
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePolymeric ionic liquid-based formulations for the fabrication of highly stable perovskite nanocrystal composites for photocatalytic applications†
Sara
Miralles-Comins
a,
Marcileia
Zanatta
a,
Andrés F.
Gualdrón-Reyes
ab,
Jhonatan
Rodriguez-Pereira
 cd,
Iván
Mora-Seró
*a and
Víctor
Sans
*a
cd,
Iván
Mora-Seró
*a and
Víctor
Sans
*a
aInstitute of Advanced Materials (INAM), Universitat Jaume I (UJI), Avenida de Vicent Sos Baynat, s/n, 12071 Castelló de la Plana, Castellón, Spain. E-mail: sans@uji.es; sero@uji.es
bFacultad de Ciencias, Instituto de Ciencias Químicas, Isla Teja, Universidad Austral de Chile, 5090000, Valdivia, Chile
cCenter of Materials and Nanotechnologies, Faculty of Chemical Technology, University of Pardubice, Nam. Cs. Legii 565, 53002 Pardubice, Czech Republic
dCentral European Institute of Technology, Brno University of Technology, Purkyňova, 123,612 00 Brno, Czech Republic
First published on 9th February 2023
Abstract
Halide perovskite nanocrystals (PNCs) have emerged as potential visible-light photocatalysts because of their outstanding intrinsic properties, including high absorption coefficient and tolerance to defects, which reduces non-radiative recombination, and high oxidizing/reducing power coming from their tuneable band structure. Nevertheless, their sensitivity to humidity, light, heat and water represents a great challenge that limits their applications in solar driven photocatalytic applications. Herein, we demonstrate the synergistic potential of embedding PNCs into polymeric ionic liquids (PILs@PS) to fabricate suitable composites for photodegradation of organic dyes. In this context, the stability of the PNCs after polymeric encapsulation was enhanced, showing better light, moisture, water and thermal stability compared to pristine PNCs for around 200 days.
Introduction
The chemical industry is responsible for the production of chemicals and materials underpinning modern societies. Nevertheless, it is also a major polluter, which needs to be addresses to become a sustainable manufacturing industry. Within this remit, organic dye emissions are a major cause of pollution in the environment.1,2 Photocatalytic degradation is a promising route to convert organic waste into harmless substances. The photocatalytic activity is determined by the catalyst's capacity to form electron–hole pairs, which produces free radicals capable of undergoing secondary reactions. Most investigations have focused on the use of TiO2 as photocatalyst because its high photocatalytic performance, low cost, chemical stability and non-toxicity.3–5 Nevertheless, it can only absorb UV light (λ < 387 nm) due to its wide band gap (3.2 eV) and high recombination of electron–hole pairs, thus only 3–5% of solar light is used.6–9 Therefore, to maximize the utilization of sunlight, it is critical to employ an adequate material capable of absorbing the whole spectrum of visible light in order to improve solar conversion efficiency. Several visible-light catalysts have been used such as organic conjugated polymers,10 inorganic semiconductors,11 and transition metal complexes.12 However, those materials are difficult to prepare and expensive. Halide perovskite nanocrystals (PNCs) have appeared as promising photocatalysts in the entire UV-visible-NIR region. They can be synthesized by facile and low-cost methods with precise control over their size and composition, offering high photoluminescence quantum yields (PLQY) up to 100%, high absorption coefficient, tolerance to defects, narrowband emission, tuneable band gap and good light-trapping ability.13–22 To date, the photocatalytic activity of all-inorganic perovskites has covered a broad range of applications: CO2 reduction,23,24 hydrogen production,25 organic chemical reactions26–32 or pollutant degradation.33–36The most extensively PNCs studied have been inorganic CsPbX3 PNCs, where X represents a halogen anion, I−, Br−, Cl− or a combination of them. Nevertheless, the relatively low stability of PNCs to light, heat, oxygen and water limit their use in large-scale applications.37–39 Currently, extensive research has been conducted to overcome the chemical and physical issues, mostly due to their ionic nature and low melting temperatures. For example, embedding them into glass by the melt-quenching method and subsequent crystallization process has been a solution, but some aggregation and clustering issues appeared.40,41 Another way to enhance the stability of PNCs is to encapsulate them within a stable sol or polymer.42–44 Incorporating PNCs into macroscale polymer matrices to form thin films has been demonstrated to be an efficient methodology to protect semiconductor devices against environmental degradation.45 Some recent reports show the benefits of encapsulating hydrophobic PNCs into water-repelling hydrophobic bulk polymers, enhancing light stability and retention of quantum yield upon water exposure by preventing access of water to the PNCs surface.46–48 The polymeric matrix employed has a direct influence on the level of encapsulation achieved. However, some drawbacks including easy leakage of PNCs from the polymer microspheres or uneven distribution can arise because PNCs are only physically bounded to the polymer.49,50 In this regard, direct polymerization of monomers in the presence of PNCs can be a fast and effective way to solve the above-mentioned problems. The in situ encapsulation can be carried out by functionalizing and polymerizing the PNCs ligands,51 the perovskite itself52,53 or by adding a monomer.13,54 This is still an underexplored field since most of the methods involve polar reaction medium and relatively high polymerization temperature (above 60 °C).55,56 Additionally, there is an actual interest to find polymeric matrices with tailored hydrophobicity and with improved ligand–polymer interactions.47
Recent works have demonstrated that polymerizable ionic liquids (PILs) are ideal materials to stabilize molecular and nanostructured materials as a result of their ionic and supramolecular interactions. PILs are high-molecular-mass compounds containing monomer units of typical ionic liquids (IL), that combine attractive IL properties with polymer features.57 Another unique characteristic of PILs is that the physico-chemical properties of the polymers can be controlled by changing the corresponding ions. Hence, PILs have found a broad range of uses in multiple fields, such as electrochemical energy materials, optoelectronic, stimuli-responsive, antimicrobials among others.58,59 Several articles have reported the use of PILs/ILs together with PNCs in order to improve the applicability of their systems, for instance as gating material for electronic applications,60 as hole/electron transport material in perovskite solar cells,61–63 as candidate for absorbing CO2,64,65 as perovskite/ionic liquid interfaces for transient opto-electronic conversion in photovoltaic devices.66 However, in those examples PILs are not used to stabilize perovskite. Xia et al. has shown the advantages of protecting hybrid perovskites from decomposition in humid environments with in situ ionic liquid polymerization.67 Furthermore, the development of these polymers can be especially designed to be printed by additive manufacturing techniques, thus being of huge interest since traditional polymerisation techniques limit the geometries that can be potentially generated. This sophisticated technology enables to directly transfer the unique properties of molecular materials to macroscopic functional devices, rapidly closing the loop between design, manufacture and application at a relatively low cost.68–71 Although organic room-temperature phosphorescent luminogens72 and fluorescent quantum dots73 have been added as additives into 3D printing materials, perovskites are not as common yet because of its instability, and there are just a few examples reported.74–77
Therefore, combinations based on PILs can represent a great chance to design photoactive 3D printed devices for photocatalysis, optoelectronic, conductive, electrochemical and redox applications amongst others. To the best of our knowledge, the development of advanced formulations stabilizing photoluminescence (PL) all-inorganic perovskites with polymerizable ionic liquid especially designed for additive manufacturing has not been widely exploited yet.
Here, we have developed for the first-time a novel photocatalytic material capable to adsorb and degrade methyl red (MR) as a target molecule. The monomeric ionic liquid [2-(acryloyloxy)ethyl]trimethyl-ammonium bis(trifluoromethylsulfonyl)imide (AcrEMA-TFSI) was able to effectively stabilize all-inorganic PNCs based on CsPbX3 structure (X = Br, and Cl![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Br combinations) by in situ photopolymerization forming insoluble and hydrophobic cross-linked films. These solid films showed impressive quantum yields and photoluminescence with enhanced water, air, light and heat resistance. The synergistic union between AcrEMA-TFSI and PNCs is attributed to the strong coordination interactions between Pb ions on the surface of PNCs and the carbonyl groups (C
:Br combinations) by in situ photopolymerization forming insoluble and hydrophobic cross-linked films. These solid films showed impressive quantum yields and photoluminescence with enhanced water, air, light and heat resistance. The synergistic union between AcrEMA-TFSI and PNCs is attributed to the strong coordination interactions between Pb ions on the surface of PNCs and the carbonyl groups (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) from AcrEMA-TFSI promoting a surface passivation in the photomaterial. Moreover, these PNCs were specifically formulated to be 3D-printed in tailored geometries, thus directly translating the perovskite properties to the additively manufactured objects.
O) from AcrEMA-TFSI promoting a surface passivation in the photomaterial. Moreover, these PNCs were specifically formulated to be 3D-printed in tailored geometries, thus directly translating the perovskite properties to the additively manufactured objects.
Experimental
General experimental information
Unless otherwise stated, all reagents were purchased from Sigma-Aldrich and used without further purification. Lithium bis(trifluoromethylsulfonyl)imide (LiTFSI, 99%) was purchased from IOLITEC GmbH and used as received.UV–vis spectra were collected using a Jasco V-780 UV–vis spectrophotometer. 1H NMR spectra and 13C NMR spectra were recorded on a Bruker ASCEND™ 400 (400 MHz) spectrometer at ambient temperature. FT-IR spectroscopy was measured using a FT/IR-6200 from Jasco, equipped with an ATR accessory (ATR Pro One, Jasco). The spectra were recorded with a resolution of 4 cm−1 in the wavelength range between 650 and 4000 cm−1 averaging 16 scans for each spectrum. Thermal gravimetric analysis was performed on TGA-DSC3 analyzer from Mettler Toledo. Samples for TGA analysis were heated in an inert atmosphere up to 800 °C with a heating rate of 10 °C min−1. Additionally, a fluorometer and an absolute-PLQY were used to measure the photoluminescence of the samples. Photoluminescence (PL) spectra of the CsPbBr3-based films were recorded on a HORIBA photoluminescence spectrometer from 450 to 575 nm with an integration time of 1 nm s−1 under an excitation wavelength of 420 nm. While photoluminescence (PL) spectra of the materials containing CsPbX3 structure with X = Cl:Br combinations were recorded from 395 to 500 nm with an integration time of 1 nm s−1 under an excitation wavelength of 380 nm.
Synthesis of ionic liquids
Full synthesis details and characterisation data are given at the end of the ESI.†Preparation of the polymerized films
All the films were prepared with the corresponding molar concentrations of [2-(acryloyloxy)ethyl]trimethyl-ammonium bis(trifluoromethylsulfonyl)imide (AcrEMA-TFSI, 100–0 mol%), butyl acrylate (BA, 0–95 mol%) and 1,4-butanediol diacrylate (BDA, 0; 5 mol%), depending on the formulation used and mixed with 1 wt% of diphenyl(2,4,6-trimethylbenzoyl)phosphine oxide (TPO) and 2 wt% of PNCs. The liquid mixture was illuminated upon UV light during 20 minutes at 40 °C. After that time, solid films were obtained with a tailored geometry. The films were characterised by FT-IR, DSC-TGA and UV-Vis (see ESI, Fig. S1–S9†).3D-printing process
The printing process was done by a masked stereolithography printer using UV light for photopolymerizing (Elegoo Mars 2 Pro). The Elegoo Mars 2 Pro printer was equipped with a 2560 × 1620 mono liquid crystal display (LCD) and an LED light source illuminating at 405 nm. The CAD files were designed and converted to STL-type files using SolidWorks CAD software. The fabrication process began by slicing the 3D CAD model into individual 2D images for projecting onto the photocurable liquid. The slicer software employed was ChituBox. The printing parameters were adjusted for the formulation regardless of the presence of perovskite nanocrystals, as their aggregation did not significantly affect the printing conditions. The exposure time of light for the layers were 6 seconds, and 8 seconds for the bottom layers (i.e., the first 6 layers attached to the platform; all layers had 0.1 mm of thickness). The light power was fixed at 40 W. This light power refers to the power of the UV lamp that is used to cure the resin during the 3D printing process. The UV light is responsible for hardening the resin in the thin layers that are deposited on the printing platform, thus forming the printed object. Then, the part was washed in hexane during 5 minutes to remove any excess of unreacted monomers and post-cured using an UV light for 20 min at 40 °C.Stability tests of the PNCs
Four different studies were performed to compare the stability of pristine CsPbBr3 with PNCs encapsulated with polymeric ionic liquids using the above-mentioned formulations. For the test in the dark the samples were covered with aluminium and kept in a drawer. Simultaneously, for the air stability test, the samples were left on the laboratory bench open to atmospheric conditions (air, light and humidity). The aqueous stability tests were done immersing the samples in deionised water in a closed 5 mL vial and stored under day light conditions. The temperature stability experiments were conducted in open 5 mL vials placed in an oven at 70 °C without light irradiation.XPS analysis
Surface chemical composition and electronic state of the materials were determined by X-ray photoelectron spectroscopy (XPS, ESCA-2R, Scienta-Omicron). Spectra were recorded using monochromatic Al Kα = 1486.6 eV. The following sequence of spectra were recorded: survey spectra, C 1s, Cs 3d, Pb 4f, Br 3d, O 1s, N 1s, F 1s, S 2p and C 1s again to verify the stability of the charge as the function of time. The survey and high-resolution spectra were recorded at a pass energy of 150 and 20 eV, respectively. Binding energy scale was referenced to adventitious carbon (284.8 eV). CasaXPS processing software (Casa software Ltd) was used to analyze the data and the quantitative analysis was made using sensitivity factors provided by the manufacturer.Set-up for the photodegradation experiments
A 30 μM solution of methyl red (MR) in hexane was prepared and introduced into fluorescent cuvettes together with the corresponding film. All the solution were prepared under aerobic conditions. The system was properly closed to avoid the evaporation of hexane. Then, the cuvettes were kept under visible light during 1–22 hours using a xenon lamp at 100 mW cm−2. An UV filter was employed to avoid the effect of UV light for degrading the dyes. The UV-Vis absorbance of the methyl red was measured at different times to study the kinetics of the degradation reaction. All the experiments were repeated at least twice. For the recycle test, the films were removed from the solution and used in the next cycle with a new MR solution without any additional treatment.Results and discussion
The polymerizations of [2-(acryloyloxy)ethyl]trimethyl-ammonium (AcrEMA) salts using different anions (Cl−, I−, N(CN)2−, BF4−, TFSI) were performed mixing a cross-linker (1,4-butanediol diacrylate (BDA), 5 mol%) and a photoinitiator (diphenyl(2,4,6-trimethylbenzoyl)phosphine oxide (TPO), 1 wt%). Only two of the monomers produced consistent films, and only one of them (AcrEMA-TFSI) was able to stabilize the perovskite into the monomer and after polymerization without quenching the PNCs (further details in the ESI, Table S2†).According to the literature, butyl acrylate (BA) is commonly used to increase the flexibility of polymeric matrices, since it reduces the glass transition temperature (Tg) of the material.78–82 Thus, different formulations using AcrEMA-TFSI (100–0 mol%) and butyl acrylate (BA, 0–95 mol%) were prepared in order to obtain adequate mechanical and optical properties for the desired application, see Fig. 1A. Additionally, different perovskites (PS) have been evaluated with the set of formulations (PILX@PSY), where PILX is referring to the used formulation (X = A–H), see Fig. 1B, and PSY, the perovskite (Y = 1–3): CsPbBr3 (PS1), CsPbCl1.5Br1.5 (PS2) and CsPbCl2.25Br0.75 (PS3). All the combinations were efficient matrices for stabilizing the PNCs, see Fig. 1B and D. A-1 was sticky and fragile, which shows that the cross-linker (BDA) was necessary to obtain resistant films. On the other hand, the neutral polymer butyl acrylate needed 5 mol% of cross-linker to obtain a film and still, that formulation (H-1) formed the most fragile and brittle films. That is why A-1 and H-1 even being fluorescent are not considered in this study. Interestingly, the fluorescence of the films decreases with increasing mol% of BA, and above 15 mol% of additive (BA) the films were not fluorescent (E-1, F-1, G-1). For these reasons, the films B-1, C-1, D-1 were selected to the following experiments. In all samples, the amount of perovskite used was 2 wt% with respect to the weight of the formulation, since it was shown to be the optimum concentration with a PLQY around 45%. Lower concentrations reported poorer PL, see Fig. S10,† while using higher concentrations (5 wt%) the PLQY was notably decreased to 26%.
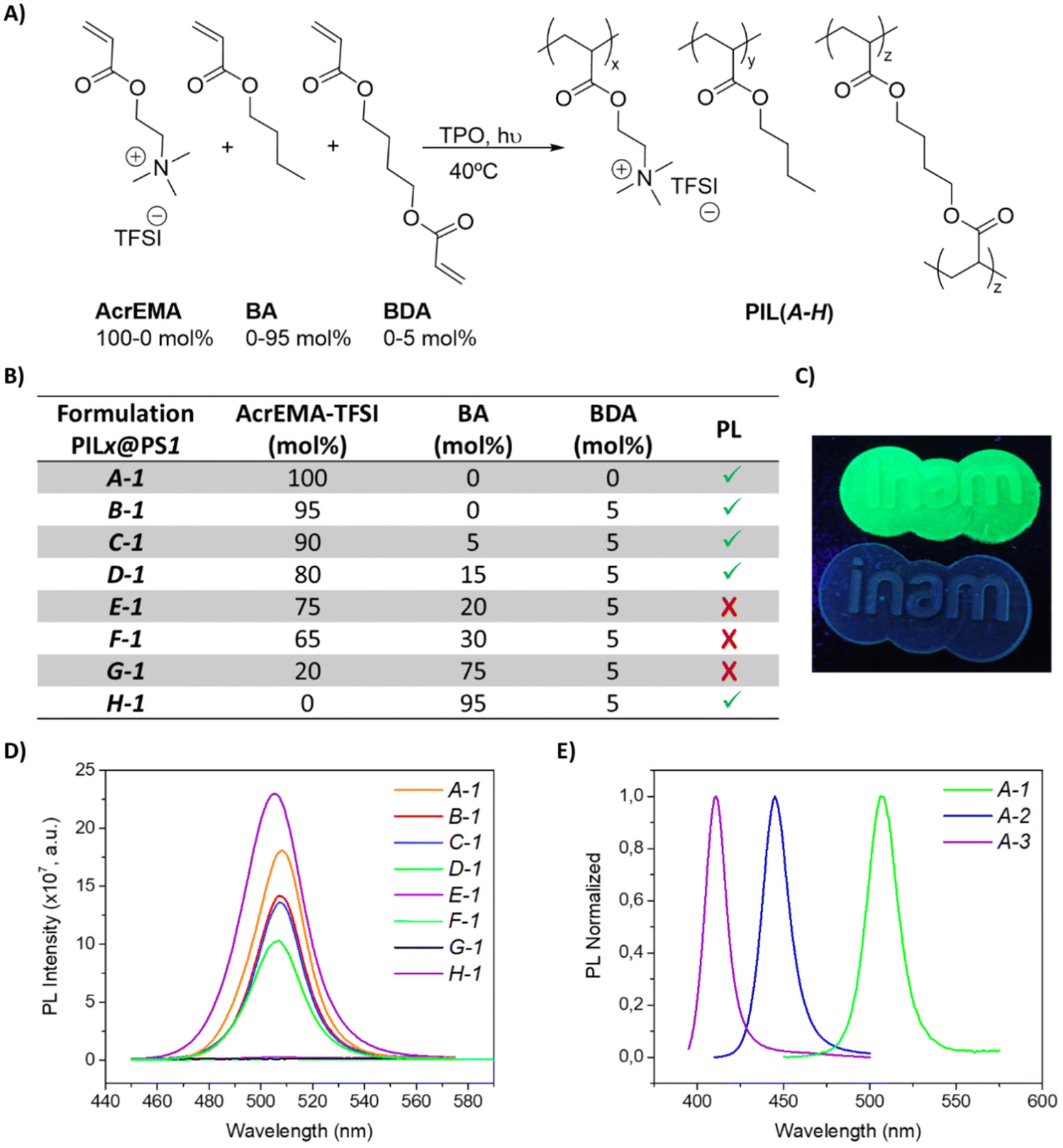 | ||
| Fig. 1 (A) Components used in the polymerization reaction; (B) different formulations prepared for encapsulating CsPbBr3 PNCs; (C) printed logo of INAM using B-1 formulation (up) and without CsPbBr3 PNCs (down) under UV light; (D) photoluminescence spectra of the formulations with CsPbBr3; (E) normalized photoluminescence spectra using different PNCs: CsPbBr3 (PS1), CsPbCl1.5Br1.5 (PS2) and CsPbCl2.25Br0.75 (PS3). | ||
The optimized formulations were able to be 3D printed, thus transferring the PL properties of the perovskite to the printed solid, as shows Fig. 1C. In addition, the absorption wavelength of the perovskite was modified changing the halides, obtaining films with different colours (see Fig. S11†): CsPbCl1.5Br1.5 (PS2) and CsPbCl2.25Br0.75 (PS3) (Fig. 1E). At this point, the mixture of PNCs and PILs is suitable to fabricate highly emitting 3D solids which improved intrinsic properties (as we will show below).
The different PNCs were stabilized into the formulation B keeping the PL properties over time for more than one year in the dark, see Fig. S12,† pointing the interest of the system for anticounterfeiting applications. In particular, CsPbBr3 (PS1) stability was further evaluated using all the different formulations. In Fig. S13,† it can be observed the high PLQY achieved using A, B and C formulations, however A formulation was not further studied since the material was fragile. According to the results, B-1 and C-1 films stored under aerobic atmosphere in the dark, at room temperature with a relative humidity of 60% kept almost constant PLQY, around 50%, during more than one year, see Fig. 2A. In this context, those two formulations were employed to study the stability of the PNCs at different conditions: exposed to air and light at 25 °C, see Fig. 2B, immersed in liquid water and stored under day light conditions at 25 °C, see Fig. 2C, and at 70 °C without light irradiation, see Fig. 2D.
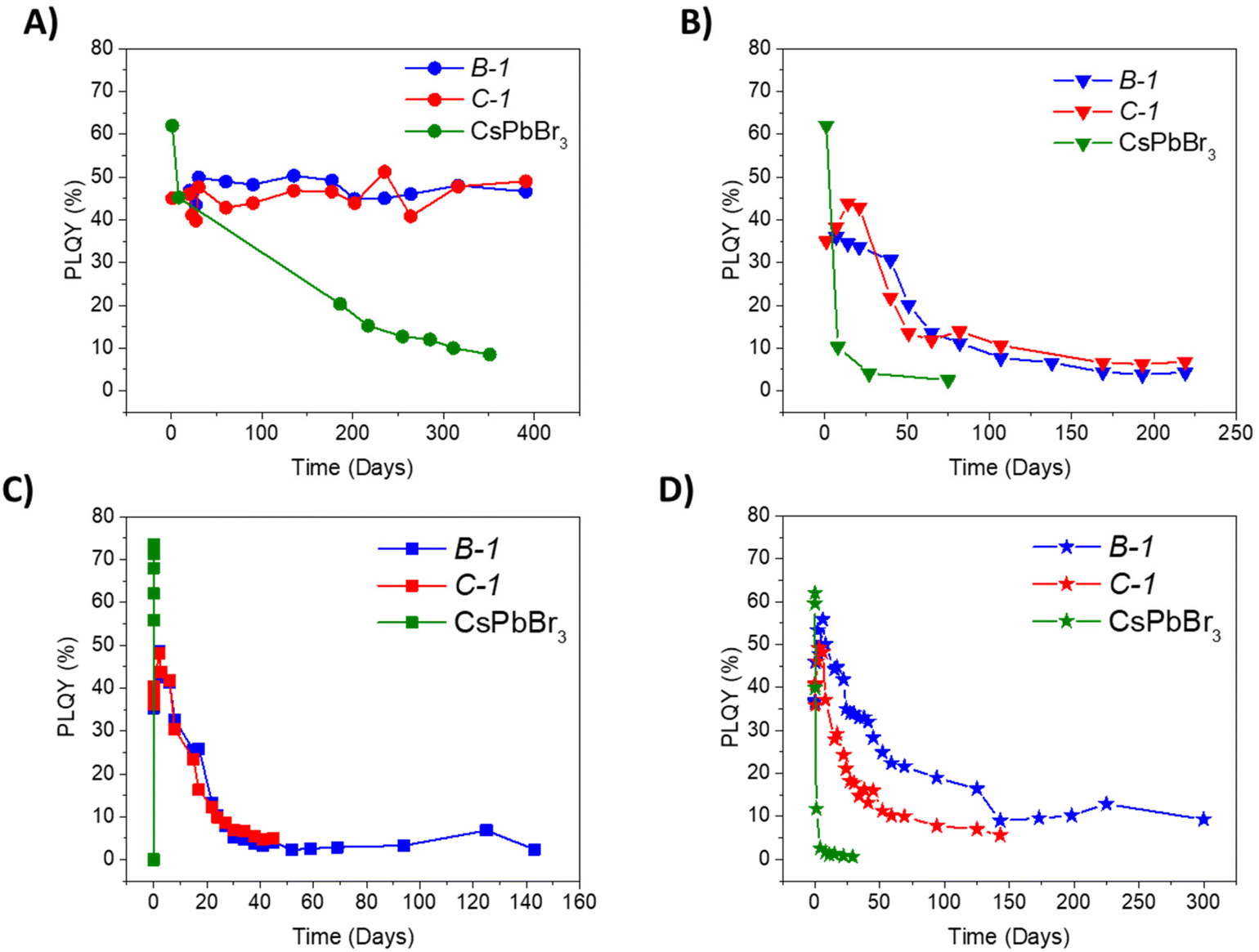 | ||
| Fig. 2 Spectra of PLQY for different formulations (B-1 and C-1) compared with pure CsPbBr3-NCs being stored (A) in a drawer in the dark at 25 °C; (B) opened vial (expose to atmospheric air); (C) closed vial in liquid water at 25 °C, and (D) opened vial inside an oven at temperature (70 °C) without light irradiation. | ||
Under extreme conditions, pristine PNCs lost more than 90% of their PLQY in less than 5 days, and upon immersion in water, the PNCs degraded within minutes. However, when they were encapsulated PNCs (B-1), stored under ambient conditions (oxygen, light and humidity) and exposed to 70 °C, they only exhibited a decrease of the PLQY around 15% after the first 40 days. In both cases, the PLQY was measured for more than 200 days, reporting still some photoluminescence. In an aqueous environment, B-1 formulation was able to stabilize CsPbBr3, and their PLQY was stable for almost 150 days. The high stabilization observed with B formulation was obtained due to the material's hydrophobicity as TFSI is a hydrophobic anion.83 In addition, the films were not hygroscopic and did not present porosity, as shown by SEM and profilometry analyses (Fig. S14†).
XPS analyses were performed to elucidate the influence of B-1 on the composition and chemical environment of the PNCs. The presence of Cs, Pb, Br, N, in addition to the co-existence of F and S from the AcrEMA-TFSI monomer was observed in the XPS spectra, see Fig. S15A.† The corresponding chemical composition of the PNCs in absence and presence of B is summarized in Table S3.†Fig. 3A exhibits the high-resolution (HR) XPS Cs 3d spectra of the samples, where a characteristic doublet located at ∼725/738 eV was observed. This doublet represents the Cs 3d5/2 and Cs 3d3/2 core levels, indicating as expected the presence of Cs+ in the CsPbBr3 lattice.84 Then, we acquired a HR-XPS Br 3d spectra of PNCs with and without the monomer, and we observed a doublet located at ∼68/69 eV, see Fig. 3B. These peaks, attributed to the Br 3d5/2 and Br 3d3/2 core levels, are representative of the formation of Pb–Br bonds coming from the [PbBr6] octahedra contained into the PNCs.85Fig. 3C shows the HR-XPS Pb 4f spectra of the samples, observing the characteristic Pb 4f7/2 and Pb 4f5/2 core levels ∼138/143 eV, respectively.86 These signals correspond to the presence of Pb2+ contained into the PNC lattice. Last, Fig. 3D displays the HR-XPS N 1s spectra of the pristine and modified PNCs with monomer, noting some differences in the chemical contributions from the main N 1s signal. In the case of pristine PNCs, two signals are evidenced ∼400 and 402 eV, ascribed to C-NH2 and C-NH3+ species, respectively. These species are attributed to the attached oleylammonium cations covering the PNCs surface.87 Interestingly, after the addition of the polymeric formulation, the abovementioned signals were displaced to 399 and 403 eV, respectively. We attributed these peaks to the emergence of C–N and S–N coming from the AcrEMA cation and TFSI anion, respectively. The shift of the characteristic bands from the Pb indicates that PNCs are covered and stabilised by the monomer.
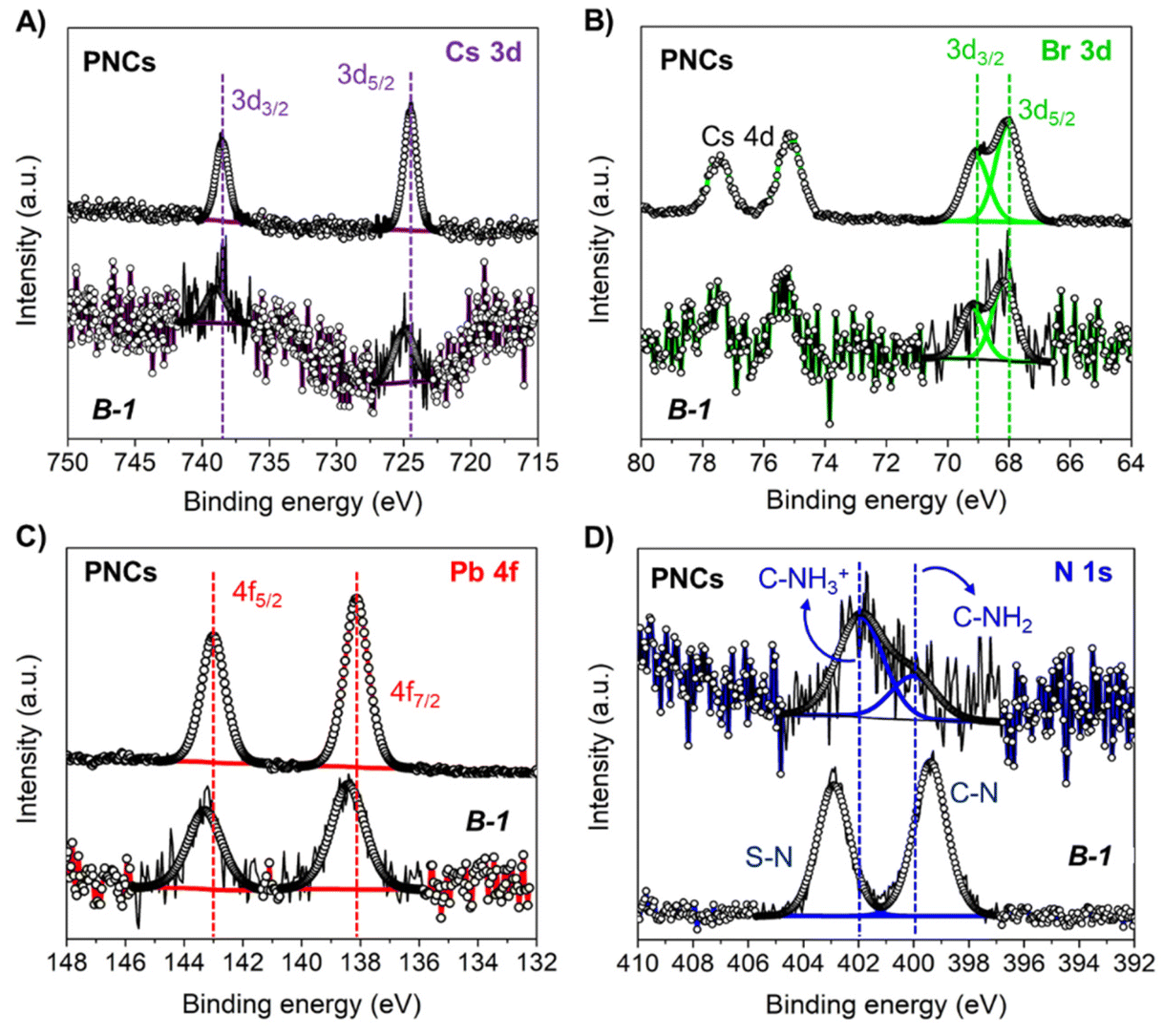 | ||
| Fig. 3 HR-XPS (A) Cs 3d; (B) Br 3d; (C) Pb 4f; and (D) N 1s spectra for pristine CsPbBr3 PNCs and B-1 samples. | ||
The stoichiometry of the pristine PNCs, see Table S3,† are far from the typical Cs:Pb:Br molar ratio = 1:1:3, suggesting that the material shows a high density of surface defects. This is probably due to the PNC synthesis and purification methodologies.88 However, after encapsulating the PNCs with the PIL formulation, the Cs+ deficiency is compensated, while the Br content is lower. Furthermore, it was observed that the stability of the PNCs is extended in the presence of the PILs, see Fig. 2. We attributed this improvement to the incorporation of fluorine and sulfur species coming from the monomeric ionic liquid, replacing/filling halide sites in the PNCs. We identified the presence of C–F (689 eV), and SOx bonds (doublet at 169/170 eV), respectively, from the bis(trifluoromethanesulfonyl)imide ion (TFSI) through HR-XPS F 1s and S 2p spectra obtained from the PNCs-PIL combination, see Fig. S15B and 15C.†89 In this context, this species could mediate the PNCs surface complexation through the sulfonyl/fluoride-metal surface interaction. This hypothesis is reinforced by the shift of BEs of the Cs 3d, Br 3d and Pb 4f doublets to higher values. This suggests the introduction of a more electronegative element into the perovskite structure, like F or O atoms from TFSI. In this way, we can conclude that PILs can favor the surface defect passivation of PNCs, which is a key factor to promote a long-term stability and good optical properties such as PLQY, into the PIL matrix under ambient conditions and high relative humidity.
The resultant B-1 film was tested for the photodegradation of organic dyes. In this experiment PILs might act as good stabilizers and also as adsorbent materials. As a proof of concept, the bifunctional smart films were employed to separate and photodegrade methyl red (MR) as a benchmark dye. Control experiments in aqueous solution showed no activity. Therefore, the experiment consisted in introducing the optimized B-1 film into a 30 μM solution of MR in an organic solvent like hexane, illuminating the sample with 1 sun of visible light, using an UV filter. MR dye solution showed two absorption bands with a λmax at 450 nm, responsible for the yellowish colour. According to the literature, the visible absorption bands are attributed to the n → π* transition associated to the chromophoric –NN– group found in azo dyes, while the UV bands are attributed to the benzene rings, see Fig. 4A.90,91 During the photodegradation, both bands decreased, indicating that the decomposition of MR involved the cleavage of benzene rings and the chromophore groups. After 22 h of reaction, the decolorization is nearly completed when B-1 is into the solution, see Fig. 4A. The control experiment (blank) was performed under same conditions without introducing the composite film, see Fig. S16A.† The effect of the film B without PNCs was also evaluated, see Fig. S16B.†Fig. 4D shows that the kinetics of the three samples fits well to a pseudo-first order model. The rate constant related to the B-1 experiment is the highest one (Ks = 0.06 h−1) compared to B (Ks = 0.02 h−1) and to the blank (Ks = 0.01 h−1). The irradiating light has also an effect on the MR degradation, as can be seen in the blank sample, although it is almost negligible compared to the photodegradation promoted by the PNCs. Differences between the MR concentration using B and the blank can be attributed to the adsorption of MR onto the surface of the film. The decolorization of MR is notable after comparing the initial colour, dark yellow (Fig. 4B-1) and the final colour, almost transparent, after 22 h of reaction by using the film B-1 (Fig. 4B-2). In contrast, when B was employed, the solution remained yellowish (Fig. 4B-3). The colour of the films also changed after the experiments. B-1 depicts a dark yellow colour both at the beginning (Fig. 4E-1) and after 22 h of MR degradation (Fig. 4E-2). The film B, without PNCs, ended reddish due to the adsorption of MR in the film, see Fig. 4E-3. This fact suggests that the photodegradation in case of B-1 was completed and no MR was left either in solution or adsorbed to the surface. The total degradation of MR was also confirmed by ESI-MS, where the peak related to MR completely disappeared, see Fig. S17.† Nevertheless, the polymer matrix of B and B-1 was not degraded during the photocatalysis reaction, as it can be observed from the FT-IR spectra shown in Fig. S18.†
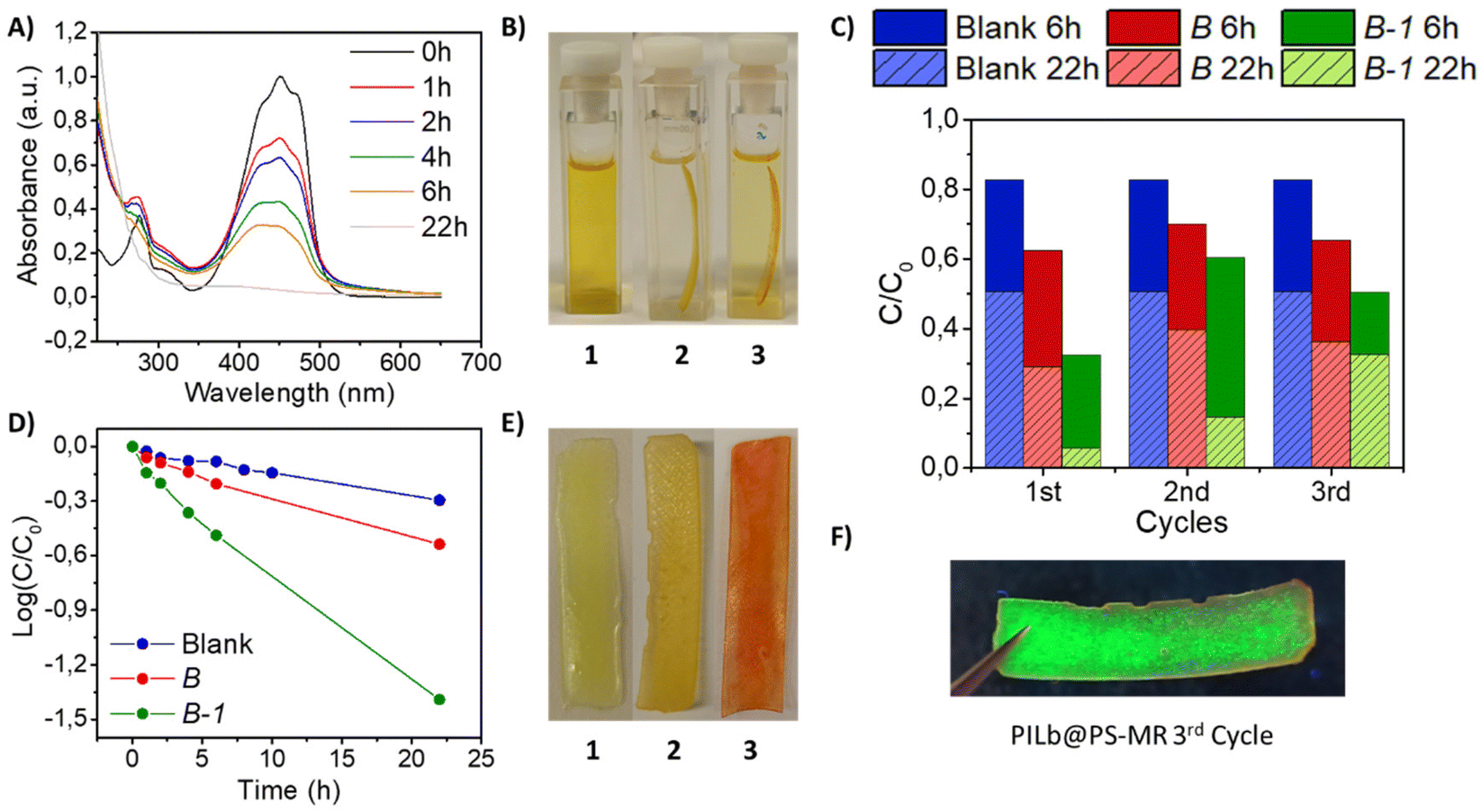 | ||
| Fig. 4 (A) The absorption spectra of MR during the photocatalytic degradation using B-1 film for 22 h under visible light irradiation; (B) (1) initial colour of MR solution; (2) MR solution colour after 22 h with a B-1 film; (3) MR solution colour after 22 h with a B film; (C) normalised concentration of dye remaining in solution after 6 h and 22 h for the blank, B and B-1 films as photocatalysts for 3 cycles; (D) the logarithm of the ratio between the concentration after photocatalytic degradation and the initial concentration of the dye showing the kinetics of the blank, using a B film and a B-1 film; (E) (1) initial colour of B-1 film; (2) B-1 film after 22 h into MR solution; (3) B film after 22 h into MR solution; (F) film B-1 after the third cycle, under UV light. | ||
The films were recycled without any further treatment for three times with new MR solution, see Fig. 4C. Clearly, the experiment with B-1 was the most effective system over the three cycles assayed. Here, the number of cycles is defined as number of photocatalytic reactions conducted by the same composite. After 22 h, in the third cycle, the degradation is far from completing so B-1 film presents a reddish appearance (Fig. S19,† left), similar to B film (Fig. S19,† right). But there are still PNCs remaining in the interior of the film after the third cycle as shows Fig. 4F, and more details in Fig. S20.† Therefore, embedding the perovskite into a polymeric matrix allows to recycle it for several reactions, different from the material without encapsulation, which is deactivated after the first cycle, see Fig. S21.†
Similarly to the well-known semiconductor based-photocatalysts, PNCs can absorb visible-light photons to promote the generation of electron–hole pairs, which are the key factor to trigger oxidation–reduction reactions, respectively.36,92 In order to determine whether the photodegradation in our system took place by the electrons or the holes, a hole scavenger (MeOH, 1 mM) was introduced. In presence of this alcohol, the kinetics of MR photodegradation slowed down, becoming very similar to that of the film without PNCs (Fig. S22†). This suggests that the film B-1 with a hole scavenger was acting only as an adsorbent material with lower photocatalytic activity. Therefore, the experiment confirms that the photoexcited holes generated in CsPbBr3 PNCs upon illumination are responsible of the decomposition of the dye. We can suggest that the photogenerated holes in the PNCs can migrate to the surface film to be in contact with MR and carry out the oxidation of the dye. Here, the luminescent composite shows a decrease of PLQY from 40% to 15%. However, the film is still photoluminescent, so the photoexcited holes come from the PNCs remaining in the core of the film. This explains why the efficiency of the system is reduced and needs longer times for degrading MR, in the posterior cycles, see Fig. 4C. In conclusion, the advanced material B-1 is acting as a bifunctional advanced film capable to adsorb and, simultaneously, to photodegrade organic dyes.
Conclusions
In this work, we have embedded perovskite nanocrystals in polymeric ionic liquid matrices exploiting their well-established ability to stabilize molecular and nanostructured materials. A variety of PNCs were evaluated and effectively encapsulated. The ionic and supramolecular interactions of PILs with the PNCs ions enable the stabilization the nanocrystals before and after the polymerization. We have optimized a 3D printable formulation that stabilizes PS1 under different conditions (air, light, oxygen, humidity, temperature and water) observing a significant increase in stability compared to the PNCs in solution. Furthermore, it has been demonstrated the possibility to embed PNCs into PILs to form solid photocatalysts capable to adsorb and degrade organic dyes. We have successfully used the B-1 films to increase the rate of decomposition of methyl red under visible light irradiation. The film was recycled for three cycles, with higher activity than the control experiment. The mechanism of photodegradation proposed through photoexcited holes, since a hole scavenger reduced the photodegradation to the level of the control experiment. Although future works should be focused to optimize the reaction conditions for improving the chemical stability of the composites (for instance, facilitate the electron–hole separation), these 3D-printed materials are good candidates to conduct efficient photocatalytic reactions. These findings represent a novel and promising future path in the implementation of solid photoactive perovskite nanocrystal materials based on 3D printing PILs.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by Generalitat Valenciana via Prometeo Grant Q-Solutions (CIPROM/2021/078). MZ and VS acknowledges the European Union's Horizon 2020 under the Marie Skłodowska-Curie Individual Fellowships (GA no. 101026335). VS thanks Generalitat Valenciana (CIDEGENT 2018/036) for funding. SM thanks Generalitat Valenciana (ACIF/2020/338) for a PhD Scholarship. The authors also thank to Ministry of Education, Youth and Sports of the Czech Republic for the financial support of XPS measurements using CEMNAT infrastructure (project LM 2018103).References
- F. Lu and D. Astruc, Coord. Chem. Rev., 2020, 408, 213180 CrossRef CAS.
- C. A. Martínez-Huitle and E. Brillas, Appl. Catal., B, 2009, 87, 105–145 CrossRef.
- U. I. Gaya and A. H. Abdullah, J. Photochem. Photobiol., C, 2008, 9, 1–12 CrossRef CAS.
- I. K. Konstantinou and T. A. Albanis, Appl. Catal., B, 2004, 49, 1–14 CrossRef CAS.
- M. Dang, Q.-L. Deng, G.-Z. Fang, D.-D. Zhang, H.-J. Li, J.-M. Liu and S. Wang, J. Colloid Interface Sci., 2018, 530, 302–311 CrossRef CAS.
- M. Y. Abdelaal and R. M. Mohamed, J. Alloys Compd., 2013, 576, 201–207 CrossRef CAS.
- X. Pu, D. Zhang, Y. Gao, X. Shao, G. Ding, S. Li and S. Zhao, J. Alloys Compd., 2013, 551, 382–388 CrossRef CAS.
- F.-t. Li, Y. Liu, R.-h. Liu, Z.-m. Sun, D.-s. Zhao and C.-g. Kou, Mater. Lett., 2010, 64, 223–225 CrossRef CAS.
- X. Niu, H. Li and G. Liu, J. Mol. Catal. A: Chem., 2005, 232, 89–93 CrossRef CAS.
- J. Xiao, X. Liu, L. Pan, C. Shi, X. Zhang and J.-J. Zou, ACS Catal., 2020, 10, 12256–12283 CrossRef CAS.
- S. Subudhi, S. P. Tripathy and K. Parida, Catal. Sci. Technol., 2021, 11, 392–415 RSC.
- M. Grübel, I. Bosque, P. J. Altmann, T. Bach and C. R. Hess, Chem. Sci., 2018, 9, 3313–3317 RSC.
- L. Protesescu, S. Yakunin, M. I. Bodnarchuk, F. Krieg, R. Caputo, C. H. Hendon, R. X. Yang, A. Walsh and M. V. Kovalenko, Nano Lett., 2015, 15, 3692–3696 CrossRef CAS PubMed.
- G. Zhang, G. Liu, L. Wang and J. T. S. Irvine, Chem. Soc. Rev., 2016, 45, 5951–5984 RSC.
- W. Wang, M. O. Tadé and Z. Shao, Chem. Soc. Rev., 2015, 44, 5371–5408 RSC.
- M. V. Kovalenko, L. Protesescu and M. I. Bodnarchuk, Science, 2017, 358, 745–750 CrossRef CAS PubMed.
- Q. A. Akkerman, G. Rainò, M. V. Kovalenko and L. Manna, Nat. Mater., 2018, 17, 394–405 CrossRef CAS PubMed.
- A. Dey, J. Ye, A. De, E. Debroye, S. K. Ha, E. Bladt, A. S. Kshirsagar, Z. Wang, J. Yin, Y. Wang, L. N. Quan, F. Yan, M. Gao, X. Li, J. Shamsi, T. Debnath, M. Cao, M. A. Scheel, S. Kumar, J. A. Steele, M. Gerhard, L. Chouhan, K. Xu, X. G. Wu, Y. Li, Y. Zhang, A. Dutta, C. Han, I. Vincon, A. L. Rogach, A. Nag, A. Samanta, B. A. Korgel, C. J. Shih, D. R. Gamelin, D. H. Son, H. Zeng, H. Zhong, H. Sun, H. V. Demir, I. G. Scheblykin, I. Mora-Seró, J. K. Stolarczyk, J. Z. Zhang, J. Feldmann, J. Hofkens, J. M. Luther, J. Pérez-Prieto, L. Li, L. Manna, M. I. Bodnarchuk, M. V. Kovalenko, M. B. J. Roeffaers, N. Pradhan, O. F. Mohammed, O. M. Bakr, P. Yang, P. Müller-Buschbaum, P. V. Kamat, Q. Bao, Q. Zhang, R. Krahne, R. E. Galian, S. D. Stranks, S. Bals, V. Biju, W. A. Tisdale, Y. Yan, R. L. Z. Hoye and L. Polavarapu, ACS Nano, 2021, 15, 10775–10981 CrossRef CAS PubMed.
- Y. Zhou, F. Lu, T. Fang, D. Gu, X. Feng, T. Song and W. Liu, J. Alloys Compd., 2022, 911, 165062 CrossRef CAS.
- H. Huang, M. I. Bodnarchuk, S. V. Kershaw, M. V. Kovalenko and A. L. Rogach, ACS Energy Lett., 2017, 2, 2071–2083 CrossRef CAS PubMed.
- G. Rainò, N. Yazdani, S. C. Boehme, M. Kober-Czerny, C. Zhu, F. Krieg, M. D. Rossell, R. Erni, V. Wood, I. Infante and M. V. Kovalenko, Nat. Commun., 2022, 13, 2587 CrossRef PubMed.
- X. Zhang, H. Lin, H. Huang, C. Reckmeier, Y. Zhang, W. C. H. Choy and A. L. Rogach, Nano Lett., 2016, 16, 1415–1420 CrossRef CAS.
- J. Wang, H. Li, P. Gao, Y. Peng, S. Cao and M. Antonietti, Chem. Eng. J., 2022, 443, 136447 CrossRef CAS.
- D. Laishram, S. Zeng, K. M. Alam, A. P. Kalra, K. Cui, P. Kumar, R. K. Sharma and K. Shankar, Appl. Surf. Sci., 2022, 592, 153276 CrossRef CAS.
- C. Cai, Y. Teng, J.-H. Wu, J.-Y. Li, H.-Y. Chen, J.-H. Chen and D.-B. Kuang, Adv. Funct. Mater., 2020, 30, 2001478 CrossRef CAS.
- A. Shi, K. Sun, X. Chen, L. Qu, Y. Zhao and B. Yu, Org. Lett., 2022, 24, 299–303 CrossRef CAS.
- T. Shi, K. Sun, X.-L. Chen, Z.-X. Zhang, X.-Q. Huang, Y.-Y. Peng, L.-B. Qu and B. Yu, Adv. Synth. Catal., 2020, 362, 2143–2149 CrossRef.
- Q. Fan, L. Zhu, X. Li, H. Ren, H. Zhu, G. Wu and J. Ding, New J. Chem., 2021, 45, 13317–13322 RSC.
- X. Zhu, Y. Lin, Y. Sun, M. C. Beard and Y. Yan, J. Am. Chem. Soc., 2019, 141, 733–738 CrossRef CAS PubMed.
- M. Corti, R. Chiara, L. Romani, B. Mannucci, L. Malavasi and P. Quadrelli, Photochem. Photobiol. Sci., 2022, 21, 613–624 CrossRef CAS.
- M. Liu, P. Xia, G. Zhao, C. Nie, K. Gao, S. He, L. Wang and K. Wu, Angew. Chem., Int. Ed., 2022, 61, e202208241 CAS.
- Y. Zhang, M. Xia, M. Li, Q. Ping, Z. Yuan, X. Liu, H. Yin, S. Huang and Y. Rao, J. Org. Chem., 2021, 86, 15284–15297 CrossRef CAS PubMed.
- M. Karami, M. Ghanbari, O. Amiri and M. Salavati-Niasari, Sep. Purif. Technol., 2020, 253, 117526 CrossRef CAS.
- X. Feng, H. Ju, T. Song, T. Fang, W. Liu and W. Huang, ACS Sustainable Chem. Eng., 2019, 7, 5152–5156 CrossRef CAS.
- X. Qian, Z. Chen, X. Yang, W. Zhao, C. Liu, T. Sun, D. Zhou, Q. Yang, G. Wei and M. Fan, J. Cleaner Prod., 2020, 249, 119335 CrossRef CAS.
- D. Cardenas-Morcoso, A. F. Gualdrón-Reyes, A. B. Ferreira Vitoreti, M. García-Tecedor, S. J. Yoon, M. S. de la Fuente, I. Mora-Seró and S. Gimenez, J. Phys. Chem. Lett., 2019, 10, 630–636 CrossRef CAS PubMed.
- H. Cho, Y.-H. Kim, C. Wolf, H.-D. Lee and T.-W. Lee, Adv. Mater., 2018, 30, 1704587 CrossRef.
- S. Huang, Z. Li, B. Wang, N. Zhu, C. Zhang, L. Kong, Q. Zhang, A. Shan and L. Li, ACS Appl. Mater. Interfaces, 2017, 9, 7249–7258 CrossRef CAS.
- J. De Roo, M. Ibáñez, P. Geiregat, G. Nedelcu, W. Walravens, J. Maes, J. C. Martins, I. Van Driessche, M. V. Kovalenko and Z. Hens, ACS Nano, 2016, 10, 2071–2081 CrossRef CAS PubMed.
- P. Li, Y. Lu, Y. Duan, S. Xu and J. Zhang, J. Phys. Chem. C, 2021, 125, 2382–2392 CrossRef CAS.
- S. Li, Y. Pan, W. Wang and Y. Li, Chem. Eng. J., 2022, 434, 134593 CrossRef CAS.
- T. Song, X. Feng, H. Ju, T. Fang, F. Zhu, W. Liu and W. Huang, J. Alloys Compd., 2020, 816, 152558 CrossRef CAS.
- Z.-J. Li, E. Hofman, J. Li, A. H. Davis, C.-H. Tung, L.-Z. Wu and W. Zheng, Adv. Funct. Mater., 2018, 28, 1704288 CrossRef.
- W. Chen, H. Liu, R. Fan, P. Wang, T. Sun and Y. Yang, ACS Appl. Mater. Interfaces, 2020, 12, 9851–9857 CrossRef CAS PubMed.
- N. Kinjo, Speciality Polymers-Polymer Physics, Akademie Verlag, 1989 Search PubMed.
- S. N. Raja, Y. Bekenstein, M. A. Koc, S. Fischer, D. Zhang, L. Lin, R. O. Ritchie, P. Yang and A. P. Alivisatos, ACS Appl. Mater. Interfaces, 2016, 8, 35523–35533 CrossRef CAS PubMed.
- G. Raino, A. Landuyt, F. Krieg, C. Bernasconi, S. T. Ochsenbein, D. N. Dirin, M. I. Bodnarchuk and M. V. Kovalenko, Nano Lett., 2019, 19, 3648–3653 CrossRef CAS.
- Y. Wang, J. He, H. Chen, J. Chen, R. Zhu, P. Ma, A. Towers, Y. Lin, A. J. Gesquiere, S. T. Wu and Y. Dong, Adv. Mater., 2016, 28, 10710–10717 CrossRef CAS.
- X. Yang, T. Xu, Y. Zhu, J. Cai, K. Gu, J. Zhu, Y. Wang, J. Shen and C. Li, J. Mater. Chem. C, 2018, 6, 7971–7975 RSC.
- C. Zhang, Z. He, H. Chen, L. Zhou, G. Tan, S.-T. Wu and Y. Dong, J. Mater. Chem. C, 2019, 7, 6527–6533 RSC.
- L. Wang, Y. Zhu, H. Liu, J. Gong, W. Wang, S. Guo, Y. Yu, H. Peng and Y. Liao, ACS Appl. Mater. Interfaces, 2019, 11, 35270–35276 CrossRef CAS PubMed.
- Y. Cai, Y. Li, L. Wang and R. J. Xie, Adv. Opt. Mater., 2019, 7, 1901075 CrossRef CAS.
- A. Pan, J. Wang, M. J. Jurow, M. Jia, Y. Liu, Y. Wu, Y. Zhang, L. He and Y. Liu, Chem. Mater., 2018, 30, 2771–2780 CrossRef CAS.
- Z. Zhang, L. Li, L. Liu, X. Xiao, H. Huang and J. Xu, J. Phys. Chem. C, 2020, 124, 22228–22234 CrossRef CAS.
- L. Liu, Y. Meng, X. Dai, K. Chen and Y. Zhu, Food Bioprocess Technol., 2019, 12, 267–279 CrossRef CAS.
- D. Yuan, L. Chen, X. Xiong, L. Yuan, S. Liao and Y. Wang, Chem. Eng. J., 2016, 285, 358–367 CrossRef CAS.
- N. Nishimura and H. Ohno, Polymer, 2014, 55, 3289–3297 CrossRef CAS.
- J. Lu, F. Yan and J. Texter, Prog. Polym. Sci., 2009, 34, 431–448 CrossRef CAS.
- W. Qian, J. Texter and F. Yan, Chem. Soc. Rev., 2017, 46, 1124–1159 RSC.
- S. Chen, F. Frenzel, B. Cui, F. Gao, A. Campanella, A. Funtan, F. Kremer, S. S. P. Parkin and W. H. Binder, J. Mater. Chem. C, 2018, 6, 8242–8250 RSC.
- C. Geffroy, E. Grana, T. Bessho, S. Almosni, Z. Tang, A. Sharma, T. Kinoshita, F. Awai, E. Cloutet, T. Toupance, H. Segawa and G. Hadziioannou, ACS Appl. Energy Mater., 2020, 3, 1393–1401 CrossRef CAS.
- D. Yang, X. Zhou, R. Yang, Z. Yang, W. Yu, X. Wang, C. Li, S. Liu and R. P. H. Chang, Energy Environ. Sci., 2016, 9, 3071–3078 RSC.
- X. Zhou, M. Hu, C. Liu, L. Zhang, X. Zhong, X. Li, Y. Tian, C. Cheng and B. Xu, Nano Energy, 2019, 63, 103866 CrossRef CAS.
- J. Tang, H. Tang, W. Sun, M. Radosz and Y. Shen, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 5477–5489 CrossRef CAS.
- M. Rebber, C. Willa and D. Koziej, Nanoscale Horiz., 2020, 5, 431–453 RSC.
- S. Karak, C. Nanjo, M. Odaka, K. Yuyama, G. Masuda, M. M. Matsushita and K. Awaga, J. Mater. Chem. A, 2016, 4, 9019–9028 RSC.
- R. Xia, X. X. Gao, Y. Zhang, N. Drigo, V. I. E. Queloz, F. F. Tirani, R. Scopelliti, Z. Huang, X. Fang, S. Kinge, Z. Fei, C. Roldan-Carmona, M. K. Nazeeruddin and P. J. Dyson, Adv. Mater., 2020, 32, e2003801 CrossRef PubMed.
- D. J. Wales, Q. Cao, K. Kastner, E. Karjalainen, G. N. Newton and V. Sans, Adv. Mater., 2018, 30, 1800159 CrossRef PubMed.
- T. D. Ngo, A. Kashani, G. Imbalzano, K. T. Q. Nguyen and D. Hui, Composites, Part B, 2018, 143, 172–196 CrossRef CAS.
- M. O. F. Emon, F. Alkadi, D. G. Philip, D.-H. Kim, K.-C. Lee and J.-W. Choi, Addit. Manuf., 2019, 28, 629–638 CAS.
- S. Sevilia, M. Yong, D. Grinstein, L. Gottlieb and Y. Eichen, Macromol. Mater. Eng., 2019, 304, 1900018 CrossRef.
- J. Wang, X. Gu, H. Ma, Q. Peng, X. Huang, X. Zheng, S. H. P. Sung, G. Shan, J. W. Y. Lam, Z. Shuai and B. Z. Tang, Nat. Commun., 2018, 9, 2963 CrossRef PubMed.
- C. D. Brubaker, T. M. Frecker, J. R. McBride, K. R. Reid, G. K. Jennings, S. J. Rosenthal and D. E. Adams, J. Mater. Chem. C, 2018, 6, 7584–7593 RSC.
- I. Recalde, A. F. Gualdrón-Reyes, C. Echeverría-Arrondo, A. Villanueva-Antolí, J. Simancas, J. Rodriguez-Pereira, M. Zanatta, I. Mora-Seró and V. Sans, Adv. Funct. Mater., 2022, 2210802 CrossRef.
- C.-L. Tai, W.-L. Hong, Y.-T. Kuo, C.-Y. Chang, M.-C. Niu, M. Karupathevar Ponnusamythevar Ochathevar, C.-L. Hsu, S.-F. Horng and Y.-C. Chao, ACS Appl. Mater. Interfaces, 2019, 11, 30176–30184 CrossRef CAS.
- G. C. Adhikari, S. Thapa, H. Zhu and P. Zhu, Adv. Opt. Mater., 2019, 7, 1900916 CrossRef CAS.
- F. Zhao, X. Luo, C. Gu, J. Chen, Z. Hu and Y. Peng, Adv. Mater. Technol., 2022, 2200521 CrossRef CAS.
- L. A. Connal, R. Vestberg, P. A. Gurr, C. J. Hawker and G. G. Qiao, Langmuir, 2008, 24, 556–562 CrossRef CAS PubMed.
- R. J. Parod, in Encyclopedia of Toxicology, ed. P. Wexler, Academic Press, Oxford, 3rd edn, 2014, pp. 578–580, DOI:10.1016/B978-0-12-386454-3.01108-8.
- Y. Wu, R. Cao, G. Wu, W. Huang, Z. Chen, X. Yang and Y. Tu, Composites, Part A, 2016, 88, 156–164 CrossRef CAS.
- T. Song, J. Deng, L. Deng, L. Bai, X. Zhang, S. Zhang, P. Szabo and A. E. Daugaard, Polymer, 2019, 160, 223–230 CrossRef CAS.
- Y. M. Kim, W. Y. Lee, W. Y. Choi and H. C. Moon, J. Ind. Eng. Chem., 2020, 90, 341–350 CrossRef CAS.
- O. Green, S. Grubjesic, S. Lee and M. A. Firestone, Polym. Rev., 2009, 49, 339–360 CrossRef CAS.
- M. Deepa, M. Salado, L. Calio, S. Kazim, S. M. Shivaprasad and S. Ahmad, Phys. Chem. Chem. Phys., 2017, 19, 4069–4077 RSC.
- A. F. Gualdrón-Reyes, J. Rodríguez-Pereira, E. Amado-González, J. Rueda-P, R. Ospina, S. Masi, S. J. Yoon, J. Tirado, F. Jaramillo, S. Agouram, V. Muñoz-Sanjosé, S. Giménez and I. Mora-Seró, ACS Appl. Mater. Interfaces, 2020, 12, 914–924 CrossRef PubMed.
- C. Lee, Y. Shin, A. Villanueva-Antolí, S. Das Adhikari, J. Rodriguez-Pereira, J. M. Macak, C. A. Mesa, S. Giménez, S. J. Yoon, A. F. Gualdrón-Reyes and I. Mora-Seró, Chem. Mater., 2021, 33, 8745–8757 CrossRef CAS.
- L. Qiu, H. Yang, Z. Dai, F. Sun, J. Hao, M. Guan, P. Dang, C. Yan, J. Lin and G. Li, Inorg. Chem. Front., 2020, 7, 2060–2071 RSC.
- Y. Zhang, T. D. Siegler, C. J. Thomas, M. K. Abney, T. Shah, A. De Gorostiza, R. M. Greene and B. A. Korgel, Chem. Mater., 2020, 32, 5410–5423 CrossRef CAS.
- F. Wu, G.-T. Kim, T. Diemant, M. Kuenzel, A. R. Schür, X. Gao, B. Qin, D. Alwast, Z. Jusys, R. J. Behm, D. Geiger, U. Kaiser and S. Passerini, Adv. Energy Mater., 2020, 10, 2001830 CrossRef CAS.
- M. R. Plutino, E. Guido, C. Colleoni and G. Rosace, Sens. Actuators, B, 2017, 238, 281–291 CrossRef CAS.
- M. Zhou, Q. Yu, L. Lei and G. Barton, Sep. Purif. Technol., 2007, 57, 380–387 CrossRef CAS.
- S. M. Gupta and M. Tripathi, High Energy Chem., 2012, 46, 1–9 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2nr07254h |
| This journal is © The Royal Society of Chemistry 2023 |