD of the average value. | ||
It should be emphasized here that the size value delivered by the manufacturer was determined directly by TEM and DLS measurements, as opposed to sp-ICP-MS, which is not an imaging technique. In sp-ICP-MS, the counts produced by each NP are recorded and converted to mass using adequate calibration46 and subsequently to particle diameter (i.e. size), using two non-negligible assumptions. The first is that the NPs are of spherical shape, and the second is that the density of the NPs is equal to that of the bulk material (see eqn (1)). Consequently, the interpretation of the analytical results regarding a potential underestimation of the particle mass and size with respect to the expected values has two implications: (1) either the particles are porous, and thus the assumption that the density of the NPs is equal to that of the bulk material does not apply, or (2) the method under investigation, i.e. sp-ICP-TOFMS, is not suited for mass and size determination of Pt NPs.
To explain the systematic underestimation of the Pt NP sizes by sp-ICP-TOFMS as compared to the nominal values reported by the manufacturer, the apparent densities of the NPs were calculated by solving eqn (1) for ρ. The determined densities were 14.5 and 12.8 g cm−3 for the 50 and 70 nm NPs, respectively, as opposed to the bulk density of Pt (21.45 g m−3). While this may appear to be unlikely at first sight, seven out of nine laboratories reported size diameters within 0.5 SD of the average size. The two outlying laboratories reported values far above or far below the measured average (see Fig. 1). The reported values were consistently different from the nominal values, which were known to the operators, but consistent among the participating laboratories. Therefore, there is no indication of operator bias. It has been reported that while the density assumption has been shown to be reliable for NPs composed of gold, it may not apply for all other types of particles.4 For example, for silicon dioxide the density may range from 1.9 g cm−3 to 2.6 g cm−3 depending on its crystal structure. Previous ILCs have demonstrated that the true particle size can be calculated quite accurately from the mass determined via sp-ICP-MS.9,10 Furthermore, the relative mass concentrations reported in the isotope ratio analysis were exact to <1%, which would suggest that most of the calibrations in the lab were incorrect in the same direction and magnitude, which is very unlikely. Subsequently, the relative deviation from the nominal value should be similar for the 50 and 70 nm Pt NPs, which is not the case.
Further inspection of the TEM images from the manufacturer clearly showed that the Pt NPs under investigation were not solid, bulk-like materials but presented a rather porous character and appeared to be formed from agglomerates of smaller particles. Consequently, the actual density must be lower than that of the solid particle density, and lighter masses can be expected. The work of Sikder et al. further supports this observation as they synthesized Pt NPs of different sizes based on the agglomeration of smaller Pt NPs.50 Bolea-Fernandez et al. collected TEM images from the same type of particles (same manufacturer and size, different batch) and also concluded that in contrast to solid Ag and Au NPs these Pt particles are aggregates composed out of much smaller primary particles.51 Consequently based on previous work, the hypothesis of lower density has been verified and can be explained by physicochemical properties as the particles are formed from aggregates of smaller particles.
3.2. Particle number concentration
The determination of the PNC for both NPs samples showed more variability between laboratories than the particle masses or sizes. Fig. 3 shows the PNC reported by the participants for the nominal 50 nm (subplot a) and 70 nm Pt NPs (subplot b), where the blue line represents the value provided by the manufacturer, i.e. 4.6 × 1010 particles per mL and 1.3 × 1010 particles per mL, respectively. The values reported by lab 8 are several orders of magnitude lower than those reported by the other labs and could be the raw values without taking into account the dilution factor. Reported results vary over six orders of magnitude and a similar pattern could be observed for both samples, indicating a laboratory-dependent bias but no dependence on instrument type. After excluding the outlier, the between-laboratory RSD remains high at 53% (see Table 2). This finding is yet consistent with results from previous ILCs, where PNC values are less in line between laboratories than particle mass or size values.8–10,52 The average values were determined to be 4.6 × 1010 ± 2.5 × 1010 particles per mL for the nominal 50 nm NPs, and 1.5 × 1010 ± 7.9 × 109 particles per mL for the nominal 70 nm NPs, as opposed to the expected 4.6 × 1010 particles per mL and 1.3 × 1010 particles per mL, respectively.| LAB no. | PNC nominal 50 nm NPs (particles per mL) | PNC nominal 70 nm NPs (particles per mL) |
|---|---|---|
| 1 | 7.20 × 1010 | 1.50 × 1010 |
| 2 | 1.48 × 1010 | 9.54 × 109 |
| 3 | 7.70 × 1010 | 2.27 × 1010 |
| 4 | 5.62 × 1010 | 2.36 × 1010 |
| 5 | 3.13 × 1010 | 8.99 × 109 |
| 6 | 1.21 × 1010 | 8.68 × 109 |
| 7 | 4.67 × 1010 | 2.68 × 1010 |
| 8 | 2.59 × 105 | 2.68 × 105 |
| 9 | 5.75 × 1010 | 7.58 × 109 |
| PNC mean | 4.60 × 1010 | 1.54 × 1010 |
| PNC SD | 2.45 × 1010 | 7.86 × 109 |
| PNC RSD | 53% | 51% |
 | ||
| Fig. 3 PNC reported by the participants for (a) the nominal 50 nm Pt NPs and (b) the nominal 70 nm Pt NPs, where the blue line represents the value reported by the manufacturer, i.e. 4.6 × 1010 particles per mL and 1.3 × 1010 particles per mL respectively. The values reported by lab 8 are several orders of magnitude lower than those reported by the other labs and could be the raw values without taking into account of the dilution factor. | ||
PNC are typically underestimated in sp-ICP-MS, and this observation has several potential causes.52,53 A prime cause is particle loss due to the NPs sticking onto the walls of the sample vial and the walls of the sample introduction system. A second contributing factor is the settling of NPs over time, but no dynamic study was performed in this case to verify this phenomenon. The accuracy of the TE can be considered as a third parameter as this one will substantially influence the results. Indeed, by preparing ionic Au calibration solutions in MilliQ water rather than stabilizing them with HCl, a loss of ionic Au may be observed and lead to an apparent lower TE, which subsequently results in a PNC overestimation with a size underestimation. It should be noted that aged ionic standards may also impact the accuracy of the TE. Additionally, it cannot be excluded that the initial PNC was altered during the division into multiple aliquots. Last but not least, the reliability of the expected value is questionable. NP suspensions provided by commercial manufacturers such as NanoComposix are not characterized to the same extent as NIST materials, i.e., the NP size distributions are based on TEM measurement of roughly 100 NPs. The PNC is not measured by counting particles, but rather calculated based on the total mass concentration (g mL−1) and the average size determined by TEM, relying on the assumption that the NPs have the same density as the bulk material. However, as discussed previously, this last assumption is likely not valid. If this assumption were wrong, then the calculated average mass per particle would be incorrect as well as the “specified” PNC. These conclusions are consistent with results reported by Minelli et al.,52 where PNC determination was compared between population-averaging methods such as small angle X-ray scattering (SAXS), centrifugal liquid sedimentation (CLS) and ultraviolet-visible spectroscopy (UV-VIS) and particle-counting methods such as particle tracking analysis (PTA) and sp-ICP-MS. Results showed that PNCs determined by particle-counting methods, such as sp-ICP-MS, are more accurate than population-averaging methods, which are based on a theoretical model with various parameters requiring additional information (i.e., NPs morphology and density).
However, a closer look into the measured results revealed in some cases an overestimation of the PNC. As the expected value is determined from the mass concentration, the observed overestimation could be explained by a higher measured number of smaller particles or particles with a lower density, as previously discussed. While the TEM images provided by the manufacturer, suggest that the size appears correct, the particles show undefined edges with a “fluffy” character. Consequently, using the previously determined densities for both samples, namely 14.5 and 12.8 g cm−3, new PNCs were calculated resulting in values of 8 × 1010 and 2 × 1010 particles per mL.
3.3. Isotope ratios
Typically, high-precision isotope ratio measurements are performed with TIMS (Thermal Ionization Mass Spectrometry), which provides an isotope ratio precision down to 0.005 to 0.001%, or MC-ICP-MS, which yields a similar precision, but is more user-friendly.54,55 These methods require long measurement times and sufficient ion-beam currents to acquire data for which the precision is not limited by counting statistics. Hence, they are not suited for isotope ratio measurements of short transient signals, such as those produced by NPs. Recently, a MC-ICP-MS study showed the general applicability of this technique for isotope ratio determinations for NPs, but reaching a precision of 10–20% (at 2 SD) for 187Os/188Os only.20 A subsequent study compared the performance of three ICP-QMS, two ICP-TOFMS and one MC-ICP-MS for multi-element/isotope analysis in a particle/cell basis.56 The determining factor for the acquired precision was the limited instrument sensitivity (or small particle size) and the relatively large difference in abundance of the isotopes involved. As previously discussed, ICP-MS instrumentation equipped with a QMS or ICP-SFMS, possess a single detector only and thus, does not allow for simultaneous multi-element analysis at the time scale of NP events. Because of the time delay in measuring a pair of isotopes, isotope ratio measurements will be skewed. Since spectral intensity skew errors can be overcome with the simultaneous analysis of different m/zs, rapid and simultaneous multi-element detection capabilities are not only advantageous for multi-element fingerprinting but also for isotope ratio determination. Consequently, ICP-TOFMS holds great promise for determining isotope ratios on a per-particle basis. For reference, the performance of icpTOF instruments in terms of isotope ratio precision has been investigated for liquid samples,23,57,58 μm-sized particles,59 as well as Ag NPs and Ag-exposed cells.56In addition to the particle mass, size, and PNC, the participants were asked to report the isotope ratios relative to the most abundant isotope 195Pt (33.8%) for 4 different Pt isotopes. The results are presented in Table 3. While the results for the higher abundant isotopes 194Pt (32.8%) and 196Pt (25.3%) were reported by all laboratories for both particle sizes, the ratios involving the lower abundant isotopes 192Pt (0.8%) and 198Pt (7.3%) were not reported by all laboratories. Reporting of the single particle based SD values was not required according to the ILC data reporting template. Seven laboratories voluntarily provided SD data from which the RSD was calculated and reported (see Table 3). As no reference material was available for isotope ratio measurements, it was assumed here that the Pt used in the production of the NPs presents the same isotopic composition as that of the IRMM-010 standard and that the production of the NPs does not induce additional isotope fractionation.†† However, minor deviations in the isotopic composition from the IRMM-010 standard cannot be excluded, as already reported for environmental samples.60
| Lab no. | 192Pt/195Pt | 194Pt/195Pt | 196Pt/195Pt | 198Pt/195Pt | ||||
|---|---|---|---|---|---|---|---|---|
| 50 nm Pt | 70 nm Pt | 50 nm Pt | 70 nm Pt | 50 nm Pt | 70 nm Pt | 50 nm Pt | 70 nm Pt | |
| 1 | n.a | n.a | 48% | 29% | 51% | 29% | n.a | n.a |
| 2 | n.a | n.a | n.a | n.a | n.a | n.a | n.a | n.a |
| 3 | 53% | 30% | 28% | 23% | 30% | 16% | 37% | 29% |
| 4 | 55% | 38% | 37% | 19% | 39% | 21% | 44% | 32% |
| 5 | 75% | 41% | 44% | 27% | 48% | 31% | 53% | 44% |
| 6 | n.a | n.a | 43% | 22% | 46% | 24% | 47% | 34% |
| 7 | n.a | 46% | 43% | 21% | 41% | 21% | 57% | 31% |
| 8 | n.a | n.a | n.a | n.a | n.a | n.a | n.a | n.a |
| 9 | n.a | 37% | 42% | 24% | 42% | 26% | 47% | n.a |
Table 4 and Fig. 4 show the deviations of the different isotope ratios 194Pt/195Pt, 196Pt/195Pt and 198Pt/195Pt for the nominal 50 nm and 70 nm Pt NPs, in percent with respect to the corresponding values in the IRMM-010 reference material in a graphical manner. As per the instructions, a minimum of 1000 particles was recommended to be measured, hence the determined isotope ratios are based on the average of a minimum of 1000 particles. From the reported data, the isotope ratios were determined from ∼100 events for the 198Pt/195Pt and up to ∼4700 for the 194Pt/195Pt and showed good precision when averaged. Overall, the deviations of the measured isotope ratio for the higher abundant isotopes 194 and 196 are below ±1% and can be regarded as in excellent agreement considering that the ratios were determined on extremely short transient signals (below 1 ms) and a single isotope mass below 1 fg. Generally, results show more variations in the measured ratios with smaller particle size and lower relative abundance of the isotopes. These observations can be explained by the fact that smaller mass fractions in the particles produce signal intensities closer to the limits of detection, making them challenging for accurate quantification. Indeed at low count rates, the precision of the isotope ratio measurements is governed by counting statistics as illustrated in Fig. 5.29 Hence, as for spherical particles the mass scales with the third order of particle size, the precision of single particle determinations improves accordingly with increase of particle size: for the 50 nm NPs ratio RSDs between 28 and 48% were obtained for 194Pt/195Pt, which decreased to 19 to 29% for the 70 nm NPs. With this in mind, the poor results obtained for the less abundant isotopes 192Pt and 198Pt (between −0.83% and +252%) are expected to improve for larger particle sizes, namely from at least ∼100 nm for 198Pt and ∼200 nm for 192Pt.
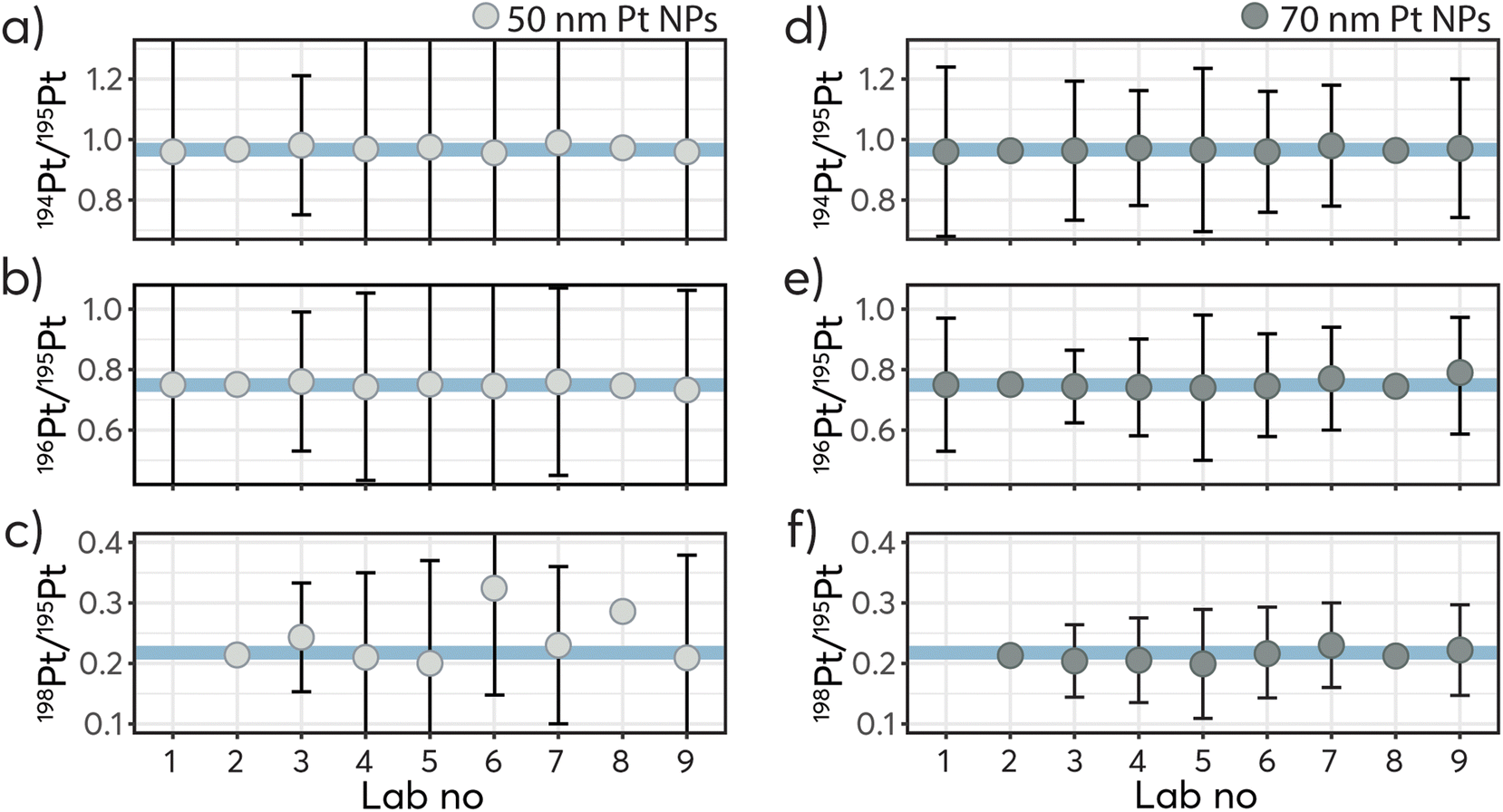 | ||
| Fig. 4 Measured 194Pt/195Pt, 196Pt/195Pt and 198Pt/195Pt ratios for the nominal 50 nm Pt NPs (a), (b) and (c) and nominal 70 nm Pt NPs (d), (e) and (f). The blue line represents the expected values: 0.976, 0.746 for and 0.022. The error bars represent ± 1 SD, which were reported by seven of the laboratories. Since the single particle signal obtained is low in intensity (close to the detection limit) and short-lived (<1 ms) the particle-to-particle variations are of course higher, increasing with decreasing particle size and decreasing abundance of the isotope. | ||
 | ||
| Fig. 5 Overview of the 194Pt/195Pt signal ratio from 70 nm Pt NPs with Poisson-Normal Confidence Bands (95% CI) demonstrating that the RSD is limited by counting statistics. | ||
| Lab no. | 192Pt/195Pt | 194Pt/195Pt | 196Pt/195Pt | 198Pt/195Pt | ||||
|---|---|---|---|---|---|---|---|---|
| 50 nm Pt | 70 nm Pt | 50 nm Pt | 70 nm Pt | 50 nm Pt | 70 nm Pt | 50 nm Pt | 70 nm Pt | |
| 1 | n.a | n.a | 38% | −0.71% | −29.0% | 0.09% | n.a | n.a |
| 2 | 9.85% | 67.0% | 0.02% | −0.40% | 0.22% | 0.22% | −1.70% | −2.16% |
| 3 | 471% | 119% | 1.46% | −0.40% | 1.56% | −0.71% | 11.6% | −6.29% |
| 4 | 495% | 205% | 0.20% | 0.53% | −0.77% | −1.06% | 33.9% | −5.79% |
| 5 | 676% | 180% | 0.89% | −0.12% | 0.30% | −1.21% | 29.1% | −8.48% |
| 6 | n.a | n.a | −1.06% | −0.76% | −0.44% | −0.43% | 49.0% | −0.83% |
| 7 | 252% | 252% | 2.39% | 1.36% | 1.42% | 2.76% | 1.06% | 5.65% |
| 8 | 493% | 104% | 0.64% | −0.30% | −0.31% | −0.58% | 31.4% | −2.62% |
| 9 | n.a | n.a | −0.77% | 0.46% | −2.25% | 4.04% | −4.11% | n.a |
| Average | 385% | 162% | 4.64% | −0.04% | −3.25% | 0.35% | 18.8% | −2.93% |
| Stand. dev. | 259% | 75.1% | 12.6% | 0.7% | 9.7% | 1.8% | 19.7% | 4.6% |
4. Conclusion
The results obtained during this ILC demonstrate that all laboratories are capable of determining the particle mass and mass equivalent spherical size, and the PNC of the test samples using the developed SOP and dedicated particle workflow within TOFpilot. Excellent data agreement among seven of the nine independent laboratory measurements was achieved using the developed SOP and particle workflow, regardless of the experience of the participants with the software or the icpTOF model used. For particle mass and MESS determination, results showed <16% RSD within each lab and <4% RSD among all participating labs. For PNC, results were more scattered and yielded 53% RSD between all labs. These findings are consistent with results obtained with conventional sp-ICP-QMS, where particle mass and size are determined accurately and a larger variation is observed for PNC. Consequently, similarly to results obtained using sp-ICP-QMS, these results show that sp-ICP-TOFMS allows accurate characterization of metal-containing NPs in simple matrices. Furthermore, because sp-ICP-MS and sp-ICP-TOFMS determine particle masses on a particle-to-particle basis rather than based on an average measurement, this information combined with morphological information acquired via TEM would allow the determination of the density of the particles, rather than assuming the same density as that of the bulk material. Indeed, from the total mass determined by bulk ICP-MS and the PNC determined by sp-ICP-MS, combined with TEM information on morphology, the density of the NPs can be effectively determined. Moreover, sp-ICP-MS and sp-ICP-TOFMS can be very useful in directly measuring disparity (size, composition, concentration) within and among samples by analyzing individual nanoparticles, rather than determining the average characteristics of a bulk sample. Additionally, accurate isotope ratios of the test samples were determined with precisions <1% for those involving the most abundant isotopes 194Pt/195Pt, which further underlines the potential of quasi-simultaneous multi-nuclide and multi-element detection of ICP-TOFMS instruments for NPs analysis in various applications. It should however be noted that the particle-to-particle variations, especially with small particles and for low abundant isotopes, can be larger than 20% RSD. This needs to be accounted for, when particles/isotopes close to the detection limit of the instrument are analysed. Precise isotope ratio determination on an individual particle basis is becoming particularly relevant for toxicology and risk assessment studies, where isotope labeling of ENPs is gaining popularity, as it provides clearer means to distinguish the effect of ENPs from NNPs and the background.61–63 Other areas of impact include material sciences and ENPs manufacturing, geochronology, and archaeometry, among others. However, it should be noted that for archaeometric purposes, where isotope ratio analysis combined with trace element analyses are a common approach to study the provenance of raw material present in ancient objects, and to determine their source, an isotope ratio precision better than 1% is required.Recent work by Gundlach-Graham et al. have provided deeper insight and comprehension into the noise of ICP-TOFMS instrument, and have led to the development of new tailored thresholds for sp-ICP-TOFMS particle identification.64–67 Consequently, future improvements to the workflow include direct calculation of the MESSs and respective standard error, as well as the implementation of a tailored algorithm, such as the Compound Poisson Thresholding, for more robust particle detection.
Finally, because the data presented here demonstrate that sp-ICP-TOFMS can successfully be employed to determine both the NP mass and size, PNC and isotope ratios of constituent elements, it is our opinion that the developed SOP for sp-ICP-TOFMS using the particle workflow in TOFpilot has been validated and that the methodology of sp-ICP-TOFMS deserves its place in the analytical toolbox for NP analysis. This ILC also had the benefit of providing a platform to the participating laboratories to compare and benchmark their measurement capabilities.
Author contributions
FvdK, RB and ST contributed to the conceptualization of the ILC study, organization of the samples including aliquot preparation and shipping, and participated as an independent lab in the study. FdvK was responsible for data collection and conducted the statistical evaluation and validation as well as contributed to the writing of the original draft, review and editing of the manuscript. LH contributed to software support during the study, analysis of the sent samples, writing of the original draft, review and editing of the manuscript. All authors contributed to the investigation, through data measurement and analysis, followed by review and editing of the manuscript.Conflicts of interest
One participating lab and first author Lyndsey Hendriks are affiliated with TOFWERK AG, the manufacturer of the instrument used in this study. However, to avoid any bias, a clear separation of the tasks was respected as described above.Acknowledgements
This interlaboratory comparison was performed within the frame of ACEnano, and has received funding from European Union Horizon 2020 Programme (H2020) under grant agreement no. 720952. The authors thank all participants from the Swiss Federal Institute of Technology in Zurich (ETHZ), Carnegie Mellon University, Ghent University, University of South Carolina, Federal Institute for Materials Research and Testing (BAM, Berlin), Helmholtz-Centre for Environmental Research in Leipzig (UFZ), and Iowa State University. E. B. F. and F. V. thank Thibaut Van Acker for his assistance in optimizing the instrument. JK and BH thank Kamyar Mehrabi for his assistance in setting up the instrument. The authors thank all persons that were not listed in the list of authors but were involved in the data acquisition and evaluation. Without their time and efforts, the entire study would not have been possible.References
- J. E. Hulla, S. C. Sahu and A. W. Hayes, Hum. Exp. Toxicol., 2015, 34, 1318–1321 CrossRef CAS PubMed.
- J. Jeevanandam, A. Barhoum, Y. S. Chan, A. Dufresne and M. K. Danquah, Beilstein J. Nanotechnol., 2018, 9, 1050–1074 CrossRef CAS PubMed.
- C. Degueldre and P. Y. Favarger, Colloids Surf., A, 2003, 217, 137–142 CrossRef CAS.
- M. D. Montaño, J. W. Olesik, A. G. Barber, K. Challis and J. F. Ranville, Anal. Bioanal. Chem., 2016, 408, 5053–5074 CrossRef PubMed.
- B. Meermann and V. Nischwitz, J. Anal. At. Spectrom., 2018, 33, 1432–1468 RSC.
- D. Mozhayeva and C. Engelhard, J. Anal. At. Spectrom., 2020, 35, 1740–1783 RSC.
- M. Resano, M. Aramendía, E. García-Ruiz, A. Bazo, E. Bolea-Fernandez and F. Vanhaecke, Chem. Sci., 2022, 13, 4437–4473 RSC.
- T. P. J. Linsinger, R. Peters and S. Weigel, Anal. Bioanal. Chem., 2014, 406, 3835–3843 CrossRef CAS PubMed.
- A. R. Montoro Bustos, E. J. Petersen, A. Possolo and M. R. Winchester, Anal. Chem., 2015, 87, 8809–8817 CrossRef CAS PubMed.
- S. Weigel, R. Peters, K. Loeschner, R. Grombe and T. P. J. Linsinger, Anal. Bioanal. Chem., 2017, 409, 4839–4848 CrossRef CAS PubMed.
- S. Gschwind, L. Flamigni, J. Koch, O. Borovinskaya, S. Groh, K. Niemax and D. Günther, J. Anal. At. Spectrom., 2011, 26, 1166–1174 RSC.
- J. W. Olesik and P. J. Gray, J. Anal. At. Spectrom., 2012, 27, 1143–1155 RSC.
- M. D. Montaño, H. R. Badiei, S. Bazargan and J. F. Ranville, Environ. Sci. Nano, 2014, 1, 338–346 RSC.
- F. von der Kammer, P. L. Ferguson, P. A. Holden, A. Masion, K. R. Rogers, S. J. Klaine, A. A. Koelmans, N. Horne and J. M. Unrine, Environ. Toxicol. Chem., 2012, 31, 32–49 CrossRef CAS PubMed.
- M. D. Montaño, G. V. Lowry, F. Von Der Kammer, J. Blue and J. F. Ranville, Environ. Chem., 2014, 11, 351–366 CrossRef.
- A. Praetorius, A. Gundlach-Graham, E. Goldberg, W. Fabienke, J. Navratilova, A. Gondikas, R. Kaegi, D. Günther, T. Hofmann and F. Von Der Kammer, Environ. Sci. Nano, 2017, 4, 307–314 RSC.
- L. Hendriks and L. M. Skjolding, Spectroscopy, 2020, 35, 9–16 Search PubMed.
- M. D. Montaño, C. W. Cuss, H. M. Holliday, M. B. Javed, W. Shotyk, K. L. Sobocinski, T. Hofmann, F. Von Der Kammer and J. F. Ranville, ACS Earth Space Chem., 2022, 6, 943–952 CrossRef PubMed.
- Y. Huang, A. A. Keller, P. Cervantes-Avilés and J. Nelson, ACS ES&T Water, 2021, 1, 205–213 Search PubMed.
- T. Hirata, S. Yamashita, M. Ishida and T. Suzuki, Mass Spectrom., 2020, 9, 1–8 Search PubMed.
- D. A. Solyom, O. A. Groøn, J. H. Barnes IV, G. M. Hieftje, M. Herzog, D. A. Solyom, O. A. Grøn, J. H. B. Iv and G. M. H. U, Spectrochim. Acta, Part B, 2001, 56, 1717–1729 CrossRef.
- O. Borovinskaya, B. Hattendorf, M. Tanner, S. Gschwind and D. Günther, J. Anal. At. Spectrom., 2013, 28, 226–233 RSC.
- L. Hendriks, A. Gundlach-Graham, B. Hattendorf and D. Günther, J. Anal. At. Spectrom., 2017, 32, 548–561 RSC.
- M. D. Montaño, F. Von Der Kammer, C. W. Cuss and J. F. Ranville, J. Anal. At. Spectrom., 2019, 34, 1768–1772 RSC.
- A. Gundlach-Graham, Compr. Anal. Chem., 2021, 93, 69–101 Search PubMed.
- S. Naasz, S. Weigel, O. Borovinskaya, A. Serva, C. Cascio, A. K. Undas, F. C. Simeone, H. J. P. Marvin and R. J. B. Peters, J. Anal. At. Spectrom., 2018, 33, 835–845 RSC.
- O. Borovinskaya, S. Gschwind, B. Hattendorf, M. Tanner and D. Günther, Anal. Chem., 2014, 86, 8142–8148 CrossRef CAS PubMed.
- G. D. Bland, M. Battifarano, A. E. Pradas del Real, G. Sarret and G. V. Lowry, Environ. Sci. Technol., 2022, 56, 2990–3001 CrossRef CAS PubMed.
- S. E. Szakas, R. Lancaster, R. Kaegi and A. Gundlach-Graham, Environ. Sci. Nano, 2022, 9, 1627–1638 RSC.
- A. Gondikas, F. Von Der Kammer, R. Kaegi, O. Borovinskaya, E. Neubauer, J. Navratilova, A. Praetorius, G. Cornelis and T. Hofmann, Environ. Sci. Nano, 2018, 5, 313–326 RSC.
- F. Loosli, J. Wang, S. Rothenberg, M. Bizimis, C. Winkler, O. Borovinskaya, L. Flamigni and M. Baalousha, Environ. Sci. Nano, 2019, 6, 763–777 RSC.
- M. M. Nabi, J. Wang and M. Baalousha, Chemosphere, 2021, 263, 128261 CrossRef CAS PubMed.
- K. Mehrabi, R. Kaegi, D. Günther and A. Gundlach-Graham, Environ. Sci. Nano, 2021, 8, 1211–1225 RSC.
- M. Baalousha, J. Wang, M. Erfani and E. Goharian, Sci. Total Environ., 2021, 792, 148426 CrossRef CAS PubMed.
- K. K. Norrfors, V. Micic, O. Borovinskaya, T. Hofmann and G. Cornelis, Environ. Sci.: Nano, 2021, 8, 1801–1814 RSC.
- S. Taskula, L. Stetten, F. von der Kammer and T. Hofmann, Nanomaterials, 2022, 12, 3307 CrossRef CAS PubMed.
- R. Brünjes, J. Schüürman, F. von der Kammer and T. Hofmann, Forensic Sci. Int., 2022, 332, 111202 CrossRef PubMed.
- S. E. Szakas, K. Menking-Hoggatt, T. Trejos and A. Gundlach-Graham, Appl. Spectrosc., 2022, 1–12 Search PubMed.
- S. Harycki and A. Gundlach-Graham, J. Anal. At. Spectrom., 2022, 111–120 Search PubMed.
- L. Hendriks and D. M. Mitrano, Environ. Sci. Technol., 2023, 57, 7263–7272 CrossRef CAS PubMed.
- T. Erhardt, C. M. Jensen, O. Borovinskaya and H. Fischer, Environ. Sci. Technol., 2019, 53, 13275–13283 CrossRef PubMed.
- L. G. Jahn, G. D. Bland, L. W. Monroe, R. C. Sullivan and M. E. Meyer, Aerosol Sci. Technol., 2021, 55, 571–585 CrossRef CAS.
- TOFWERK, TOFpilot – Integrated Control Software for the icpTOF, 2020.
- NanoComposix, NanoXact Platinum Nanoparticles – Bare (Citrate), https://nanocomposix.com/products/nanoxact-platinum-nanoparticles-bare-citrate?_pos=2&_sid=ceb6ecb67&_ss=r&variant=39660083445849 (date accessed 27.10.2022).
- TOFWERK, TOFWERK icpTOF Specifications, https://www.tofwerk.com/icptof-specifications/ (date accessed 27.10.2022).
- H. E. Pace, N. J. Rogers, C. Jarolimek, V. A. Coleman, C. P. Higgins and J. F. Ranville, Anal. Chem., 2011, 83, 9361–9369 CrossRef CAS PubMed.
- ACEnano, D5.1 Guidelines for interlaboratory comparisons (ILC) for validation of harmonised SOPs, https://ec.europa.eu/research/participants/documents/downloadPublic?documentIds=080166e5bbd6dad0&appId=PPGMS (date accessed 23.11.2022).
- C. and R. M. JOINT RESEARCH CENTRE Directorate F – Health, CERTIFICATE OF ANALYSIS IRMM-010, 1999.
- L. Hendriks, A. Gundlach-Graham and D. Günther, Chimia, 2018, 72, 221–226 CrossRef CAS PubMed.
- M. Sikder, J. Wang, G. T. Chandler, D. Berti and M. Baalousha, J. Colloid Interface Sci., 2019, 540, 330–341 CrossRef CAS PubMed.
- E. Bolea-Fernandez, D. Leite, A. Rua-Ibarz, T. Liu, G. Woods, M. Aramendia, M. Resano and F. Vanhaecke, Anal. Chim. Acta, 2019, 1077, 95–106 CrossRef CAS PubMed.
- C. Minelli, M. Wywijas, D. Bartczak, S. Cuello-Nuñez, H. G. Infante, J. Deumer, C. Gollwitzer, M. Krumrey, K. E. Murphy, M. E. Johnson, A. R. Montoro Bustos, I. H. Strenge, B. Faure, P. Høghøj, V. Tong, L. Burr, K. Norling, F. Höök, M. Roesslein, J. Kocic, L. Hendriks, V. Kestens, Y. Ramaye, M. C. Contreras Lopez, G. Auclair, D. Mehn, D. Gilliland, A. Potthoff, K. Oelschlägel, J. Tentschert, H. Jungnickel, B. C. Krause, Y. U. Hachenberger, P. Reichardt, A. Luch, T. E. Whittaker, M. M. Stevens, S. Gupta, A. Singh, F. Lin, Y.-H. Liu, A. L. Costa, C. Baldisserri, R. Jawad, S. E. L. Andaloussi, M. N. Holme, T. G. Lee, M. Kwak, J. Kim, J. Ziebel, C. Guignard, S. Cambier, S. Contal, A. C. Gutleb, J. “Kuba” Tatarkiewicz, B. J. Jankiewicz, B. Bartosewicz, X. Wu, J. A. Fagan, E. Elje, E. Rundén-Pran, M. Dusinska, I. P. Kaur, D. Price, I. Nesbitt, S. O'Reilly, R. J. B. Peters, G. Bucher, D. Coleman, A. J. Harrison, A. Ghanem, A. Gering, E. McCarron, N. Fitzgerald, G. Cornelis, J. Tuoriniemi, M. Sakai, H. Tsuchida, C. Maguire, A. Prina-Mello, A. J. Lawlor, J. Adams, C. L. Schultz, D. Constantin, N. T. K. Thanh, L. D. Tung, L. Panariello, S. Damilos, A. Gavriilidis, I. Lynch, B. Fryer, A. Carrazco Quevedo, E. Guggenheim, S. Briffa, E. Valsami-Jones, Y. Huang, A. A. Keller, V.-T. Kinnunen, S. Perämäki, Z. Krpetic, M. Greenwood and A. G. Shard, Nanoscale, 2022, 14, 4690–4704 RSC.
- S. Gschwind, M. de L. Aja Montes and D. Günther, Anal. Bioanal. Chem., 2015, 407, 4035–4044 CrossRef CAS PubMed.
- D. Weis, B. Kieffer, C. Maerschalk, J. Barling, J. De Jong, G. A. Williams, D. Hanano, W. Pretorius, N. Mattielli, J. S. Scoates, A. Goolaerts, R. M. Friedman and J. B. Mahoney, Geochem., Geophys., Geosyst., 2006, 7, 1–30 CrossRef.
- O. Hanousek, M. Brunner, D. Pröfrock, J. Irrgeher and T. Prohaska, Anal. Methods, 2016, 8, 7661–7672 RSC.
- X. Tian, H. Jiang, M. Wang, W. Cui, Y. Guo, L. Zheng, L. Hu, G. Qu, Y. Yin, Y. Cai and G. Jiang, Anal. Chim. Acta, 2023, 1240, 340756 CrossRef CAS PubMed.
- M. Ohata and H. Hagino, Int. J. Mass Spectrom., 2018, 430, 31–36 CrossRef CAS.
- S. Faßbender, M. von der Au, M. Koenig, J. Pelzer, C. Piechotta, J. Vogl and B. Meermann, Anal. Bioanal. Chem., 2021, 413, 5279–5289 CrossRef PubMed.
- A. L. Ronzani, F. Pointurier, M. Rittner, O. Borovinskaya, M. Tanner, A. Hubert, A. C. Humbert, J. Aupiais and N. Dacheux, J. Anal. At. Spectrom., 2018, 33, 1892–1902 RSC.
- J. Creech, J. Baker, M. Handler, M. Schiller and M. Bizzarro, J. Anal. At. Spectrom., 2013, 28, 853–865 RSC.
- F. Larner, Y. Dogra, A. Dybowska, J. Fabrega, B. Stolpe, L. J. Bridgestock, R. Goodhead, D. J. Weiss, J. Moger, J. R. Lead, E. Valsami-Jones, C. R. Tyler, T. S. Galloway and M. Rehkämper, Environ. Sci. Technol., 2012, 46, 12137–12145 CrossRef CAS PubMed.
- F. Larner, B. Gulson, M. McCall, Y. Oytam and M. Rehkämper, J. Anal. At. Spectrom., 2014, 29, 471–477 RSC.
- A. Laycock, M. Diez-Ortiz, F. Larner, A. Dybowska, D. Spurgeon, E. Valsami-Jones, M. Rehkämper and C. Svendsen, Environ. Sci. Technol., 2016, 50, 412–419 CrossRef CAS PubMed.
- A. Gundlach-Graham, L. Hendriks, K. Mehrabi and D. Günther, Anal. Chem., 2018, 90, 11847–11855 CrossRef CAS PubMed.
- L. Hendriks, A. Gundlach-Graham and D. Günther, J. Anal. At. Spectrom., 2019, 34, 1900–1909 RSC.
- A. Gundlach-Graham and K. Mehrabi, J. Anal. At. Spectrom., 2020, 35, 1727–1739 RSC.
- A. Gundlach-Graham and R. Lancaster, J. Anal. At. Spectrom., 2023, 38, 1244–1252 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3nr00435j |
| ‡ Current address: Lonza DPS, Basel, Switzerland. |
| § Current address: Department of Obstetrics, Gynecology and Reproductive Sciences, Program on Reproductive Health and the Environment, University of California San Francisco, San Francisco, California, USA. |
| ¶ Current address: University of Zaragoza, Aragón Institute of Engineering Research (I3A), Department of Analytical Chemistry, Pedro Cerbuna 12, 50009 Zaragoza, Spain. |
| || Current address: Friedrich-Schiller-Universität Jena, Institut für Physikalische Chemie, Jena, Germany. |
| ** Current address: Hochschule Fresenius gem. Trägergesellschaft mbH, Institute for Analytical Research, Germany. |
| †† The validity of this assumption was confirmed by NanoComposix. |
| This journal is © The Royal Society of Chemistry 2023 |