
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBilayer lipid membrane formation on surface assemblies with sparsely distributed tethers†
Martynas
Gavutis‡
 a,
Eric
Schulze-Niemand‡
bc,
Hung-Hsun
Lee§
d,
Bo
Liedberg
d,
Matthias
Stein
*b and
Ramūnas
Valiokas
*a
a,
Eric
Schulze-Niemand‡
bc,
Hung-Hsun
Lee§
d,
Bo
Liedberg
d,
Matthias
Stein
*b and
Ramūnas
Valiokas
*a
aDepartment of Nanoengineering, Center for Physical Sciences and Technology, Savanorių 231, 02300 Vilnius, Lithuania. E-mail: valiokas@ftmc.lt
bMolecular Simulations and Design Group, Max Planck Institute for Dynamics of Complex Technical System, Magdeburg, Germany
cInstitute of Experimental Internal Medicine, Medical Faculty, Otto von Guericke University, 39120 Magdeburg, Germany
dDivision of Molecular Physics, Department of Physics, Chemistry and Biology, Linköping University, 58183 Linköping, Sweden
First published on 2nd May 2023
Abstract
A combined computational and experimental study of small unilamellar vesicle (SUV) fusion on mixed self-assembled monolayers (SAMs) terminated with different deuterated tether moieties (–(CD2)7CD3 or –(CD2)15CD3) is reported. Tethered bilayer lipid membrane (tBLM) formation of synthetic 1-stearoyl-2-oleoyl-sn-glycero-3-phosphocholine was initially probed on SAMs with controlled tether (d-alkyl tail) surface densities and lateral molecular packing using quartz crystal microbalance with dissipation monitoring (QCM-D). Long time-scale coarse-grained molecular dynamics (MD) simulations were then employed to elucidate the mechanisms behind the interaction between the SUVs and the different phases formed by the –(CD2)7CD3 and –(CD2)15CD3 tethers. Furthermore, a series of real time kinetics was recorded under different osmotic conditions using QCM-D to determine the accumulated lipid mass and for probing the fusion process. It is shown that the key factors driving the SUV fusion and tBLM formation on this type of surfaces involve tether insertion into the SUVs along with vesicle deformation. It is also evident that surface densities of the tethers as small as a few mol% are sufficient to obtain stable tBLMs with a high reproducibility. The described “sparsely tethered” tBLM system can be advantageous in studying different biophysical phenomena, such as membrane protein insertion, effects of receptor clustering, and raft formation.
1. Introduction
Surface-supported synthetic lipid membranes are convenient and reliable model systems for biophysical studies of the complex processes occurring on the surface and across native cell membranes.1–3 Also, this platform has become the core of powerful technologies, for example, biosensing,4,5 real-time bioanalysis,6 protein arrays,5,7 drug screening8 and design.9Despite the broad variety of the supported membrane architectures reported in the literature, the majority of them can be arbitrarily classified into the three main groups: floating, hybrid or tethered bilayer lipid membranes (tBLMs).10,11 The latter system offers an important advantage over the first two: the substrate-bound tethers physically integrate and keep the lipid membrane in a close proximity to the supporting surface yet providing an aqueous interfacial layer between the substrate and the bottom lipid leaflet. Therefore, tBLMs display an improved stability in comparison to the floating (non-tethered) membranes.12,13 This strategy also may facilitate incorporation of membrane proteins into the tBLM.1,14 Previous studies reported on proteins,15–17 oligonucleotides,18 chemically modified lipids13,19,20 and synthetic molecules21 as efficient tethers.
The mechanism of lipid bilayer formation on solid substrates by means of lipid vesicle fusion is well understood both from the experimental11,22,23 and theoretical24,25 points of view. The general picture is that the vesicles adsorb on the surface and stay intact until a critical vesicle density is reached. Subsequently, they become destabilized by the increasing surface–vesicle and inter–vesicle interactions and finally rupture to re-assemble into a continuous (“floating”) bilayer. In such a process, the kinetics is dependent on the chemical structure of the lipids as well as on the net surface charge and ionic composition in the aqueous solution.
In the case of the tBLMs, the structure of the tethering assemblies becomes the major factor of the vesicle fusion process.12 Despite of that, the literature in general is lacking systematic investigations on the detailed mechanisms of structure-guided tBLM formation. One possible reason is insufficient characterization of the surface density of the tethers, their orientation, conformation, nature of defects and phase behavior. Second, the insights obtained from one tethering system are not always directly transferable to other types of molecular tethers due to significant differences in their chemical and physical structures. Only a few previous studies26–32 related the tether length, phase formation and distribution to vesicle fusion kinetics and the equilibrium properties of the formed tBLM. They suggest that higher surface densities of the tethers (in the range of 20–30 mol%) are needed to ensure the overall lipid bilayer stability.
Previously, we introduced a series of mixed self-assembled monolayers (SAMs) on gold that are suitable for tBLM formation.33 Such model assemblies consisted of an ethylene glycol-terminated matrix compound mixed with similar compounds providing protruding deuterated alkyl (d-alkyl) chain tethers. Our detailed studies have shown that this type of SAM architecture is highly ordered, and it is stabilized by lateral hydrogen bonding.34,35 Also, deuteration provided the possibility to discriminate d-alkyl tethers from the matrix monolayer spectroscopically. We have investigated in detail the structure of the designed tethering systems by combining experimental and modeling techniques.33,36 We observed that, depending on the chain length of the aliphatic tether (8 or 16 methyl units), they are either homogenously distributed (randomly mixed) on the gold substrate or they phase-separate into clusters of at least 40 molecules. Therefore, such SAMs allow the control of several parameters that potentially may influence the tBLM formation, e.g. the surface densities of the tethers, the length of the d-alkyl chains tethering the lipid bilayer and their conformation and phase.
The aim of the present study is to elucidate the interactions between lipid vesicles and the linear tethers in the process of tBLM formation. For this purpose, we chose a combination of two complementary techniques: experimental quartz crystal microbalance with dissipation monitoring (QCM-D)37 and computational molecular dynamics (MD)38,39 simulations. The first technique reveals the events related to the mass of the adsorbed material and probes the lipid phases on the substrate. While QCM-D allows monitoring real time events at a scale of seconds and minutes, the molecular level information about the immediate tether–vesicle interactions at shorter timescales can only be obtained from MD simulations. When combined, the developed experimental model surface and the two characterization techniques elucidate the major difference of tBLM formation by small unilamellar vesicle (SUV) fusion compared to floating (tetherless) membranes.
2. Methods
2.1 Chemicals and substrates
All ω-substituted deuterated alkanethiol compounds (Scheme 1) were synthesized in our laboratory.40 All analytical grade solvents were obtained from commercial suppliers and were used without further purification. The 1-stearoyl-2-oleoyl-sn-glycero-3-phosphocholine (SOPC) was purchased from Avanti Polar Lipids. Commercially available quartz sensors with a 100 nm-thick gold layer were employed for SUV fusion real-time analysis (QSense, Biolin Scientific). | ||
| Scheme 1 Chemical structures of the compounds used for formation of supporting self-assembled monolayers on gold and for small unilamellar vesicles, respectively: a matrix compound (EG1H), two tether compounds (EG6AC8D and EG6AC16D) and the synthetic SOPC lipid. | ||
2.2 Surface modifications
The details on the SAM formation and characterization have been described elsewhere.33 Briefly, the gold-coated QCM-D sensors were cleaned in the SC-1 solution (5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1 mixture of deionized water (Milli-Q), 25% hydrogen peroxide, and 30% ammonia, for 5 min at 80 °C, followed by rinsing with deionized water). The thiol compounds were dissolved in ethanol (99.5%) and mixed at several molar ratios (see the Results section) to the final 20 μM concentration. After 24 h incubation, the SAM-coated substrates were rinsed in deionized water, washed in an ultrasonic bath in ethanol for 3–5 min and blown dry with nitrogen gas.
:1:1 mixture of deionized water (Milli-Q), 25% hydrogen peroxide, and 30% ammonia, for 5 min at 80 °C, followed by rinsing with deionized water). The thiol compounds were dissolved in ethanol (99.5%) and mixed at several molar ratios (see the Results section) to the final 20 μM concentration. After 24 h incubation, the SAM-coated substrates were rinsed in deionized water, washed in an ultrasonic bath in ethanol for 3–5 min and blown dry with nitrogen gas.
2.3 Lipid adsorption
Pre-adsorption of the synthetic SOPC compounds on the SAM surface was made by dissolving the SOPC powder in hexane (0.5 mg ml−1) and then immersing the SAM samples into the lipid solution for 30 minutes. The samples were rinsed in hexane three times, dried in the nitrogen gas flow before placing them into a QCM-D flow cell (see below for details).2.4 Lipid vesicle preparation
Small unilamellar vesicles (SUVs) were prepared adopting the previously published protocol.41,42 Briefly, the SOPC powder was dissolved in chloroform. After removing the solvent in vacuum, the lipids were resuspended in HEPES buffer (20 mM HEPES, 150 mM NaCl, pH 7.5) to the total lipid concentration of 250 μM. To form the SUVs the lipid solution was treated by probe sonication, at the same time cooling the vial on ice. After the ultrasound treatment, the SOPC stock solution was diluted in HEPES buffers made either with 150 mM or 1 M NaCl to achieve neutral (isotonic solution) and positive (SUVs in hypertonic buffer) osmotic pressure, respectively, to the final lipid concentration of 25 μM.2.5 QCM-D kinetics recording and data analysis
A quartz crystal microbalance instrument (Q-sense E4) equipped with a dissipation monitoring (QCM-D) unit, a flow cell and a peristaltic pump was employed to record sensorgrams of SOPC vesicle adsorption on the sensor surfaces modified with SAMs (see Sections 2.1 and 2.2 above). All measurement runs were performed at 23 °C and the 0.1 ml min−1 flow rate. The signal of the 7th harmonic was selected for analysis, because of the lowest signal-to-noise ratio. The experimental data was analyzed using the software package QSense DFind provided by the equipment vendor. For data presentation, the MS Excel and Origin (OriginLab Corporation) software packages were used.2.6 Computational methods
| ID | Name | Force constant/kJ mol−1 nm−1 | No. of matrix molecules | No. of EG6AC8D tether molecules | No. of EG6AC16D tether molecules | No. of SOPC molecules in vesicle | Simulation time/ns |
|---|---|---|---|---|---|---|---|
| 1 | Tetherless | 100 | 7020 | 0 | 0 | 7532 | 3 × 500 |
| 2 | Tetherless | 1000 | 7020 | 0 | 0 | 7532 | 3 × 500 |
| 3 | 5% EG6AC8D | 100 | 6669 | 351 | 0 | 7532 | 3 × 500 |
| 4 | 5% EG6AC8D | 1000 | 6669 | 351 | 0 | 7532 | 3 × 500 |
| 5 | 5% EG6AC16D | 100 | 6669 | 0 | 351 | 7532 | 3 × 500 |
| 6 | 5% EG6AC16D | 1000 | 6669 | 0 | 351 | 7532 | 3 × 500 |
000000 steps (with a time step of 0.025 ps) were simulated. Pressure coupling was realized with the Parrinello–Rahman47 method using a 12 ps coupling constant. Finally, the external force was released, and the vesicle was allowed to relax for additional 500 ns (20000000 steps with a 0.025 ps time step). All simulations were carried out with GROMACS version 5.1.5.48–53
where σ is the standard deviation, μ the mean, and rPO4 are the radial distances of the leaflet MARTINI particles comprising the phosphate groups, i.e. the distance from the center of the vesicle. In this way, the value would become one if all such particles were perfectly spherically distributed.

As distance and interaction criteria for the vesicle approaching the SAM, two descriptors of vesicle–surface contacts were monitored. The first set corresponds to the hydrophilic vesicle–SAM contacts and is defined as the number of interactions between the SOPC head group MARTINI particles (phosphate group and choline group, see Scheme 1) and the terminal particles of the matrix compound molecules. The second set is a measure for hydrophobic vesicle–SAM interactions and is calculated as the number of contacts between the SOPC phospholipid tail particles and the tether alkyl chain particles. The number of contacts were computed using the GROMACS tool gmx_mindist with a distance cutoff of 0.6 nm. Visualization and rendering were performed with VMD v. 1.9.3.54
3. Results
The experimental part of the present study was undertaken in parallel with a detailed structural investigation of mixed SAMs containing either –(CD2)7CD3 or –(CD2)15CD3 deuterated tethers.33 Thus, the combined QCM-D kinetics and MD simulation work presented herein can be directly related to the structural details about the SAMs, including the tether densities and phases obtained from contact angle goniometry, null ellipsometry and infrared reflection-absorption spectroscopy. For convenience, hereafter we refer to the surface densities of the two types of tethers in the SAM as χEG6AC8D and χEG6AC16D, respectively.3.1 Monitoring of lipid vesicle adsorption, rupture and bilayer formation
Fig. 1 shows typical QCM-D kinetics (sensorgrams) recorded upon multiple SUV injection cycles, at a positive (SUVs in hypertonic buffer) or neutral (isotonic solution) osmotic pressure by employing quartz sensors functionalized with the mixed SAMs. Both types of surfaces had a relatively low and similar density of the tethers, χEG6AC8D = (3.6 ± 3.3) mol% and χEG6AC16D = (3.5 ± 2.1) mol% respectively. Note that for all experiments presented in this study, we initially pre-incubated the SAM-modified sensors with SOPC dissolved in hexane prior to vesicle injection. This treatment was introduced in the experimental study to elucidate any eventual differences in SUV fusion on bare SAM or lipid exposed surfaces, as suggested by Munro et al.55 The frequency and dissipation signals were subsequently obtained under equilibrium, upon exposure to a constant flow. | ||
| Fig. 1 Typical QCM-D sensorgrams of multiple cycles of small unilamellar vesicle (SUV) injection and fusion in HEPES buffer on sensor surfaces with low tether densities. Panels (a and b) are QCM-D sensorgrams recorded for the sensor modified by mixed SAM terminated with EG6AC8D, where the tether density χEG6AC8D = 3.6 ± 3.3 mol%. In (c and d) the mixed SAM contained the tether EG6AC16D at χEG6AC16D = 3.5 ± 2.1 mol% (see text for details). At the start of the experiment the sensor surfaces contained pre-adsorbed SOPC lipids. The first four SUV deposition and removal cycles consisted of repetitive injections marked I, II and III, which correspond to 1 M NaCl, SUVs in 1M NaCl solution and ethanol–water mixture, respectively. Injection IV was deionized water. The last cycle we repeated at a lower ionic strength, where injections V and VI were 150 mM NaCl and SUVs in 150 mM NaCl solution, respectively. | ||
In principle, the QCM-D kinetics obtained for our tethering system resemble typical sensorgrams reported for vesicle fusion on solid unmodified surfaces37 (Fig. 1, injections II). We related the recorded kinetic curves to the following stages of the tBLM formation on the surface.33 First, the initial decrease of the frequency and increase in dissipation signals upon the SUV injection are indicative of rapid vesicle adsorption and accumulation on the SAM surfaces. Then, the appearance of the distinct peaks in the frequency and dissipation signals, respectively, are attributed to the SUV fusion step that includes both vesicle rupture and the loss of excess lipids and water initially trapped inside the vesicles.37 Subsequently, the equilibrium value reached for the frequency signal can be related to a certain mass of lipids (see the Discussion section below), i.e. the density of the tBLM formed. The equilibrium frequency and dissipation values remained stable during the following buffer washing step (Fig. 1, injections I).
After recording the sensorgram comprising the above-listed steps, we fully removed the lipids from the surface by injecting 50% ethanol/water solution. The viscosity of the washing solution differed from the running buffer; therefore it induced large changes in the frequency and dissipation signals: −130 Hz (Fig. 1a and c, injections III) and 52 × 10−6, respectively (see also ESI† for details). Note that during the first two minutes of the ethanol solution injection the Δfrequency and Δdissipation values underwent a jump, which we explain as the lipid removal from the surface. Finally, we exchanged ethanol/water solution by the running buffer (injections I). In comparison to the start of the sensorgram, the frequency and dissipation values were higher and lower, respectively. We interpret the observed signal differences as the removal of any preadsorbed lipids, which were innitially present on the substrate.
In the second injection cycle of the SOPC vesicles (starting at around 5000 s, see Fig. 1), the QCM-D signals revealed a similar bilayer formation kinetics via superficial vesicle accumulation and rupture. This happened despite the surfaces contained no pre-adsorbed lipids. At least three subsequent cycles of tBLM formation and removal indicated good reproducibility (see ESI Fig. S1.2 and S1.9† for the overlays of the QCM-D sensorgrams). After the fourth tBLM formation cycle, we injected deionized water to probe the lipid phase (vesicles vs. tBLM) on the surface, see Fig. 1, injections IV. The change in the dissipation signal during the injection could be related to the decreased viscosity of the running solution and an increase of trapped water inside the vesicles due to the osmotic stress. Indeed, we observed the increase of the dissipation signal of the vesicle layer adsorbed on the tetherless surface during injection of deionized water (see ESI Fig. S1.1†). The dissipation signal decreased similarly upon injecting deionized water on the lipid-free, or tBLM surfaces, respectively, thus indirectly confirming the fusion of the vesicles and tBLM formation.
Before the last SUV solution injection we adjusted the ionic strength of the running buffer and the SUVs solution to be the same (Fig. 1, injections V and VI respectively), thus achieving a neutral osmotic pressure (isotonic solution). The change in the buffer ionic strength caused an increase in the frequency signal and a decrease in the dissipation signal values (this change is visible just before the injection cycle V, Fig. 1). Notably, the last stage of the SUV injection kinetics (Fig. 1, injection VI) looks very different for EG6AC8D and EG6AC16D, despite the very close surface density of the tethers estimated for both types of mixed SAMs. On the surface terminated with EG6AC16D the vesicle injection resembled that of tBLM formation, while in the case of the EG6AC8D one could interpret the signal as vesicle accumulation on the surface, without rupture.
3.2 Effect of the surface density of the tethers
Fig. 2 shows the kinetics analysis of SUV injection sensorgrams at the positive osmotic stress (SUVs in hypertonic buffer). In these experiments, the sensors were modified with the mixed SAMs, containing different surface densities of the tethers EG6AC8D (Fig. 2a and c) or EG6AC16D (Fig. 2e and g), respectively. We present the plots calculated from the recorded raw sensorgrams, with the peak and equilibrium Δfrequency (Fig. 2b and f) and Δdissipation (Fig. 2d and h) values plotted versus the tether surface molar density χ (color coding is provided in panels d, h in Fig. 2). The corresponding raw sensorgrams are provided in the ESI.† One can group the kinetics into three groups, suggesting three distinctive vesicle adsorption regimes. The first regime is seen on the SAM lacking any tethering groups (χ = 0 mol%) as a steadily decreasing frequency and increasing dissipation during the SUV injection.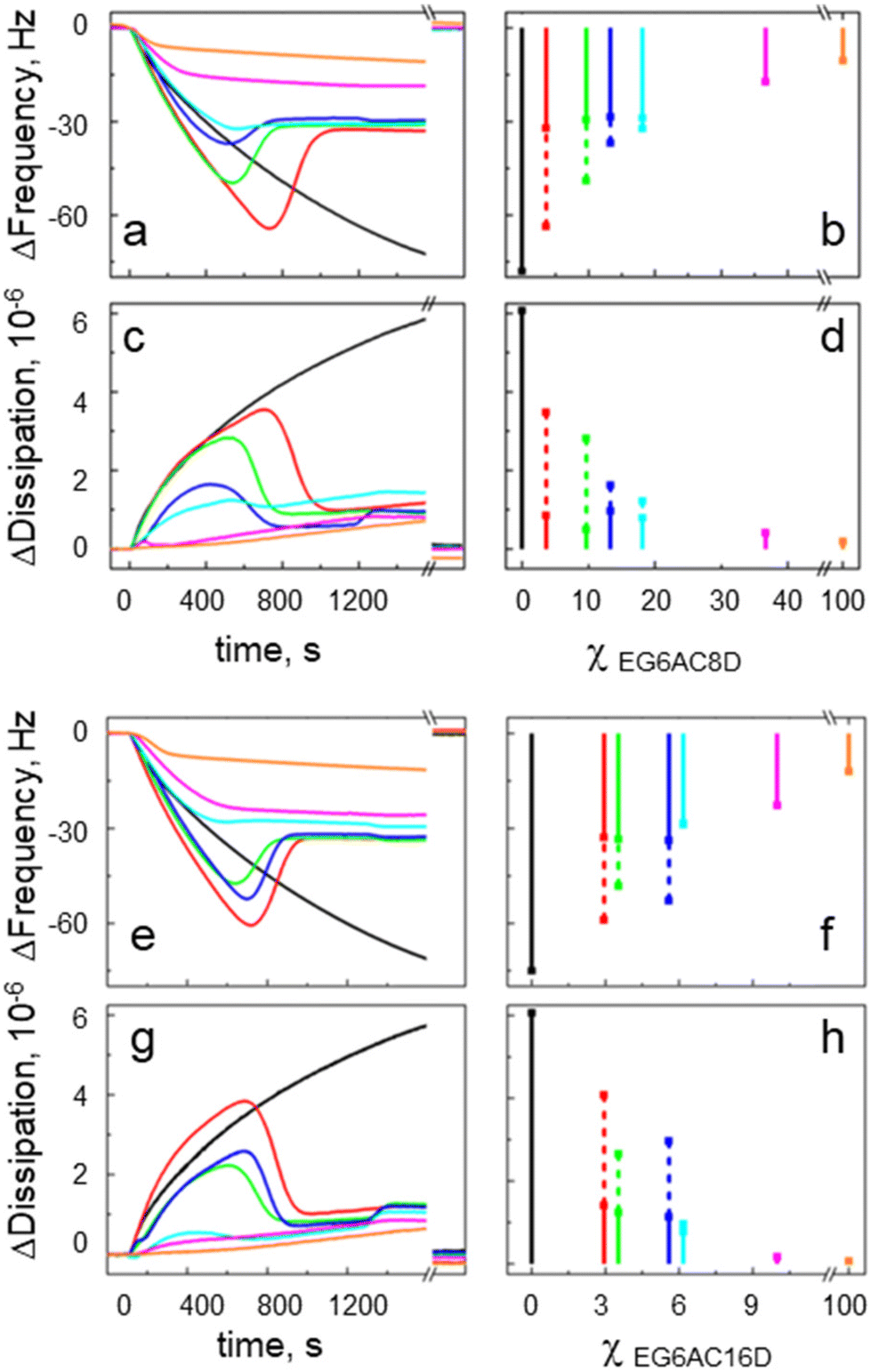 | ||
| Fig. 2 Analysis of QCM-D kinetics showing lipid bilayer formation on mixed SAMs containing either EG6AC8D (a and c) or EG6AC16D (e and g) tethers. The series of kinetics were recorded for different tether surface molar densities χ. In the bar charts, Δfrequency (b and f) and Δdissipation (d and h) signal peak values (dotted bars), and equilibrium values (solid bars) are also plotted for different χ. Note, that curve sections shown after the time axis break correspond to the signals after the lipid removal ethanol solution. | ||
For EG6AC8D, the second regime can be seen from the sensorgrams recorded on the SAMs presenting the tether densities up to χEG6AC8D = (18 ± 3) mol%. For increasing χEG6AC8D, the peak Δfrequency value changes from −78 Hz to −32 Hz, while at the same time the peak Δdissipation value decreases from 6 × 10−6 to 1 × 10−6 (Fig. 2b and d). Moreover, the equilibrium signal values were around −29 Hz and 0.8 × 10−6 for the Δfrequency and Δdissipation, respectively.
The third regime is related to the sensorgrams recorded for χEG6AC8D = (37 ± 3) mol% and 100 mol%. In this case, the signals are monotonically converging to their equilibrium values at −17 Hz and −10 Hz, respectively. The corresponding Δdissipation values converge to 0.4 × 10−6 and 0.2 × 10−6 respectively. The frequency signal decreases rapidly during the first five minutes of SUV injection, followed by a slower phase. This occurs in parallel with a slow increase in the corresponding dissipation signal. The signal values for χEG6AC8D = (37 ± 3) mol% and 100 mol% presented in Fig. 2b and d were estimated from the early stage of the process that corresponds to formation of a rigid (dense) lipid layer (at ∼1200 s).
Similarlly, one can group the sensorgrams also of SUV injections on the SAMs terminated with the EG6AC16D tether. We attribute to the second regime the sensorgrams recorded for χEG6AC16D in the range from (3 ± 2) mol% to (6 ± 2) mol%. In this case, the peak Δfrequency values change from −59 Hz to −29 Hz, while at the same time the peak Δdissipation values decrease from 4 × 10−6 to 1 × 10−6, respectively. The equilibrium signal values are around −32 Hz and 1.1 × 10−6 for Δfrequency and Δdissipation, respectively. In the third regime ( χEG6AC16D = 10.0 ± 3.0 mol% and 100 mol%) the sensorgrams monotonically converge to the equilibrium Δfrequency values, which are at −23 Hz and −12 Hz, respectively. The corresponding Δdissipation values converge to 0.2 × 10−6 and 0.1 × 10−6 respectively. The signal values for χEG6AC16D = (10 ± 3) mol% and 100 mol% presented in Fig. 2f and h were taken at 1200 s after the SUV injection.
3.3 The effect of osmotic pressure on vesicle–surface interactions
To estimate the effect of osmotic pressure, we chose to analyze QCM-D sensorgrams of the SUV injection on sensors with tether densities of χEG6AC8D = 9.7 ± 2.4 mol% for EG6AC8D (Fig. 3a and b) and χEG6AC16D = 5.6 ± 3.6 mol% for EG6AC16D (Fig. 3c and d), respectively. We probed, for comparison, surface interactions with the SUVs having a positive (SUVs in hypertonic buffer) and neutral (isotonic solution) osmotic pressure and observed certain differences in the frequency and dissipation sensorgrams.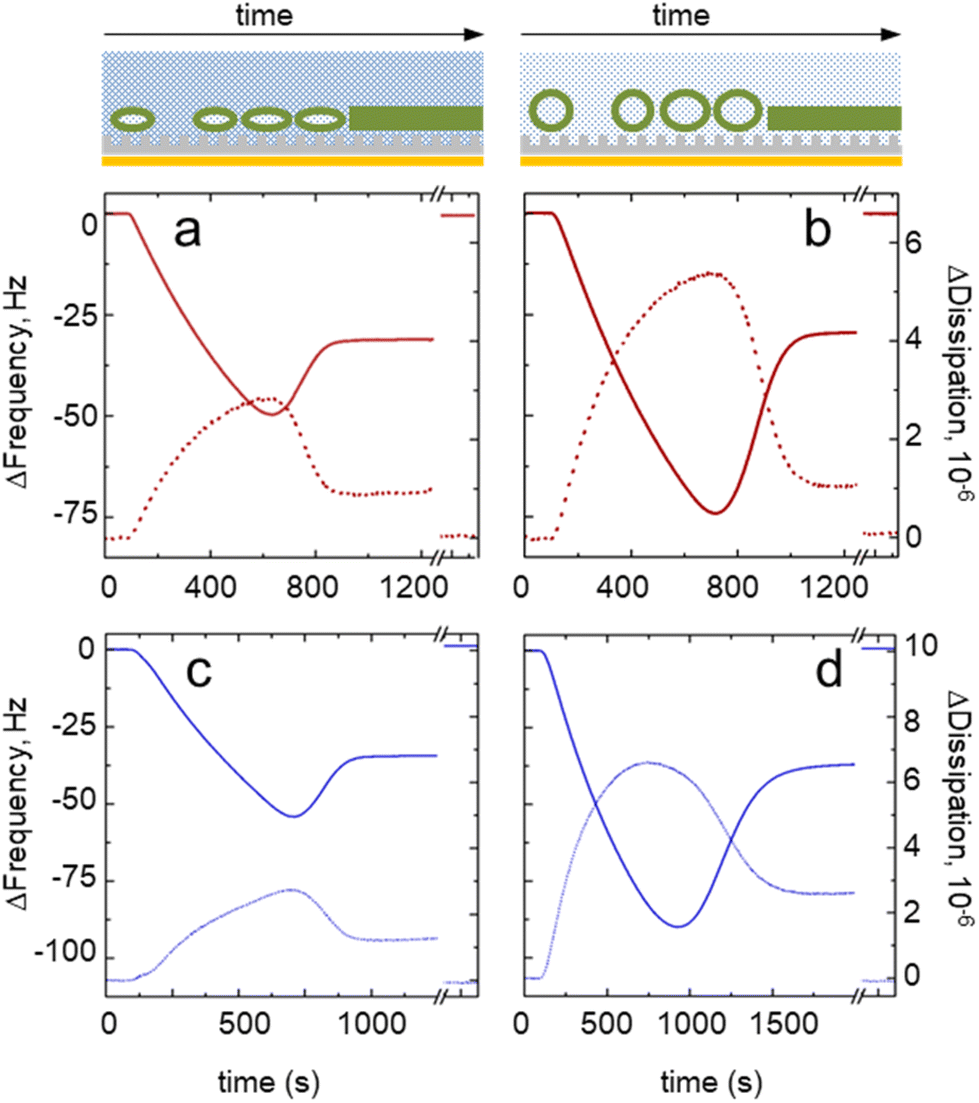 | ||
| Fig. 3 QCM-D sensorgrams of small unilamellar vesicle (SUV) fusion recorded at a positive (SUVs in hypertonic buffer) and neutral (isotonic solution) osmotic pressure. The cartoons (top panel) illustrate the expected lipid phases on the surface (a deformed or relaxed vesicle, or a bilayer) on the mixed SAMs surface (not to scale). The QCM-D sensorgrams were recorded on the mixed SAMs at a tether density of χEG6AC8D = 9.7 ± 2.4 mol% for EG6AC8D (red curves, a and b) and χEG6AC16D = 5.6 ± 3.6 mol% (blue curves, c and d), respectively. The Δfrequency signal is represented by solid lines and Δdissipation by dotted lines. The injected SUVs had either positive (a and c) or neutral (b and d) osmotic pressure. The curve sections shown after the time axis break correspond to the signals after the lipid removal by ethanol solution. | ||
For the EG6AC8D tether, we obtained similar equilibrium Δfrequency values, which were around −30 Hz, regardless of the pressure. The peak value of Δfrequency (i.e., the critical vesicle density) decreased from −50 Hz to −74 Hz for vesicles experiencing the positive and neutral osmotic pressure, respectively. The peak value of Δdissipation increased from 3 × 10−6 to 5 × 10−6, while at the equilibrium it was at around 1 × 10−6. Both the frequency and dissipation signals reached their peak and equilibrium values simultaneously.
In the case of the EG6AC16D tether, the peak Δfrequency decreased from −54 Hz to −90 Hz. Notably, the bilayer formation time increased from ∼900 s (Fig. 3c) to >1500 s (Fig. 3d) for the vesicles experiencing positive and neutral osmotic pressure, respectively. As for the EG6AC8D tether, the Δdissipation signal did not reach zero after the bilayer formation stage. However, for the EG6AC8D tether, the dissipation and frequency reached their peak values simultaneously at positive osmotic pressure, whereas the dissipation signal peaked before the frequency signal for the vesicles at neutral osmotic pressure. The same trend was also visible for the other surface concentrations of the tethers (see ESI† for details).
3.4 Molecular dynamics simulation and analysis of the vesicle and self-assembled monolayer interactions
We performed molecular dynamics (MD) simulations to rationalize the experimental observations regarding the effect of the presence and length of molecular tethers on the vesicle rupturing kinetics. We thus selected a set of experimental compositions to investigate the effects of different molecular tethers on tBLM formation. For reasons of computational efficiency for these large molecular systems with around 2 million particles, we chose to concentrate our efforts to the low tether density regime (5%). Further, we developed a steered MD workflow, in which the equilibrium state of a tethered vesicle on the SAM is independent of simulation scenarios. The steady-state corresponds to a situation when a constant number of the tethering alkyl chains of the SAM are incorporated into the SUV. In order to investigate two feasible scenarios of tBLM (spontaneous vs. forced), we chose two limiting cases of vesicle attachment with two different force constants.In one set of the simulations, the vesicle is only weakly pulled towards the surface of the SAM (with a steered MD force constant of 100 kJ mol−1 nm−1), so that the insertion of the tether must occur spontaneously during the subsequent equilibrium MD simulations in the absence of an external force (k = 0 kJ mol−1 nm−1). In the second set of the simulations, the steering force is increased to 1000 kJ mol−1 nm−1. Thus, the SUV is squeezed onto the SAM, so that a larger number of tethers are inserting into the SUV than in the equilibrium state (Fig. 4). The chosen force constants of 100 kJ mol−1 nm−1 and 1000 kJ mol−1 nm−1 are in the order of MARTINI bond force constants or that of positional restraints and thus shown to be numerically stable. During the following equilibrium MD simulations, a certain number of tethers must leave the vesicle spontaneously. After infinite sampling, both approaches should, in principle, converge to the same steady state and yield identical numbers of hydrophobic and hydrophilic SUV–SAM interactions. However, as some of these transitions are extremely slow, such a perfect sampling cannot be reached in practice.
 | ||
| Fig. 4 Details of small unilamellar vesicle (SUV) and mixed self-assembled monolayer (SAM) interactions. The matrix EG1H molecules are colored in gray, the shorter EG6AC8D tethers in red, and the longer EG6AC16D tethers in blue. (a) Different SAM starting configurations (pure matrix molecules, EG6AC8D tether randomly distributed in the SAM, and self-clustered EG6AC16D tethers). In (b) and (c) phospholipid (SOPC) molecules forming the SUV are shown in green. Steered MD simulation of vesicles approaching the SAM with force constants of 100 kJ mol−1 nm−1 and 1000 kJ mol−1 nm−1, respectively. Side-view snapshots are shown at the initial (t = 0 ns) and final (t = 500 ns) moment of MD simulation. Top: pure EG1H, middle: the χEG6AC8D = 5 mol%, bottom: the χEG6AC16D = 5 mol%. As compared to the experiments, the modeled tether alkyls are hydrogenous and not deuterated. | ||
During both the steered MD SUV–SAM adsorption and the subsequent equilibrium MD phases, we continuously monitored the shape of the vesicle by means of the sphericity as well as the number and type (hydrophilic and hydrophobic) of contacts between the vesicle and SAM (see above).
In the case of the tetherless SAM, the SOPC vesicle stays in a close proximity to the SAM surface after adsorption, independently of the attachment force constant. Upon the release of the force, it quickly relaxes to a nearly perfect sphericity (Fig. 5a and d). At equilibrium, the SOPC vesicle adsorbs only weakly to the tetherless SAM surface as shown by only few and transient polar interactions mediated by the headgroup of the lipids and SAM surface (Fig. 6a and d).
 | ||
| Fig. 5 Monitoring of the lipid vesicle sphericity upon approaching different molecular tethering phases in steered MD simulations. Black lines (a and d) correspond to the reference system in absence of tethering compounds (the EG1H SAM), red lines (b and e) to the surface density χEG6AC8D = 5 mol% of short EG6AC8D, and blue lines (e and f) to the χEG6AC16D = 5 mol% of longer EG6AC16D tethers. Panels (a–c) correspond to a pulling force constant of 100 kJ mol−1 nm−1 and panels (d–f) to a force constant of 1000 kJ mol−1 nm−1. Each panel presents the data of three independent replica of simulations with different initial conditions (see Methods). The moving averages (N = 10) and the individual data points are superimposed. | ||
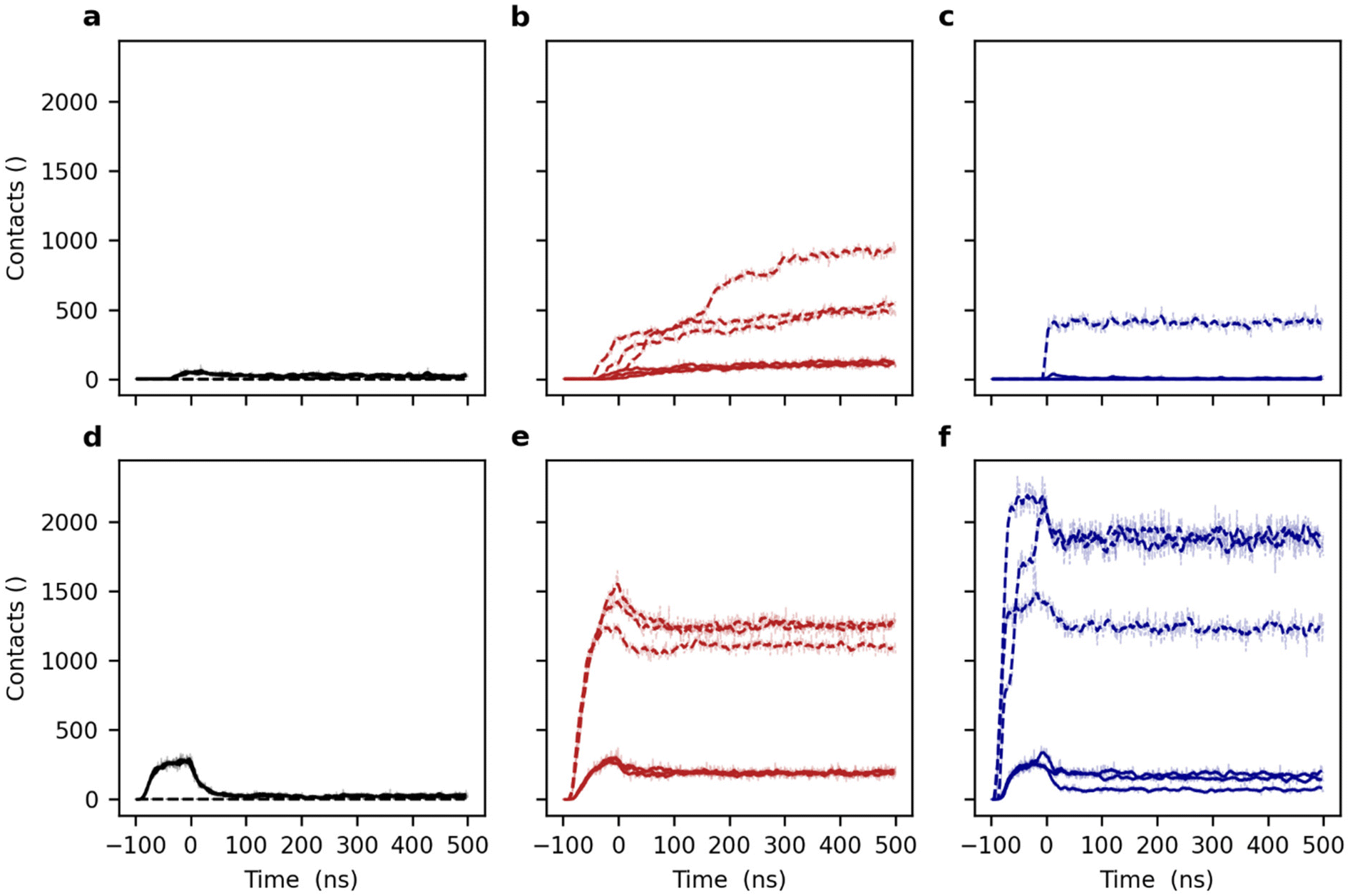 | ||
| Fig. 6 Monitoring of lipid vesicle and self-assembled monolayer (SAM) interactions upon approaching different molecular tethering phases in steered MD simulations. Black lines (a and d) correspond to the reference system in the absence of tethering compounds (the EG1H SAM), red lines (b and e) to the surface density χEG6AC8D = 5 mol% of short EG6AC8D, and blue lines (e and f) to the χEG6AC16D = 5 mol% of longer EG6AC16D tethers. Solid lines correspond to hydrophilic interactions between the hydroxy group, amide group and oligo(ethylene glycol) particles of the SAMs and the headgroup particles of the vesicles. Dashed lines correspond to the hydrophobic interactions between the alkyl carbon particles of the SAMs and lipid vesicles. Panels (a–c) correspond to a pulling force constant of 100 kJ mol−1 nm−1 and panels (d–f) to a constant of 1000 kJ mol−1 nm−1. Each panel presents the data of three independent replica simulations with different initial conditions (see Methods). The moving averages (N = 10) and the individual data points are overlaid. | ||
Previously, we were able to show that the EG6AC8D alkyl tethers tend to be isolated and adopt a random spatial distribution in a matrix of shorter compounds (e.g. EG1H) in mixed SAMs on Au(111) surfaces.33,36 Upon adsorption of a vesicle on a SAM with randomly distributed short EG6AC8D tethers (χEG6AC8D = 5 mol%), the initial deformation is similar to that observed for the tetherless SAM. The same applies for the EG6AC16D tethers (Fig. 5a–f).
Using the weak force constant for the vesicular attachment to the EG6AC8D containing SAM, only few hydrophobic tethers are incorporating into the lipid tail region of the vesicle (Fig. 6b). With the larger force constant, however, a total of 1500 pairwise hydrophobic and hydrophilic interactions are detected during the attachment step alone (Fig. 6e). Upon the release of the force, the weakly attached (k = 100 kJ mol−1 nm−1) vesicle begins to deform because of spontaneous incorporation of an increasing number of the EG6AC8D tethers (Fig. 5b and 6b). The sphericity converges to a value of 0.96 while the number of hydrophobic contacts increases to 500–900 depending on the replicate. Similarly, the number of hydrophilic contacts between the SAM and the SOPC vesicle increases to a value of 140. Thus, the insertion of a larger number of the tethers drives a deformation of the vesicle only until equilibrium is reached. At this point, there is an equilibrium between favorable energy contributions from desolvation of the hydrophobic tethers and unfavorable energy contributions from the induced vesicle shape distortion.
Equilibrium MD simulations following the stronger SUV adsorption (k = 1000 kJ mol−1 nm−1) yield an even more deformed vesicle with a sphericity of only 0.94 which remains stable during the course of the simulation (Fig. 5e). The number of polar interactions immediately decreases after releasing the attachment force and reaches a value of 200. The number of hydrophobic interactions, which is a measure of the number of inserted tether moieties, slowly decreases upon force relaxation to a value of 1300 (Fig. 6e). Thus, relaxation of SUV structures adsorbed on a SAM with different steering force constants leads to consistent equilibrium structures.
The absolute numbers are only slightly varying (sphericity 0.96 vs. 0.94, number of polar contacts 140 vs. 200, number of hydrophobic contacts 900 vs. 1300) and thus correspond to the same overall state and present a consistent picture. We conclude that insertion and extraction of the randomly distributed shorter EG6AC8D tethers would most likely happen spontaneously and are only limited by the surface tension of the spherical vesicle itself.
Based on our previous studies,33,36 the longer EG6AC16D tethers tend to form nanometer-scale clusters in the mixed SAM. Thus, during SUV association to a clustered surface arrangement of tethers (for example, at χEG6AC16D = 5 mol%), the local surface density differs significantly from that of the short tether that is homogeneously distributed. After relaxation of a weak steering force for the initial attachment, a vesicle quickly re-adopts its spherical shape (Fig. 5c). Notably, the number of polar interactions between vesicle and bilayer stays low – even lower than in the case of the tetherless SAM (Fig. 6c).
Towards the end of the simulation with vesicular approach with a low force, in one single simulation replicate, one of the EG6AC16D tether nano-clusters inserted into the vesicle, yielding 400 hydrophobic contacts between the alkyl chains of the tether and the SOPC vesicle. This number of contacts remains stable until the end of the trajectory, which shows that there is no further tether insertion into the vesicle. The large difference between the number of the hydrophobic and hydrophilic contacts can only lead to the conclusion that the vesicle position is elevated by interacting with the long EG6AC16D tethers along the z-axis, so that it remains at a certain distance from the SAM surface. Additional polar vesicle–surface interactions between the SAM and vesicle are avoided. In the two further replicates, no tether insertion was observed (Fig. 6c).
Upon application of a larger force, the vesicle–EG6AC16D SAM system shows features similar to those of the shorter EG6AC8D tether SAM. The number of the inserted hydrophobic EG6AC16D tether molecules is large at the beginning of the equilibrium MD simulations, and then decreases as the force is relaxed (Fig. 6f). To be more specific, one peripheral EG6AC16D cluster is released from the vesicle as the force is relaxed. The numerical difference of the hydrophilic and hydrophobic contacts between the SUV and the SAM for different MD pulling forces remains larger in the case of the longer EG6AC16D tether. The three simulation replicates show largely similar outcomes, however, the total number of inserted domains is varying as reflected by the number of interactions. Two replicates exhibit around 1800 initial hydrophobic interactions, and the third one only 1200. A higher number of polar interactions also reflects the stronger adsorption by more tether insertions in these two replicates.
In summary, the above observations suggest that the energy barrier for insertion of the long chain hydrophobic and clustered tether EG6AC16D into the hydrophilic SOPC head group region of the vesicle is much higher as compared to short-chain tethers. This can be rationalized by the fact that the ratio between the solvent accessible surface area and volume of a cluster is lower than that of the individually solvated short tethers. Thus, the overall energy gain from desolvation is lower as compared to the EG6AC8D tethers.
4. Discussion
We have shown that tBLM forms on binary SAMs on gold that are terminated with different linear molecular tethers (EG6AC8D and EG6AC16D). The distinctive feature of our tethering system is that the supporting alkylthiolate portion of the SAM is highly ordered and stable, independently of the conformational/phase states of the upper portion comprising the ethylene glycol spacer and terminal d-alkyls.33,36,40 We stress again, that such an architecture allows for not only tuning the surface density of the tethers, but also their phase (homogeneously distributed, conformationally disordered tether tails vs. nanoscopic clusters of relatively densely packed aliphatic chains).To the best of our knowledge, previous studies that addressed the influence of the tether density on vesicle fusion focused only on the properties of an equilibrium tBLM. They were based on electrochemical impedance spectroscopy, neutron reflectometry or atomic force microscopy techniques that lack the real time information about tBLM formation. On the other hand, QCM-D has become the standard technique for monitoring and analyzing the main steps of the lipid vesicle–solid surface interactions and lipid membrane formation.37 Molecular dynamics simulations are by now an essential tool for investigating surface-induced lipid phase reorganization and membrane formation. The accessibility of microsecond long trajectories for a large number of particles using coarse-grained molecular dynamics simulations provides additional insight into self-assembled monolayers and their functionalization.29,56,57
We recently developed a semi-automated protocol to model mixed monolayers with tethers of varying length, concentration and spatial distribution, which helped to elucidate the structural information previously obtained from experimentally determined infrared spectra.33,36 It is now possible to build upon these results and elucidate the adsorption and attachment of spherical unilamellar vesicles to solid surfaces functionalized with SAMs.
Therefore, the rationale of our study was to obtain as complete view of the tBLM formation by SUV fusion as possible: from the first instances of the vesicle–tethering surface interactions to the resulting equilibrium lipid bilayer. When taken together, the MD and QCM-D techniques reveal the full sequence of the process of tBLM formation by vesicle fusion on SAMs. Based on the presented data, the main steps of this process are the following: (i) tether insertion into an adsorbed SUV; (ii) mechanical deformation of the SUV that becomes partially tethered; (iii) continuous accumulation of the SUVs at the solid-liquid interface (overall mass increase); (iv) rupture of the surface bound-SUVs transitioning into a tBLM state. Below we briefly discuss the main aspects of each step.
(i) Tether insertion into the SUV. Close-up molecular snapshots of the interface regions between the vesicle and tetherless (ethylene glycol-terminated) and tether-displaying substrates, respectively, reveal the different molecular adsorption mechanisms (Fig. 7). For the tetherless SAM, hydrogen bonds are the predominant interaction type. They are mostly formed between the terminal hydroxyl groups of the SAM and the phosphate group of the lipids. The transient interactions lead to local deformation of the vesicle (flattening) at only the points of contact. The vesicle is separated from the SAM by a continuous water layer, which is locally interrupted by a small number of direct SAM–vesicle contacts (Fig. 7a).
 | ||
| Fig. 7 Density profiles and snapshots of molecular interface between the SOPC vesicles and mixed self-assembled monolayers. The normal directed density profiles and simulated images for (a) the SAMs that are tetherless (black lines and black image frame), (b) functionalized with χEG6AC8D = 5 mol% EG6AC8D tethers (red lines and red image frame), and (c) functionalized with χEG6AC16D = 5 mol% EG6AC16D tethers (blue lines and blue image frame) are shown. Cyan lines display the density of the aqueous solvent particles, green lines the density of the lipid vesicle. The density profiles are aggregated over the final 200 ns and individually drawn for the three replicates. The images show the final snapshot after steered MD simulations plus 500 ns of relaxation. All panels correspond to simulations using strong initial pulling with a force constant of 1000 kJ mol−1 nm−1. In panel (a) the differences between the three replicates are too small to be seen by eye. | ||
Incorporation of the shorter EG6AC8D tether moieties leads to increased SAM–vesicle contacts and a more pronounced planar distortion of the vesicle. The tether alkyl chains penetrate the outer leaflet of the local phospholipid bilayer zone, but they do not advance into the inner layer. The tether amide groups largely intercalate with the phosphate groups of the lipids due to favorable polar interactions (hydrogen bonds, not explicitly modeled). In total, the number of polar interactions between the SAM and vesicle is higher when compared to the tetherless SAM. The thickness and continuity of the interfacial water layer are slightly decreased compared to the tetherless SAM. The interfacial water layer appears to be thinner and less continuous compared to the tetherless SAM (Fig. 7b).
The longer EG6AC16D alkyl chain tethers penetrate much deeper into the phospholipid bilayer and locally interfere with the outer and inner leaflets. The vesicle–SAM interface area is comparable to that on the EG6AC8D tether SAM. The water layer is of a similar thickness and more continuous (Fig. 7c). The insertion of multiple nanosized EG6AC16D tether clusters was observed for large external forces. In the weakly forced attachment simulations, insertion of a single cluster was observed once. We thus expect that upon extended simulation time, insertion of clusters would also happen spontaneously. This is in line with the QCM-D findings, which clearly show that cluster incorporation does occur (see below). We note however, that this process is slow because it requires concomitant displacement of a large number of lipids from the membrane.
Usually, the penetration depth and vertical architecture is obtained from the normal direction density profiles (Fig. 7a–c). In ordered systems, the density profile allows the assessment of the partial density of distinct molecules in different slabs along a coordinate (e.g. surface normal) of the simulation box. However due to the spherical shape of the vesicles, the density profiles are slightly misleading and must be analyzed with care. The density profile in surface-normal (z-coordinate) direction of the untethered vesicle gradually increases in a distance of 2 to 7 nm relative from the surface (Fig. 7a). At distances above 7 nm, the vesicle density corresponds to the density of circular segments of the vesicle which is mostly constant. The short tethers penetrate the vesicle up to 2 nm (Fig. 7b), whereas the long tethers up to 4 nm (Fig. 7c). Furthermore, there are two layers of the Martini water particles. The first one occurs at about 1 nm distance from the hydroxyl terminal groups of the SAM (center-of-mass distance) and is well pronounced. It exhibits the highest density above the tetherless SAM and is less prominent for the EG6AC8D tether and the EG6AC16D tether SAMs. The second and third water layers are less apparent above the tetherless SAM and instead well pronounced above the two tether containing SAMs (Fig. 7a–c).
In the case of uniformly distributed EG6AC8D tethers, the MD simulations reveal their insertion into the vesicle upon contact formation. The numerous insertions that are observed during the course of the simulation are suggesting that the energy of the thermal motion is sufficient for the insertion to happen. It is noteworthy that in biological membranes the docking step of a post-translationally palmitoylated protein is guided by enzymes and requires a driving force to overcome an energy barrier. For example, experimental studies showed that each methylene group of a lipidated peptide contributes 3.3 kJ mol−1 to the free energy of membrane binding.58 Thus, palmitoylated peptides (linear, saturated fatty acid with 16 carbons) would exhibit a free energy of binding of approximately 52 kJ mol−1. Correspondingly, for the EG6AC8D tethers (8 carbons), a binding free energy of 26 kJ mol−1 and for the EG6AC16D tethers (16 carbons) of 52 kJ mol−1 can be expected. In comparison, a geranylgeranyl peptide (branched, unsaturated modification of 20 carbons) showed a free energy difference of 50 kJ mol−1.59 Previous full atomistic MD simulations of a single and double geranylgeranylated peptide yielded bilayer extraction free energies of 69 kJ mol−1 and 119 kJ mol−1, respectively60 The corresponding coarse-grained model slightly underestimated the experimental results and yielded 30 kJ mol−1 for the single geranylgeranyl peptide.60 Thus, our coarse-grained model might as well slightly underestimate the insertion free energy of the tether molecules into the vesicle bilayer. Such discrepancy is known and the accuracy of partition energies in MARTINI is discussed in several articles, e.g. ref. 61–63.
(ii) Mechanical deformation of the SUV. The insertion of the EG6AC8D tethers decreases the sphericity of the tethered SUV (Fig. 5a and e) from 0.98 to 0.94 (Fig. 5b and f). This decrease in sphericity can also be also treated as an increase in rigidity of the tethered SUV. Based on QCM-D, the SUV deformation due to penetration of EG6AC8D tethers is fast and almost reaches equilibrium during the course of the MD simulation (500 ns).
As stated before, EG6AC16D cluster incorporation does only occur when an external force is applied and is significantly less likely during the weakly forced attachment. Thus, EG6AC16D cluster insertion happens on much slower timescale than incorporation of EG6AC8D tethers. A comparison of the frequency and dissipation sensorgrams recorded during tBLM formation at neutral osmotic pressure, confirms that the cluster incorporation is feasible, and most likely occurring on a longer timescale (Fig. 3). For the SUVs interacting with the EG6AC16D tether at the neutral osmotic pressure (isotonic solution), the peak dissipation value precedes the frequency peak (Fig. 3d). After reaching its peak value the dissipation signal starts to decrease while the SUVs are still accumulating as seen from the frequency signal. This behavior suggests that the SUVs become more rigid after adsorption and that this transition happens at the timescale of seconds. Based on the analogy with EG6AC8D, we interpret it as a gradual insertion of EG6AC16D tether clusters, which is either spontaneous or is promoted by continuous vesicle accumulation and/or by mechanical forces acting in the microfluidic setup. For comparison, in the case of EG6AC8D, tether penetration is faster and therefore both signals reach their peak and equilibrium values simultaneously (Fig. 3a and b).
(iii) Accumulation and critical surface density of the SUVs at the liquid solid interface. The most evident effect of the tether density on tBLM formation is observed at this stage. The QCM-D data clearly show that the critical vesicle density (seen as the peak frequency and dissipation values) is decreasing with the increasing surface density (χ) of the homogeneously distributed EG6AC8D (which corresponds to an increasing exposure of hydrophobic d-alkyl chains). Ultimately, for χEG6AC8D = 20 mol% the adsorbed individual SUVs rupture without interaction with neighboring vesicles, i.e. the vesicle density is not the critical factor at high χEG6AC8D (Fig. 2b and d).
Similarly, the increasing EG6AC16D tether density also lowers the critical vesicle density for rupture (Fig. 2f and g). However, for this longer tether, the tendency is less pronounced, probably due to nanoscopic cluster presentation posing additional energetic barrier, as estimated from MD. However, already at around χEG6AC16D = 10 mol% the fusion process enters the regime of single vesicle rupture.
The experimentally observed decrease of the critical vesicle density with increasing surface tether density can be explained by the complementary MD data presented above. Tether penetration into the SUV results in deformation of the latter. Based on the comparison of the SUV shape on the tetherless and 5 mol% tether surfaces, respectively, we extrapolate that the increasing surface density of both tethers further distort the adsorbed SUVs (decrease their sphericity) this way promoting their rupture.
(iv) Rupture of the surface bound SUVs and transition into a tBLM state. In the recorded QCM-D sensorgrams, the increasing frequency and decreasing dissipation signals after reaching their corresponding peak values are attributed to the SUVs rupture and correspond to the loss of excess lipids and the trapped water. Equilibration of these signals can be related to formation of the equilibrium lipid bilayer (tBLM) on the surface.37 The equilibrium Δfrequency and Δdissipation values for both tethers are in the range of −29 to −32 Hz and (1 to 2) × 10−6, respectively (see ESI† for detailed comparison). For the structurally similar DOPC lipid64 and for mixed DOPC/DOPS bilayers37 formed on silicon dioxide substrates the Δfrequency was around −25 Hz and Δdissipation was close to zero. We attribute the observed deviations of our measured equilibrium QCM-D signal values from these literature values to elastic contribution from the tethering assemblies that also display certain dissipative properties. Indeed, the equilibration of the Δdissipation signal at certain values also has been observed in other tBLM systems.27,65 The exact dissipative pathways of our tBLM system, that likely would relate to the molecular hydration states and the structure of aqueous layer formed inbetween the lipid bilayer and the SAM surface, are complex and beyond the scope of the present study. For that purpose, detailed resolution of the structure of the formed tBLM is important. Such a structural information can be obtained by combining QCM-D, MD and neutron reflectometry techniques together with analysis of membrane protein incorporation and will be the topic of a follow-up article.
Previously published SAM-based tBLM systems reported on tether densities falling in the range of 20–30 mol%26,27,65 and considered them as being “sparsely” tethered membranes. Therefore, a rather surprising observation of the present study is that surfaces with tether presentation in the range of 2–3 mol% are already sufficient to initiate spontaneous tBML formation from SOPC SUVs, at least under the microfluidic conditions. Unfortunately, the minimum surface density required to trigger the SUV fusion was not possible to determine in our study because of the limitations of the ellipsometry and contact angle techniques previously employed33 for characterization of the SAMs used on this study.
Finally, we would like to note that although the coarse-grained MD simulations using the MARTINI force field and the experimental insights are in full agreement with each other, it is important to recognize the limitations of the model. Bases on the four-to-one mapping in CG, subtle chemical differences between similar lipid molecules such as SOPC or POPC cannot be considered. Thus, for a more accurate representation of the interactions at the SAM–vesicle interface, resolution transformation would be beneficial and should be included in future simulation frameworks. This would also allow an explicit description of directed hydrogen bonds, which are only implicitly modeled in the CG MARTINI mapping. The simplified representation of the molecular interactions smoothens the free energy landscape is a compromise between accuracy and sampling efficacy. This has enabled to sample the spontaneous insertion of a large EG6AC16D tether cluster, which would have been more unlikely to occur in full-atomistic simulations, even at similar time-scales. Nonetheless, the MARTINI force field has been carefully parametrized on partitioning free energies to reliably capture the thermodynamic properties of various systems including small molecules, lipids, artificial polymers, as well as proteins and nucleic acids. In our opinion, the MARTINI force field excels in large assemblies of mostly linear molecules with predominantly hydrophobic intermolecular interactions. It was therefore our method of choice for modelling the SAM–vesicle interaction. Furthermore, we note that we have intentionally refrained from calculating absolute macroscopic properties from our coarse-grained MD simulations. Instead, we use the simulations as a semi-quantitative means to be able to compare the experimentally investigated systems among each other. Extrapolation or generalization of the MD results to other, similar systems should be considered with care.
Nevertheless, based on the coarse-grained MD simulations we have successfully created an in silico analog of a rather complex experimental tBLM system. In the future studies, it can help to predict its behavior and various parameters facilitating development of physical models and prototypes of various nanoengineered devices. In turn, such a combined computational and experimental strategy could enhance the performance of quantitative real-time analysis of different interaction phenomena that are supposed to reflect as precisely as possible those taking place in the native membranes of the cell. The need for better designed membrane interaction studies that would ultimately translate better in vivo is known in the broad field of model membrane-based biophysical and pharmacological studies.66
5. Conclusions
We have addressed the role of the tether surface density and phase on the process of the tBLM formation by SUV fusion on self-assembled monolayers. By employing the combination of state-of-the-art MD simulations and QCM-D as a real time experimental technique, we were able cover all the stages of the process, from the initial intermolecular and surface interactions in the vesicle–surface contact zone, to the resulting equilibrium lipid bilayer.Our data demonstrates that either homogeneously mixed or nanoscopic cluster-forming tethers equally well promote spontaneous formation of the tBLM on SAMs. When combined, experimental and simulation results reveal that the key factors driving the vesicle fusion on this type of molecular surfaces is the tether insertion into SUVs attaching to the surface, along with the onset of SUV deformation (decrease in sphericity).
Furthermore, the detailed analysis of the process showed different molecular pathways of the interactions occurring in the contact (adsorption) zone of an SUV and the tether-terminated SAM. For the homogeneously distributed EG6AC8D tethers the thermal motion was sufficient for their insertion into the adjacent lipid bilayer of the SUV, whereas the clusters formed by EG6AC16D posed an additional energetic barrier for achieving surface tethering. Upon increasing the surface density of either tether, the critical surface density required for vesicle rupture and tBLM phase formation decreased. This effect could be related to a decreasing sphericity of adsorbed SUVs.
From the application point of view, the benefit of our tethering system is that the surface densities of the tethers as small as a few mol% are sufficient to reproducibly obtain stable tBLMs. The possibility to select from an optimal surface density and/or phase of the tethers can be important in designing tBLMs for efficiently mimicking different diffusion, transport and interaction phenomena occurring in the cell membrane, as well as for membrane protein incorporation.
Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
The authors report no conflicts of interest.Acknowledgements
M. G., H.-H. L., B. L., and R. V. are grateful to the Swedish Institute's Visby program for financial support. E. S.-N. and M. S. are grateful to the Max Planck Society for the Advancement of Science for financial support. This work was also supported by the International Max Planck Research School ‘Advanced Methods in Process and Systems Engineering’ and the European Regional Development Fund (ERDF) of the Ministry of Economy, Science and Digitalization in Saxony Anhalt within the Center of Dynamic Systems, ZS/2016/04/78155.References
- T. Penkauskas and G. Preta, Biological Applications of Tethered Bilayer Lipid Membranes, Biochimie, 2019, 157, 131–141 CrossRef CAS PubMed.
- A. Alghalayini, A. Garcia, T. Berry and C. G. Cranfield, The Use of Tethered Bilayer Lipid Membranes to Identify the Mechanisms of Antimicrobial Peptide Interactions with Lipid Bilayers, Antibiotics, 2019, 8(1), 12 CrossRef CAS PubMed.
- L. A. Clifton, R. A. Campbell, F. Sebastiani, J. Campos-Terán, J. F. Gonzalez-Martinez, S. Björklund, J. Sotres and M. Cárdenas, Design and Use of Model Membranes to Study Biomolecular Interactions Using Complementary Surface-Sensitive Techniques, Adv. Colloid Interface Sci., 2020, 277, 102118 CrossRef CAS PubMed.
- T. Ma, M. Sato, M. Komiya, X. Feng, D. Tadaki and A. Hirano-Iwata, Advances in Artificial Bilayer Lipid Membranes as a Novel Biosensing Platform: From Drug-Screening to Self-Assembled Devices, Chem. Lett., 2021, 50(3), 418–425, DOI:10.1246/cl.200764.
- F. Mazur, M. Bally, B. Städler and R. Chandrawati, Liposomes and Lipid Bilayers in Biosensors, Adv. Colloid Interface Sci., 2017, 249, 88–99 CrossRef CAS PubMed.
- S. G. Patching, Surface Plasmon Resonance Spectroscopy for Characterisation of Membrane Protein–Ligand Interactions and Its Potential for Drug Discovery, Biochim. Biophys. Acta, Biomembr., 2014, 1838(1), 43–55 CrossRef CAS PubMed.
- M. Zagnoni, Miniaturised Technologies for the Development of Artificial Lipid Bilayer Systems, Lab Chip, 2012, 12(6), 1026–1039 RSC.
- C. Peetla, A. Stine and V. Labhasetwar, Biophysical Interactions with Model Lipid Membranes: Applications in Drug Discovery and Drug Delivery, Mol. Pharm., 2009, 6(5), 1264–1276 CrossRef CAS PubMed.
- J. Payandeh and M. Volgraf, Ligand Binding at the Protein–Lipid Interface: Strategic Considerations for Drug Design, Nat. Rev. Drug Discovery, 2021, 20(9), 710–722 CrossRef CAS PubMed.
- S. Damiati, Can We Rebuild the Cell Membrane?Biological, Physical and Technical Basics of Cell Engineering, ed. G. Artmann, A. Artmann, A. Zhubanova and I. Digel, Springer, Singapore, 2018 Search PubMed.
- R. Tero, Substrate Effects on the Formation Process, Structure and Physicochemical Properties of Supported Lipid Bilayers, Materials, 2012, 5(12), 2658–2680 CrossRef CAS.
- J. Andersson, I. Köper and W. Knoll, Tethered Membrane Architectures—Design and Applications, Front. Mater., 2018, 55 CrossRef.
- W. Hoiles, V. Krishnamurthy and B. Cornell, Formation of Engineered Tethered Membranes, In Dynamics of Engineered Artificial Membranes and Biosensor, Cambridge University Press, Cambridge, 2018, pp. 61–82 Search PubMed.
- R. Veneziano, C. Rossi, A. Chenal, C. Brenner, D. Ladant and J. Chopineau, Synthesis and Characterization of Tethered Lipid Assemblies for Membrane Protein Reconstitution (Review), Biointerphases, 2017, 12(4), 04E301 CrossRef PubMed.
- V. Proux-Delrouyre, C. Elie, J.-M. Laval, J. Moiroux and C. Bourdillon, Formation of Tethered and Streptavidin-Supported Lipid Bilayers on a Microporous Electrode for the Reconstitution of Membranes of Large Surface Area, Langmuir, 2002, 18(8), 3263–3272 CrossRef CAS.
- K. Ataka, F. Giess, W. Knoll, R. Naumann, S. Haber-Pohlmeier, B. Richter and J. Heberle, Oriented Attachment and Membrane Reconstitution of His-Tagged Cytochrome c Oxidase to a Gold Electrode: In Situ Monitoring by Surface-Enhanced Infrared Absorption Spectroscopy, J. Am. Chem. Soc., 2004, 126(49), 16199–16206 CrossRef CAS PubMed.
- M. K. Sharma and M. L. Gilchrist, Templated Assembly of Biomembranes on Silica Microspheres Using Bacteriorhodopsin Conjugates as Structural Anchors, Langmuir, 2007, 23(13), 7101–7112 CrossRef CAS PubMed.
- L. D. Hughes and S. G. Boxer, DNA-Based Patterning of Tethered Membrane Patches, Langmuir, 2013, 29(39), 12220–12227 CrossRef CAS PubMed.
- N. Boden, R. J. Bushby, S. Clarkson, S. D. Evans, P. F. Knowles and A. Marsh, The Design and Synthesis of Simple Molecular Tethers for Binding Biomembranes to a Gold Surface, Tetrahedron, 1997, 53(31), 10939–10952 CrossRef CAS.
- N. Boden, R. J. Bushby, Q. Liu, S. D. Evans, A. Toby, A. Jenkins, P. F. Knowles and R. E. Miles, N,N′-Disuccinimidyl Carbonate as a Coupling Agent in the Synthesis of Thiophospholipids Used for Anchoring Biomembranes to Gold Surfaces, Tetrahedron, 1998, 54(38), 11537–11548 CrossRef CAS.
- G. Priske, Z. F. Su, F. Abbasi, J. Lipkowski and F. I. Auzanneau, Synthesis and Electrochemical Characterization of 4-Thio Pseudo-Glycolipids as Candidate Tethers for Lipid Bilayer Models, Electrochim. Acta, 2019, 298, 150–162 CrossRef CAS.
- E. Reimhult, B. Kasemo and F. Höök, Rupture Pathway of Phosphatidylcholine Liposomes on Silicon Dioxide, Int. J. Mol. Sci., 2009, 10(4), 1683–1696 CrossRef CAS PubMed.
- R. P. Richter, R. Bérat, A. R. Brisson, R. P. Richter, R. Bérat and A. R. Brisson, Formation of Solid-Supported Lipid Bilayers: An Integrated View, Langmuir, 2006, 22(8), 3497–3505 CrossRef CAS PubMed.
- M. Fuhrmans and M. Müller, Mechanisms of Vesicle Spreading on Surfaces: Coarse-Grained Simulations, Langmuir, 2013, 29(13), 4335–4349 CrossRef CAS PubMed.
- X. Kong, D. Lu, J. Wu and Z. Liu, Spreading of a Unilamellar Liposome on Charged Substrates: A Coarse-Grained Molecular Simulation, Langmuir, 2016, 32(15), 3785–3793 CrossRef CAS PubMed.
- F. Heinrich, T. Ng, D. J. Vanderah, P. Shekhar, M. Mihailescu, H. Nanda and M. Lösche, A New Lipid Anchor for Sparsely Tethered Bilayer Lipid Membranes, Langmuir, 2009, 25(7), 4219–4229 CrossRef CAS PubMed.
- H. Basit, A. Van der Heyden, C. Gondran, B. Nysten, P. Dumy and P. Labbé, Tethered Bilayer Lipid Membranes on Mixed Self-Assembled Monolayers of a Novel Anchoring Thiol: Impact of the Anchoring Thiol Density on Bilayer Formation, Langmuir, 2011, 27(23), 14317–14328 CrossRef CAS PubMed.
- J. Andersson and I. Köper, Tethered and Polymer Supported Bilayer Lipid Membranes: Structure and Function, Multidisciplinary Digital Publishing Institute, 2016, vol. 6, p. 30 Search PubMed.
- W. Hoiles, R. Gupta, B. Cornell, C. Cranfield and V. Krishnamurthy, The Effect of Tethers on Artificial Cell Membranes: A Coarse-Grained Molecular Dynamics Study, PLoS ONE, 2016, 11(10), e0162790 CrossRef PubMed.
- R. Budvytyte, G. Valincius, G. Niaura, V. Voiciuk, M. Mickevicius, H. Chapman, H. Z. Goh, P. Shekhar, F. Heinrich and S. Shenoy, et al., Structure and Properties of Tethered Bilayer Lipid Membranes with Unsaturated Anchor Molecules, Langmuir, 2013, 29(27), 8645–8656 CrossRef CAS PubMed.
- A. Erbe, R. J. Bushby, S. D. Evans and L. J. C. Jeuken, Tethered Bilayer Lipid Membranes Studied by Simultaneous Attenuated Total Reflectance Infrared Spectroscopy and Electrochemical Impedance Spectroscopy, J. Phys. Chem. B, 2007, 111(13), 3515–3524 CrossRef CAS PubMed.
- L. J. C. Jeuken, N. N. Daskalakis, X. Han, K. Sheikh, A. Erbe, R. J. Bushby and S. D. Evans, Phase Separation in Mixed Self-Assembled Monolayers and Its Effect on Biomimetic Membranes, Sens. Actuators, B, 2007, 124(2), 501–509 CrossRef CAS.
- H.-H. Lee, M. Gavutis, Ž Ruželė, R. Valiokas and B. Liedberg, Mixed Self-Assembled Monolayers with Terminal Deuterated Anchors: Characterization and Probing of Model Lipid Membrane Formation, J. Phys. Chem. B, 2018, 122(34), 8201–8210 CrossRef CAS PubMed.
- R. Valiokas, M. Östblom, S. Svedhem, S. C. T. Svensson and B. Liedberg, Thermal Stability of Self-Assembled Monolayers: Influence of Lateral Hydrogen Bonding, J. Phys. Chem. B, 2002, 106(40), 10401–10409 CrossRef CAS.
- R. Valiokas, L. Malysheva, A. Onipko, H.-H. Lee, Ž Ruželė, S. Svedhem, S. C. T. Svensson, U. Gelius and B. Liedberg, On the Quality and Structural Characteristics of Oligo(Ethylene Glycol) Assemblies on Gold: An Experimental and Theoretical Study, J. Electron Spectrosc. Relat. Phenom., 2009, 172(1–3), 9–20 CrossRef CAS.
- E. Schulze and M. M. Stein, Simulation of Mixed Self-Assembled Monolayers on Gold: Effect of Terminal Alkyl Anchor Chain and Monolayer Composition, J. Phys. Chem. B, 2018, 122(31), 7699–7710 CrossRef CAS PubMed.
- N.-J. J. Cho, C. W. Frank, B. Kasemo and F. Höök, Quartz Crystal Microbalance with Dissipation Monitoring of Supported Lipid Bilayers on Various Substrates, Nat. Protoc., 2010, 5(6), 1096–1106 CrossRef CAS PubMed.
- M. Karplus and J. A. McCammon, Molecular Dynamics Simulations of Biomolecules, Nat. Struct. Biol., 2002, 9(9), 646–652 CrossRef CAS PubMed.
- M. P. Allen and D. J. Tildesley, Computer Simulation of Liquids, 2nd edn, 2017, pp. 1–626 Search PubMed.
- H.-H. Lee, Ž Ruželė, L. Malysheva, A. Onipko, A. Gutés, F. Björefors, R. Valiokas and B. Liedberg, Long-Chain Alkylthiol Assemblies Containing Buried In-Plane Stabilizing Architectures, Langmuir, 2009, 25(24), 13959–13971 CrossRef CAS PubMed.
- M. Gavutis, S. Lata, P. Lamken, P. Müller and J. Piehler, Lateral Ligand-Receptor Interactions on Membranes Probed by Simultaneous Fluorescence-Interference Detection, Biophys. J., 2005, 88(6), 4289–4302 CrossRef CAS PubMed.
- A. A. Brian and H. M. McConnell, Allogeneic Stimulation of Cytotoxic T Cells by Supported Planar Membranes, Proc. Natl. Acad. Sci. U. S. A., 1984, 81(19 I), 6159–6163 CrossRef CAS PubMed.
- P. C. Hsu, B. M. H. Bruininks, D. Jefferies, P. C. T. Souza, J. Lee, D. S. Patel, S. J. Marrink, Y. Qi, S. Khalid and W. Im, CHARMM-GUI Martini Maker for Modeling and Simulation of Complex Bacterial Membranes with Lipopolysaccharides, J. Comput. Chem., 2017, 38(27), 2354–2363 CrossRef CAS PubMed.
- S. O. Yesylevskyy, L. V. Schäfer, D. Sengupta and S. J. Marrink, Polarizable Water Model for the Coarse-Grained MARTINI Force Field, PLoS Comput. Biol., 2010, 6(6), e1000810 CrossRef PubMed.
- S. J. Marrink, H. J. Risselada, S. Yefimov, D. P. Tieleman and A. H. De Vries, The MARTINI Force Field: Coarse Grained Model for Biomolecular Simulations, J. Phys. Chem. B, 2007, 111(27), 7812–7824 CrossRef CAS PubMed.
- T. A. Wassenaar, H. I. Ingólfsson, R. A. Böckmann, D. P. Tieleman and S. J. Marrink, Computational Lipidomics with Insane: A Versatile Tool for Generating Custom Membranes for Molecular Simulations, J. Chem. Theory Comput., 2015, 11(5), 2144–2155 CrossRef CAS PubMed.
- M. Parrinello and A. Rahman, Crystal Structure and Pair Potentials: A Molecular-Dynamics Study, Phys. Rev. Lett., 1980, 45(14), 1196 CrossRef CAS.
- H. J. C. Berendsen, D. van der Spoel and R. van Drunen, GROMACS: A Message-Passing Parallel Molecular Dynamics Implementation, Comput. Phys. Commun., 1995, 91(1–3), 43–56 CrossRef CAS.
- E. Lindahl, B. Hess and D. van der Spoel, GROMACS 3.0: A Package for Molecular Simulation and Trajectory Analysis, Mol. Model. Annu., 2001, 7(8), 306–317 CrossRef CAS.
- B. Hess, C. Kutzner, D. Van Der Spoel and E. Lindahl, GROMACS 4: Algorithms for Highly Efficient, Load-Balanced, and Scalable Molecular Simulation, J. Chem. Theory Comput., 2008, 4(3), 435–447 CrossRef CAS PubMed.
- S. Pronk, S. Páll, R. Schulz, P. Larsson, P. Bjelkmar, R. Apostolov, M. R. Shirts, J. C. Smith, P. M. Kasson and D. Van Der Spoel, et al., GROMACS 4.5: A High-Throughput and Highly Parallel Open Source Molecular Simulation Toolkit, Bioinformatics, 2013, 29(7), 845–854 CrossRef CAS PubMed.
- M. J. Abraham, T. Murtola, R. Schulz, S. Páll, J. C. Smith, B. Hess and E. Lindah, GROMACS: High Performance Molecular Simulations through Multi-Level Parallelism from Laptops to Supercomputers, SoftwareX, 2015, 1–2, 19–25 CrossRef.
- P. Szilárd, M. J. Abraham, C. Kutzner, B. Hess and E. Lindahl, Tackling Exascale Software Challenges in Molecular Dynamics Simulations with GROMACS, ed. S. Markidis and E. Laure, Solving Software Challenges for Exascale, Springer, Cham, 2015.
- W. Humphrey, A. Dalke and K. Schulten, VMD: Visual Molecular Dynamics, J. Mol. Graphics, 1996, 14(1), 33–38 CrossRef CAS PubMed.
- J. C. Munro and C. W. Frank, In Situ Formation and Characterization of Poly(Ethylene Glycol)-Supported Lipid Bilayers on Gold Surfaces, Langmuir, 2004, 20(24), 10567–10575 CrossRef CAS PubMed.
- S. Wang and R. G. Larson, Coarse-Grained Molecular Dynamics Simulation of Tethered Lipid Assemblies, Soft Matter, 2012, 9(2), 480–486 RSC.
- A. Koutsioubas, Combined Coarse-Grained Molecular Dynamics and Neutron Reflectivity Characterization of Supported Lipid Membranes, J. Phys. Chem. B, 2016, 120(44), 11474–11483 CrossRef CAS PubMed.
- R. M. Peitzsch and S. McLaughlin, Binding of Acylated Peptides and Fatty Acids to Phospholipid Vesicles: Pertinence to Myristoylated Proteins, Biochemistry, 2002, 32(39), 10436–10443 CrossRef PubMed.
- J. R. Silvius and F. l'Heureux, Fluorimetric Evaluation of the Affinities of Isoprenylated Peptides for Lipid Bilayers, Biochemistry, 1994, 33(10), 3014–3022 CrossRef CAS PubMed.
- E. Edler, E. Schulze and M. Stein, Membrane Localization and Dynamics of Geranylgeranylated Rab5 Hypervariable Region, Biochim. Biophys. Acta, Biomembr., 2017, 1859(8), 1335–1349 CrossRef CAS PubMed.
- S. Genheden, Solvation Free Energies and Partition Coefficients with the Coarse-Grained and Hybrid All-Atom/Coarse-Grained MARTINI Models, J. Comput.-Aided Mol. Des., 2017, 31(10), 867–876 CrossRef CAS PubMed.
- T. Taddese and P. Carbone, Effect of Chain Length on the Partition Properties of Poly(Ethylene Oxide): Comparison between MARTINI Coarse-Grained and Atomistic Models, J. Phys. Chem. B, 2017, 121(7), 1601–1609 CrossRef CAS PubMed.
- R. Alessandri, P. C. T. Souza, S. Thallmair, M. N. Melo, A. H. De Vries and S. J. Marrink, Pitfalls of the Martini Model, J. Chem. Theory Comput., 2019, 15(10), 5448–5460 CrossRef CAS PubMed.
- T. J. Zwang, W. R. Fletcher, T. J. Lane and M. S. Johal, Quantification of the Layer of Hydration of a Supported Lipid Bilayer, Langmuir, 2010, 26(7), 4598–4601 CrossRef CAS PubMed.
- S. Park, S. Yorulmaz Avsar, B. Cornell, A. R. Ferhan, W. Y. Jeon, M. Chung and N. J. Cho, Probing the Influence of Tether Density on Tethered Bilayer Lipid Membrane (TBLM)-Peptide Interactions, Appl. Mater. Today, 2020, 18, 100527 CrossRef.
- A.-I. Bunea, S. Harloff-Helleberg, R. Taboryski and H. M. Nielsen, Membrane Interactions in Drug Delivery: Model Cell Membranes and Orthogonal Techniques, Adv. Colloid Interface Sci., 2020, 281, 102177 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: (a) Raw QCM-D sensorgrams with additional analysis plots; (b) molecular dynamics video of SOPC vesicle attachment to the SAM with sparsely distributed tethers. See DOI: https://doi.org/10.1039/d2nr07069c |
| ‡ Dr Martynas Gavutis and Dr Eric Schulze-Niemand contributed equally to this article. |
| § Present address: Swedish National Forensic Centre, Brigadgatan 13, 58758 Linköping, Sweden. |
| This journal is © The Royal Society of Chemistry 2023 |