
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Molecular dynamics study on evaporation of metal nitrate-containing nanodroplets in flame spray pyrolysis†
Dingyu
Hou‡
,
Geng
Wang
,
Jingqi
Gao
and
Kai H.
Luo
 *
*
Department of Mechanical Engineering, University College London, Torrington Place, London, WC1E 7JE, UK. E-mail: k.luo@ucl.ac.uk
First published on 1st March 2023
Abstract
Flame spray pyrolysis (FSP) provides an advantageous synthetic route for LiNi1−x−yCoxMnyO2 (NCM) materials, which are one of the most practical and promising cathode materials for Li-ion batteries. However, a detailed understanding of the NCM nanoparticle formation mechanisms through FSP is lacking. To shed light on the evaporation of NCM precursor droplets in FSP, in this work, we employ classical molecular dynamics (MD) simulations to explore the dynamic evaporation process of nanodroplets composed of metal nitrates (including LiNO3, Ni(NO3)2, Co(NO3)2, and Mn(NO3)2 as solutes) and water (as solvent) from a microscopic point of view. Quantitative analysis on the evaporation process has been performed by tracking the temporal evolution of key features including the radial distribution of mass density, the radial distribution of number density of metal ions, droplet diameter, and coordination number (CN) of metal ions with oxygen atoms. Our MD simulation results show that during the evaporation of an MNO3-containing (M = Li, Ni, Co, or Mn) nanodroplet, Ni2+, Co2+, and Mn2+ will precipitate on the droplet surface, forming a solvent–core–solute–shell structure; whereas the distribution of Li+ within the evaporating LiNO3-containing droplet is more even due to the high diffusivity of Li+ compared with other metal ions. For the evaporation of a Ni(NO3)2- or Co(NO3)2-containing nanodroplet, the temporal evolution of the CN of M–OW (M = Ni or Co; OW represents O atoms from water) suggests a “free H2O” evaporation stage, during which both CN of M–OW and CN of M–ON are unchanged with time. Evaporation rate constants at various conditions are extracted by making analogy to the classical D2 law for droplet evaporation. Unlike Ni or Co, CN of Mn–OW keeps changing with time, yet the temporal evolution of the squared droplet diameter indicates the evaporation rate for a Ni(NO3)2-, Co(NO3)2-, or Mn(NO3)2-containing droplet is hardly affected by the different types of the metal ions.
1. Introduction
LiNi1−x−yCoxMnyO2 (NCM) is one of the most promising cathode materials for Li-ion batteries (LIBs) today.1 Currently, commercial NCM materials for LIB cathodes are produced mainly by the solid-state method or the co-precipitation method.2,3 However, both of these synthetic routes require complex mixing, drying, and high-temperature (>750 °C) sintering process, making it hard to reduce the cathode material production cost, thus impeding the expansion of commercial markets of Electric Vehicles (EVs). In contrast, flame spray pyrolysis (FSP) provides an alternative and promising synthetic route for NCM materials.3,4 The advantages of FSP include controllable particle morphology with specifically engineered functionalities, wide particle sizes ranging from the nanoscale to several microns, a reduced number of processing steps, the capability to produce particles with desirable uniformity of different components, and the potential for scalable synthesis.5–10 In addition, it is more environmentally friendly than the traditional methods that can result in large volumes of ion-containing liquid wastes. Furthermore, a recent techno-economic analysis of cathode material production pointed out the minimum cathode material selling price (MCSP) of FSP is 17% lower than the traditional co-precipitation pathway, and could be further decreased if the process is integrated with in situ sintering.2In the synthesis of NCM materials by FSP, the first step is to atomize the precursor solution into precursor droplets. Metal nitrates dissolved in deionized water are one of the most common choices for the precursor solutions.3,11 After atomization, the metal nitrates-containing precursor droplets usually enter a preheat zone (∼450 K) first before they are delivered to the high-temperature flame region (can reach ∼2000 K), where they undergo a series of processes including evaporation of the solvent, thermal decomposition of the metal nitrates, particle inception, coagulation, and sintering before they are eventually transformed into the desired NCM materials.3 Although FSP has been demonstrated to be a promising approach to synthesizing NCM cathode materials in lab-scale experiments, a detailed understanding of the NCM particle formation mechanisms in FSP is still lacking, which can hinder the further development of the FSP technology. Knowing how particles form from liquid precursor through flames is of vital importance for active control/design of the properties of the flame synthesized products, otherwise the optimal operating conditions to obtain the desired products have to be determined by engineers through trial and error.
Among the aforementioned FSP processes, the very first step, i.e., solvent evaporation, which is always accompanied by solute diffusion and precipitation within the droplet, can have a significant effect on both the size and morphology of the final product. According to a comprehensive review article,12 by carefully choosing the types and concentrations of precursors, designing the drying condition, and adding certain supporting materials, particles in various morphology can be obtained, such as dense spherical particles, porous particles, hollow particles, encapsulated particles, or even doughnut-shaped particles. A detailed understanding of the solvent evaporation, as well as the simultaneous solute diffusion inside the droplet, is beneficial to design the process of synthesizing NCM materials via FSP. However, the typical time scale of droplet-to-particle conversion in high-temperature flames can be extremely short (usually on the order microseconds), making it difficult to investigate the evaporation process merely based on experimental instruments.8,13 Besides, in an FSP experiment, it is challenging to isolate the droplet evaporation process for examination, as the solvent evaporation may be coupled with other processes during the droplet-to-particle conversion, such as collisions of the droplets, thermal decomposition of the solute, particle inception, etc. In this regard, as a complement to current experimental techniques, the molecular dynamics (MD) simulation method presents to be a powerful tool to provide atomic insights into the droplet evaporation process in the context of FSP synthesized NCM materials. In MD simulations, besides intermolecular interaction potentials, no prior assumptions (such as the absence of temperature gradient within the droplet) are required to describe the droplet evaporation.14,15 In fact, MD simulations have already been successfully deployed to investigate the evaporation of nanodroplets in various scenarios from a microscopic point of view. For example, Gong et al.16 and Xiao et al.17 studied the evaporation of a single hydrocarbon fuel droplet under both sub- and super-critical nitrogen environments, and proposed the criterion to determine the transition of the dominant mixing mode from evaporation to diffusion during fuel-ambient gas mixing. Wu et al.18 simulated the dynamic evaporation process of a difluoromethane (CH2F2, also called “R32”) nanodroplet under different conditions to analyze the effects of different parameters on the evaporation rate. More complicated conditions such as evaporation of water droplets with dissolved salt (LiCl, NaCl, and KCl) in the presence of an electric field19,20 or the evaporation of a nanodroplet with net charges21–25 have also been investigated, as such knowledge is important for understanding the electrospinning and electrospray mass spectrometry experiments.
However, the evaporation of droplets composed of transition metal nitrates (i.e., Ni(NO3)2, Co(NO3)2, Mn(NO3)2) as solute and water as solvent is less explored both experimentally and computationally. One of the reasons for the lack of research in such area may be that FSP synthesis of NCM materials using metal nitrates solutions as the precursor is just emerging. To the best of the authors’ knowledge, the earliest report of FSP synthesized LiNi0.8Co0.1Mn0.1O2 (NCM811) particles dates back to the work of Abram et al.3 in 2019. The nickel-rich cathode material NCM811 has been regarded as the cathode materials for next-generation LIBs mainly owing to its higher capacity and lower cost compared with its preceding counterpart—LiNi0.33Co0.33Mn0.33O2 (NCM111). In the aforementioned pioneering experimental study of Abram et al.,3 besides NCM811, the authors also synthesized NCM111 for comparison. The precursor solutions for NCM811 and NCM111 were prepared by dissolving Ni(NO3)2, Co(NO3)2, Mn(NO3)2, and LiNO3 in deionized water in the required proportions (molar ratios of Ni2+![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Co2+:Mn2+ = 8:1:1 for NCM811; 1:1:1 for NCM111), while the total concentration of transition metal ions (Ni2+, Co2+, Mn2+) and lithium ions (Li+) were kept the same. Interestingly, the morphology of the FSP synthesized NCM811 and NCM111 particles is quite different. Most as-synthesized NCM111 particles are spherical in shape while NCM811 particles appear much irregular. However, no conclusive explanations exist for the distinct morphology of the FSP synthesized NMC811 and NMC111 particles, although the authors suspected that it might be caused by the different evaporation behavior of the NCM811 and NCM111 precursor droplets in flame.
:Co2+:Mn2+ = 8:1:1 for NCM811; 1:1:1 for NCM111), while the total concentration of transition metal ions (Ni2+, Co2+, Mn2+) and lithium ions (Li+) were kept the same. Interestingly, the morphology of the FSP synthesized NCM811 and NCM111 particles is quite different. Most as-synthesized NCM111 particles are spherical in shape while NCM811 particles appear much irregular. However, no conclusive explanations exist for the distinct morphology of the FSP synthesized NMC811 and NMC111 particles, although the authors suspected that it might be caused by the different evaporation behavior of the NCM811 and NCM111 precursor droplets in flame.
In this work, we employ MD simulations to explore the dynamic evaporation process of nanodroplets composed of transition metal nitrates as solute and water as solvent from a microscopic point of view. Since this is the first step towards providing a fundamental understanding of the evaporation of NCM precursor droplets, we examine the dynamic evaporation process of nanodroplets containing only one metal nitrate, i.e., a nanodroplet with Ni(NO3)2, Co(NO3)2, Mn(NO3), or LiNO3 being the solute and water being the solvent, because such knowledge serves as building blocks for understanding the evaporation of droplets containing multiple metal nitrates. More specifically, in this work, we investigate the evaporation of a nanodroplet composed of metal nitrates (M(NO3), M = Li, Co, Mn, Ni) and water suspended in ambient nitrogen in a temperature range of 1000–3000 K using classical MD simulations. The evaporation rate of the solute-containing nanodroplets is compared with that of a pure water droplet. The effects of temperature, solution concentration, and metal ion type on the dynamic evaporation process of a nanodroplet are determined based on the simulation results.
2. Simulation methods
2.1. Simulation setup
Fig. 1 illustrates the initial configuration for MD simulation of the evaporation of a nanodroplet with Ni(NO3)2 being the solute and H2O being the solvent suspending in the ambient nitrogen. The size of the simulation box is 500 × 500 × 500 Å3, and periodic boundary conditions are used in all three dimensions. The initial diameter of the nanodroplet is ∼10 nm and the nanodroplet is placed at the center of the simulation box. The initial atom coordinates of the central nanodroplet shown in Fig. 1 are generated in two steps: (1) Randomly pack 315 Ni2+, 630 NO3−, and 17518 H2O within a confined spherical region with diameter = 11 nm using the PACKMOL software.26 This corresponds to a solution concentration of 1 mole solute per liter solvent, i.e., H2O. For solutions of other concentrations (see Table 1 for all simulation conditions), different number of metal ions and nitrate ions are used to build the initial structure in the same manner. (2) Equilibrate this nanodroplet in a vacuum simulation box at 300 K for 500 ps. Fig. S4 in the ESI† shows the potential energy of the system versus time during equilibration, suggesting 200 ps is enough for the Ni(NO3)2 nanodroplet to reach an equilibrium state at 300 K. Hence, three configurations after 200 ps are randomly selected (in uniform distribution) from the equilibration trajectory to serve as the initial configurations for the evaporation simulation. The canonical ensemble (i.e., controlled particle number, volume and temperature, NVT) is used during the equilibration process with a 1.0 fs time step. Constant temperature is maintained by the Nosé–Hoover thermostat with a damping constant of 100 fs.
 | ||
| Fig. 1 Initial configuration for evaporation simulation of a 10 nm nickel nitrate (Ni(NO3)2)-containing nanodroplet suspended in the nitrogen ambient. The thermostat region is placed outside the 30 nm sphere as illustrated by the blue dashed circle (a); Zoomed-in orthographic view of the nanodroplet (b); Zoomed-in view of the nanodroplet with water molecules (c1) and without (c2). Ni atoms are in reddish-orange; N atoms in NO3− are in yellow; O atoms in NO3− are in orange; H and O atoms in water are in cyan; N atoms in N2 are in light yellow. | ||
To simulate the evaporation process, the equilibrated Ni(NO3)2 nanodroplet is then surrounded by hot ambient gas. The initial configuration of the ambient nitrogen is generated by randomly packing 30580 N2 molecules in the region outside the nanodroplet and inside the simulation box using the PACKMOL software.26 This corresponds to an ambient pressure of 10 atm which is higher than that of the FSP experiment condition (usually 1 atm), however, increasing the number of the surrounding N2 molecules can enhance heat transfer from the ambient N2 to the nanodroplet by increasing the collision frequency between N2 molecules and the central droplet, so that the dynamic evaporation of the nanodroplet can be observed within a reasonable amount of computational time. In fact, this is a common technique used in MD studies to accelerate the process that is concerned as the typical time scale that MD simulations can handle currently is on the order of nanosecond. For example, Zhan et al.27 investigated the evaporation of a 6 nm single wastewater droplet surrounded by N2 using MD simulations, in which the ambient pressure of N2 was set at ∼15 atm instead of 1 atm.
For each evaporation case, we start the MD simulation with potential energy minimization using the conjugated gradient (CG) algorithm to eliminate possible irrational initial geometries, which may be induced by assembling the nanodroplet with the surrounding N2 molecules. As mentioned earlier, the heating method is used to initiate the evaporation of the nanodroplet. As shown in Table 1, the temperatures of the ambient N2 are in the range of 1000 K–3000 K, which cover the typical flame temperature in FSP; while the initial temperature of the droplet is 300 K. The evaporation simulations are performed using the micro-canonical ensemble (i.e., controlled particle number, volume and energy, NVE). The region outside of the sphere with a radius of 15 nm as shown by the dashed blue circle in Fig. 1 is the “thermostat region”. The translational velocities of the N2 molecules located in this region are rescaled every time step according to
 | (1) |
518 to a value that is equal to the number of Me(NO3)2 (Me = Ni/Co/Mn) or LiNO3 within the droplet, i.e., the water–salt-ratio (WSR) reduces to 1. For post-processing, atomic positions are collected every 10 ps from the MD simulation trajectories. MD simulations in this work are performed using the Large-scale Atomic/Molecular Massively Parallel Simulator (LAMMPS) package.28 The MOLTEMLATE software is used to facilitate generating the LAMMPS data file.29 Snapshots are prepared by OVITO.30
2.2. Force field parameters
In MD simulations, positions and velocities of the atoms are updated according to the interatomic potential and the 2nd Newton's Law. Table 2 lists the parameters of the intermolecular potential used in this work for describing the dynamic evolution of various aqueous metal nitrates solution. The non-bonded interaction between a pair of atoms i, j, is described by the Lennard–Jones (L–J) 12–6 potential and the coulombic potential: | (2) |
 | (3) |
| Atom type | Charge | ε ii (kcal mol−1) | σ ii (Å) | Ref. |
|---|---|---|---|---|
| Li+ | 1.0 | 5.950 × 10−3 | 2.343 | 31 |
| Ni2+ | 2.0 | 1.179 × 10−2 | 2.446 | 32 |
| Mn2+ | 2.0 | 2.960 × 10−2 | 2.614 | 32 |
| Co2+ | 2.0 | 1.636 × 10−2 | 2.502 | 32 |
| N(in NO3−) | 0.8603 | 2.001 × 10−1 | 3.9 | 33–35 |
| O(in NO3−) | −0.6201 | 1.551 × 10−1 | 3.154 | 33–35 |
| O(in H2O) | −0.8476 | 1.553 × 10−1 | 3.166 | 36 |
| H(in H2O) | 0.4238 | 0.0 | 0.0 | 36 |
| N(in N2) | 0.0 | 7.239 × 10−2 | 3.32 | 16 |
2.3. Force field validation
Before simulating the evaporation of a solute-containing nanodroplet, two simpler cases are carried out to verify the force field parameters. We first simulated the evaporation of a pure water droplet, the initial diameter of which is 10 nm, and it is suspended in 10 atm ambient N2 at T = 1000 K. Fig. S1(a)† shows the temporal evolution of the droplet temperature and the number of water molecules within the droplet during the evaporation process. According to Fig. S1(a),† the evaporation of the water droplet achieves a quasi-equilibrium state at ∼1500 ps, after when the droplet temperature stabilized at ∼450 K, i.e., the boiling point (Tbp) of water in 10 atm ambient N2 is ∼450 K based on our MD simulations, which is in good agreement with Tbp = 452.9 K for saturated water at 10 atm based on theoretical calculations43 (since the droplet is evaporating, it is at the saturation state). Fig. S1(b)† illustrates the radial distribution of mass density of the water droplet after it enters the quasi-equilibrium evaporation state. Based on Fig. S1(b),† the mass density inside the droplet is around 0.88 g cm−3, which is close to the calculated mass density of saturated water at 10 atm: ρ = 0.887 g cm−3.We also calculated the diffusion coefficient of Ni2+ (DNi) in bulk Ni(NO3)2 solution with a concentration of 1.1 mol L−1 at 300 K using MD simulations. Detailed procedures of calculating DNi are provided in the ESI.†DNi = (5.45 ± 0.457) × 10−10 m2 s−1 is obtained based on five replica MD runs, which is comparable to the experimental data for the diffusion coefficient of Ni2+ in unbounded aqueous solution: DNi = 6.61 × 10−10 m2 s−1.44–46 Note that in MD simulations DNi is calculated in 1.1 mol L−1 Ni(NO3)2 solution while the available experimental data is for Ni2+ diffusion in Ni(NO3)2 solution of infinite dilution. The different solution concentration between MD simulations and experimental conditions could lead to slight deviation of the MD simulated DNi from the experimental value, however, it is less practical to simulate Ni(NO3)2 solution in infinite dilution.
3. Results and discussion
3.1. Dynamic process of evaporation of a nickel nitrate droplet
Fig. 2 illustrates the dynamic evaporation process of a 10 nm Ni(NO3)2 nanodroplet suspended in ambient N2 at 2500 K. As the solvent H2O molecules are evaporated from the droplet, the droplet diameter is decreasing with time, until most of the H2O molecules are evaporated. The snapshots in Fig. 2 clearly show that the Ni(NO3)2 solute tends to precipitate on the droplet surface during evaporation, forming a solvent(H2O)–core–solute(Ni(NO3)2)–shell structure, as demonstrated by the slice view of the droplet at ∼2180 ps. Complete animations (ortho view and slice view; each animation contains 400 frames in total, which are extracted every 10 ps along the simulated trajectory) of the dynamic evaporation process of the Ni(NO3)2-containing nanodroplets are provided in the ESI.† | ||
| Fig. 2 Snapshots (slice view) at several typical time during the evaporation of a 10 nm Ni(NO3)2-containing nanodroplet. The initial solution concentration is 5.4 mol L−1. Ambient nitrogen is at 2500 K. Ni atoms are in reddish-orange; N atoms in NO3− are in yellow; O atoms in NO3− are in orange; H and O atoms in water are in cyan; N atoms in N2 are in light yellow. | ||
To quantitatively describe the evaporation process, the droplet is divided into a series of concentric spherical shells with a thickness δ = 1.0 Å as illustrated in Fig. 3. By calculating the mass density of each shell, the radial distribution of mass density of the droplet can be obtained. Fig. 3 shows the radial distribution of mass density at different times during the evaporation of a 10 nm Ni(NO)3 droplet with an initial solution concentration of 5.4 mol L−1 in 2500 K N2. Before 1040 ps, mass density of the entire droplet is almost at a constant value of ∼1.5 g cm−3 except for the interface region between the droplet surface and ambient N2. However, the mass density shows an obvious gradient versus radial position at 2010 ps and 2180 ps, suggesting an inhomogeneous distribution of the solute (Ni(NO3)2, molar mass M = 182.7 g mol−1) and solvent molecules (H2O, M = 18.0 g mol−1) within the droplet. As the evaporation continue, the mass density gradient along the radial direction disappears as illustrated by the radial distribution of ρ at 3440 ps in Fig. 3. This is because at this stage, almost all of the solvent H2O molecules are evaporated, leaving the dehydrated Ni(NO3)2 solutes. The radial distribution of mass density is also useful to estimate the droplet radius. Take the initial state of evaporation (t = 0) as an example. The mass density in the inner region of the droplet (i.e., at radial position r ≈ 20 Å) can be regarded as the bulk density of Ni(NO3)2 solution (ρ = 1.48 g cm−3). In the interface region between the droplet and ambient N2, mass density of the spherical shells decreases rapidly with increasing r. The radial position at which the mass density decreases to half of the bulk density is estimated as the radius of the droplet (r0 = 55 Å at t = 0). In this way, droplet radius is calculated every 1 ps during evaporation, so that the evolution of droplet size during evaporation can be tracked and analyzed as suggested in Fig. 5.
 | ||
| Fig. 3 Radial distribution of mass density during the evaporation of a 10 nm Ni(NO3)2-containing nanodroplet at different times. The initial solution concentration is 5.4 mol L−1. Ambient nitrogen is at 2500 K. | ||
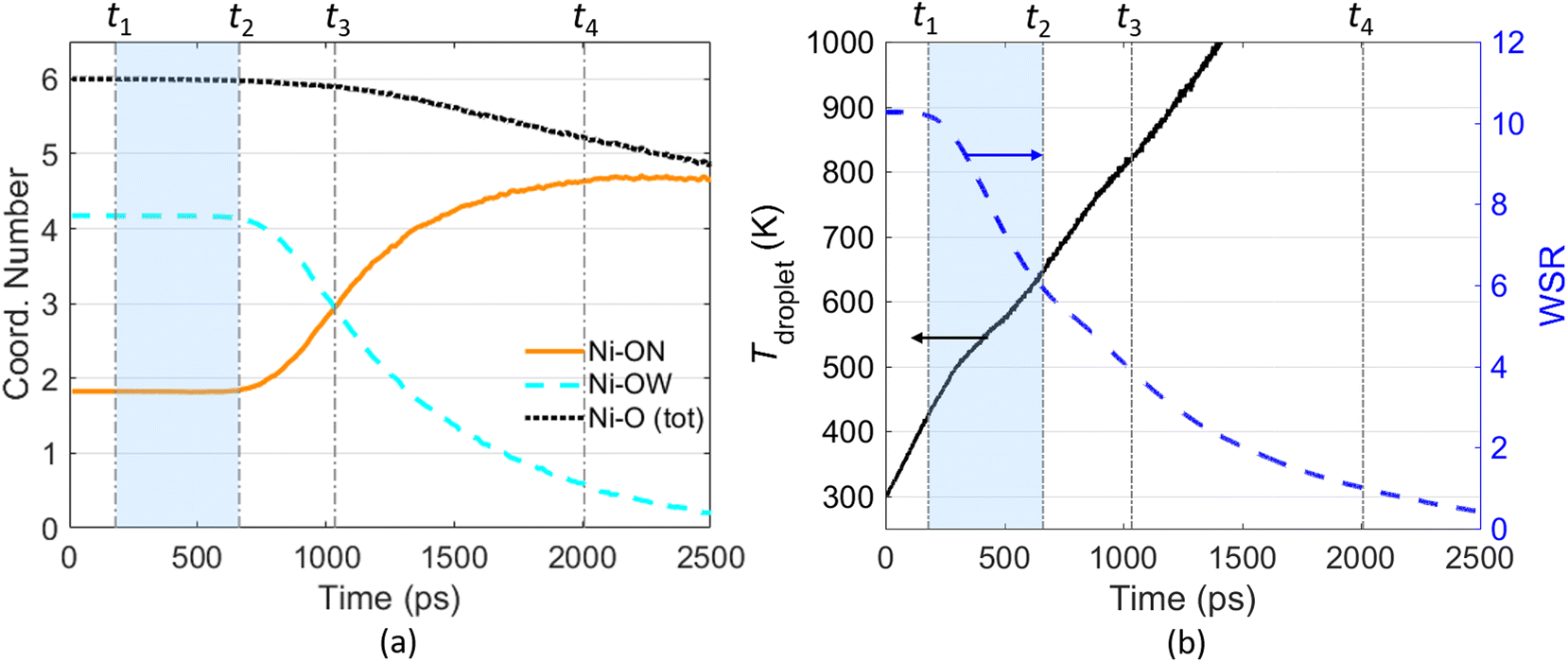 | ||
| Fig. 4 Temporal evolution of the Ni–O coordination number during the evaporation of a 10 nm Ni(NO3)2 nanodroplet (a); temporal evolution of the droplet temperature (left y-axis) and the water–salt-ratio (WSR; right y-axis) during the evaporation of a 10 nm Ni(NO3)2 nanodroplet (b). The initial solution concentration is 5.4 mol L−1. Ambient nitrogen is at 2500 K. | ||
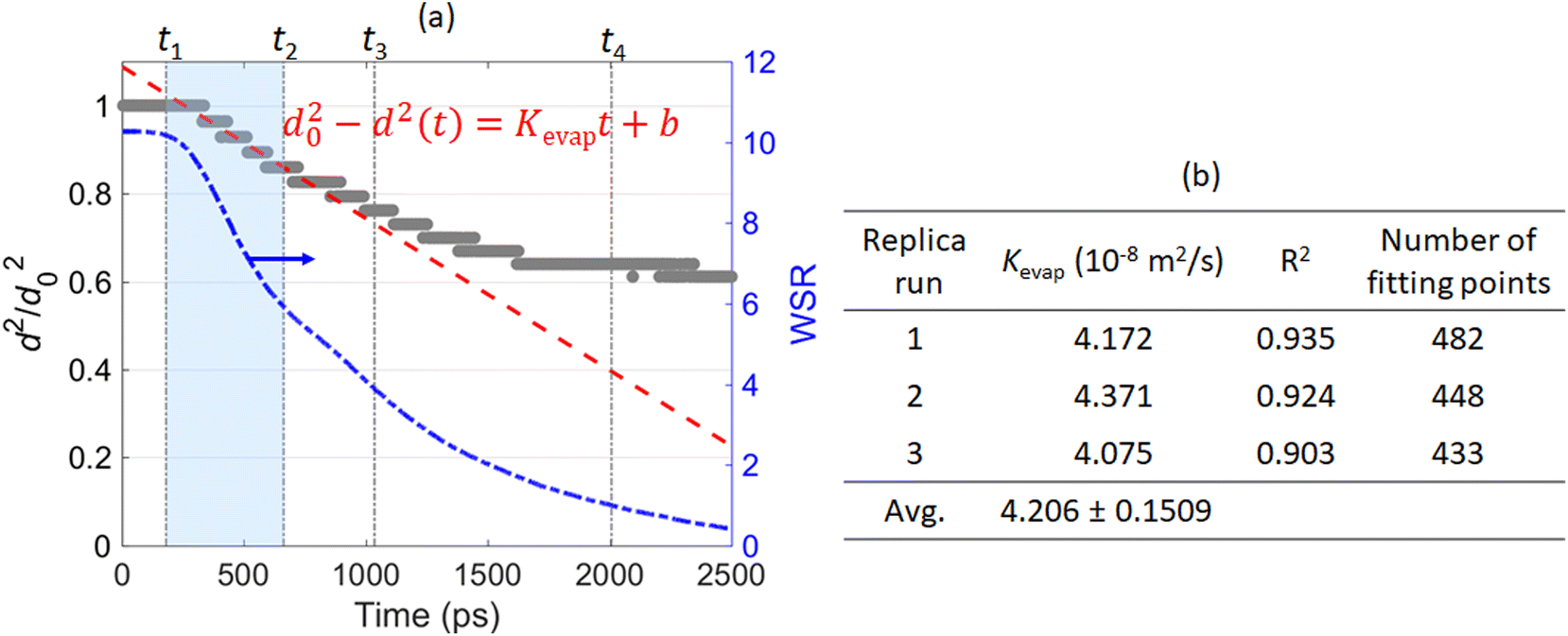 | ||
| Fig. 5 Temporal evolution of normalized square droplet diameter (left y-axis) versus time; temporal evolution of water–salt-ratio (WSR; right y-axis) during evaporation of a 10 nm Ni(NO3)2 droplet (a); The fitted evaporation rate constant Kevap from three replica simulations (b). The droplet initial solution concentration is 5.4 mol L−1. Ambient N2 is at 2500 K. | ||
Fig. 4(a) illustrates the temporal evolution of the micro-structure, i.e., Ni–O atom pair, during evaporation of a Ni(NO3)2-containing nanodroplet. The coordination number (CN) shown in Fig. 4(a) measures the number of O atoms around a Ni2+ ion within a predefined cutoff distance, which is averaged over all Ni2+ ions within the droplet. In this work, an atomic distance cutoff 2.5 Å is employed to perform the coordination analysis based on the radial distribution function (RDF) of the Ni–O atom pair within the droplet. Fig. S5† illustrates the Ni–O RDF diagram at different time during the evaporation of a Ni(NO3)2-containing nanodroplet, suggesting the location of the first peak of gNi–O(r) is almost independent with the solution concentration inside the droplet, i.e., the water–salt-ratio (WSR, which can be calculated as nH2O/nNi(NO3)2 with nH2O being the number of H2O molecules and nNi(NO3)2 being the number of solute molecules, i.e., Ni(NO3)2, within the droplet). The coordinated O atoms are divided into two groups according to whether it is from a nitrate ion (denoted as ON) or a solvent water molecule (denoted as OW). As suggested by Fig. 4(a), at the beginning stage of droplet evaporation (before time t2), both the coordination number (CN) of Ni–ON and Ni–OW are constant. On average, one Ni2+ is coordinated with ∼4 O atoms from H2O and ∼2 O atoms from NO3−, leading to a total Ni–O coordination number equal to 6. As the evaporation process proceeds, Ni2+ will be gradually dehydrated from water molecules and will interact with the NO3− ions more frequently, which can be reflected by the decreasing CN of Ni–OW and increasing CN of Ni–ON. The total CN of Ni–O keeps decreasing with time. After time t4 (corresponds to WSR = 1), when most of the H2O molecules are evaporated, CN of Ni–OW is close to zero, while CN of Ni–ON is basically stable over time at some value below 5. The snapshots in Fig. 2 clearly show the formation of Ni(NO3)2 clusters/shells at this stage.
Fig. 4(b) shows the temporal evolution of the water–salt-ratio (WSR) during the evaporation process. On the basis of Fig. 4(a) and (b), the evaporation of a Ni(NO3)2 droplet can be divided into four stages as illustrated by the vertical dash-dotted lines labeled from t1 through t4 to assist quantitative analysis of the evaporation process. The first stage starts from the very beginning to t1, which can be regarded as the preheating stage. t1 is determined as the time when 1% of the water molecules are evaporated. During the preheating stage, WSR of the droplet is nearly unchanged while the droplet temperature rises from 300 K to around 400 K due to the heat transfer from the ambient N2 molecules to the droplet through collisions between the molecules. From t1 to t2, as water molecules keep evaporating from the droplet, WSR of the droplet decreases from ∼10 to ∼6. However, during this stage, the coordination number of both Ni–OW and Ni–ON are unchanged, meaning that the micro-structure of Ni2+–H2O–NO3− cluster is hardly changed. Therefore, the t1–t2 stage is named as the “free H2O” evaporation stage. t3 denotes the time when the coordination number of Ni–OW is equal to that of Ni–ON. It marks the time when the micro-structure of the Ni2+–H2O–NO3− cluster is converted. Before t3, Ni2+ is coordinated with more H2O molecules than NO3−. After t3, Ni2+ is coordinated with more NO3− than H2O molecules. t4 corresponds to the time when WSR of the droplet decreases to 1, i.e., the number of H2O molecules equals to the number of metal ions within the droplet. At t4, evaporation almost finishes as most of the water molecules have been lost, hence t4 can also be regarded as the lifetime of the droplet during evaporation.
Besides WSR, Fig. 4(b) also shows the temporal evolution of the temperature of the Ni(NO3)2 droplet during evaporation. As the interface between the droplet and ambient gas can be ambiguous during droplet evaporation, here the droplet temperature is estimated based on the kinetic energy of all atoms of H2O molecules, Ni2+ ions, and NO3− ions within the D = 30 nm spherical region. With the solvent H2O molecules being evaporated constantly, the droplet temperature increases rapidly. It is worth mentioning that the thermal decomposition temperature of Ni(NO3)2 is around 600 K based on experimental measurements,47,48 while our MD simulation results show that the droplet temperature reaches ∼1000 K when WSR decreases to ∼2. This indicates that the evaporation of the solvent H2O molecules and the thermal decomposition of the solute Ni(NO3)2 may be coupled with each other during the evaporation of a Ni(NO3)2-containing nanodroplet, causing the problem more complicated. Further investigations using reactive molecular dynamics simulations are to be conducted.
To make comparison with the classical droplet evaporation model, i.e., the D2 law,49,50 the temporal evolution of the squared droplet diameter (normalized by d20, where d0 is the initial droplet diameter) during the evaporation of a 10 nm Ni(NO3)2-containing droplet is illustrated in Fig. 5. According to the D2 law, the square of droplet diameter will decrease linearly with the time supposing the droplet is in the quasi-equilibrium evaporation state:
| d20 − d2(t) = Kevapt + b | (4) |
3.2. Effect of initial solution concentration and ambient gas temperature on droplet evaporation
To study the effect of initial solution concentration and ambient gas temperature on the dynamic evaporation process of a solute-containing nanodroplet, the evaporation of Ni(NO3)2-containing nanodroplets with various initial solution concentrations suspended in ambient N2 at different temperatures ranging from 1000 K to 3000 K has been investigated through MD simulations. Fig. 6 illustrates the temporal evolution of the radial distribution of mass density during the evaporation of a 10 nm Ni(NO3)2-containing droplet, the initial solution concentration of which is 1.0 mol L−1 (more dilute than the case discussed in section 3.1) and the ambient N2 is at 1500 K (cooler than the case discussed in section 3.1). In contrast with the droplet evaporation starting from a much higher solution concentration and being surrounded by much hotter ambient N2, no obvious mass density gradient along the radial direction within the droplet can be observed throughout the evaporation process, suggesting that the solute (Ni2+ and NO3− ions) and solvent (H2O molecules) are evenly distributed inside the droplet as the evaporation proceeds. | ||
| Fig. 6 Radial distribution of mass density during the evaporation of a 10 nm Ni(NO3)2-containing nanodroplet at different times. The initial solution concentration is 1.0 mol L−1 and the ambient N2 is at 1500 K. The snapshots below are slice view of the droplet at different times during evaporation. Refer to the legend of Fig. 2 for correspondence between colors and atom types. | ||
To systematically investigate the effect of the initial solution concentration on the dynamic evaporation process of a salt-containing nanodroplet, we have carried out MD simulations on the evaporation of a Ni(NO3)2-containing nanodroplet of three different initial solution concentrations, namely 1, 3, and 5.4 mol L−1. The temperature of the ambient N2 is kept the same at 2500 K. Fig. 7 shows the radial distribution of mass density when water–salt-ratio (WSR) of the droplet decreases to 1. The highest initial solution concentration, i.e., 5.4 mol L−1, leads to the most obvious H2O–core–Ni(NO3)2–shell configuration, as suggested by the inserted snapshots of the slice view of the droplet and also the mass density gradient along the radial direction from r = 10 to 40 Å. Although for lower initial solution concentrations, the solute Ni(NO3)2 also tends to precipitate on the droplet surface, the H2O–core–Ni(NO3)2–shell structure is less evident.
 | ||
| Fig. 7 Radial distribution of mass density during evaporation of a 10 nm Ni(NO3)2-containing nanodroplet with different initial solution concentrations (c = 1, 3, 5.4 mol L−1) at the time when water–salt-ratio (WSR) decreases to 1. The ambient N2 is at 2500 K. Inserted snapshots are slice view of the droplet at WSR = 1. Refer to the legend of Fig. 2 for correspondence between colors and atom types. | ||
Fig. 8 demonstrates the effect of ambient N2 temperature on the radial distribution of mass density at the end stage of evaporation of a 10 nm Ni(NO3)2-containing droplet, i.e., at the time when water–salt-ratio (WSR) decreases to 1. According to Fig. 8, at WSR = 1, the radius of the remaining nanodroplet is similar for all five cases, which is ∼45 Å. However, the distribution of the solute (Ni2+ and NO3− ions) and solvent (H2O molecules) inside the droplet is very different as indicated by the distinct radial distribution of mass density under different evaporation temperatures. For droplet evaporating in ambient N2 at 3000 K, the slice view of the droplet clearly shows a H2O–core–Ni(NO3)2–shell structure at WSR = 1. As the temperature of ambient N2 decreases from 3000 K to 1000 K, the H2O–core–Ni(NO3)2–shell structure becomes less evident. In obvious contrast to evaporation at 3000 K, if the same droplet is evaporating in much cooler ambient N2 at 1000 K, the H2O molecules and the Ni(NO3)2 solute are almost evenly distributed inside the droplet. This is because at 3000 K, the solvent, i.e., H2O molecules evaporate much faster than the solute ions, i.e., Ni2+ and NO3− diffuse within the nanodroplet. Hence in a very short time, a large number of H2O molecules leave the droplet entering the gas-phase, leaving the Ni2+ and NO3− precipitate on the droplet surface and forming a solute shell. By contrast, at 1000 K, the solvent H2O molecules evaporate at a much lower rate due to reduced heat transfer between the ambient N2 and the suspending nanodroplet, so that the solute – Ni2+ and NO3− have enough time to redistribute themselves within the droplet as H2O molecules are constantly leaving the droplet. Therefore, the slice view of the droplet evaporating in 1000 K ambient gas shows almost evenly distributed solute and solvent.
 | ||
| Fig. 8 Radial distribution of mass density during evaporation of a 10 nm Ni(NO3)2-containing nanodroplet suspended in ambient N2 of different temperatures (1000–3000 K) at the time when water–salt-ratio (WSR) decreases to 1. The initial solution concentration is 5.4 mol L−1. Inserted snapshots are slice view of the droplet. Refer to the legend of Fig. 2 for correspondence between colors and atom types. | ||
Fig. 7 and 8 suggest that both the initial solution concentration of the droplet and the ambient gas temperature can influence the distribution of the solute ions – Ni2+ and NO3− and the solvent – H2O molecules within the nanodroplet during evaporation. To draw some more quantitative conclusions, we calculated the evaporation rate constant Kevap of a 10 nm Ni(NO3)2-containing droplet with different initial solution concentrations evaporating at different ambient N2 temperatures. To achieve results with statistical significance, for each evaporation condition, three replica MD runs are performed with different initial configurations of the Ni(NO3)2-containing nanodroplet. Then the evaporation rate constant Kevap is obtained by fitting d2 and time t into eqn (4) using the data points during the “free H2O” evaporation stage, i.e., from t1 to t2. The averaged Kevap of the three replica runs is finally taken as the Kevap for the certain ambient temperature and initial solution concentration. The evaporation rate constant Kevap of a pure H2O nanodroplet evaporating in ambient N2 at of different temperatures can be obtained following the same manner. Fig. 9 summarizes the calculated ![[K with combining macron]](https://www.rsc.org/images/entities/i_char_004b_0304.gif) evap of a 10 nm Ni(NO3)2-containing droplet evaporating from different initial solution concentrations at various temperatures. To clearly show the effect of solute on the evaporation rate of a nanodroplet, in Fig. 9 the evaporation rate constants are normalized by Kevap of a pure water droplet (d0 ≈ 10 nm) at corresponding temperatures, which is equivalent to an initial solution concentration being 0. The originally calculated Kevap (before normalization) as well as the standard deviation based on three replica runs are provided in Table S1 in ESI.†Fig. 9 shows that compared with a pure water droplet, addition of Ni(NO3)2 solute decreases the evaporation rate constant. For a given temperature, the higher the solution concentration, the lower the Kevap. This is reasonable because Ni2+ and NO3− within the droplet tend to attract H2O molecules around them, thus the higher the concentrations of Ni2+ and NO3−, the larger the resistance H2O molecules would need to overcome for evaporation. Fig. 9 also suggests that the effect of the initial solution concentration on Kevap at a lower temperature such as 1000 K is less significant than its effect on Kevap at a higher temperature such as 3000 K. This may be explained as follows. When the ambient N2 is not hot enough, the rate-limiting process for droplet evaporation is the heat transfer between the ambient N2 and the droplet. Therefore, the solution concentration of the droplet only has limited effect on its evaporation rate. In contrast, if the ambient temperature is very high, heat transfer between the ambient N2 and the droplet is no longer the rate-limiting step for droplet evaporation. Instead, the interaction between the solute ions (Ni2+ and NO3−) and H2O molecules within the droplet becomes more dominant on the evaporation rate. Another interesting finding is that for a given solution concentration, evap decreases linearly with increasing temperature. The fitted parameters (m and n) for the excellent linear relationship between evap and temperature T are also presented in the table beside Fig. 9.
evap of a 10 nm Ni(NO3)2-containing droplet evaporating from different initial solution concentrations at various temperatures. To clearly show the effect of solute on the evaporation rate of a nanodroplet, in Fig. 9 the evaporation rate constants are normalized by Kevap of a pure water droplet (d0 ≈ 10 nm) at corresponding temperatures, which is equivalent to an initial solution concentration being 0. The originally calculated Kevap (before normalization) as well as the standard deviation based on three replica runs are provided in Table S1 in ESI.†Fig. 9 shows that compared with a pure water droplet, addition of Ni(NO3)2 solute decreases the evaporation rate constant. For a given temperature, the higher the solution concentration, the lower the Kevap. This is reasonable because Ni2+ and NO3− within the droplet tend to attract H2O molecules around them, thus the higher the concentrations of Ni2+ and NO3−, the larger the resistance H2O molecules would need to overcome for evaporation. Fig. 9 also suggests that the effect of the initial solution concentration on Kevap at a lower temperature such as 1000 K is less significant than its effect on Kevap at a higher temperature such as 3000 K. This may be explained as follows. When the ambient N2 is not hot enough, the rate-limiting process for droplet evaporation is the heat transfer between the ambient N2 and the droplet. Therefore, the solution concentration of the droplet only has limited effect on its evaporation rate. In contrast, if the ambient temperature is very high, heat transfer between the ambient N2 and the droplet is no longer the rate-limiting step for droplet evaporation. Instead, the interaction between the solute ions (Ni2+ and NO3−) and H2O molecules within the droplet becomes more dominant on the evaporation rate. Another interesting finding is that for a given solution concentration, evap decreases linearly with increasing temperature. The fitted parameters (m and n) for the excellent linear relationship between evap and temperature T are also presented in the table beside Fig. 9.
 | ||
| Fig. 9 Evaporation rate constant of a 10 nm Ni(NO3)2 droplet with different initial solution concentrations in ambient N2 at different temperatures. Kevap is normalized by the evaporation rate constant of a pure H2O nanodroplet at corresponding temperatures. | ||
3.3. Effect of metal ions on droplet evaporation
When synthesizing NCM materials through flame spray pyrolysis, the precursor solution is usually prepared by dissolving LiNO3, Ni(NO3)2, Co(NO3)2, and Mn(NO3)2 at desired proportions into deionized water. In Sections 3.1 and 3.2, the evaporation of a Ni(NO3)2-containing nanodroplet with different initial solution concentrations suspended in ambient N2 of different temperatures has been examined in detail. The evaporation process of other metal nitrates besides Ni(NO3)2 is also worth examination. To study the effect of different metal ions on nanodroplet evaporation, we simulated the evaporation of a nanodroplet with LiNO3, Co(NO3)2, or Mn(NO3)2 being the solute respectively. For all of the four evaporation cases, the ambient N2 is at 2000 K and the initial solution concentration is 5.4 mol L−1. Fig. 10 illustrates the radial distribution of mass density (a) as well as the radial distribution of the number density of metal ions (b) during evaporation of a 10 nm LiNO3, Ni(NO3)2, Co(NO3)2, and Mn(NO3)2 nanodroplet at the time when water–salt-ratio (WSR) decreases to 1. As indicated by Fig. 10, except for LiNO3, other three metal nitrates – Ni(NO3)2, Co(NO3)2, and Mn(NO3)2 will precipitate on droplet surface during evaporation, forming a solvent–core–solute–shell configuration at WSR = 1. Although Fig. 10(b) also suggests a slight Li+ number density gradient along the radial direction, the tendency of Li+ precipitating on the droplet surface is much weaker compared with other three metal ions. The more even distribution of solute ions and solvent H2O molecules within a LiNO3-containing nanodroplet during its evaporation can be attributed to the much higher diffusivity of Li+ compared with other metal ions, as Li is the lightest metal (the molar mass of Li, Ni, Co, Mn are 6.94, 58.693, 58.933, and 54.938 g mol−1 respectively). | ||
| Fig. 10 Radial distribution of mass density (a); and radial distribution of the number density of metal ions (b) during evaporation of a 10 nm LiNO3-, Ni(NO3)2-, Co(NO3)2-, and Mn(NO3)2-containing nanodroplet at the time when water–salt-ratio (WSR) decreases to 1. The ambient N2 is at 2000 K and the initial solution concentration is 5.4 mol L−1. The snapshots below are slice view of the droplet at WSR = 1. Ni atoms are in reddish-orange; Li atoms are in purple; Co atoms are in green; Mn atoms are in magenta; N atoms in NO3− are in yellow; O atoms in NO3− are in orange; H and O atoms in water are in cyan; N atoms in N2 are in light yellow. | ||
To further reveal the evolution of the micro-structure of the solute ions and the solvent H2O molecules during droplet evaporation, the coordination number (CN) of metal ions with oxygen atoms from nitrate ions (denoted as ON) and water molecules (denoted as OW) has been tracked. Fig. 11 shows the temporal evolution of CN of M–OW and CN of M–ON (M = Li, Ni, Co, or Mn) during evaporation of a nanodroplet with LiNO3, Ni(NO3)2, Co(NO3)2, or Mn(NO3)2 being the solute and H2O being the solvent. According to Fig. 11(a) and (b), the temporal evolution of CN of Ni–O and CN of Co–O are very similar to each other. Initially, both Ni2+ and Co2+ are surrounded by ∼4 O atoms from H2O and ∼2 O atoms from NO3−. Both CN of Ni–O (including Ni–ON and Ni–OW) and CN of Co–O (including Co–ON and Co–OW) keep unchanged with time during droplet evaporation until ∼800 ps. As mentioned previously in Section 3.1, the stage with a constant CN of M–OW (M = Ni2+ or Co2+) can be regarded as the “free H2O” evaporation stage, which has also been denoted by the semi-transparent blue area in Fig. 11(a) and (b). In contrast with CN of Ni–O and CN of Co–O, CN of Li–O and Mn–O evolve in different manners during droplet evaporation. For evaporation of a droplet containing LiNO3 or Mn(NO3)2, CN of Li–OW and CN of Mn–OW keep changing with time, so it is hard to define a “free H2O” evaporation stage. According to Fig. 11, the total CN of Li–O decreases from ∼4.5 to ∼4 at WSR = 1, while for the other three metal ions, the total CN of M–O (M = Ni2+, Co2+, Mn2+) is 6 initially and decrease to ∼5. Li+ is coordinated by fewer O atoms because it is monovalent, hence the electrostatic interaction between Li+ and O atoms is weaker compared with the electrostatic interaction between O atoms and Ni2+, Co2+, or Mn2+, which are divalent ions.
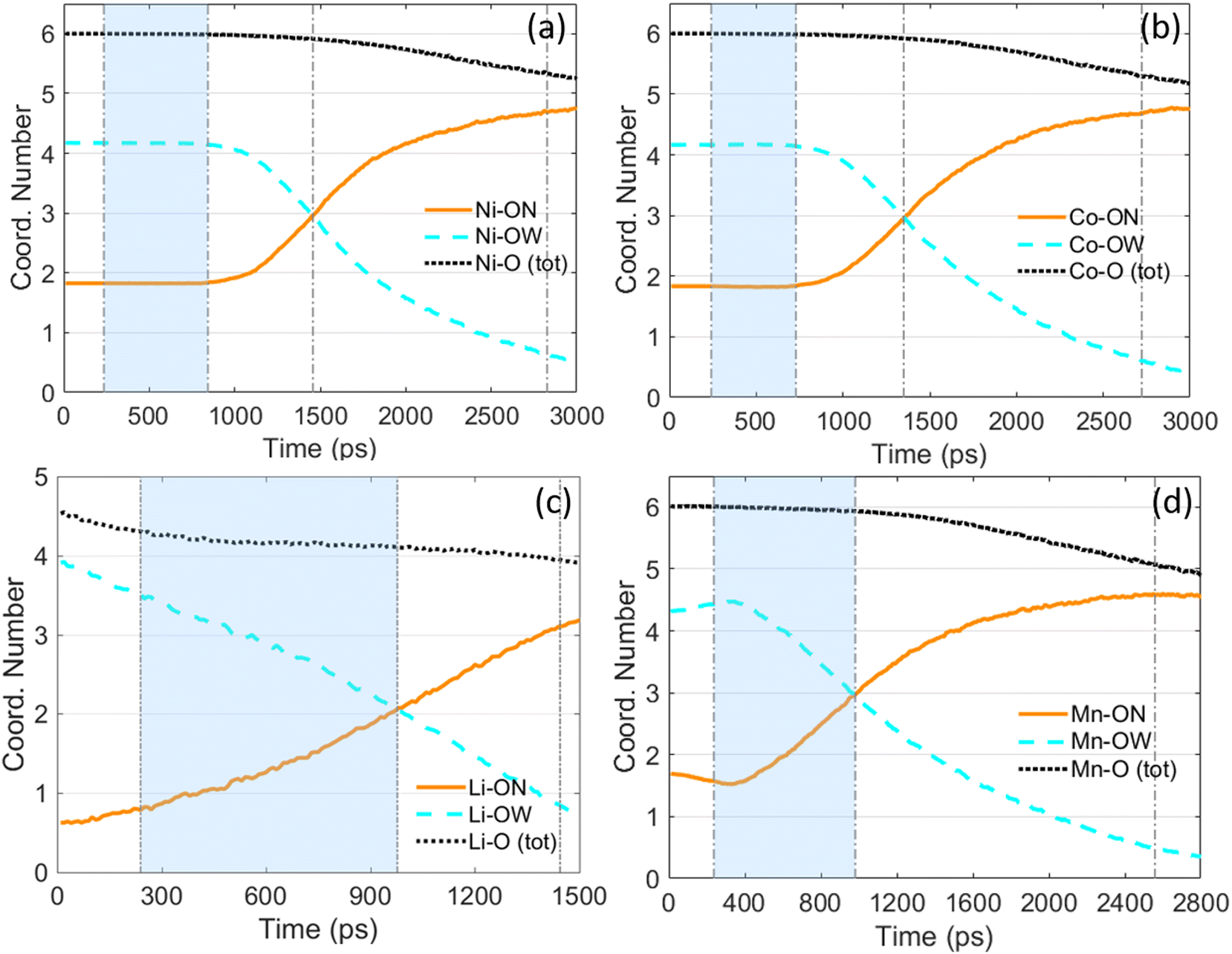 | ||
| Fig. 11 Temporal evolution of the coordination number (CN) of M–O (M = Ni, Co, Li, or Mn) during evaporation of a 10 nm Ni(NO3)2 (a); Co(NO3)2 (b); LiNO3 (c); and Mn(NO3)2 (d) nanodroplet. For all four droplets containing different metal nitrates as the solutes, the initial solution concentration is 5.4 mol L−1 and the ambient N2 is at 2000 K. ON represents O atoms from nitrate ions while OW represents O atoms from water molecules. | ||
Fig. 12 illustrates the temporal evolution of squared droplet diameter (normalized by the initial droplet diameter d0) during evaporation of a 10 nm droplet containing four different metal nitrates, namely LiNO3, Ni(NO3)2, Co(NO3)2, and Mn(NO3)2 respectively. For the evaporation of a nanodroplet with Ni(NO3)2 or Co(NO3)2 being the solute, a “free H2O” evaporation stage exists as suggested by Fig. 11(a) and (b). Although the evaporation of a Mn(NO3)2-containing nanodroplet demonstrates no “free H2O” evaporation stage based on Fig. 11(d), scatters of d2/d20 during the evaporation of a Mn(NO3)2 nanodroplet almost coincide with those of a Ni(NO3)2 or Co(NO3)2 nanodroplet, suggesting similar evaporation rate of a MNO3-containing (M = Ni, Co, or Mn) nanodroplet. By contrast, the evaporation rate of a LiNO3-containing nanodroplet is significantly larger than that of a Ni(NO3)2-, Co(NO3)2-, or Mn(NO3)2-containing nanodroplet. This is reasonable because the electrostatic interaction between Li+ and H2O molecules is weaker than that between those divalent metal ions and H2O molecules, causing the solvent H2O molecules more easily to escape/evaporate from a LiNO3-containing nanodroplet. Since Fig. 12 suggests similar evaporation rate for a droplet with either Ni(NO3)2, Co(NO3)2, or Mn(NO3)2 being the solute, we further suspect that the dynamic evaporation process of an NCM811 precursor droplet should be similar to that of an NCM111 precursor droplet, if the total concentrations of the metal nitrates and the ambient gas temperature are kept the same for evaporation. (The precursor solutions for NCM811 and NCM111 in flame spray pyrolysis (FSP) can be prepared by dissolving Ni(NO3)2, Co(NO3)2, Mn(NO3)2, and LiNO3 in deionized water in the required proportions (molar ratios of Ni2+:Co2+:Mn2+:Li+ = 8:1:1:1.05 for NCM811; 1:1:1:1.05 for NCM111), while the total concentration of transition metal ions (Ni2+, Co2+, Mn2+) are kept the same.3) Such an inference requires an additional assumption that different metal nitrates have little influence on each other within the same droplet during the evaporation process, which will be examined in our future study. In the pioneering experimental study of Abram et al.,3 both NCM811 and NCM111 nanoparticles were synthesized successfully through FSP. Although the total concentration of transition metal ions is the same in precursor solutions for NCM811 and NCM111, the as-synthesized NCM811 nanoparticles were found to be much irregular in shape while most NCM111 products were spherical. The authors attributed the difference between the morphology of the as-prepared NCM111 and NCM811 particles to variation in solubility of those transition metal nitrates and their relative solution saturation. Specifically, they suspected that the relative solution saturation of Ni(NO3)2 is lower in 811 solutions, which can promote earlier precipitation of the salts, forming a shell. This can inhibit evaporation of the remaining water through the partially dried layer. As the particle continues going through the flame, this layer can buckle and collapse, explaining the irregular shape of the obtained NCM811 particles in comparison to the spherical shape of the as-prepared NCM111 particles. However, this explanation is not supported by our MD simulation results, which show similar evaporation rate (see Fig. 12) and similar tendency of forming a solute shell (see Fig. 10) during evaporation of a nanodroplet with Ni(NO3)2, Co(NO3)2, or Mn(NO3)2 being the solute, suggesting the much irregular shape of the as-synthesized NCM811 nanoparticles compared with the spherical NCM111 nanoparticles is less likely to be caused by any difference during the evaporation stage of the precursor droplets. This leads to another possible explanation that it may be the different thermal decomposition properties of different metal nitrates that are responsible for the different morphology of the as-prepared NCM811 and NCM111 nanoparticles.
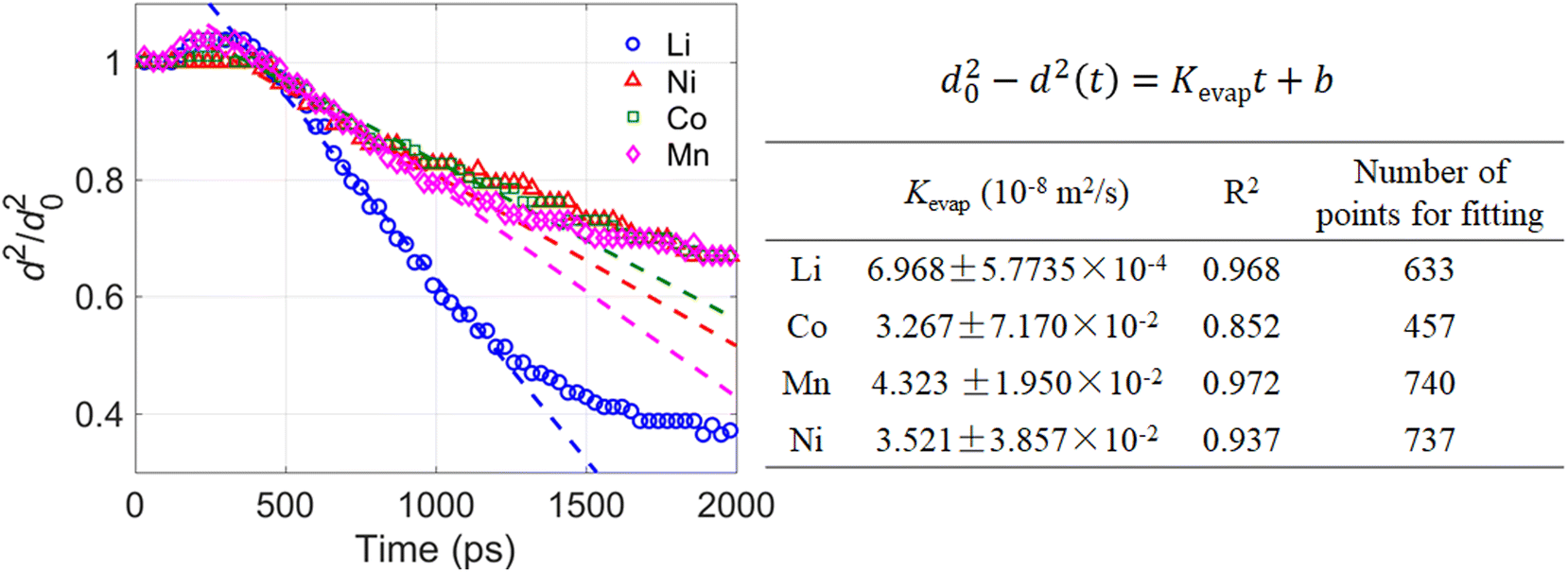 | ||
| Fig. 12 Temporal evolution of squared droplet diameter during the evaporation of a 10 nm droplet with LiNO3, Ni(NO3)2, Co(NO3)2, or Mn(NO3)2 being the solute respectively. For all four droplets containing different metal nitrates, the initial solution concentration is 5.4 mol L−1 and the ambient N2 is at 2000 K. | ||
4. Conclusions
In this work, the evaporation of nanodroplets with metal nitrates (including LiNO3, Ni(NO3)2, Co(NO3)2, and Mn(NO3)2) being the solute and water being the solvent has been investigated using molecular dynamics simulation. To quantitatively describe the droplet evaporation process, the temporal evolution of a variety of key features including the radial distribution of mass density, the radial distribution of metal ion number density, droplet diameter, and coordination number (CN) of metal ions with oxygen atoms have been tracked. Evaporation rate constant is obtained by making analogy to the classical D2 law for droplet evaporation. The effect of initial solution concentration of the droplet, ambient gas temperature, and metal ion type on the dynamic evaporation process is examined in detail. The main conclusions are as follows:(1) During the evaporation process of a nanodroplet with Ni(NO3)2 being the solute and H2O being the solvent, Ni(NO3)2 tends to precipitate on the droplet surface, forming a solute–core–solvent–shell structure. The Ni(NO3)2–shell–H2O–core configuration forms more easily for those droplets with high initial solution concentration (e.g., >5 mol L−1) evaporating in ambient N2 at a high temperature (e.g., ≥2500 K). This is because under such conditions, the evaporation rate of H2O is much higher than the diffusion rate of Ni2+ and NO3− ions within the droplet. Therefore, in a very short period of time, a large number of H2O molecules will leave the droplet entering the gas-phase, leaving Ni2+ and NO3− ions precipitated on the droplet surface, forming a solute shell.
(2) The temporal evolution of the CN of Ni–OW (O atoms from water) and Ni–ON (O atoms from nitrate ions) during the evaporation of a Ni(NO3)2-containing nanodroplet suggests a “free H2O” evaporation stage, during which both CN of Ni–OW and CN of Ni–ON are unchanged with time. The squared droplet diameter (d2) is found to be linearly decreasing with time in this stage. To make analogy to the classical D2 law for droplet evaporation, the evaporation rate constant Kevap for a Ni(NO3)2-containing nanodroplet is obtained by fitting d2 and time t during the “free H2O” evaporation stage into the linear relationship d20 − d2 = Kevapt + b, where d0 is the initial droplet diameter.
(3) Compared with a pure water droplet, addition of the Ni(NO3)2 solute will decrease the evaporation rate constant Kevap. Kevap decreases with increasing initial solution concentration due to the stronger interaction between the solute ions (Ni2+ and NO3−) and the H2O molecules at a high solution concentration. Kevap increases with increasing ambient gas temperature due to enhanced heat transfer.
(4) By tracking the radial distribution of the number density of metal ions during the evaporation of four nanodroplets with four different metal nitrates namely LiNO3, Ni(NO3)2, Co(NO3)2, and Mn(NO3)2 being the solute respectively, it is found that Ni2+, Co2+, and Mn2+ will precipitate on the droplet surface, forming a metal nitrates shell. In contrast, the distribution of Li+ within the evaporating droplet is more even due to the high diffusivity of Li+ compared with other metal ions.
(5) The temporal evolutions of CN of Ni–O and CN of Co–O are similar to each other, both of which suggest a “free H2O” evaporation stage when CN Ni–OW and CN of Co–OW are constant with time. Although the coordination analysis for Mn–O suggests CN of Mn–OW keeps changing with time, the evaporation rate for a Ni(NO3)2, Co(NO3)2, or Mn(NO3)2-containing droplet is hardly affected by the different types of metal ions if the initial solution concentration and the ambient gas temperature are kept the same.
Author contributions
Dingyu Hou: conceptualization, methodology, software, investigation, data curation, visualization, writing – original draft; Geng Wang: methodology, software, investigation, writing – review & editing; Jingqi Gao: methodology, software, investigation, writing – review & editing; Kai H. Luo: conceptualization, supervision, funding acquisition, resources, investigation, project administration, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the UK Engineering and Physical Sciences Research Council (EPSRC) under Grant No. EP/T015233/1, EP/R029598 and EP/X035875/1. This work made use of computational support by CoSeC, the Computational Science Centre for Research Communities, through UKCOMES.References
- A. Chakraborty, S. Kunnikuruvan, S. Kumar, B. Markovsky, D. Aurbach, M. Dixit and D. T. Major, Chem. Mater., 2020, 32, 915–952 CrossRef CAS.
- G. Zang, J. Zhang, S. Xu and Y. Xing, Energy, 2021, 218, 119504 CrossRef CAS.
- C. Abram, J. Shan, X. Yang, C. Yan, D. Steingart and Y. Ju, ACS Appl. Energy Mater., 2019, 2, 1319–1329 CrossRef CAS.
- J. Zhang, V. L. Muldoon and S. Deng, J. Power Sources, 2022, 528, 231244 CrossRef CAS.
- D. S. Jung, Y. N. Ko, Y. C. Kang and S. B. Park, Adv. Powder Technol., 2014, 25, 18–31 CrossRef CAS.
- L. Mädler, H. K. Kammler, R. Mueller and S. E. Pratsinis, J. Aerosol Sci., 2002, 33, 369–389 CrossRef.
- S. Li, Y. Ren, P. Biswas and D. T. Stephen, Prog. Energy Combust. Sci., 2016, 55, 1–59 CrossRef.
- F. Meierhofer and U. Fritsching, Energy Fuels, 2021, 35, 5495–5537 CrossRef CAS.
- W. Y. Teoh, R. Amal and L. Mädler, Nanoscale, 2010, 2, 1324–1347 RSC.
- J.-S. Park, J. K. Kim, J. H. Hong, J. S. Cho, S.-K. Park and Y. C. Kang, Nanoscale, 2019, 11, 19012–19057 RSC.
- J. E. Madero, J. Li, K.-Y. Shen, J. Wojtak and R. L. Axelbaum, Appl. Energy Combust. Sci., 2021, 5, 100020 Search PubMed.
- A. B. D. Nandiyanto and K. Okuyama, Adv. Powder Technol., 2011, 22, 1–19 CrossRef CAS.
- D. Hardy, J. Archer, P. Lemaitre, R. Vehring, J. Reid and J. Walker, Phys. Chem. Chem. Phys., 2021, 23, 18568–18579 RSC.
- M. Farid, Chem. Eng. Sci., 2003, 58, 2985–2993 CrossRef CAS.
- M. Rezaei and R. R. Netz, Phys. Fluids, 2021, 33, 091901 CrossRef CAS PubMed.
- Y. Gong, G. Xiao, X. Ma, K. H. Luo, S. Shuai and H. Xu, Fuel, 2021, 287, 119516 CrossRef CAS.
- G. Xiao, K. H. Luo, X. Ma and S. Shuai, Proc. Combust. Inst., 2019, 37, 3219–3227 CrossRef CAS.
- X. Wu, Z. Yang and Y. Duan, Int. J. Heat Mass Transfer, 2020, 158, 120024 CrossRef CAS.
- B.-B. Wang, X.-D. Wang, Y.-Y. Duan and M. Chen, Int. J. Heat Mass Transfer, 2014, 73, 533–541 CrossRef CAS.
- B.-B. Wang, X.-D. Wang and T.-H. Wang, Appl. Phys. Lett., 2014, 105, 121906 CrossRef.
- H. Higashi, T. Tokumi, C. J. Hogan, H. Suda, T. Seto and Y. Otani, Phys. Chem. Chem. Phys., 2015, 17, 15746–15755 RSC.
- L. Konermann, R. G. McAllister and H. Metwally, J. Phys. Chem. B, 2014, 118, 12025–12033 CrossRef CAS PubMed.
- R. G. McAllister, H. Metwally, Y. Sun and L. Konermann, J. Am. Chem. Soc., 2015, 137, 12667–12676 CrossRef CAS PubMed.
- C. Caleman and D. van der Spoel, Phys. Chem. Chem. Phys., 2007, 9, 5105–5111 RSC.
- E. Ahadi and L. Konermann, J. Am. Chem. Soc., 2010, 132, 11270–11277 CrossRef CAS PubMed.
- L. Martínez, R. Andrade, E. G. Birgin and J. M. Martínez, J. Comput. Chem., 2009, 30, 2157–2164 CrossRef PubMed.
- L. Zhan, H. Chen, H. Zhou, Q. Feng, L. Gu, L. Yang and Z. Sun, Appl. Therm. Eng., 2022, 118752 CrossRef CAS.
- S. Plimpton, J. Comput. Phys., 1995, 117, 1–19 CrossRef CAS.
- A. I. Jewett, D. Stelter, J. Lambert, S. M. Saladi, O. M. Roscioni, M. Ricci, L. Autin, M. Maritan, S. M. Bashusqeh and T. Keyes, et al. , J. Mol. Biol., 2021, 433, 166841 CrossRef CAS PubMed.
- A. Stukowski, Modell. Simul. Mater. Sci. Eng., 2009, 18, 015012 CrossRef.
- P. Li, L. F. Song and K. M. Merz Jr., J. Chem. Theory Comput., 2015, 11, 1645–1657 CrossRef CAS PubMed.
- P. Li, B. P. Roberts, D. K. Chakravorty and K. M. Merz Jr., J. Chem. Theory Comput., 2013, 9, 2733–2748 CrossRef CAS PubMed.
- Y. Wang, L. Song, G. Wang, H. Liu, Z. Jing, Y. Zhou, F. Zhu and Y. Zhang, Spectrochim. Acta, Part A, 2022, 267, 120478 CrossRef CAS PubMed.
- H. Krienke and D. Opalka, J. Phys. Chem. C, 2007, 111, 15935–15941 CrossRef CAS.
- S. Díaz-Moreno, S. Ramos and D. T. Bowron, J. Phys. Chem. A, 2011, 115, 6575–6581 CrossRef PubMed.
- H. Berendsen, J. Grigera and T. Straatsma, J. Phys. Chem., 1987, 91, 6269–6271 CrossRef CAS.
- M. P. Allen and D. J. Tildesley, Computer Simulation of Liquids, Oxford University Press, 1989 Search PubMed.
- T. Darden, D. York and L. Pedersen, J. Chem. Phys., 1993, 98, 10089–10092 CrossRef CAS.
- D. Zong, Z. Yang and Y. Duan, Appl. Therm. Eng., 2017, 122, 71–79 CrossRef CAS.
- G. Qiao, M. Lasfargues, A. Alexiadis and Y. Ding, Appl. Therm. Eng., 2017, 111, 1517–1522 CrossRef CAS.
- S. Jayaraman, A. P. Thompson, O. A. von Lilienfeld and E. J. Maginn, Ind. Eng. Chem. Res., 2010, 49, 559–571 CrossRef CAS.
- J.-P. Ryckaert, G. Ciccotti and H. J. Berendsen, J. Comput. Phys., 1977, 23, 327–341 CrossRef CAS.
- W. Berndt, CalcSteam, https://www.peacesoftware.de/einigewerte/wasser_dampf_e.html, 2007 Search PubMed.
- R. Takahashi, S. Sato, T. Sodesawa and Y. Kamomae, Phys. Chem. Chem. Phys., 2000, 2, 1199–1204 RSC.
- R. Takahashi, S. Sato, T. Sodesawa and H. Nishida, Phys. Chem. Chem. Phys., 2002, 4, 3800–3805 RSC.
- D. Lide and Boca Raton, CalcSteam, 2004, 8–141 Search PubMed.
- W. Brockner, C. Ehrhardt and M. Gjikaj, Thermochim. Acta, 2007, 456, 64–68 CrossRef CAS.
- T. Segawa, K. Kawaguchi, K. Ishii, M. Suzuki, N. Arimitsu, H. Yoshida and K. Fukui, Adv. Powder Technol., 2015, 26, 983–990 CrossRef CAS.
- S. R. Turns et al. , Introduction to combustion, McGraw-Hill Companies New York, NY, USA, 1996, vol. 287 Search PubMed.
- F. Dalla Barba, J. Wang and F. Picano, Phys. Fluids, 2021, 33, 051701 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3nr00060e |
| ‡ Present address: Beijing National Laboratory for Molecular Sciences, State Key Laboratory of Polymer Physics and Chemistry, Institute of Chemistry, Chinese Academy of Sciences, Beijing 100190, People's Republic of China. |
| This journal is © The Royal Society of Chemistry 2023 |